


O-GlycNacylation Remission Retards the Progression of Non-Alcoholic Fatty Liver Disease

 , ,
, ,  , ,
, ,
Abstract
1. Introduction
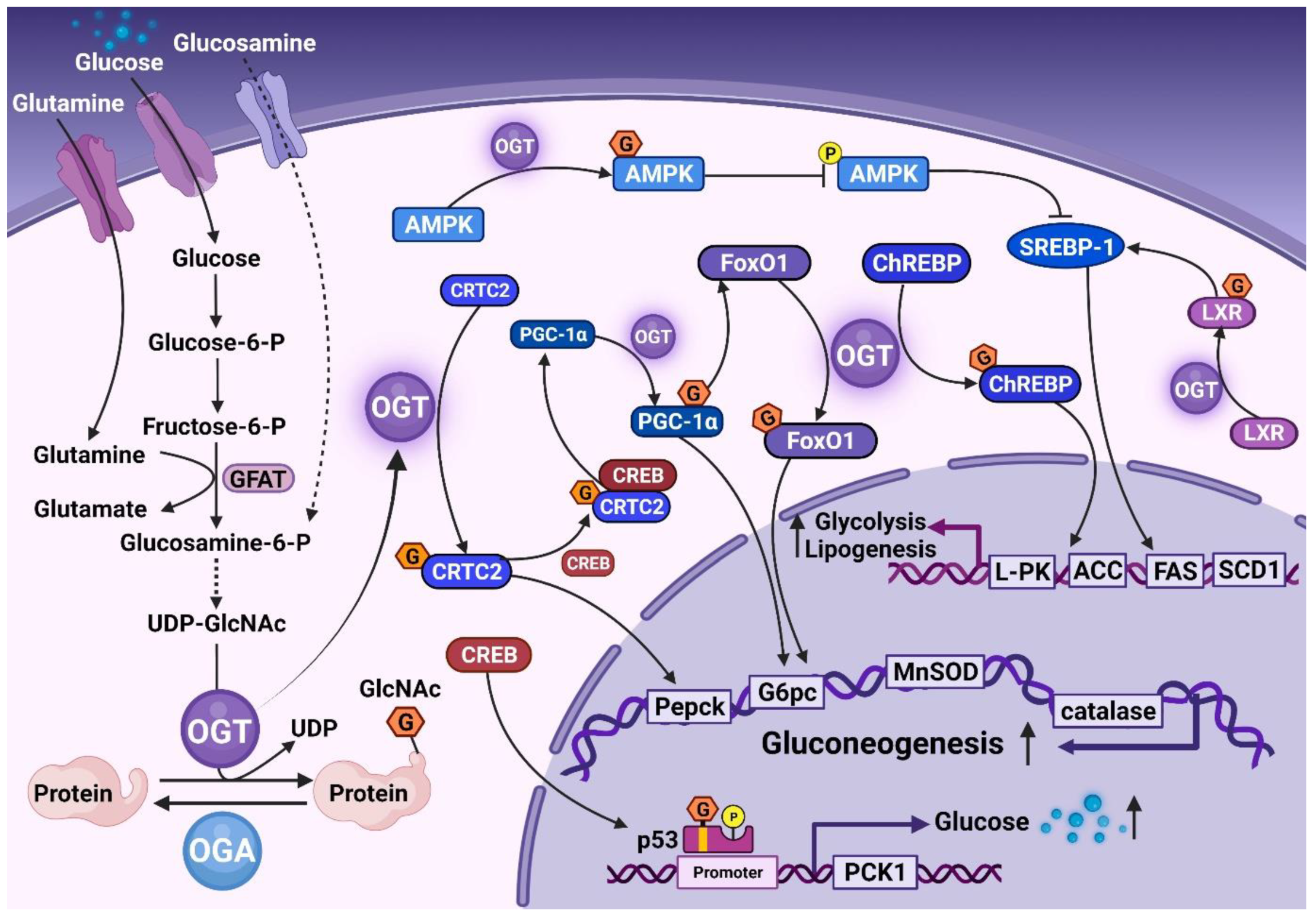
2. Role of O-GlcNAc in Normal Liver Tissue
3. O-GlcNAcylation Contributes to IR
4. Association of O-GlcNAc with NAFLD Process
4.1. O-GlcNAc and NAFL
4.2. O-GlcNAc and NASH
4.3. O-GlcNAc and Hepatic Fibrosis
4.4. O-GlcNAc and NAFLD-HCC
5. Drugs Ameliorates NAFLD through Inhibition of O-GlcNAcylation
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Ang | angiotensin |
| APAP | acetaminophen |
| ALF | acute liver failure |
| ALA | alpha-lipoic acid |
| ApoA5 | apolipoprotein A5 |
| ACC | acetyl-CoA carboxylase |
| AMP | adenosine monophosphate |
| AMPK | AMP-activated protein kinase |
| AKT | serine/threonine-protein kinase B |
| ACEI | Angiotensin-converting enzyme inhibitors |
| CAT | catalase |
| CRTC2 | cAMP-regulated transcriptional co-activator 2 |
| CREB | cyclic AMP-responsive element-binding protein |
| CREBH | cyclic AMP-responsive element-binding protein H |
| ChREBP | carbohydrate-responsive element-binding protein |
| DCM | diabetic cardiomyopathy |
| EMPA | empagliflozin |
| ER | endoplasmic reticulum |
| FAS | fatty acid synthase |
| FBPase | fructose-1,6-bisphosphatase |
| GSH | glutathione |
| GS | glycogen synthase |
| GLP-1 | glucagon-like peptide-1 |
| GSK3β | glycogen synthase kinase 3 beta |
| GFAT | glutamine fructose-6-phosphate amidotransferase |
| HCV | hepatitis C virus |
| HSP | heat shock protein |
| HCF-1 | host cell factor C1 |
| HSCs | hepatic stellate cells |
| HCD | high-carbohydrate diet |
| HCC | hepatocellular carcinoma |
| HBP | hexosamine biosynthetic pathway |
| IL | interleukin |
| IR | insulin resistance |
| IRS-1 | insulin receptor substrate 1 |
| IRE1α | inositol requiring enzyme 1α |
| IP6K1 | inositol hexakisphosphate kinases 1 |
| JNK | Jun N-terminal kinases |
| LXRs | liver X receptors |
| L-PK | liver pyruvate kinase |
| MET | metformin |
| MCD | methionine-choline deficient |
| mTOR | mammalian target of rapamycin |
| MnSOD | manganese superoxide dismutase |
| MAFLD | metabolic dysfunction-associated fatty liver disease |
| NASH | non-alcoholic steatohepatitis |
| NAFLD | non-alcoholic fatty liver disease |
| NLRP3 | NLR family, pyrin domain containing 3 |
| OGA | GlcNAcase |
| OGT | O-GlcNAc transferase |
| O-GlcNAc | O-linked β-N-acetylglucosamine |
| PI3K | phosphatidylinositol-3-kinase |
| PTP1B | protein tyrosine phosphatase 1B |
| PDGFRβ | platelet-derived growth factor receptorβ |
| PDK1 | phosphoinositide-dependent protein kinase 1 |
| PGC1α | peroxisome proliferator-activated receptor-γ co-activator1α |
| ROS | reactive oxygen species |
| RAS | renin-angiotensin system |
| RIPK3 | receptor-interacting protein kinase 3 |
| SRF | serum response factor |
| SOD | superoxide dismutase |
| SCD1 | stearoyl-CoA desaturase1 |
| SGLT-2i | sodium-glucose cotransporter 2 inhibitor |
| SREBP-1 | sterol regulatory element-binding protein 1 |
| TG | thyroglobulin |
| TFF2 | trefoil factor 2 |
| T2DM | type 2 diabetes mellitus |
| TXNIP | thioredoxin interacting protein |
| XBP1 | X-box-binding protein 1 |
References
- Angulo, P. Nonalcoholic fatty liver disease. N. Engl. J. Med. 2002, 346, 1221–1231. [Google Scholar] [CrossRef] [PubMed]
- Khamphaya, T.; Chukijrungroat, N.; Saengsirisuwan, V.; Mitchell-Richards, K.A.; Robert, M.E.; Mennone, A.; Ananthanarayanan, M.; Nathanson, M.H.; Weerachayaphorn, J. Nonalcoholic fatty liver disease impairs expression of the type II inositol 1,4,5-trisphosphate receptor. Hepatology 2018, 67, 560–574. [Google Scholar] [CrossRef] [PubMed]
- Allen, A.M.; Therneau, T.M.; Larson, J.J.; Coward, A.; Somers, V.K.; Kamath, P.S. Nonalcoholic fatty liver disease incidence and impact on metabolic burden and death: A 20 year-community study. Hepatology 2018, 67, 1726–1736. [Google Scholar] [CrossRef] [PubMed]
- Cotter, T.G.; Rinella, M. Nonalcoholic Fatty Liver Disease 2020: The State of the Disease. Gastroenterology 2020, 158, 1851–1864. [Google Scholar] [CrossRef] [PubMed]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wai-Sun Wong, V.; Dufour, J.F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef]
- Gawrieh, S.; Marion, M.C.; Komorowski, R.; Wallace, J.; Charlton, M.; Kissebah, A.; Langefeld, C.D.; Olivier, M. Genetic variation in the peroxisome proliferator activated receptor-gamma gene is associated with histologically advanced NAFLD. Dig. Dis. Sci. 2012, 57, 952–957. [Google Scholar] [CrossRef]
- Shao, M.; Ye, Z.; Qin, Y.; Wu, T. Abnormal metabolic processes involved in the pathogenesis of non-alcoholic fatty liver disease (Review). Exp. Ther. Med. 2020, 20, 26. [Google Scholar] [CrossRef]
- Lim, J.S.; Mietus-Snyder, M.; Valente, A.; Schwarz, J.M.; Lustig, R.H. The role of fructose in the pathogenesis of NAFLD and the metabolic syndrome. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 251–264. [Google Scholar] [CrossRef]
- Jensen, T.; Abdelmalek, M.F.; Sullivan, S.; Nadeau, K.J.; Green, M.; Roncal, C.; Nakagawa, T.; Kuwabara, M.; Sato, Y.; Kang, D.H.; et al. Fructose and sugar: A major mediator of non-alcoholic fatty liver disease. J. Hepatol. 2018, 68, 1063–1075. [Google Scholar] [CrossRef]
- Browning, J.D.; Horton, J.D. Molecular mediators of hepatic steatosis and liver injury. J. Clin. Investig. 2004, 114, 147–152. [Google Scholar] [CrossRef]
- Park, J.S.; Lee, D.H.; Lee, Y.S.; Oh, E.; Bae, K.H.; Oh, K.J.; Kim, H.; Bae, S.H. Dual roles of ULK1 (unc-51 like autophagy activating kinase 1) in cytoprotection against lipotoxicity. Autophagy 2020, 16, 86–105. [Google Scholar] [CrossRef]
- Navarro, L.A.; Wree, A.; Povero, D.; Berk, M.P.; Eguchi, A.; Ghosh, S.; Papouchado, B.G.; Erzurum, S.C.; Feldstein, A.E. Arginase 2 deficiency results in spontaneous steatohepatitis: A novel link between innate immune activation and hepatic de novo lipogenesis. J. Hepatol. 2015, 62, 412–420. [Google Scholar] [CrossRef]
- Kim, S.H.; Kim, G.; Han, D.H.; Lee, M.; Kim, I.; Kim, B.; Kim, K.H.; Song, Y.M.; Yoo, J.E.; Wang, H.J.; et al. Ezetimibe ameliorates steatohepatitis via AMP activated protein kinase-TFEB-mediated activation of autophagy and NLRP3 inflammasome inhibition. Autophagy 2017, 13, 1767–1781. [Google Scholar] [CrossRef]
- Lin, Y.; Zhu, J.; Pan, L.; Zhang, J.; Tan, Z.; Olivares, J.; Singal, A.G.; Parikh, N.D.; Lubman, D.M. A Panel of Glycopeptides as Candidate Biomarkers for Early Diagnosis of NASH Hepatocellular Carcinoma Using a Stepped HCD Method and PRM Evaluation. J. Proteome Res. 2021, 20, 3278–3289. [Google Scholar] [CrossRef]
- Zhang, N.; Wang, Y.; Zhang, J.; Liu, B.; Deng, X.; Xin, S.; Xu, K. N-glycosylation of CREBH improves lipid metabolism and attenuates lipotoxicity in NAFLD by modulating PPARalpha and SCD-1. FASEB J. 2020, 34, 15338–15363. [Google Scholar] [CrossRef]
- Zhang, K.; Yin, R.; Yang, X. O-GlcNAc: A Bittersweet Switch in Liver. Front. Endocrinol. 2014, 5, 221. [Google Scholar] [CrossRef]
- Mukherjee, S.; Chakraborty, M.; Ulmasov, B.; McCommis, K.; Zhang, J.; Carpenter, D.; Msengi, E.N.; Haubner, J.; Guo, C.; Pike, D.P.; et al. Pleiotropic actions of IP6K1 mediate hepatic metabolic dysfunction to promote nonalcoholic fatty liver disease and steatohepatitis. Mol. Metab. 2021, 54, 101364. [Google Scholar] [CrossRef]
- Lee, S.J.; Nam, M.J.; Lee, D.E.; Park, J.W.; Kang, B.S.; Lee, D.S.; Lee, H.S.; Kwon, O.S. Silibinin Ameliorates O-GlcNAcylation and Inflammation in a Mouse Model of Nonalcoholic Steatohepatitis. Int. J. Mol. Sci. 2018, 19, 2165. [Google Scholar] [CrossRef]
- Lee, D.E.; Lee, S.J.; Kim, S.J.; Lee, H.S.; Kwon, O.S. Curcumin Ameliorates Nonalcoholic Fatty Liver Disease through Inhibition of O-GlcNAcylation. Nutrients 2019, 11, 2702. [Google Scholar] [CrossRef]
- Prakoso, D.; Lim, S.Y.; Erickson, J.R.; Wallace, R.S.; Lees, J.G.; Tate, M.; Kiriazis, H.; Donner, D.G.; Henstridge, D.C.; Davey, J.R.; et al. Fine-tuning the cardiac O-GlcNAcylation regulatory enzymes governs the functional and structural phenotype of the diabetic heart. Cardiovasc. Res. 2022, 118, 212–225. [Google Scholar] [CrossRef]
- Qin, L.; Wang, J.; Zhao, R.; Zhang, X.; Mei, Y. Ginsenoside-Rb1 Improved Diabetic Cardiomyopathy through Regulating Calcium Signaling by Alleviating Protein O-GlcNAcylation. J. Agric. Food Chem. 2019, 67, 14074–14085. [Google Scholar] [CrossRef] [PubMed]
- Olivier-Van Stichelen, S.; Hanover, J.A. You are what you eat: O-linked N-acetylglucosamine in disease, development and epigenetics. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Peterson, S.B.; Hart, G.W. New insights: A role for O-GlcNAcylation in diabetic complications. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 150–161. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.N.; Collins, H.E.; Wende, A.R.; Chatham, J.C. O-GlcNAcylation and cardiovascular disease. Biochem. Soc. Trans. 2017, 45, 545–553. [Google Scholar] [CrossRef]
- Bond, M.R.; Hanover, J.A. A little sugar goes a long way: The cell biology of O-GlcNAc. J. Cell Biol. 2015, 208, 869–880. [Google Scholar] [CrossRef]
- Hardiville, S.; Hart, G.W. Nutrient regulation of gene expression by O-GlcNAcylation of chromatin. Curr. Opin. Chem. Biol. 2016, 33, 88–94. [Google Scholar] [CrossRef]
- Yang, X.; Qian, K. Protein O-GlcNAcylation: Emerging mechanisms and functions. Nat. Rev. Mol. Cell Biol. 2017, 18, 452–465. [Google Scholar] [CrossRef]
- Pekkurnaz, G.; Trinidad, J.C.; Wang, X.; Kong, D.; Schwarz, T.L. Glucose regulates mitochondrial motility via Milton modification by O-GlcNAc transferase. Cell 2014, 158, 54–68. [Google Scholar] [CrossRef]
- Hart, G.W. Dynamic O-linked glycosylation of nuclear and cytoskeletal proteins. Annu. Rev. Biochem. 1997, 66, 315–335. [Google Scholar] [CrossRef]
- Kaasik, K.; Kivimae, S.; Allen, J.J.; Chalkley, R.J.; Huang, Y.; Baer, K.; Kissel, H.; Burlingame, A.L.; Shokat, K.M.; Ptacek, L.J.; et al. Glucose sensor O-GlcNAcylation coordinates with phosphorylation to regulate circadian clock. Cell Metab. 2013, 17, 291–302. [Google Scholar] [CrossRef]
- Li, M.D.; Ruan, H.B.; Hughes, M.E.; Lee, J.S.; Singh, J.P.; Jones, S.P.; Nitabach, M.N.; Yang, X. O-GlcNAc signaling entrains the circadian clock by inhibiting BMAL1/CLOCK ubiquitination. Cell Metab. 2013, 17, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Housley, M.P.; Udeshi, N.D.; Rodgers, J.T.; Shabanowitz, J.; Puigserver, P.; Hunt, D.F.; Hart, G.W. A PGC-1alpha-O-GlcNAc transferase complex regulates FoxO transcription factor activity in response to glucose. J. Biol. Chem. 2009, 284, 5148–5157. [Google Scholar] [CrossRef] [PubMed]
- Housley, M.P.; Rodgers, J.T.; Udeshi, N.D.; Kelly, T.J.; Shabanowitz, J.; Hunt, D.F.; Puigserver, P.; Hart, G.W. O-GlcNAc regulates FoxO activation in response to glucose. J. Biol. Chem. 2008, 283, 16283–16292. [Google Scholar] [CrossRef] [PubMed]
- Ruan, H.B.; Han, X.; Li, M.D.; Singh, J.P.; Qian, K.; Azarhoush, S.; Zhao, L.; Bennett, A.M.; Samuel, V.T.; Wu, J.; et al. O-GlcNAc transferase/host cell factor C1 complex regulates gluconeogenesis by modulating PGC-1alpha stability. Cell Metab. 2012, 16, 226–237. [Google Scholar] [CrossRef] [PubMed]
- Herzig, S.; Long, F.; Jhala, U.S.; Hedrick, S.; Quinn, R.; Bauer, A.; Rudolph, D.; Schutz, G.; Yoon, C.; Puigserver, P.; et al. CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature 2001, 413, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Dominy, J.E., Jr.; Lee, Y.; Jedrychowski, M.P.; Chim, H.; Jurczak, M.J.; Camporez, J.P.; Ruan, H.B.; Feldman, J.; Pierce, K.; Mostoslavsky, R.; et al. The deacetylase Sirt6 activates the acetyltransferase GCN5 and suppresses hepatic gluconeogenesis. Mol. Cell 2012, 48, 900–913. [Google Scholar] [CrossRef]
- Wu, Z.; Jiao, P.; Huang, X.; Feng, B.; Feng, Y.; Yang, S.; Hwang, P.; Du, J.; Nie, Y.; Xiao, G.; et al. MAPK phosphatase-3 promotes hepatic gluconeogenesis through dephosphorylation of forkhead box O1 in mice. J. Clin. Investig. 2010, 120, 3901–3911. [Google Scholar] [CrossRef]
- Dentin, R.; Hedrick, S.; Xie, J.; Yates, J., 3rd; Montminy, M. Hepatic glucose sensing via the CREB coactivator CRTC2. Science 2008, 319, 1402–1405. [Google Scholar] [CrossRef]
- Li, M.D.; Ruan, H.B.; Singh, J.P.; Zhao, L.; Zhao, T.; Azarhoush, S.; Wu, J.; Evans, R.M.; Yang, X. O-GlcNAc transferase is involved in glucocorticoid receptor-mediated transrepression. J. Biol. Chem. 2012, 287, 12904–12912. [Google Scholar] [CrossRef]
- Efeyan, A.; Serrano, M. p53: Guardian of the genome and policeman of the oncogenes. Cell Cycle 2007, 6, 1006–1010. [Google Scholar] [CrossRef]
- Gonzalez-Rellan, M.J.; Fondevila, M.F.; Fernandez, U.; Rodriguez, A.; Varela-Rey, M.; Veyrat-Durebex, C.; Seoane, S.; Bernardo, G.; Lopitz-Otsoa, F.; Fernandez-Ramos, D.; et al. O-GlcNAcylated p53 in the liver modulates hepatic glucose production. Nat. Commun. 2021, 12, 5068. [Google Scholar] [CrossRef]
- Anthonisen, E.H.; Berven, L.; Holm, S.; Nygard, M.; Nebb, H.I.; Gronning-Wang, L.M. Nuclear receptor liver X receptor is O-GlcNAc-modified in response to glucose. J. Biol. Chem. 2010, 285, 1607–1615. [Google Scholar] [CrossRef]
- Sodi, V.L.; Bacigalupa, Z.A.; Ferrer, C.M.; Lee, J.V.; Gocal, W.A.; Mukhopadhyay, D.; Wellen, K.E.; Ivan, M.; Reginato, M.J. Nutrient sensor O-GlcNAc transferase controls cancer lipid metabolism via SREBP-1 regulation. Oncogene 2018, 37, 924–934. [Google Scholar] [CrossRef]
- Guinez, C.; Filhoulaud, G.; Rayah-Benhamed, F.; Marmier, S.; Dubuquoy, C.; Dentin, R.; Moldes, M.; Burnol, A.F.; Yang, X.; Lefebvre, T.; et al. O-GlcNAcylation increases ChREBP protein content and transcriptional activity in the liver. Diabetes 2011, 60, 1399–1413. [Google Scholar] [CrossRef]
- Robarts, D.R.; McGreal, S.R.; Umbaugh, D.S.; Parkes, W.S.; Kotulkar, M.; Abernathy, S.; Lee, N.; Jaeschke, H.; Gunewardena, S.; Whelan, S.A.; et al. Regulation of Liver Regeneration by Hepatocyte O-GlcNAcylation in Mice. Cell. Mol. Gastroenterol. Hepatol. 2022, 13, 1510–1529. [Google Scholar] [CrossRef]
- Ruan, H.B.; Ma, Y.; Torres, S.; Zhang, B.; Feriod, C.; Heck, R.M.; Qian, K.; Fu, M.; Li, X.; Nathanson, M.H.; et al. Calcium-dependent O-GlcNAc signaling drives liver autophagy in adaptation to starvation. Genes Dev. 2017, 31, 1655–1665. [Google Scholar] [CrossRef]
- Tan, E.P.; McGreal, S.R.; Graw, S.; Tessman, R.; Koppel, S.J.; Dhakal, P.; Zhang, Z.; Machacek, M.; Zachara, N.E.; Koestler, D.C.; et al. Sustained O-GlcNAcylation reprograms mitochondrial function to regulate energy metabolism. J. Biol. Chem. 2017, 292, 14940–14962. [Google Scholar] [CrossRef]
- Wang, X.; Lin, Y.; Liu, S.; Zhu, Y.; Lu, K.; Broering, R.; Lu, M. O-GlcNAcylation modulates HBV replication through regulating cellular autophagy at multiple levels. FASEB J. 2020, 34, 14473–14489. [Google Scholar] [CrossRef]
- Tolman, K.G.; Fonseca, V.; Dalpiaz, A.; Tan, M.H. Spectrum of liver disease in type 2 diabetes and management of patients with diabetes and liver disease. Diabetes Care 2007, 30, 734–743. [Google Scholar] [CrossRef]
- Choudhury, J.; Sanyal, A.J. Insulin resistance and the pathogenesis of nonalcoholic fatty liver disease. Clin. Liver Dis. 2004, 8, 575–594. [Google Scholar] [CrossRef]
- Bugianesi, E.; McCullough, A.J.; Marchesini, G. Insulin resistance: A metabolic pathway to chronic liver disease. Hepatology 2005, 42, 987–1000. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.; Cabrera, D.; Arrese, M.; Feldstein, A.E. Triggering and resolution of inflammation in NASH. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Whelan, S.A.; Dias, W.B.; Thiruneelakantapillai, L.; Lane, M.D.; Hart, G.W. Regulation of insulin receptor substrate 1 (IRS-1)/AKT kinase-mediated insulin signaling by O-Linked beta-N-acetylglucosamine in 3T3-L1 adipocytes. J. Biol. Chem. 2010, 285, 5204–5211. [Google Scholar] [CrossRef] [PubMed]
- Whelan, S.A.; Lane, M.D.; Hart, G.W. Regulation of the O-linked beta-N-acetylglucosamine transferase by insulin signaling. J. Biol. Chem. 2008, 283, 21411–21417. [Google Scholar] [CrossRef]
- Wang, S.; Huang, X.; Sun, D.; Xin, X.; Pan, Q.; Peng, S.; Liang, Z.; Luo, C.; Yang, Y.; Jiang, H.; et al. Extensive crosstalk between O-GlcNAcylation and phosphorylation regulates Akt signaling. PLoS ONE 2012, 7, e37427. [Google Scholar] [CrossRef]
- Wang, Z.; Pandey, A.; Hart, G.W. Dynamic interplay between O-linked N-acetylglucosaminylation and glycogen synthase kinase-3-dependent phosphorylation. Mol. Cell. Proteom. 2007, 6, 1365–1379. [Google Scholar] [CrossRef]
- Zhao, Y.; Tang, Z.; Shen, A.; Tao, T.; Wan, C.; Zhu, X.; Huang, J.; Zhang, W.; Xia, N.; Wang, S.; et al. The Role of PTP1B O-GlcNAcylation in Hepatic Insulin Resistance. Int. J. Mol. Sci. 2015, 16, 22856–22869. [Google Scholar] [CrossRef]
- Parker, G.J.; Lund, K.C.; Taylor, R.P.; McClain, D.A. Insulin resistance of glycogen synthase mediated by o-linked N-acetylglucosamine. J. Biol. Chem. 2003, 278, 10022–10027. [Google Scholar] [CrossRef]
- Yang, W.H.; Park, S.Y.; Nam, H.W.; Kim, D.H.; Kang, J.G.; Kang, E.S.; Kim, Y.S.; Lee, H.C.; Kim, K.S.; Cho, J.W. NFkappaB activation is associated with its O-GlcNAcylation state under hyperglycemic conditions. Proc. Natl. Acad. Sci. USA 2008, 105, 17345–17350. [Google Scholar] [CrossRef]
- Ramakrishnan, P.; Clark, P.M.; Mason, D.E.; Peters, E.C.; Hsieh-Wilson, L.C.; Baltimore, D. Activation of the transcriptional function of the NF-kappaB protein c-Rel by O-GlcNAc glycosylation. Sci. Signal. 2013, 6, ra75. [Google Scholar] [CrossRef]
- Fan, X.; Chuan, S.; Hongshan, W. Protein O glycosylation regulates activation of hepatic stellate cells. Inflammation 2013, 36, 1248–1252. [Google Scholar] [CrossRef]
- Li, R.; Ong, Q.; Wong, C.C.; Chu, E.S.H.; Sung, J.J.Y.; Yang, X.; Yu, J. O-GlcNAcylation inhibits hepatic stellate cell activation. J. Gastroenterol. Hepatol. 2021, 36, 3477–3486. [Google Scholar] [CrossRef]
- Vucur, M.; Reisinger, F.; Gautheron, J.; Janssen, J.; Roderburg, C.; Cardenas, D.V.; Kreggenwinkel, K.; Koppe, C.; Hammerich, L.; Hakem, R.; et al. RIP3 inhibits inflammatory hepatocarcinogenesis but promotes cholestasis by controlling caspase-8- and JNK-dependent compensatory cell proliferation. Cell Rep. 2013, 4, 776–790. [Google Scholar] [CrossRef]
- Zhang, B.; Li, M.D.; Yin, R.; Liu, Y.; Yang, Y.; Mitchell-Richards, K.A.; Nam, J.H.; Li, R.; Wang, L.; Iwakiri, Y.; et al. O-GlcNAc transferase suppresses necroptosis and liver fibrosis. JCI Insight 2019, 4, e127709. [Google Scholar] [CrossRef]
- Dentin, R.; Benhamed, F.; Hainault, I.; Fauveau, V.; Foufelle, F.; Dyck, J.R.; Girard, J.; Postic, C. Liver-specific inhibition of ChREBP improves hepatic steatosis and insulin resistance in ob/ob mice. Diabetes 2006, 55, 2159–2170. [Google Scholar] [CrossRef]
- Lane, E.A.; Choi, D.W.; Garcia-Haro, L.; Levine, Z.G.; Tedoldi, M.; Walker, S.; Danial, N.N. HCF-1 Regulates De Novo Lipogenesis through a Nutrient-Sensitive Complex with ChREBP. Mol. Cell 2019, 75, 357–371.e7. [Google Scholar] [CrossRef]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Investig. 2002, 109, 1125–1131. [Google Scholar] [CrossRef]
- Kawano, Y.; Cohen, D.E. Mechanisms of hepatic triglyceride accumulation in non-alcoholic fatty liver disease. J. Gastroenterol. 2013, 48, 434–441. [Google Scholar] [CrossRef]
- Knebel, B.; Haas, J.; Hartwig, S.; Jacob, S.; Kollmer, C.; Nitzgen, U.; Muller-Wieland, D.; Kotzka, J. Liver-specific expression of transcriptionally active SREBP-1c is associated with fatty liver and increased visceral fat mass. PLoS ONE 2012, 7, e31812. [Google Scholar] [CrossRef]
- Gorgani-Firuzjaee, S.; Meshkani, R. SH2 domain-containing inositol 5-phosphatase (SHIP2) inhibition ameliorates high glucose-induced de-novo lipogenesis and VLDL production through regulating AMPK/mTOR/SREBP1 pathway and ROS production in HepG2 cells. Free Radic. Biol. Med. 2015, 89, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Sage, A.T.; Walter, L.A.; Shi, Y.; Khan, M.I.; Kaneto, H.; Capretta, A.; Werstuck, G.H. Hexosamine biosynthesis pathway flux promotes endoplasmic reticulum stress, lipid accumulation, and inflammatory gene expression in hepatic cells. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E499–E511. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Lee, Y.; Jung, E.H.; Kim, S.M.; Cho, H.; Han, I.O. Glucosamine regulates hepatic lipid accumulation by sensing glucose levels or feeding states of normal and excess. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158764. [Google Scholar] [CrossRef] [PubMed]
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef] [PubMed]
- Elsharkawy, A.M.; Mann, D.A. Nuclear factor-kappaB and the hepatic inflammation-fibrosis-cancer axis. Hepatology 2007, 46, 590–597. [Google Scholar] [CrossRef]
- Malhi, H.; Kaufman, R.J. Endoplasmic reticulum stress in liver disease. J. Hepatol. 2011, 54, 795–809. [Google Scholar] [CrossRef]
- Lake, A.D.; Novak, P.; Hardwick, R.N.; Flores-Keown, B.; Zhao, F.; Klimecki, W.T.; Cherrington, N.J. The adaptive endoplasmic reticulum stress response to lipotoxicity in progressive human nonalcoholic fatty liver disease. Toxicol. Sci. 2014, 137, 26–35. [Google Scholar] [CrossRef]
- Baudoin, L.; Issad, T. O-GlcNAcylation and Inflammation: A Vast Territory to Explore. Front. Endocrinol. 2014, 5, 235. [Google Scholar] [CrossRef]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Ni, M.; Zhang, Y.; Lee, A.S. Beyond the endoplasmic reticulum: Atypical GRP78 in cell viability, signalling and therapeutic targeting. Biochem. J. 2011, 434, 181–188. [Google Scholar] [CrossRef]
- Ngoh, G.A.; Hamid, T.; Prabhu, S.D.; Jones, S.P. O-GlcNAc signaling attenuates ER stress-induced cardiomyocyte death. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1711–H1719. [Google Scholar] [CrossRef]
- So, J.S.; Hur, K.Y.; Tarrio, M.; Ruda, V.; Frank-Kamenetsky, M.; Fitzgerald, K.; Koteliansky, V.; Lichtman, A.H.; Iwawaki, T.; Glimcher, L.H.; et al. Silencing of lipid metabolism genes through IRE1alpha-mediated mRNA decay lowers plasma lipids in mice. Cell Metab. 2012, 16, 487–499. [Google Scholar] [CrossRef]
- Kim, I.; Xu, W.; Reed, J.C. Cell death and endoplasmic reticulum stress: Disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov. 2008, 7, 1013–1030. [Google Scholar] [CrossRef]
- Visinoni, S.; Khalid, N.F.; Joannides, C.N.; Shulkes, A.; Yim, M.; Whitehead, J.; Tiganis, T.; Lamont, B.J.; Favaloro, J.M.; Proietto, J.; et al. The role of liver fructose-1,6-bisphosphatase in regulating appetite and adiposity. Diabetes 2012, 61, 1122–1132. [Google Scholar] [CrossRef]
- Dulai, P.S.; Singh, S.; Patel, J.; Soni, M.; Prokop, L.J.; Younossi, Z.; Sebastiani, G.; Ekstedt, M.; Hagstrom, H.; Nasr, P.; et al. Increased risk of mortality by fibrosis stage in nonalcoholic fatty liver disease: Systematic review and meta-analysis. Hepatology 2017, 65, 1557–1565. [Google Scholar] [CrossRef]
- Hui, J.M.; Kench, J.G.; Chitturi, S.; Sud, A.; Farrell, G.C.; Byth, K.; Hall, P.; Khan, M.; George, J. Long-term outcomes of cirrhosis in nonalcoholic steatohepatitis compared with hepatitis C. Hepatology 2003, 38, 420–427. [Google Scholar] [CrossRef]
- Pinzani, M.; Rombouts, K. Liver fibrosis: From the bench to clinical targets. Dig. Liver Dis. 2004, 36, 231–242. [Google Scholar] [CrossRef]
- Adachi, M.; Osawa, Y.; Uchinami, H.; Kitamura, T.; Accili, D.; Brenner, D.A. The forkhead transcription factor FoxO1 regulates proliferation and transdifferentiation of hepatic stellate cells. Gastroenterology 2007, 132, 1434–1446. [Google Scholar] [CrossRef]
- Valenti, L.; Rametta, R.; Dongiovanni, P.; Maggioni, M.; Fracanzani, A.L.; Zappa, M.; Lattuada, E.; Roviaro, G.; Fargion, S. Increased expression and activity of the transcription factor FOXO1 in nonalcoholic steatohepatitis. Diabetes 2008, 57, 1355–1362. [Google Scholar] [CrossRef]
- Zhang, B.; Lapenta, K.; Wang, Q.; Nam, J.H.; Chung, D.; Robert, M.E.; Nathanson, M.H.; Yang, X. Trefoil factor 2 secreted from damaged hepatocytes activates hepatic stellate cells to induce fibrogenesis. J. Biol. Chem. 2021, 297, 100887. [Google Scholar] [CrossRef]
- Paik, J.M.; Golabi, P.; Younossi, Y.; Mishra, A.; Younossi, Z.M. Changes in the Global Burden of Chronic Liver Diseases from 2012 to 2017: The Growing Impact of NAFLD. Hepatology 2020, 72, 1605–1616. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.J.; Cheung, R.; Ahmed, A. Nonalcoholic steatohepatitis is the most rapidly growing indication for liver transplantation in patients with hepatocellular carcinoma in the U.S. Hepatology 2014, 59, 2188–2195. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Zhang, X.; Wu, J.L.; Fu, L.; Liu, K.; Liu, D.; Chen, G.G.; Lai, P.B.; Wong, N.; Yu, J. O-GlcNAc transferase promotes fatty liver-associated liver cancer through inducing palmitic acid and activating endoplasmic reticulum stress. J. Hepatol. 2017, 67, 310–320. [Google Scholar] [CrossRef] [PubMed]
- Gautheron, J.; Vucur, M.; Reisinger, F.; Cardenas, D.V.; Roderburg, C.; Koppe, C.; Kreggenwinkel, K.; Schneider, A.T.; Bartneck, M.; Neumann, U.P.; et al. A positive feedback loop between RIP3 and JNK controls non-alcoholic steatohepatitis. EMBO Mol. Med. 2014, 6, 1062–1074. [Google Scholar] [CrossRef] [PubMed]
- Krawczyk, M.; Jimenez-Aguero, R.; Alustiza, J.M.; Emparanza, J.I.; Perugorria, M.J.; Bujanda, L.; Lammert, F.; Banales, J.M. PNPLA3 p.I148M variant is associated with greater reduction of liver fat content after bariatric surgery. Surg. Obes. Relat. Dis. 2016, 12, 1838–1846. [Google Scholar] [CrossRef]
- Miyaaki, H.; Nakao, K. Significance of genetic polymorphisms in patients with nonalcoholic fatty liver disease. Clin. J. Gastroenterol. 2017, 10, 201–207. [Google Scholar] [CrossRef]
- Kim, M.Y.; Kim, Y.S.; Kim, M.; Choi, M.Y.; Roh, G.S.; Lee, D.H.; Kim, H.J.; Kang, S.S.; Cho, G.J.; Shin, J.K.; et al. Metformin inhibits cervical cancer cell proliferation via decreased AMPK O-GlcNAcylation. Anim. Cells Syst. 2019, 23, 302–309. [Google Scholar] [CrossRef]
- Kim, Y.S.; Kim, M.; Choi, M.Y.; Lee, D.H.; Roh, G.S.; Kim, H.J.; Kang, S.S.; Cho, G.J.; Kim, S.J.; Yoo, J.M.; et al. Metformin protects against retinal cell death in diabetic mice. Biochem. Biophys. Res. Commun. 2017, 492, 397–403. [Google Scholar] [CrossRef]
- Barbero-Becerra, V.J.; Santiago-Hernandez, J.J.; Villegas-Lopez, F.A.; Mendez-Sanchez, N.; Uribe, M.; Chavez-Tapia, N.C. Mechanisms involved in the protective effects of metformin against nonalcoholic fatty liver disease. Curr. Med. Chem. 2012, 19, 2918–2923. [Google Scholar] [CrossRef]
- Pang, Y.; Xu, X.; Xiang, X.; Li, Y.; Zhao, Z.; Li, J.; Gao, S.; Liu, Q.; Mai, K.; Ai, Q. High Fat Activates O-GlcNAcylation and Affects AMPK/ACC Pathway to Regulate Lipid Metabolism. Nutrients 2021, 13, 1740. [Google Scholar] [CrossRef]
- Lin, M.J.; Dai, W.; Scott, M.J.; Li, R.; Zhang, Y.Q.; Yang, Y.; Chen, L.Z.; Huang, X.S. Metformin improves nonalcoholic fatty liver disease in obese mice via down-regulation of apolipoprotein A5 as part of the AMPK/LXRalpha signaling pathway. Oncotarget 2017, 8, 108802–108809. [Google Scholar] [CrossRef]
- Levine, P.M.; Balana, A.T.; Sturchler, E.; Koole, C.; Noda, H.; Zarzycka, B.; Daley, E.J.; Truong, T.T.; Katritch, V.; Gardella, T.J.; et al. O-GlcNAc Engineering of GPCR Peptide-Agonists Improves Their Stability and in Vivo Activity. J. Am. Chem. Soc. 2019, 141, 14210–14219. [Google Scholar] [CrossRef]
- Cusi, K. Incretin-Based Therapies for the Management of Nonalcoholic Fatty Liver Disease in Patients with Type 2 Diabetes. Hepatology 2019, 69, 2318–2322. [Google Scholar] [CrossRef]
- Hodrea, J.; Balogh, D.B.; Hosszu, A.; Lenart, L.; Besztercei, B.; Koszegi, S.; Sparding, N.; Genovese, F.; Wagner, L.J.; Szabo, A.J.; et al. Reduced O-GlcNAcylation and tubular hypoxia contribute to the antifibrotic effect of SGLT2 inhibitor dapagliflozin in the diabetic kidney. Am. J. Physiol. Ren. Physiol. 2020, 318, F1017–F1029. [Google Scholar] [CrossRef]
- Akuta, N.; Watanabe, C.; Kawamura, Y.; Arase, Y.; Saitoh, S.; Fujiyama, S.; Sezaki, H.; Hosaka, T.; Kobayashi, M.; Kobayashi, M.; et al. Effects of a sodium-glucose cotransporter 2 inhibitor in nonalcoholic fatty liver disease complicated by diabetes mellitus: Preliminary prospective study based on serial liver biopsies. Hepatol. Commun. 2017, 1, 46–52. [Google Scholar] [CrossRef]
- Dierschke, S.K.; Toro, A.L.; Barber, A.J.; Arnold, A.C.; Dennis, M.D. Angiotensin-(1-7) Attenuates Protein O-GlcNAcylation in the Retina by EPAC/Rap1-Dependent Inhibition of O-GlcNAc Transferase. Investig. Ophthalmol. Vis. Sci. 2020, 61, 24. [Google Scholar] [CrossRef]
- Zhang, X.; Wong, G.L.; Yip, T.C.; Tse, Y.K.; Liang, L.Y.; Hui, V.W.; Lin, H.; Li, G.L.; Lai, J.C.; Chan, H.L.; et al. Angiotensin-converting enzyme inhibitors prevent liver-related events in nonalcoholic fatty liver disease. Hepatology 2022, 76, 469–482. [Google Scholar] [CrossRef]
- Chen, Y.; Zhu, G.; Liu, Y.; Wu, Q.; Zhang, X.; Bian, Z.; Zhang, Y.; Pan, Q.; Sun, F. O-GlcNAcylated c-Jun antagonizes ferroptosis via inhibiting GSH synthesis in liver cancer. Cell Signal. 2019, 63, 109384. [Google Scholar] [CrossRef]
- Honda, Y.; Kessoku, T.; Sumida, Y.; Kobayashi, T.; Kato, T.; Ogawa, Y.; Tomeno, W.; Imajo, K.; Fujita, K.; Yoneda, M.; et al. Efficacy of glutathione for the treatment of nonalcoholic fatty liver disease: An open-label, single-arm, multicenter, pilot study. BMC Gastroenterol. 2017, 17, 96. [Google Scholar] [CrossRef]
- Arambasic, J.; Mihailovic, M.; Uskokovic, A.; Dinic, S.; Grdovic, N.; Markovic, J.; Poznanovic, G.; Bajec, D.; Vidakovic, M. Alpha-lipoic acid upregulates antioxidant enzyme gene expression and enzymatic activity in diabetic rat kidneys through an O-GlcNAc-dependent mechanism. Eur. J. Nutr. 2013, 52, 1461–1473. [Google Scholar] [CrossRef]
- Mirjana, M.; Jelena, A.; Aleksandra, U.; Svetlana, D.; Nevena, G.; Jelena, M.; Goran, P.; Melita, V. Alpha-lipoic acid preserves the structural and functional integrity of red blood cells by adjusting the redox disturbance and decreasing O-GlcNAc modifications of antioxidant enzymes and heat shock proteins in diabetic rats. Eur. J. Nutr. 2012, 51, 975–986. [Google Scholar] [CrossRef] [PubMed]
- Ko, C.Y.; Lo, Y.M.; Xu, J.H.; Chang, W.C.; Huang, D.W.; Wu, J.S.; Yang, C.H.; Huang, W.C.; Shen, S.C. Alpha-lipoic acid alleviates NAFLD and triglyceride accumulation in liver via modulating hepatic NLRP3 inflammasome activation pathway in type 2 diabetic rats. Food Sci. Nutr. 2021, 9, 2733–2742. [Google Scholar] [CrossRef] [PubMed]
- Rahmanabadi, A.; Mahboob, S.; Amirkhizi, F.; Hosseinpour-Arjmand, S.; Ebrahimi-Mameghani, M. Oral alpha-lipoic acid supplementation in patients with non-alcoholic fatty liver disease: Effects on adipokines and liver histology features. Food Funct. 2019, 10, 4941–4952. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, M.J.; Gaunt, P.; Aithal, G.P.; Barton, D.; Hull, D.; Parker, R.; Hazlehurst, J.M.; Guo, K.; LEAN Trial Team; Abouda, G.; et al. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): A multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet 2016, 387, 679–690. [Google Scholar] [CrossRef]
- Newsome, P.N.; Buchholtz, K.; Cusi, K.; Linder, M.; Okanoue, T.; Ratziu, V.; Sanyal, A.J.; Sejling, A.S.; Harrison, S.A.; Investigators, N.N. A Placebo-Controlled Trial of Subcutaneous Semaglutide in Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2021, 384, 1113–1124. [Google Scholar] [CrossRef]
- Yu, X.; Hao, M.; Liu, Y.; Ma, X.; Lin, W.; Xu, Q.; Zhou, H.; Shao, N.; Kuang, H. Liraglutide ameliorates non-alcoholic steatohepatitis by inhibiting NLRP3 inflammasome and pyroptosis activation via mitophagy. Eur. J. Pharmacol. 2019, 864, 172715. [Google Scholar] [CrossRef]
- Goldberg, H.; Whiteside, C.; Fantus, I.G. O-linked beta-N-acetylglucosamine supports p38 MAPK activation by high glucose in glomerular mesangial cells. Am. J. Physiol. Endocrinol. Metab. 2011, 301, E713–E726. [Google Scholar] [CrossRef]
- Park, M.J.; Kim, D.I.; Lim, S.K.; Choi, J.H.; Han, H.J.; Yoon, K.C.; Park, S.H. High glucose-induced O-GlcNAcylated carbohydrate response element-binding protein (ChREBP) mediates mesangial cell lipogenesis and fibrosis: The possible role in the development of diabetic nephropathy. J. Biol. Chem. 2014, 289, 13519–13530. [Google Scholar] [CrossRef]
- Nakano, S.; Katsuno, K.; Isaji, M.; Nagasawa, T.; Buehrer, B.; Walker, S.; Wilkison, W.O.; Cheatham, B. Remogliflozin Etabonate Improves Fatty Liver Disease in Diet-Induced Obese Male Mice. J. Clin. Exp. Hepatol. 2015, 5, 190–198. [Google Scholar] [CrossRef]
- Qiang, S.; Nakatsu, Y.; Seno, Y.; Fujishiro, M.; Sakoda, H.; Kushiyama, A.; Mori, K.; Matsunaga, Y.; Yamamotoya, T.; Kamata, H.; et al. Treatment with the SGLT2 inhibitor luseogliflozin improves nonalcoholic steatohepatitis in a rodent model with diabetes mellitus. Diabetol. Metab. Syndr. 2015, 7, 104. [Google Scholar] [CrossRef]
- Jojima, T.; Tomotsune, T.; Iijima, T.; Akimoto, K.; Suzuki, K.; Aso, Y. Empagliflozin (an SGLT2 inhibitor), alone or in combination with linagliptin (a DPP-4 inhibitor), prevents steatohepatitis in a novel mouse model of non-alcoholic steatohepatitis and diabetes. Diabetol. Metab. Syndr. 2016, 8, 45. [Google Scholar] [CrossRef]
- Petito-da-Silva, T.I.; Souza-Mello, V.; Barbosa-da-Silva, S. Empaglifozin mitigates NAFLD in high-fat-fed mice by alleviating insulin resistance, lipogenesis and ER stress. Mol. Cell. Endocrinol. 2019, 498, 110539. [Google Scholar] [CrossRef]
- Tahara, A.; Takasu, T. SGLT2 inhibitor ipragliflozin alone and combined with pioglitazone prevents progression of nonalcoholic steatohepatitis in a type 2 diabetes rodent model. Physiol. Rep. 2019, 7, e14286. [Google Scholar] [CrossRef]
- Chiang, H.; Lee, J.C.; Huang, H.C.; Huang, H.; Liu, H.K.; Huang, C. Delayed intervention with a novel SGLT2 inhibitor NGI001 suppresses diet-induced metabolic dysfunction and non-alcoholic fatty liver disease in mice. Br. J. Pharmacol. 2020, 177, 239–253. [Google Scholar] [CrossRef]
- Nasiri-Ansari, N.; Nikolopoulou, C.; Papoutsi, K.; Kyrou, I.; Mantzoros, C.S.; Kyriakopoulos, G.; Chatzigeorgiou, A.; Kalotychou, V.; Randeva, M.S.; Chatha, K.; et al. Empagliflozin Attenuates Non-Alcoholic Fatty Liver Disease (NAFLD) in High Fat Diet Fed ApoE(-/-) Mice by Activating Autophagy and Reducing ER Stress and Apoptosis. Int. J. Mol. Sci. 2021, 22, 818. [Google Scholar] [CrossRef]
- Meng, Z.; Liu, X.; Li, T.; Fang, T.; Cheng, Y.; Han, L.; Sun, B.; Chen, L. The SGLT2 inhibitor empagliflozin negatively regulates IL-17/IL-23 axis-mediated inflammatory responses in T2DM with NAFLD via the AMPK/mTOR/autophagy pathway. Int. Immunopharmacol. 2021, 94, 107492. [Google Scholar] [CrossRef]
- Kucharewicz, I.; Pawlak, R.; Matys, T.; Pawlak, D.; Buczko, W. Antithrombotic effect of captopril and losartan is mediated by angiotensin-(1-7). Hypertension 2002, 40, 774–779. [Google Scholar] [CrossRef]
- Ishiyama, Y.; Gallagher, P.E.; Averill, D.B.; Tallant, E.A.; Brosnihan, K.B.; Ferrario, C.M. Upregulation of angiotensin-converting enzyme 2 after myocardial infarction by blockade of angiotensin II receptors. Hypertension 2004, 43, 970–976. [Google Scholar] [CrossRef]
- Xu, Y.Z.; Zhang, X.; Wang, L.; Zhang, F.; Qiu, Q.; Liu, M.L.; Zhang, G.R.; Wu, X.L. An increased circulating angiotensin II concentration is associated with hypoadiponectinemia and postprandial hyperglycemia in men with nonalcoholic fatty liver disease. Intern. Med. 2013, 52, 855–861. [Google Scholar] [CrossRef]
- Cao, X.; Yang, F.Y.; Xin, Z.; Xie, R.R.; Yang, J.K. The ACE2/Ang-(1-7)/Mas axis can inhibit hepatic insulin resistance. Mol. Cell. Endocrinol. 2014, 393, 30–38. [Google Scholar] [CrossRef]
- Nourjah, P.; Ahmad, S.R.; Karwoski, C.; Willy, M. Estimates of acetaminophen (Paracetomal)-associated overdoses in the United States. Pharmacoepidemiol. Drug Saf. 2006, 15, 398–405. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.M. Etiologies of acute liver failure. Semin. Liver Dis. 2008, 28, 142–152. [Google Scholar] [CrossRef] [PubMed]
- McGreal, S.R.; Bhushan, B.; Walesky, C.; McGill, M.R.; Lebofsky, M.; Kandel, S.E.; Winefield, R.D.; Jaeschke, H.; Zachara, N.E.; Zhang, Z.; et al. Modulation of O-GlcNAc Levels in the Liver Impacts Acetaminophen-Induced Liver Injury by Affecting Protein Adduct Formation and Glutathione Synthesis. Toxicol. Sci. 2018, 162, 599–610. [Google Scholar] [CrossRef] [PubMed]
- Filhoulaud, G.; Benhamed, F.; Pagesy, P.; Bonner, C.; Fardini, Y.; Ilias, A.; Movassat, J.; Burnol, A.F.; Guilmeau, S.; Kerr-Conte, J.; et al. O-GlcNacylation Links TxNIP to Inflammasome Activation in Pancreatic beta Cells. Front. Endocrinol. 2019, 10, 291. [Google Scholar] [CrossRef]
- Hosseinpour-Arjmand, S.; Amirkhizi, F.; Ebrahimi-Mameghani, M. The effect of alpha-lipoic acid on inflammatory markers and body composition in obese patients with non-alcoholic fatty liver disease: A randomized, double-blind, placebo-controlled trial. J. Clin. Pharm. Ther. 2019, 44, 258–267. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Experiment Type | Key Factor | Directly Modified or Not | Level of O-GlcNAc | Specific Mechanism | Final Conclusion | Ref. |
|---|---|---|---|---|---|---|
| Animal and Cell | ChREBP | Yes |  | Transcriptional activity of L-PK, ACC, FAS, and SCD1 | Hepatic TG deposition | [44] |
| Cell and Animal | SREBP-1 | No | | SREBP-1 phosphorylation and stability via AMPK signaling | TG deposition | [43] |
| Cell and Animal | IP6K1 | No | | Unclarified | Promote NASH and fibrosis | [17] |
| Cell and Animal | NF-κB | Yes | | p65 is modified to induce activation of NFκB | Inflammatory damage | [60] |
| c-Rel is modified and activated | Anti-inflammatory effect | [61] | ||||
| Cell | Collagen | Yes | | Activate HSCs | Liver fibrosis | [62] |
| Animal & Cell | SRF | Yes | | Inhibited SRF activity to induce α-SMA transcription | Prevent liver fibrosis | [63] |
| Animal & Cell | RIPK3 | Yes | |  RIPK3 stability, caspase 8 cleavage, and JNK activation RIPK3 stability, caspase 8 cleavage, and JNK activation | Promote NAFLD-HCC | [64,65] |
| Drug. | Correlation with O-GlcNAcylation | Effects on NAFLD | Ref. |
|---|---|---|---|
| MET | O-GlcNAcylation of AMP, NF-κB, and ChREBP | Liver TG accumulation and improved NAFLD | [97,98,99,101] |
| GLP-1 | O-GlcNAcylation enhance GLP-1 activity | Liver enzyme levels and liver fat | [102,103] |
| SGLT-2I | Reduced O-GlcNAcylation exerts an anti-fibrotic effect | Liver steatosis and liver fibrosis | [104,105] |
| ACEI | Enhancement of Ang1-7 axis to reduce O-GlcNAcylation | Incidence of liver cancer and cirrhosis | [106,107] |
| GSH | Positive correlation between c-Jun O-GlcNAcylation and GSH synthesis | supported liver metabolism and improved NAFLD | [108,109] |
| ALA | O-GlcNAcylation of ERK, p38, CuZnSOD, CAT, HSP70, and HSP90 | Liver TG accumulation and improved NAFLD | [110,111,112,113] |
| Curcumin | Inhibition O-GlcNAcylation and blocked NF-κB signaling pathway | Exert anti-inflammatory effect, alleviated NAFLD/NASH | [19] |
| Silibinin | Inhibition of O-GlcNAcylation and blocked NF-κB signaling pathway | Anti-inflammatory effect, alleviated NASH | [18] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Y.; Li, Z.; Xu, M.; Zhang, D.; Ling, J.; Yu, P.; Shen, Y. O-GlycNacylation Remission Retards the Progression of Non-Alcoholic Fatty Liver Disease. Cells 2022, 11, 3637. https://doi.org/10.3390/cells11223637
Zhou Y, Li Z, Xu M, Zhang D, Ling J, Yu P, Shen Y. O-GlycNacylation Remission Retards the Progression of Non-Alcoholic Fatty Liver Disease. Cells. 2022; 11(22):3637. https://doi.org/10.3390/cells11223637
Chicago/Turabian StyleZhou, Yicheng, Zhangwang Li, Minxuan Xu, Deju Zhang, Jitao Ling, Peng Yu, and Yunfeng Shen. 2022. "O-GlycNacylation Remission Retards the Progression of Non-Alcoholic Fatty Liver Disease" Cells 11, no. 22: 3637. https://doi.org/10.3390/cells11223637
APA StyleZhou, Y., Li, Z., Xu, M., Zhang, D., Ling, J., Yu, P., & Shen, Y. (2022). O-GlycNacylation Remission Retards the Progression of Non-Alcoholic Fatty Liver Disease. Cells, 11(22), 3637. https://doi.org/10.3390/cells11223637