


Lipid Dys-Homeostasis Contributes to APOE4-Associated AD Pathology

 and
and 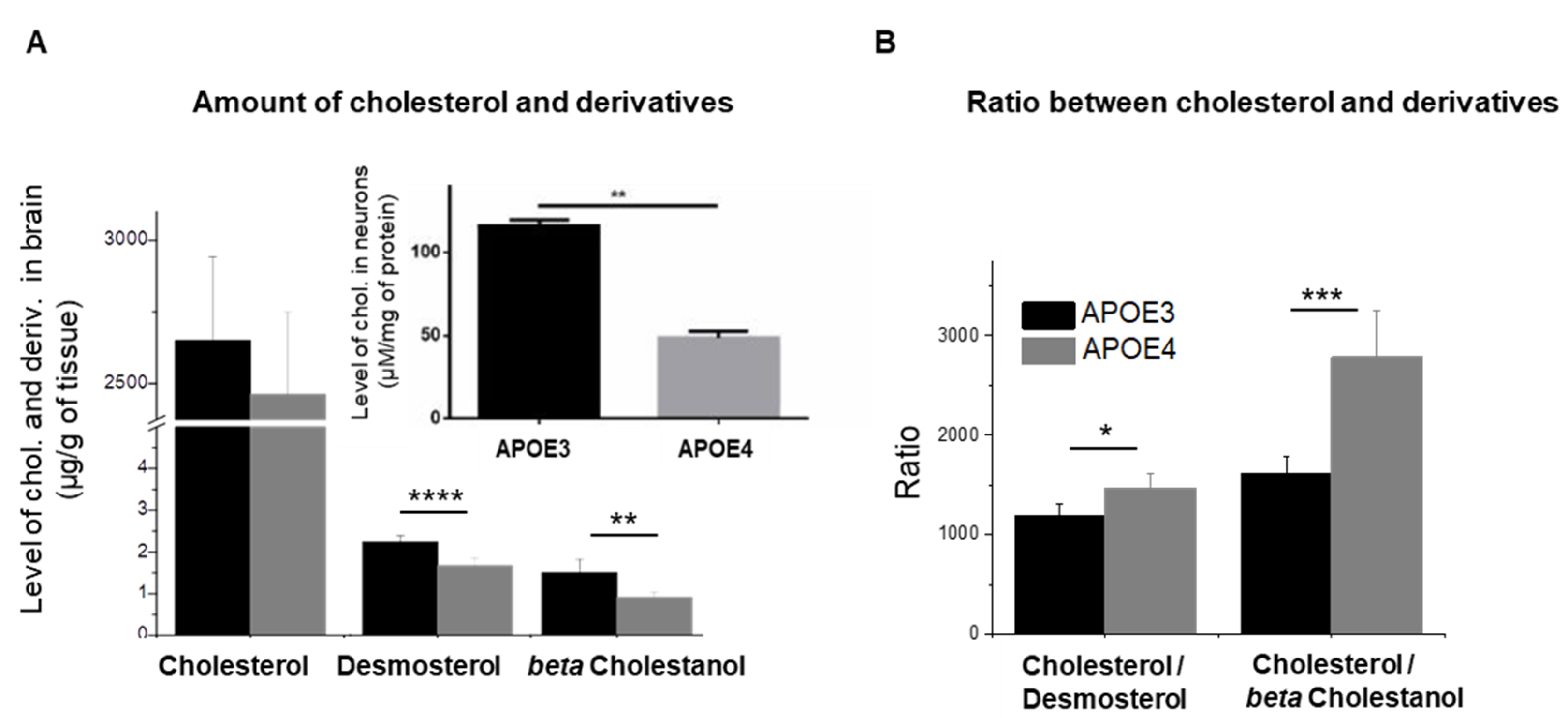{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Preparation and Lipid Extraction
2.2. Lipid Analysis
2.2.1. Analysis of Cholesterol and Derivatives by GC-MS/MS
2.2.2. Phospholipids Analysis by Electrospray Mass Spectrometry (ESI-MS)
2.2.3. Analysis of FA Composition by GC
2.3. Cultures of Primary Neurons
2.4. Aβ 38, 40 and 42 Measurements
2.5. Western Blotting
2.6. Data Processing
3. Results
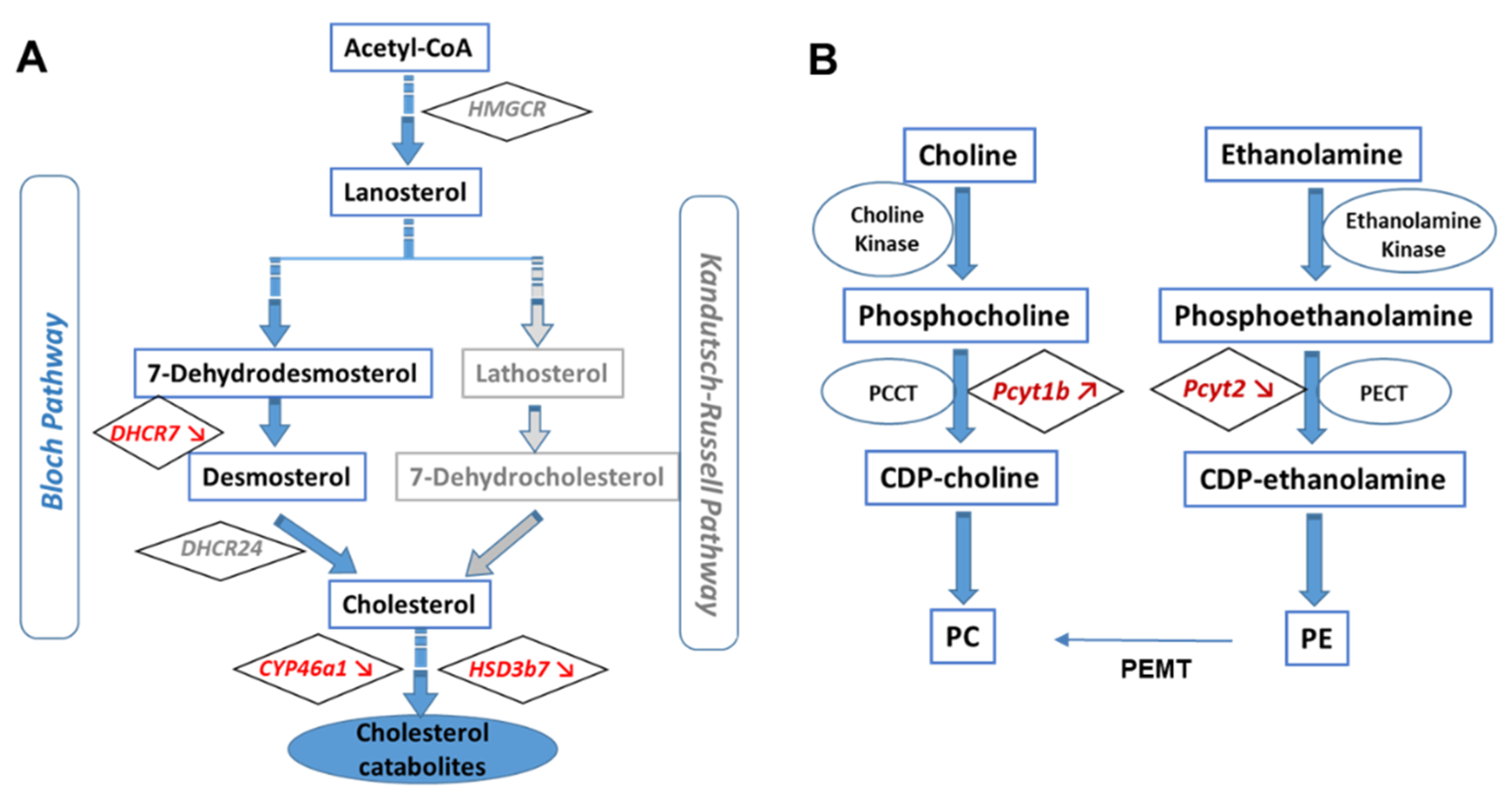
3.1. APOE Genotype Disturbs Cholesterol Turnover
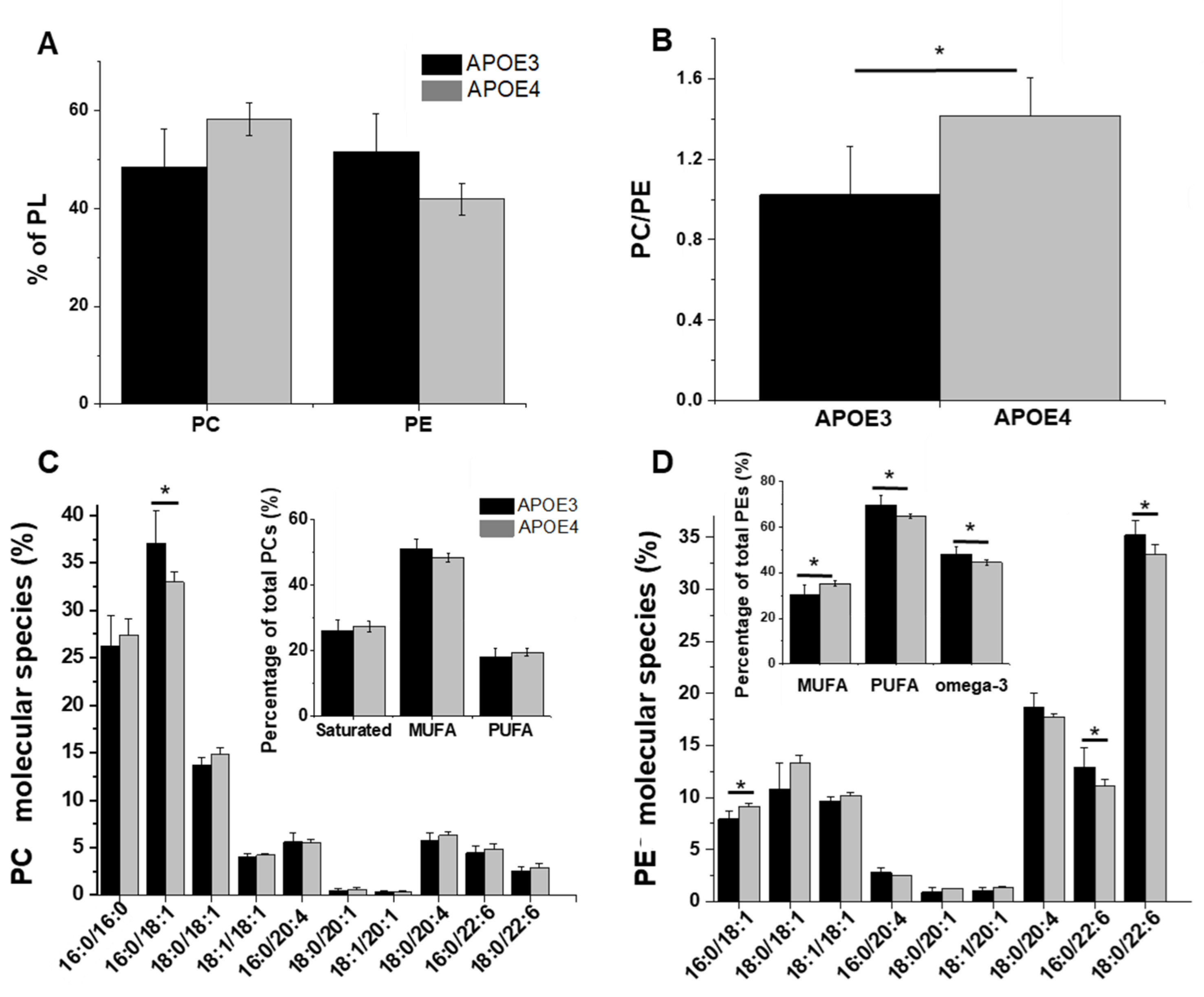
3.2. Levels of PC and PE and Changes in Their Composition in APOE4-KI Mice
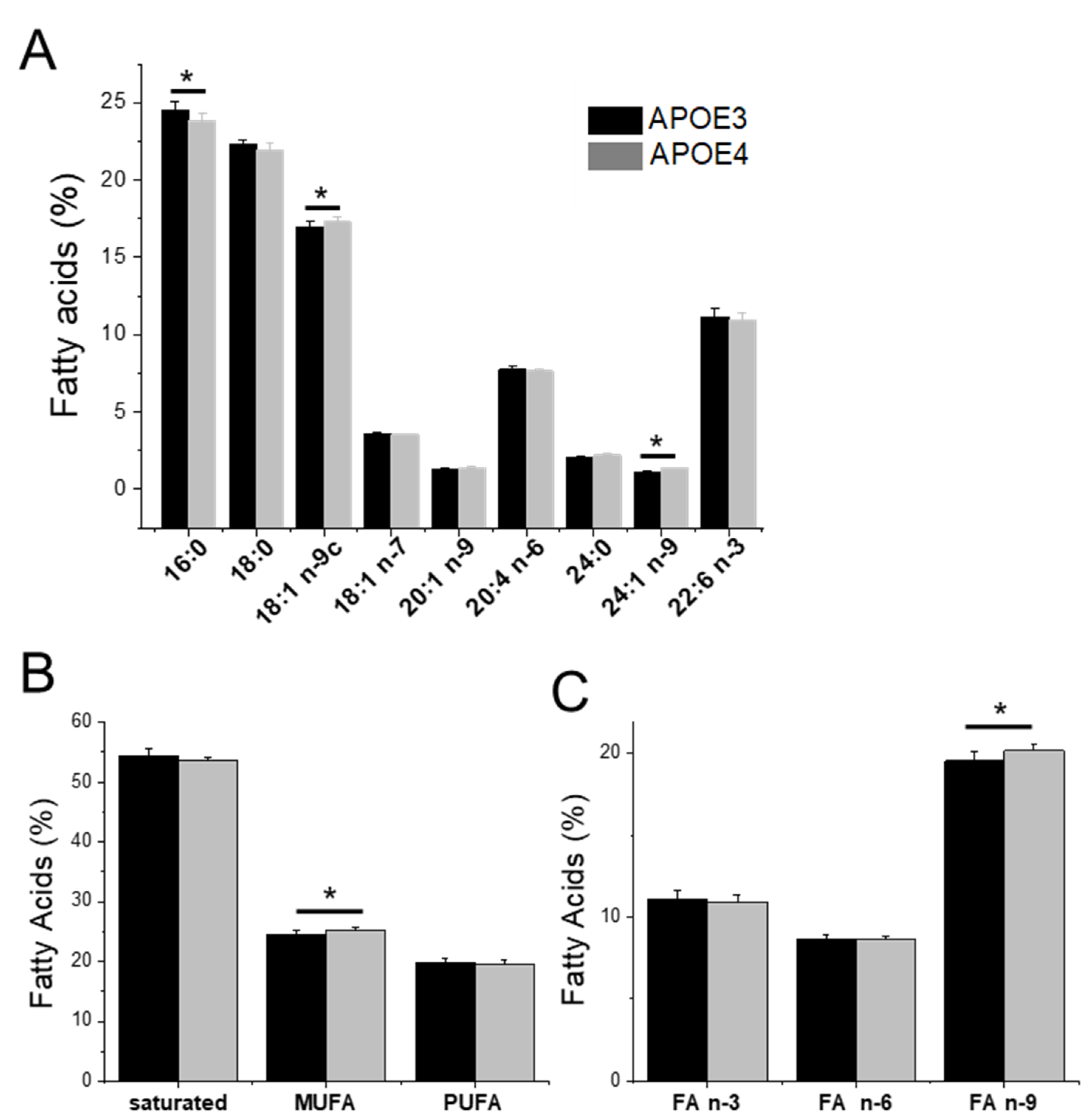
3.3. APOE Genotype and Fatty Acid Profile and Metabolism
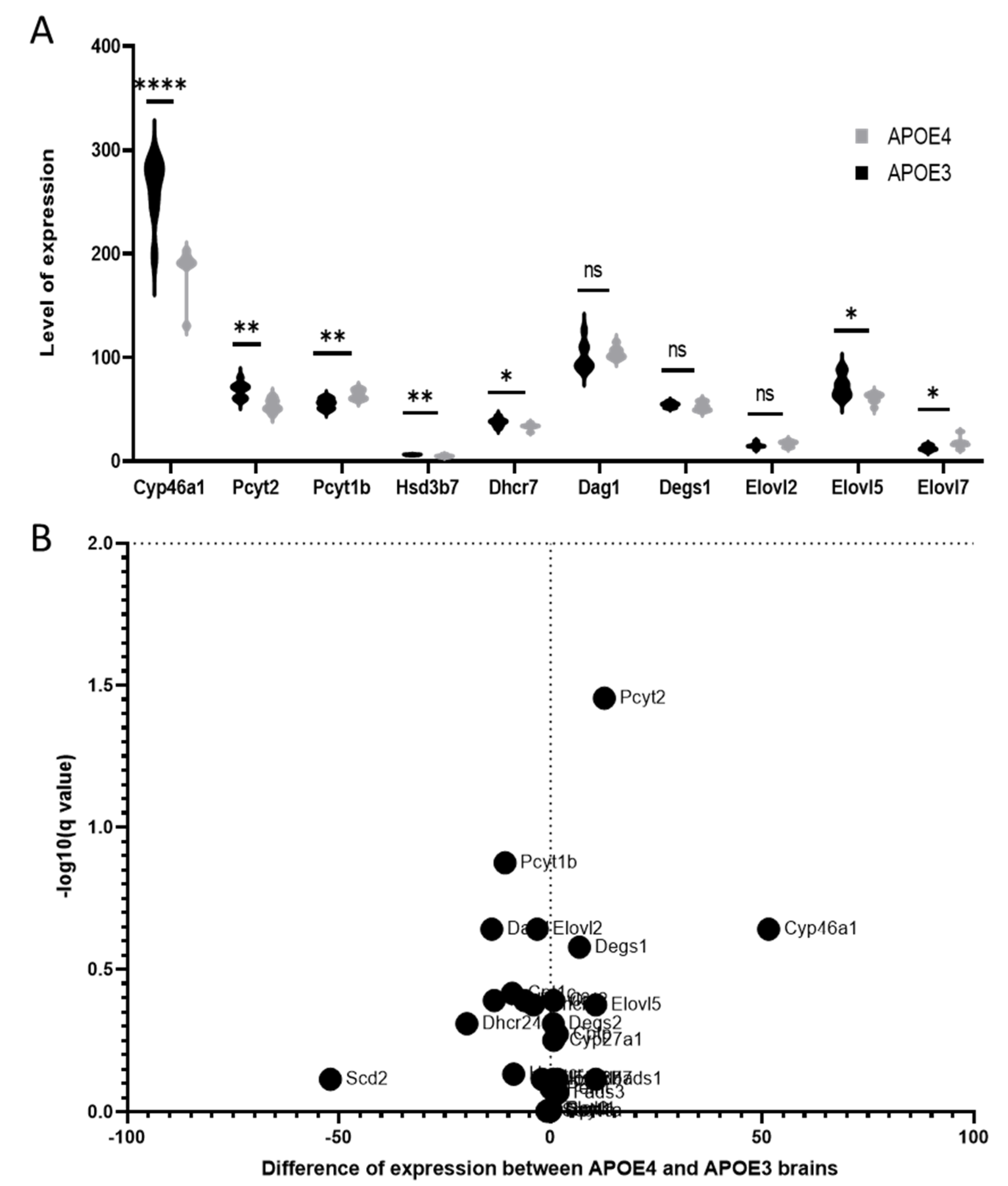
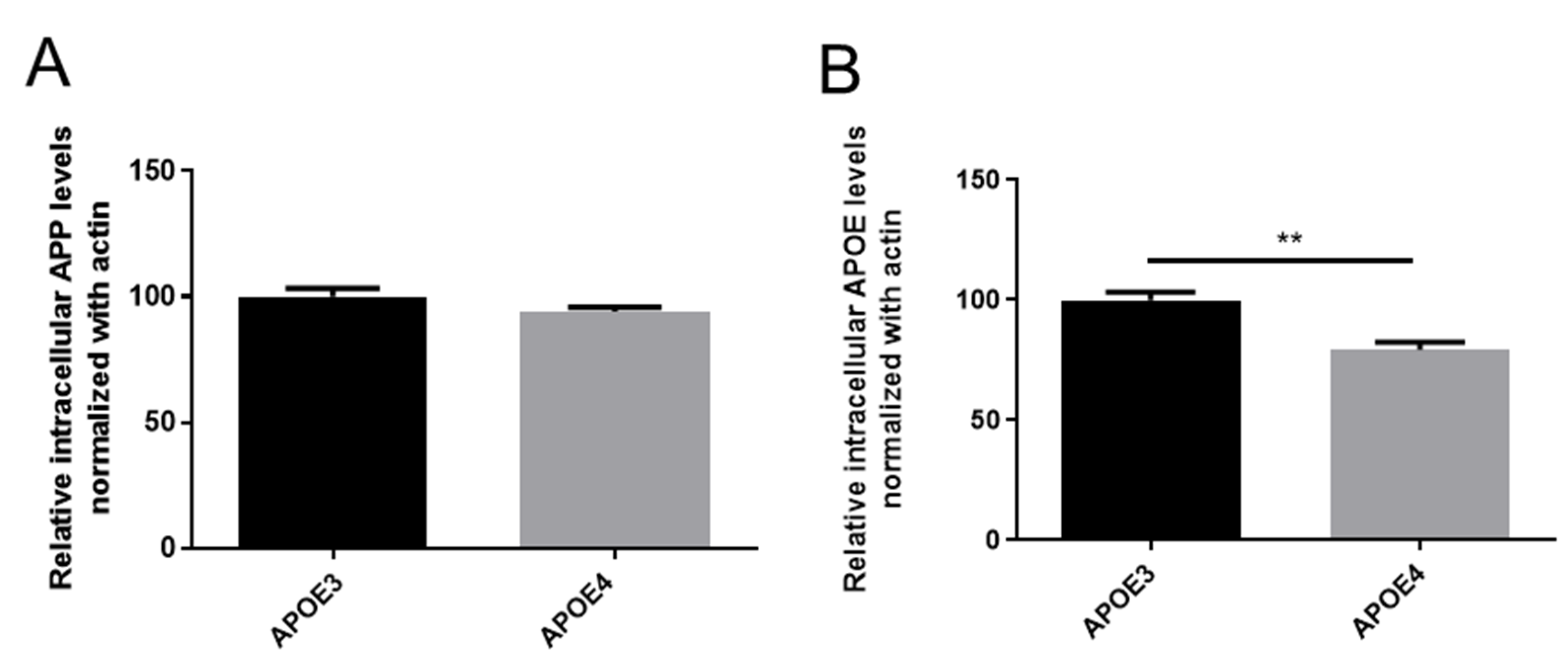
3.4. Lipid Dysregulations and Gene Expression in APOE Genotypes
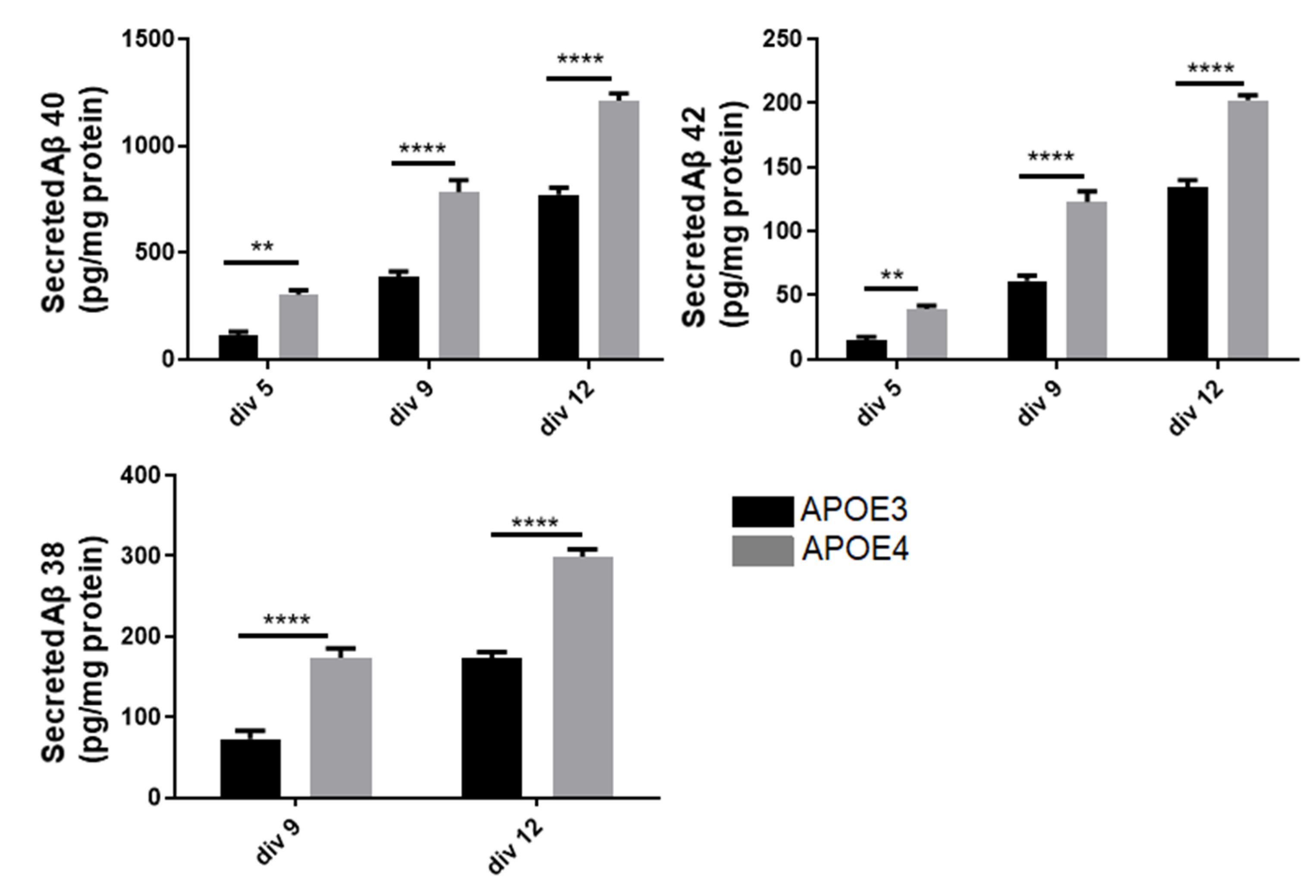
3.5. Neurons of APOE4-KI Mice Produce a Higher Amount of Aβ Peptide
4. Discussion
4.1. Altered Cholesterol Turnover in APOE4-KI Mice
4.2. APOE4 Genotype Is Associated to Lower Levels of Phosphatidylethanolamines Bearing Polyunsaturated Fatty Acids

4.3. Elevated Levels of MUFAs, Particularly Omega-9 FA, in APOE4-KI Mice
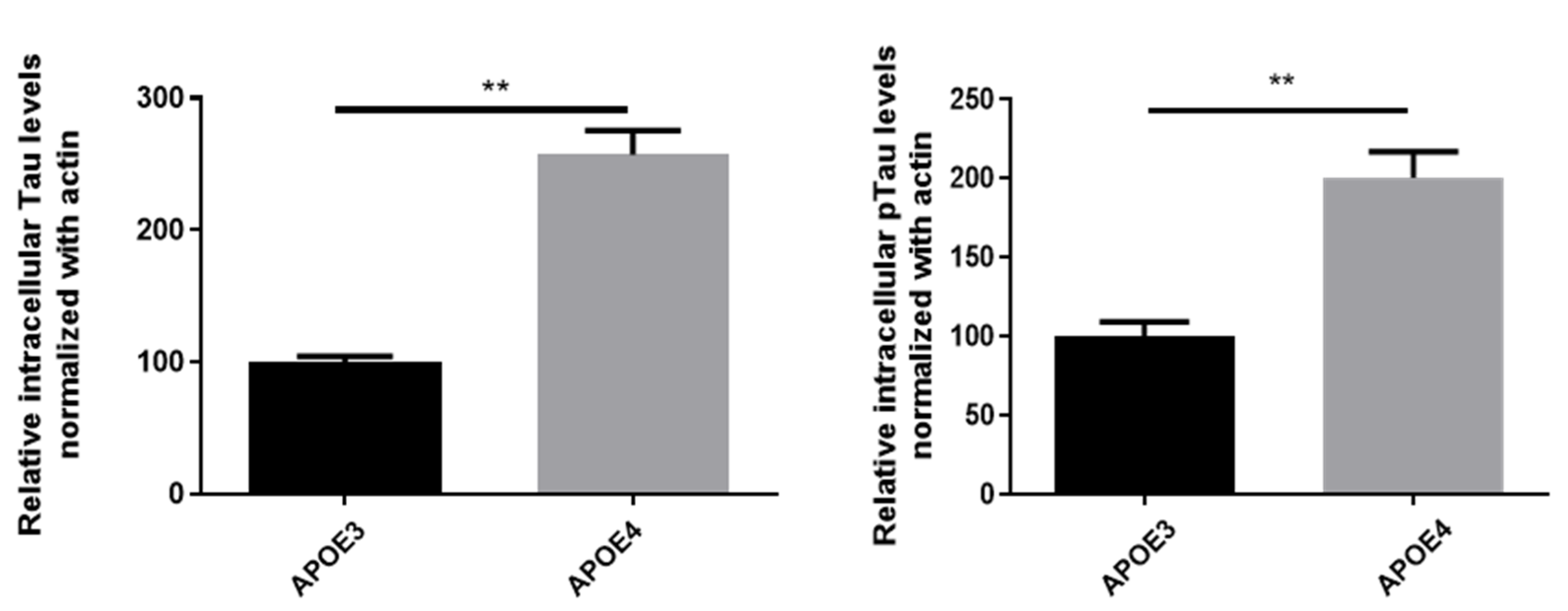
4.4. Interrelationship between Lipid Dys-Homeostasis and AD Pathological Markers
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Duyckaerts, C.; Delatour, B.; Potier, M.C. Classification and Basic Pathology of Alzheimer Disease. Acta Neuropathol. 2009, 118, 5–36. [Google Scholar] [CrossRef] [PubMed]
- Foley, P. Lipids in Alzheimer’s Disease: A Century-Old Story. Biochim. Et Biophys. Acta Mol. Cell Biol. Lipids 2010, 1801, 750–753. [Google Scholar] [CrossRef] [PubMed]
- Graeber, M.B.; Kösel, S.; Grasbon-Frodl, E.; Möller, H.J.; Mehraein, P. Histopathology and APOE Genotype of the First Alzheimer Disease Patient, Auguste D. Neurogenetics 1998, 1, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Möller, H.-J.; Graeber, M.B. The Case Described by Alois Alzheimer in 1911. Eur. Arch. Psychiatry Clin. Neurosci. 1998, 248, 111–122. [Google Scholar] [CrossRef]
- Jové, M.; Mota-Martorell, N.; Torres, P.; Ayala, V.; Portero-Otin, M.; Ferrer, I.; Pamplona, R. The Causal Role of Lipoxidative Damage in Mitochondrial Bioenergetic Dysfunction Linked to Alzheimer’s Disease Pathology. Life 2021, 11, 388. [Google Scholar] [CrossRef]
- Panchal, M.; Gaudin, M.; Lazar, A.N.; Salvati, E.; Rivals, I.; Ayciriex, S.; Dauphinot, L.; Dargère, D.; Auzeil, N.; Masserini, M.; et al. Ceramides and Sphingomyelinases in Senile Plaques. Neurobiol. Dis. 2014, 65, 193–201. [Google Scholar] [CrossRef]
- Lazar, A.N.; Bich, C.; Panchal, M.; Desbenoit, N.; Petit, V.W.; Touboul, D.; Dauphinot, L.; Marquer, C.; Laprévote, O.; Brunelle, A.; et al. Time-of-Flight Secondary Ion Mass Spectrometry (TOF-SIMS) Imaging Reveals Cholesterol Overload in the Cerebral Cortex of Alzheimer Disease Patients. Acta Neuropathol. 2013, 125, 133–144. [Google Scholar] [CrossRef]
- Shi, H.; Medway, C.; Bullock, J.; Brown, K.; Kalsheker, N.; Morgan, K. Analysis of Genome-Wide Association Study (GWAS) Data Looking for Replicating Signals in Alzheimer’s Disease (AD). Int. J. Mol. Epidemiol. Genet. 2010, 1, 53–66. [Google Scholar]
- Hao, S.; Wang, R.; Zhang, Y.; Zhan, H. Prediction of Alzheimer’s Disease-Associated Genes by Integration of GWAS Summary Data and Expression Data. Front. Genet. 2019, 10, 653. [Google Scholar] [CrossRef]
- Burlot, M.A.; Braudeau, J.; Michaelsen-Preusse, K.; Potier, B.; Ayciriex, S.; Varin, J.; Gautier, B.; Djelti, F.; Audrain, M.; Dauphinot, L.; et al. Cholesterol 24-Hydroxylase Defect Is Implicated in Memory Impairments Associated with Alzheimer-like Tau Pathology. Hum. Mol. Genet. 2015, 24, 5965–5976. [Google Scholar] [CrossRef]
- Wollmer, M.A.; Streffer, J.R.; Tsolaki, M.; Grimaldi, L.M.E.; Lütjohann, D.; Thal, D.; von Bergmann, K.; Nitsch, R.M.; Hock, C.; Papassotiropoulos, A. Genetic Association of Acyl-Coenzyme A: Cholesterol Acyltransferase with Cerebrospinal Fluid Cholesterol Levels, Brain Amyloid Load, and Risk for Alzheimer’s Disease. Mol. Psychiatry 2003, 8, 635–638. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhao, F.G.; Wang, Y.H.; Yang, J.F.; Ma, Q.L.; Tang, Z.; Dong, X.M.; Chan, P. Association between Acyl-Coenzyme A: Cholesterol Acyltransferase Gene and Risk for Alzheimer’s Disease in Chinese. Neurosci. Lett. 2005, 388, 17–20. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, Y.; Chang, C.C.Y.; Huang, L.H.; Bryleva, E.Y.; Chang, T.Y. Inhibiting ACAT1/SOAT1 in Microglia Stimulates Autophagy-Mediated Lysosomal Proteolysis and Increases Aβ1-42 Clearance. J. Neurosci. 2014, 34, 14484–14501. [Google Scholar] [CrossRef] [PubMed]
- di Paolo, G.; Kim, T.W. Linking Lipids to Alzheimer’s Disease: Cholesterol and Beyond. Nat. Rev. Neurosci. 2011, 12, 284–296. [Google Scholar] [CrossRef]
- Kim, J.; Castellano, J.M.; Jiang, H.; Basak, J.M.; Parsadanian, M.; Pham, V.; Mason, S.M.; Paul, S.M.; Holtzman, D.M. Overexpression of Low-Density Lipoprotein Receptor in the Brain Markedly Inhibits Amyloid Deposition and Increases Extracellular Aβ Clearance. Neuron 2009, 64, 632–644. [Google Scholar] [CrossRef]
- Kang, D.E.; Saitoh, T.; Chen, X.; Xia, Y.; Masliah, E.; Hansen, L.A.; Thomas, R.G.; Thal, L.J.; Katzman, R. Genetic Association of the Low-Density Lipoprotein Receptor-Related Protein Gene (LRP), an Apolipoprotein E Receptor, with Late-Onset Alzheimer’s Disease. Neurology 1997, 49, 56–61. [Google Scholar] [CrossRef]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene Dose of Apolipoprotein E Type 4 Allele and the Risk of Alzheimer’s Disease in Late Onset Families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef]
- Dubois, B.; Epelbaum, S.; Nyasse, F.; Bakardjian, H.; Gagliardi, G.; Uspenskaya, O.; Houot, M.; Lista, S.; Cacciamani, F.; Potier, M.C.; et al. Cognitive and Neuroimaging Features and Brain β-Amyloidosis in Individuals at Risk of Alzheimer’s Disease (INSIGHT-PreAD): A Longitudinal Observational Study. Lancet Neurol. 2018, 17, 335–346. [Google Scholar] [CrossRef]
- Simonovitch, S.; Schmukler, E.; Bespalko, A.; Iram, T.; Frenkel, D.; Holtzman, D.M.; Masliah, E.; Michaelson, D.M.; Pinkas-Kramarski, R. Impaired Autophagy in APOE4 Astrocytes. J. Alzheimer’s Dis. 2016, 51, 915–927. [Google Scholar] [CrossRef]
- Strittmatter, W.J.; Weisgraber, K.H.; Huang, D.Y.; Dong, L.M.; Salvesen, G.S.; Pericak-Vance, M.; Schmechel, D.; Saunders, A.M.; Goldgaber, D.; Roses, A.D. Binding of Human Apolipoprotein E to Synthetic Amyloid β Peptide: Isoform-Specific Effects and Implications for Late-Onset Alzheimer Disease. Proc. Natl. Acad. Sci. USA 1993, 90, 8098–8102. [Google Scholar] [CrossRef]
- Deane, R.; Sagare, A.; Hamm, K.; Parisi, M.; Lane, S.; Finn, M.B.; Holtzman, D.M.; Zlokovic, B.V. ApoE Isoform-Specific Disruption of Amyloid β Peptide Clearance from Mouse Brain. J. Clin. Investig. 2008, 118, 4002–4013. [Google Scholar] [CrossRef] [PubMed]
- Hori, Y.; Hashimoto, T.; Nomoto, H.; Hyman, B.T.; Iwatsubo, T. Role of Apolipoprotein E in β-Amyloidogenesis: Isoform-Specific Effects on Protofibril to Fibril Conversion of Aβ in Vitro and Brain Aβ Deposition in Vivo. J. Biol. Chem. 2015, 290, 15163–15174. [Google Scholar] [CrossRef] [PubMed]
- Cerf, E.; Gustot, A.; Goormaghtigh, E.; Ruysschaert, J.-M.; Raussens, V. High Ability of Apolipoprotein E4 to Stabilize Amyloid-β Peptide Oligomers, the Pathological Entities Responsible for Alzheimer’s Disease. FASEB J. 2011, 25, 1585–1595. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Yamada, K.; Liddelow, S.A.; Smith, S.T.; Zhao, L.; Luo, W.; Tsai, R.M.; Spina, S.; Grinberg, L.T.; Rojas, J.C.; et al. ApoE4 Markedly Exacerbates Tau-Mediated Neurodegeneration in a Mouse Model of Tauopathy. Nature 2017, 549, 523–527. [Google Scholar] [CrossRef]
- Montagne, A.; Nation, D.A.; Sagare, A.P.; Barisano, G.; Sweeney, M.D.; Chakhoyan, A.; Pachicano, M.; Joe, E.; Nelson, A.R.; D’Orazio, L.M.; et al. APOE4 Leads to Blood–Brain Barrier Dysfunction Predicting Cognitive Decline. Nature 2020, 581, 71–76. [Google Scholar] [CrossRef]
- Mahley, R.W. Central Nervous System Lipoproteins: ApoE and Regulation of Cholesterol Metabolism. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1305–1315. [Google Scholar] [CrossRef]
- Jenner, A.M.; Lim, W.L.F.; Ng, M.P.E.; Wenk, M.R.; Shui, G.; Sharman, M.J.; Gandy, S.E.; Martins, R.N. The Effect of APOE Genotype on Brain Levels of Oxysterols in Young and Old Human APOE Ε2,Ε3 and Ε4 Knock-in Mice. Neuroscience 2010, 169, 109–115. [Google Scholar] [CrossRef]
- Levi, O.; Lütjohann, D.; Devir, A.; von Bergmann, K.; Hartmann, T.; Michaelson, D.M. Regulation of Hippocampal Cholesterol Metabolism by ApoE and Environmental Stimulation. J. Neurochem. 2005, 95, 987–997. [Google Scholar] [CrossRef]
- Sharman, M.J.; Shui, G.; Fernandis, A.Z.; Lim, W.L.F.; Berger, T.; Hone, E.; Taddei, K.; Martins, I.J.; Ghiso, J.; Buxbaum, J.D.; et al. Profiling Brain and Plasma Lipids in Human Apoe Ε2, Ε3, and Ε4 Knock—In Mice Using Electrospray Ionization Mass Spectrometry. J. Alzheimer’s Dis. 2010, 20, 105–111. [Google Scholar] [CrossRef]
- Martinsen, A.; Tejera, N.; Vauzour, D.; Harden, G.; Dick, J.; Shinde, S.; Barden, A.; Mori, T.A.; Minihane, A.M. Altered SPMs and Age-Associated Decrease in Brain DHA in APOE4 Female Mice. FASEB J. 2019, 33, 10315–10326. [Google Scholar] [CrossRef]
- Fitz, N.F.; Wolfe, C.M.; Playso, B.E.; Biedrzycki, R.J.; Lu, Y.; Nam, K.N.; Lefterov, I.; Koldamova, R. Trem2 Deficiency Differentially Affects Phenotype and Transcriptome of Human APOE3 and APOE4 Mice. Mol. Neurodegener. 2020, 15, 1–21. [Google Scholar] [CrossRef] [PubMed]
- FOLCH, J.; LEES, M.; SLOANE STANLEY, G.H. A Simple Method for the Isolation and Purification of Total Lipides from Animal Tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [CrossRef]
- Boussicault, L.; Kacher, R.; Lamazière, A.; Vanhoutte, P.; Caboche, J.; Betuing, S.; Potier, M.C. CYP46A1 Protects against NMDA-Mediated Excitotoxicity in Huntington’s Disease: Analysis of Lipid Raft Content. Biochimie 2018, 153, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Michaud, L.N.; McCoy, K.F. Empirical Derivation of a Sequence of User Stereotypes for Language Learning. User Model. User-Adapt. Interact. 2004, 14, 317–350. [Google Scholar] [CrossRef]
- Brion, J.P.; Trempdagger, G.; Octavedagger, J.N. Transgenic Expression of the Shortest Human Tau Affects Its Compartmentalization and Its Phosphorylation as in the Pretangle Stage of Alzheimer’s Disease. Am. J. Pathol. 1999, 154, 255–270. [Google Scholar] [CrossRef]
- Mann, K.M.; Thorngate, F.E.; Katoh-Fukui, Y.; Hamanaka, H.; Williams, D.L.; Fujita, S.; Lamb, B.T. Independent Effects of APOE on Cholesterol Metabolism and Brain Aβ Levels in an Alzheimer Disease Mouse Model. Hum. Mol. Genet. 2004, 13, 1959–1968. [Google Scholar] [CrossRef]
- Fourrier, C.; Remus-Borel, J.; Greenhalgh, A.D.; Guichardant, M.; Bernoud-Hubac, N.; Lagarde, M.; Joffre, C.; Layé, S. Docosahexaenoic Acid-Containing Choline Phospholipid Modulates LPS-Induced Neuroinflammation in Vivo and in Microglia in Vitro. J. Neuroinflamm. 2017, 14, 170. [Google Scholar] [CrossRef]
- Hooijmans, C.R.; Pasker-De Jong, P.C.M.; de Vries, R.B.M.; Ritskes-Hoitinga, M. The Effects of Long-Term Omega-3 Fatty Acid Supplementation on Cognition and Alzheimer’s Pathology in Animal Models of Alzheimer’s Disease: A Systematic Review and Meta-Analysis. J. Alzheimer’s Dis. 2012, 28, 191–209. [Google Scholar] [CrossRef]
- lo Van, A.; Sakayori, N.; Hachem, M.; Belkouch, M.; Picq, M.; Fourmaux, B.; Lagarde, M.; Osumi, N.; Bernoud-Hubac, N. Targeting the Brain with a Neuroprotective Omega-3 Fatty Acid to Enhance Neurogenesis in Hypoxic Condition in Culture. Mol. Neurobiol. 2019, 56, 986–999. [Google Scholar] [CrossRef]
- Balbo, I.; Montarolo, F.; Boda, E.; Tempia, F.; Hoxha, E. Elovl5 Expression in the Central Nervous System of the Adult Mouse. Front. Neuroanat. 2021, 15, 669073. [Google Scholar] [CrossRef]
- Guillou, H.; Zadravec, D.; Martin, P.G.P.; Jacobsson, A. The Key Roles of Elongases and Desaturases in Mammalian Fatty Acid Metabolism: Insights from Transgenic Mice. Prog. Lipid Res. 2010, 49, 186–199. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.C.; Zhao, N.; Fu, Y.; Wang, N.; Linares, C.; Tsai, C.W.; Bu, G. ApoE4 Accelerates Early Seeding of Amyloid Pathology. Neuron 2017, 96, 1024–1032. [Google Scholar] [CrossRef]
- Sims, R.; Hill, M.; Williams, J. The Multiplex Model of the Genetics of Alzheimer’s Disease. Nat. Neurosci. 2020, 23, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Genin, E.; Hannequin, D.; Wallon, D.; Sleegers, K.; Hiltunen, M.; Combarros, O.; Bullido, M.J.; Engelborghs, S.; de Deyn, P.; Berr, C.; et al. APOE and Alzheimer Disease: A Major Gene with Semi-Dominant Inheritance. Mol. Psychiatry 2011, 16, 903–907. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y. Roles of Apolipoprotein E4 (ApoE4) in the Pathogenesis of Alzheimer’s Disease: Lessons from ApoE Mouse Models. Biochem. Soc. Trans. 2011, 39, 924–932. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Holtzman, D.M.; Bales, K.R.; Tenkova, T.; Fagan, A.M.; Parsadanian, M.; Sartorius, L.J.; Mackey, B.; Olney, J.; McKeel, D.; Wozniak, D.; et al. Apolipoprotein E Isoform-Dependent Amyloid Deposition and Neuritic Degeneration in a Mouse Model of Alzheimer’s Disease. Proc. Natl. Acad. Sci. USA 2000, 97, 2892–2897. [Google Scholar] [CrossRef]
- Lee, S.I.; Jeong, W.; Lim, H.; Cho, S.; Lee, H.; Jang, Y.; Cho, J.; Bae, S.; Lin, Y.T.; Tsai, L.H.; et al. APOE4-Carrying Human Astrocytes Oversupply Cholesterol to Promote Neuronal Lipid Raft Expansion and Aβ Generation. Stem Cell Rep. 2021, 16, 2128–2137. [Google Scholar] [CrossRef]
- Genaro-Mattos, T.C.; Anderson, A.; Allen, L.B.; Korade, Z.; Mirnics, K. Cholesterol Biosynthesis and Uptake in Developing Neurons. ACS Chem. Neurosci. 2019, 10, 3671–3681. [Google Scholar] [CrossRef]
- Mitsche, M.A.; McDonald, J.G.; Hobbs, H.H.; Cohen, J.C. Flux Analysis of Cholesterol Biosynthesis in Vivo Reveals Multiple Tissue and Cell-Type Specific Pathways. Elife 2015, 4, e07999. [Google Scholar] [CrossRef]
- Kölsch, H.; Heun, R.; Jessen, F.; Popp, J.; Hentschel, F.; Maier, W.; Lütjohann, D. Alterations of Cholesterol Precursor Levels in Alzheimer’s Disease. Biochim. Et Biophys. Acta Mol. Cell Biol. Lipids 2010, 1801, 945–950. [Google Scholar] [CrossRef]
- Wisniewski, T.; Newman, K.; Javitt, N.B. Alzheimer’s Disease: Brain Desmosterol Levels. J. Alzheimer’s Dis. 2013, 33, 881–888. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein e and Alzheimer Disease: Risk, Mechanisms and Therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef]
- Lütjohann, D.; Papassotiropoulos, A.; Björkhem, I.; Locatelli, S.; Bagli, M.; Oehring, R.D.; Schlegel, U.; Jessen, F.; Rao, M.L.; von Bergmann, K.; et al. Plasma 24S-Hydroxycholesterol (Cerebrosterol) Is Increased in Alzheimer and Vascular Demented Patients. J. Lipid Res. 2000, 41, 195–198. [Google Scholar] [CrossRef]
- Varma, V.R.; Büşra Lüleci, H.; Oommen, A.M.; Varma, S.; Blackshear, C.T.; Griswold, M.E.; An, Y.; Roberts, J.A.; O’Brien, R.; Pletnikova, O.; et al. Abnormal Brain Cholesterol Homeostasis in Alzheimer’s Disease—A Targeted Metabolomic and Transcriptomic Study. Npj Aging Mech. Dis. 2021, 7, 11. [Google Scholar] [CrossRef] [PubMed]
- Pikuleva, I.A.; Cartier, N. Cholesterol Hydroxylating Cytochrome P450 46A1: From Mechanisms of Action to Clinical Applications. Front. Aging Neurosci. 2021, 13, 696778. [Google Scholar] [CrossRef] [PubMed]
- Lund, E.G.; Xie, C.; Kotti, T.; Turley, S.D.; Dietschy, J.M.; Russell, D.W. Knockout of the Cholesterol 24-Hydroxylase Gene in Mice Reveals a Brain-Specific Mechanism of Cholesterol Turnover. J. Biol. Chem. 2003, 278, 22980–22988. [Google Scholar] [CrossRef]
- Gibellini, F.; Smith, T.K. The Kennedy Pathway-de Novo Synthesis of Phosphatidylethanolamine and Phosphatidylcholine. IUBMB Life 2010, 62, 414–428. [Google Scholar] [CrossRef]
- Ross, B.M.; Moszczynska, A.; Erlich, J.; Kish, S.J. Phospholipid-Metabolizing Enzymes in Alzheimer’s Disease: Increased Lysophospholipid Acyltransferase Activity and Decreased Phospholipase A2 Activity. J. Neurochem. 1998, 70, 786–793. [Google Scholar] [CrossRef]
- Ojo, J.O.; Algamal, M.; Leary, P.; Abdullah, L.; Mouzon, B.; Evans, J.E.; Mullan, M.; Crawford, F. Converging and Differential Brain Phospholipid Dysregulation in the Pathogenesis of Repetitive Mild Traumatic Brain Injury and Alzheimer’s Disease. Front. Neurosci. 2019, 13, 103. [Google Scholar] [CrossRef]
- Chouinard-Watkins, R.; Plourde, M. Fatty Acid Metabolism in Carriers of Apolipoprotein e Epsilon 4 Allele: Is It Contributing to Higher Risk of Cognitive Decline and Coronary Heart Disease? Nutrients 2014, 6, 4452–4471. [Google Scholar] [CrossRef]
- Pararasa, C.; Ikwuobe, J.; Shigdar, S.; Boukouvalas, A.; Nabney, I.T.; Brown, J.E.; Devitt, A.; Bailey, C.J.; Bennett, S.J.; Griffiths, H.R. Age-Associated Changes in Long-Chain Fatty Acid Profile during Healthy Aging Promote pro-Inflammatory Monocyte Polarization via PPARγ. Aging Cell 2016, 15, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Harayama, T.; Shimizu, T. Roles of Polyunsaturated Fatty Acids, from Mediators to Membranes. J. Lipid Res. 2020, 61, 1150–1160. [Google Scholar] [CrossRef] [PubMed]
- Ollila, S.; Hyvönen, M.T.; Vattulainen, I. Polyunsaturation in Lipid Membranes: Dynamic Properties and Lateral Pressure Profiles. J. Phys. Chem. B 2007, 111, 3139–3150. [Google Scholar] [CrossRef]
- Schley, P.D.; Brindley, D.N.; Field, C.J. (N-3) PUFA Alter Raft Lipid Composition and Decrease Epidermal Growth Factor Receptor Levels in Lipid Rafts of Human Breast Cancer Cells. J. Nutr. 2007, 137, 548–553. [Google Scholar] [CrossRef]
- Shaikh, S.R.; Rockett, B.D.; Salameh, M.; Carraway, K. Docosahexaenoic Acid Modifies the Clustering and Size of Lipid Rafts and the Lateral Organization and Surface Expression of MHC Class I of EL4 Cells. J. Nutr. 2009, 139, 1632–1639. [Google Scholar] [CrossRef] [PubMed]
- Ridgway, N.D.; McLeod, R.S. Biochemistry of Lipids, Lipoproteins and Membranes, 6th ed.; Elsevier science: Amsterdam, The Netherlands, 2015; eBook; ISBN 9780444634498. [Google Scholar]
- Prasinou, P.; Dafnis, I.; Giacometti, G.; Ferreri, C.; Chroni, A.; Chatgilialoglu, C. Fatty Acid-Based Lipidomics and Membrane Remodeling Induced by ApoE3 and ApoE4 in Human Neuroblastoma Cells. Biochim. Et Biophys. Acta Biomembr. 2017, 1859, 1967–1973. [Google Scholar] [CrossRef]
- López, G.H.; Ilincheta de Boschero, M.G.; Castagnet, P.I.; Giusto, N.M. Age-Associated Changes in the Content and Fatty Acid Composition of Brain Glycerophospholipids. Comp. Biochem. Physiol. Part B Biochem. 1995, 112, 331–343. [Google Scholar] [CrossRef]
- Delgado, G.E.; Krämer, B.K.; Lorkowski, S.; März, W.; von Schacky, C.; Kleber, M.E. Individual Omega-9 Monounsaturated Fatty Acids and Mortality—The Ludwigshafen Risk and Cardiovascular Health Study. J. Clin. Lipidol. 2017, 11, 126–135.e5. [Google Scholar] [CrossRef]
- Astarita, G.; Jung, K.M.; Vasilevko, V.; DiPatrizio, N.V.; Martin, S.K.; Cribbs, D.H.; Head, E.; Cotman, C.W.; Piomelli, D. Elevated Stearoyl-CoA Desaturase in Brains of Patients with Alzheimer’s Disease. PLoS ONE 2011, 6, e24777. [Google Scholar] [CrossRef]
- Kihara, A. Very Long-Chain Fatty Acids: Elongation, Physiology and Related Disorders. J. Biochem. 2012, 152, 387–395. [Google Scholar] [CrossRef]
- Yamada, K. Extracellular Tau and Its Potential Role in the Propagation of Tau Pathology. Front. Neurosci. 2017, 11, 667. [Google Scholar] [CrossRef] [PubMed]
- Laird, F.M.; Cai, H.; Savonenko, A.V.; Farah, M.H.; He, K.; Melnikova, T.; Wen, H.; Chiang, H.C.; Xu, G.; Koliatsos, V.E.; et al. BACE1, a Major Determinant of Selective Vulnerability of the Brain to Amyloid-β Amyloidogenesis, Is Essential for Cognitive, Emotional, and Synaptic Functions. J. Neurosci. 2005, 25, 11693–11709. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.T.; Seo, J.; Gao, F.; Feldman, H.M.; Wen, H.L.; Penney, J.; Cam, H.P.; Gjoneska, E.; Raja, W.K.; Cheng, J.; et al. APOE4 Causes Widespread Molecular and Cellular Alterations Associated with Alzheimer’s Disease Phenotypes in Human IPSC-Derived Brain Cell Types. Neuron 2018, 98, 1141–1154.e7. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, M.; Holm, M.L.; Liu, C.C.; Shinohara, M.; Aikawa, T.; Oue, H.; Yamazaki, Y.; Martens, Y.A.; Murray, M.E.; Sullivan, P.M.; et al. APOE4-Mediated Amyloid-β Pathology Depends on Its Neuronal Receptor LRP1. J. Clin. Investig. 2019, 129, 1272–1277. [Google Scholar] [CrossRef]
- Vincent, B.; Smith, J.D. Astrocytes Down-Regulate Neuronal β-Amyloid Precursor Protein Expression and Modify Its Processing in an Apolipoprotein E Isoform-Specific Manner. Eur. J. Neurosci. 2001, 14, 256–266. [Google Scholar] [CrossRef]
- Ye, S.; Huang, Y.; Mü llendorff, K.; Dong, L.; Giedt, G.; Meng, E.C.; Cohen, F.E.; Kuntz, I.D.; Weisgraber, K.H.; Mahley, R.W. Apolipoprotein (Apo) E4 Enhances Amyloid Peptide Production in Cultured Neuronal Cells: ApoE Structure as a Potential Therapeutic Target. Proc. Natl. Acad. Sci. USA 2005, 102, 18700–18705. [Google Scholar] [CrossRef]
- Fan, Q.W.; Yu, W.; Senda, T.; Yanagisawa, K.; Michikawa, M. Cholesterol-Dependent Modulation of Tau Phosphorylation in Cultured Neurons. J. Neurochem. 2001, 76, 391–400. [Google Scholar] [CrossRef]
- Díaz, M.; Fabelo, N.; Martín, V.; Ferrer, I.; Gómez, T.; Marín, R. Biophysical Alterations in Lipid Rafts from Human Cerebral Cortex Associate with Increased BACE1/AβPP Interaction in Early Stages of Alzheimer’s Disease. J. Alzheimer’s Dis. 2014, 43, 1185–1198. [Google Scholar] [CrossRef]
- Martín, V.; Fabelo, N.; Santpere, G.; Puig, B.; Marín, R.; Ferrer, I.; Díaz, M. Lipid Alterations in Lipid Rafts from Alzheimer’s Disease Human Brain Cortex. J. Alzheimer’s Dis. 2010, 19, 489–502. [Google Scholar] [CrossRef]
- Stillwell, W.; Shaikh, S.R.; Zerouga, M.; Siddiqui, R.; Wassal, S.R. Docosahexaenoic Acid Affects Cell Signaling by Altering Lipid Rafts. Reprod. Nutr. Dev. 2005, 45, 559–579. [Google Scholar] [CrossRef]
- Marquer, C.; Devauges, V.; Cossec, J.C.; Liot, G.; Lécart, S.; Saudou, F.; Duyckaerts, C.; Lévêque-Fort, S.; Potier, M.C. Local Cholesterol Increase Triggers Amyloid Precursor Protein-Bace1 Clustering in Lipid Rafts and Rapid Endocytosis. FASEB J. 2011, 25, 1295–1305. [Google Scholar] [CrossRef] [PubMed]
- Kalvodova, L.; Kahya, N.; Schwille, P.; Ehehalt, R.; Verkade, P.; Drechsel, D.; Simons, K. Lipids as Modulators of Proteolytic Activity of BACE: Involvement of Cholesterol, Glycosphingolipids, and Anionic Phospholipids in Vitro. J. Biol. Chem. 2005, 280, 36815–36823. [Google Scholar] [CrossRef] [PubMed]
- Lebbadi, M.; Julien, C.; Phivilay, A.; Tremblay, C.; Emond, V.; Kang, J.X.; Calon, F. Endogenous Conversion of Omega-6 into Omega-3 Fatty Acids Improves Neuropathology in an Animal Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2011, 27, 853–869. [Google Scholar] [CrossRef] [PubMed]
- Kawarabayashi, T.; Shoji, M.; Younkin, L.H.; Wen-Lang, L.; Dickson, D.W.; Murakami, T.; Matsubara, E.; Abe, K.; Ashe, K.H.; Younkin, S.G. Dimeric Amyloid β Protein Rapidly Accumulates in Lipid Rafts Followed by Apolipoprotein E and Phosphorylated Tau Accumulation in the Tg2576 Mouse Model of Alzheimer’s Disease. J. Neurosci. 2004, 24, 3801–3809. [Google Scholar] [CrossRef] [PubMed]
- Basil, M.C.; Levy, B.D. Specialized Pro-Resolving Mediators: Endogenous Regulators of Infection and Inflammation. Nat. Rev. Immunol. 2016, 16, 51–67. [Google Scholar] [CrossRef] [PubMed]
- Devassy, J.G.; Leng, S.; Gabbs, M.; Monirujjaman, M.; Aukema, H.M. Omega-3 Polyunsaturated Fatty Acids and Oxylipins in Neuroinflammation and Management of Alzheimer Disease. Adv. Nutr. 2016, 7, 905–916. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, L.; Conde-Knape, K.; Beher, D.; Shearman, M.S.; Shachter, N.S. Fatty Acids Increase Presenilin-1 Levels and γ-Secretase Activity in PSwt-1 Cells. J. Lipid Res. 2004, 45, 2368–2376. [Google Scholar] [CrossRef]
- Barracchia, C.G.; Tira, R.; Parolini, F.; Munari, F.; Bubacco, L.; Spyroulias, G.A.; D’Onofrio, M.; Assfalg, M. Unsaturated Fatty Acid-Induced Conformational Transitions and Aggregation of the Repeat Domain of Tau. Molecules 2020, 25, 2716. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lazar, A.-N.; Hanbouch, L.; Boussicaut, L.; Fourmaux, B.; Daira, P.; Millan, M.J.; Bernoud-Hubac, N.; Potier, M.-C. Lipid Dys-Homeostasis Contributes to APOE4-Associated AD Pathology. Cells 2022, 11, 3616. https://doi.org/10.3390/cells11223616
Lazar A-N, Hanbouch L, Boussicaut L, Fourmaux B, Daira P, Millan MJ, Bernoud-Hubac N, Potier M-C. Lipid Dys-Homeostasis Contributes to APOE4-Associated AD Pathology. Cells. 2022; 11(22):3616. https://doi.org/10.3390/cells11223616
Chicago/Turabian StyleLazar, Adina-Nicoleta, Linda Hanbouch, Lydie Boussicaut, Baptiste Fourmaux, Patricia Daira, Mark J. Millan, Nathalie Bernoud-Hubac, and Marie-Claude Potier. 2022. "Lipid Dys-Homeostasis Contributes to APOE4-Associated AD Pathology" Cells 11, no. 22: 3616. https://doi.org/10.3390/cells11223616
APA StyleLazar, A.-N., Hanbouch, L., Boussicaut, L., Fourmaux, B., Daira, P., Millan, M. J., Bernoud-Hubac, N., & Potier, M.-C. (2022). Lipid Dys-Homeostasis Contributes to APOE4-Associated AD Pathology. Cells, 11(22), 3616. https://doi.org/10.3390/cells11223616