Abstract
Genome editing tools based on CRISPR–Cas systems can repair genetic mutations in situ; however, off-target effects and DNA damage lesions that result from genome editing remain major roadblocks to its full clinical implementation. Protein and chemical inhibitors of CRISPR–Cas systems may reduce off-target effects and DNA damage. Here we describe the identification of several lead chemical inhibitors that could specifically inhibit the activity of Streptococcus pyogenes Cas9 (SpCas9). In addition, we obtained derivatives of lead inhibitors that could penetrate the cell membrane and inhibit SpCas9 in cellulo. Two of these compounds, SP2 and SP24, were able to improve the specificity of SpCas9 in cellulo at low-micromolar concentration. Furthermore, microscale thermophoresis (MST) assays showed that SP24 might inhibit SpCas9 activity by interacting with both the SpCas9 protein and the SpCas9–gRNA ribonucleoprotein complex. Taken together, SP24 is a novel chemical inhibitor of SpCas9 which has the potential to enhance therapies that utilize SpCas9.
1. Introduction
Most archaea and 50% of bacteria harbor CRISPR–Cas loci in their genomes [1,2,3,4,5,6,7]. Guided by the CRISPR RNA (crRNA), Cas proteins cleave exogenous DNA/RNA molecules and act as part of the organisms’ adaptive immune system [8,9,10]. The discovery of CRISPR–Cas systems and the subsequent development of genome editing tools have revolutionized genome targeting and manipulation techniques. During genome editing, cleavage by Cas nucleases results in double-stranded DNA breaks (DSBs) [11], which may be repaired through non-homologous end joining (NHEJ) or homology-directed repair (HDR) pathways [12,13,14]. Consequently, off-target cleavage and DNA damage lesions often result from CRISPR–Cas-mediated genome editing and pose significant challenges to its clinical applications. In addition, due to the high activity of the Cas protein, chromosomal translocations and genotoxicity were observed [15]. The Cas protein elicits an immune response [16], and activates the p53 signaling pathway, with a potential cancer risk [17]. Therefore, it is necessary to develop Cas nuclease inhibitors to control the activity of Cas9, thereby improving the safety of CRISPR-based gene therapy. Methods and strategies that help overcome such problems have become areas of intensive research.
About 50 different anti-CRISPR (Acr) proteins have been identified to date, which inhibit different CRISPR–Cas systems [18,19,20] and can be found in the phage genome [21]. Acr proteins inhibit CRISPR-Cas binding or cleavage of target DNA [22]. Despite their effectiveness, Acr proteins may be less than ideal for clinical applications because of their immunogenicity [18,22], slower kinetics, and difficulties in traversing the cell membrane [23]. In addition, large-scale production and storage of Acr proteins can be costly as well. Experimentally, numerous strategies have been devised to increase the specificity and reduce the off-target effects of Cas nucleases. For example, for the most widely studied type II Cas9 nuclease, light-inducible [24,25,26,27] and drug-inducible split-Cas9 systems [28,29,30] were developed to modulate the duration of Cas9 activity; however, the induction kinetics, needed for special instruments, and background activities of such systems have limited their clinical use [31,32]. Small-molecule inhibitors of Cas proteins have also been reported. For example, BRD0539 can inhibit Streptococcus pyogenes Cas9 (SpCas9) by blocking its binding to DNA [33], although its IC50 is quite high (22 μM). To date, how BRD0539 acts to modulate SpCas9 off-target effects remains unknown [33]. In addition, given the differences in affinity and mode of action of Cas proteins in targeting DNA [34], small-molecule inhibitors that can interact with and/or target Cas9 as well as other Cas proteins in different settings are sorely needed. In vitro and in cellulo methods to screen and evaluate Cas9 inhibitors are still limited.
In this study, we aimed to identify more specific and high-affinity small-molecule inhibitors of SpCas9. Furthermore, a series of assays were utilized, including in vitro cleavage, binding affinity, and cell-based fluorescence screening assays, to better evaluate and study the inhibitors. Through these approaches, we pinpointed several types of lead compound scaffolds that could inhibit SpCas9 in cellulo. Two of the derivatives of these lead compounds could effectively reduce SpCas9 off-target activity, thus improving the specificity of SpCas9. We further showed the interaction mechanisms between the inhibitor-derivative compounds and the SpCas9–gRNA complex, which should greatly benefit further explorations of modulating Cas9 activity and specificity.
2. Materials and Methods
2.1. Cell Culture
Parental and derived HEK293T cells and iCas9 U2OS cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM, Corning, New York, NY, USA) containing 10% dialyzed fetal bovine serum (FBS, Biological Industries, Beit HaEmek, Israel) in a humid environment containing 5% CO2 at 37 °C.
2.2. SpCas9 and dSpCas9 Expression and Purification
The expression and purification of SpCas9 were done as previously described [35]. BL21 Star (DE3)-competent E. coli cells (Thermo Fisher, Waltham, MA, USA) were transformed with pET28a-SpCas9 (His6-TEV-NLS tag at N-terminal and the other one NLS tag at C-terminal). A single clone was cultured in LB broth containing 50 μg/mL kanamycin at 37 °C overnight, diluted into 1 L of LB medium with a dilution ratio of 1:100, and cultured at 37 °C until the OD600 reached 0.5–0.6. The cells were cooled at 18 °C for 0.5 h and then isopropyl β-D-thiogalactoside (IPTG, Sigma-Aldrich, St. Louis, MO, USA) was added to a final concentration of 0.5 mM. The cells were continued to culture at 18 °C for 14–16 h. The following operations were carried out on the ice or at 4 °C. The cells were centrifuged and resuspended in 10 mL of binding buffer (50 mM Tris-HCl, pH 8.0, 500 mM NaCl, 5% glycerol, and 20 mM imidazole; Sigma-Aldrich) with 50 U benzonase (Sigma-Aldrich, St. Louis, MO, USA). Cells were lysed by sonication and centrifuged at 10,000× g for 20 min. The supernatant was filtered with a 0.22 μm PVDF membrane (Millipore, Billerica, MA, USA). The filtered supernatant was loaded into a column with Ni-NTA affinity chromatography column (GE Healthcare, Torrington, CT, USA) and then washed with 20 column volumes of binding buffer. The His-tagged protein was eluted with 10 mL of elution buffer (50 mM Tris-HCl, pH 8.0, 500 mM NaCl, 5% glycerol, and 300 mM imidazole). The eluent was concentrated to approximately 300 μL with Amicon ultra-4 centrifugal filters-30K (Merck Millipore, Billerica, MA, USA) by centrifugation at 4000× g and was further purified with a gel-filtration chromatography (HiLoad 10/300 Superdex200, GE Healthcare, Torrington, CT, USA). The protein was eluted with SEC buffer (50 mM Tris-HCl, pH 8.0, 500 mM NaCl, and 5% glycerol) and concentrated. Finally, the protein was filtered with a 0.22 μm PVDF membrane and stored at 4 °C.
2.3. Prokaryotic Expression and Purification of LbCas12a
The pET28a-6xHis-TEV-LbCas12a-NLS-3xHA plasmid was transformed into the Rosetta (DE3) E. coli strain and protein was induced with 500 μM IPTG at 18 °C for 18 h. Cells were collected and lysed, followed by Ni-NTA affinity chromatography column (GE Healthcare) and Superdex 200 10/300 column (GE Healthcare). The purified proteins were concentrated with Amicon ultra-4 centrifugal filters-50K (Merck Millipore) and quantitated with BCA protein assay Kit (Thermo Fisher Scientific). The proteins were stored at −80 °C.
2.4. In Vitro Transcription of crRNA and gRNA
The SpCas9 gRNA and LbCas12a crRNA were in vitro transcribed from linear DNA templates using the MEGAshortscript high-yield transcription kit (Invitrogen, Carlsbad, CA, USA) at 37 °C for 4 h. The linearized DNA templates were generated by oligo annealing (IGE Biotechnology Ltd., Guangzhou, China) (Supplementary Table S3). The transcribed RNA was purified with the RNeasy protect mini kit (QIAGEN, Hiden, Germany) and quantified by the Nanodrop 1000 spectrophotometer.
2.5. SpCas9 In Vitro Cleavage Assays
The assay was carried out in NEBuffer 3 (New England Biolabs, Ipswich, MA, USA). First, 19.2 μL of the SpCas9–gRNA (58.8 nM) mixture was incubated with 2.4 μL of various compounds (dissolved in DMSO) at desired concentrations (0.1–2 mM) at room temperature for 10 min. Then, 2.4 μL of 3.48 nM ApaI-linearized px458 plasmid was added for further incubation at 37 °C for 60 min. The reaction was terminated by adding 0.5 μL Proteinase K (20 mg/mL) (Invitrogen) and incubation at 55 °C for 15 min. The reaction products were resolved on a 0.7% TAE agarose gel, stained with Gel Red (Biotium, Fremont, CA, USA). Then the products were visualized by Chemidoc XRS plus system (Bio-Rad, Hercules, CA, USA), and band intensities were quantified using ImageJ software (National Institutes of Health developed; Bethesda, MD, USA). The inhibition efficiency in the cleavage assays was calculated using the following equation:
where E is the inhibition efficiency, and C1 and C2 are the cleavage rate of the DMSO group and compound treated group, respectively.
E = (C1 − C2)/C1 × 100%,
The cleavage rate was calculated using the following equation:
where C is the cleavage rate, B0, B1, and B2 are the band intensities of uncleaved products, upper cleaved products, and lower cleaved products, respectively.
C = (B1 + B2)/(B0 + B1 + B2) × 100%,
2.6. LbCas12a In Vitro Cleavage Assays
In vitro cleavage experiments were performed using purified recombinant LbCas12a proteins similar to SpCas9 cleavage assays. LbCas12a proteins (50 nM), crRNA (50 nM), and different concentrations of the inhibitor compounds were mixed in NEBuffer 2 (New England Biolabs). After incubation at room temperature for 10 min, ScaI-linearized pUC19 DNA was added to a final concentration of 1.52 nM. The solution was vortex mixed and centrifuged, then incubated for 45 min in a water bath at 37 °C. The reaction was quenched with 0.5 μL proteinase K for 15 min at 56 °C. The samples were mixed with DNA loading buffers and resolved by 1.2% TAE agarose gel electrophoresis. Gel bands were visualized by Chemidoc XRS plus system (Bio-Rad, Hercules, CA, USA).
2.7. NotI/XbaI In Vitro Cleavage Assays
In vitro cleavage experiments used commercial restriction enzymes (Thermo Fisher Scientific). The restriction enzyme NotI or XbaI (0.1 μL) was mixed with the compounds in a FastDigest buffer (Thermo Fisher Scientific). It was then incubated at room temperature for 10 min, and an ApaI-linearized px458 plasmid was added and reacted at 37 °C for 15 min. The samples were mixed with a DNA loading buffer. The cleavage products were electrophoresed on 1% TAE agarose gel and imaged using a Bio-Rad gel imager.
2.8. Establishing the EGFP/iCas9/gRNA-EGFP Reporter 293T Cells
The H2AX-EGFP 293T cell line was first generated using the previously reported MMEJ-mediated CRIS-PITCh system [36,37]. Briefly, we cloned two gRNAs (Supplementary Table S3) that respectively target H2AX gene and the PITCh vector into the px330 plasmid (Addgene, #42230). The PITCh vector contains a pair of 20-bp homologous sequences targeting the upstream and downstream regions of the H2AX gRNA cleavage site, and the SFB-EGFP expression cassette. The three plasmids were co-transfected into 293T cells at an equimolar ratio. GFP-positive cells were sorted by FACS and individually expanded to isolate single clones for genomic DNA extraction and PCR verification of successful knock-in (KI) of the SFB-EGFP cassette into 5′-end of first exon of the H2AX gene. The selected H2AX-EGFP 293T cell clone was subsequently infected with lentiviruses encoding tetracycline-inducible SpCas9 (iCas9). After drug selection, individual clones were again expanded and verified for inducible Cas9 expression. The selected clone was subsequently infected with lentivirus encoding a gRNA that targets EGFP. Puromycin-resistant clones (1 μg/μL, Thermo Fisher Scientific) were then isolated after >7 days for further analysis.
2.9. Lentivirus Production
Lentivirus is produced by 293T cells. Specifically, 293T cells were seeded in a 6 cm dish (Thermo Fisher Scientific) before transfection. About 24 h later, the target vector, psPAX2 (Addgene, #12260) and pMD2.G (Addgene, #12259) were transfected together at a ratio of 4:3:1. The virus supernatants were collected 48 h and 72 h after the transient and filtered with a 0.45 μm membrane (Millipore) for usage.
2.10. Inhibition of SpCas9 Nuclease Activity in Reporter 293T Cells
The reporter 293T cells were seeded into 96-well plates at a density of 10,000 cells per well and cultured overnight before the addition of DMSO alone or various compounds (in DMSO solution) at appropriate concentrations (0.125–100 μM) with or without 2 μg/mL doxycycline for an additional 72 h of incubation. The cells were then harvested for analysis with the CytoFLEX flow cytometer (Beckman Coulter, Brea, CA, USA) or analyzed by western blot with antibodies as indicated. The results were plotted in the GraphPad Prism 6.0 software or Origin 2020b (OriginLab, Northampton, MA, USA). The inhibiting efficiency in reporter 293T cells was calculated using the following equation:
where E’ is the inhibiting efficiency in reporter 293T cells, and P0, P1, and P2 are the GFP positive cell ratio of the uninduced group, Dox and DMSO treated control group, and Dox and compound treated group, respectively.
E’ = (P2 − P1)/(P0 − P1) × 100%,
2.11. High-Throughput Sequencing and Data Analysis
The iCas9-expressing U2OS cells were first through lentiviral infection. The cells were cultured with 1 μg/mL puromycin until all cells in the negative control group were dead. Monoclonal cells were then isolated and verified. Lentiviruses encoding appropriate gRNAs were used to infect the iCas9 U2OS cells, which were further selected with 10 μg/mL Blasticidin S HCL (Gibco, New York, NY, USA) until all cells in the control group were dead. The iCas9/gRNA cells were then plated in 12-well plates (Thermo Fisher Scientific) at ~40% confluence per well at 24 h before the doxycycline (2 μg/mL) addition. An individual inhibitor compound was also added at this point at the appropriate concentrations. DMSO alone served as the control. Cells cultured without doxycycline served as the SpCas9 cleavage control. After 24 h, cells were collected for genomic DNA extraction using the AxyPrep blood genomic DNA miniprep kit (Axygen, New York, NY, USA). Sequences surrounding the on- and off-target sites were amplified using barcoded primers (Supplementary Table S3) and the KOD PCR kit (Toyobo, Japan) to generate the sequencing libraries. Single-read sequencing of the pooled samples were performed on the 150 Hiseq 2000 (Illumina, San Diego, CA, USA). Each experiment was repeated twice. MATLAB was used to split the sequencing file and CRISPResso2 [38] was used to analyze the frequency of indels.
2.12. Cytotoxicity Assays
HEK293T reporter cells were plated in 96-well plates (4–6 × 103 cells/well) and cultured for 12 h before the following experiments. The medium was replaced with 100 μL fresh medium containing compounds or DMSO. The medium change was performed 72 h afterward and cytotoxicity was measured by the cell-counting kit-8 (CCK-8, Dojindo Laboratories, Japan) reagent following the manufacturer’s instructions. Graphs were created by GraphPad Prism 6.0 software (San Diego, CA, USA).
2.13. Fluorescence Polarization (FP) Analysis
The fluorescence polarization experiment was as previously reported [33]. DNA molecules (sequences are listed in Supplementary Table S3) were synthesized, one oligonucleotide contained 94 bases (3′ end is PAM (NGG) sequence). The DNA was labeled at the 3′ end with fluorescein isothiocyanate (FITC). The other one is complementary to the first 58 bases of the previous oligonucleotide. These two oligonucleotides were annealed to generate the target DNA substrate labeled with fluorescent molecules (FITC-DNA). Unlabeled DNA (PAM-DNA) was considered competitive positive controls. Experiments were performed on black 384-well plates (Corning, New York, NY, USA). 50 nM SpCas9-gRNA (1:1) and different concentrations of the compound were added to the reaction buffer (50 mM Tris-HCl, pH 8.0, 0.1 M NaCl, 10 mM MgCl2, and 1 mM DTT). PAM-DNA (1250 nM) was used as a positive control. The mixture was then incubated at room temperature for 30 min, and FITC-DNA was added to the final concentration of 25 nM in each well. Fluorescence polarization signals were measured using a multilabel reader (Victor X5, PerkinElmer, Waltham, MA, USA) within 10 min. These values were plotted using GraphPad Prism 6.0 software (San Diego, CA, USA).
2.14. Microscale Thermophoresis (MST) Assays
The MST method was carried out similar to the previous report [34,39]. The binding ability of the compounds to SpCas9 in different states was tested by Monolith NT.115 (NanoTemper Technologies, Munich, Germany). The SpCas9-EGFP fusion protein or the SpCas9-EGFP/gRNA RNP complex (protein: gRNA = 1:1) was diluted with a binding buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.05% Tween-20, and 10 mM MgCl2) to make a final concentration of 200 nM. The protein diluent was mixed with the compound diluent in equal volume. The mixture was kept at room temperature for 10 min. Then the sample was loaded into the NanoTemper standard capillaries. All experiments were carried out at 25 °C, 80% MST power, and 50% LED power. The data were analyzed with the NanoTemper MO affinity analysis software (NanoTemper Technologies, Munich, Germany). The raw data were then exported and plotted with Origin 2020b (OriginLab, Northampton, MA, USA).
2.15. Molecular Docking
We used Sybyl-X 2.1.1 [40] to conduct the docking study on the interaction region between SpCas9 and compounds. The 3D macromolecules of SpCas9 (PDB: 4ZT9) were obtained from the RCSB Protein Data Bank. All water and ions were removed from the crystal structure, and hydrogen atoms were added. Amber FF99 force field was used for the optimization of the whole protein. The Tripos force field and Gasteiger—Huckel charge were used to optimize the compounds before docking. The Surflex-dock module [41] was used for docking. The highest-scoring conformation was selected, and a 2D plot of virtual hits at SpCas9 binding sites was generated by the Pymol software (version 1.8.x, Schrödinger LLC, New York, NY, USA).
2.16. Statistical Analysis
The statistical analysis was carried out using GraphPad Prism 6.0 software (San Diego, CA, USA) to assess differences between the experimental groups. A two-tailed, unpaired t-test was used to calculate the p-value. Unless specified otherwise, ns means no significance, significance values are (*) p < 0.05, (**) p < 0.01, (***) p < 0.001, and (****) p < 0.0001.
2.17. Compound Synthesis and Characterization
See Appendix A for detailed descriptions.
3. Results
3.1. Identification of SpCas9 Inhibitors through Unbiased Screens
To identify new small-molecule inhibitors of SpCas9, we chose compounds in our laboratory for initial testing using in vitro cleavage assays. These compounds were first grouped based on structural similarities, and we chose compounds containing nitro or hydroxyl groups. Here, successful SpCas9 cleavage of the substrate DNA would result in the appearance of two smaller-sized products via agarose gel electrophoresis (Figure 1A and Supplementary Figure S1A). Of the compounds we tested, C4 and C12 were able to inhibit the activity of SpCas9 at 200 μM concentration with relative efficiencies of 60% and 84%, respectively (Figure 1B). Furthermore, their ability to inhibit SpCas9 was concentration-dependent because the inhibitory effects lessened as their concentrations decreased (Figure 1C). In contrast, the activities of recombinant-bacterial restriction endonucleases XbaI and NotI were not affected by either of these compounds at 200 μM concentration (Supplementary Figure S1B), indicating a specificity toward SpCas9. C4 and C12 are structurally different from each other (Supplementary Figure S1C), suggesting distinct mechanisms of inhibition. To better examine their effects, we established a reporter cell line for the evaluation of SpCas9 activities in live cells. An EGFP tag was first knocked into the 5′-end of the first exon of the H2AX gene in 293T cells using the CRIS-PITCh system [36,37] (Supplementary Figure S2A,B). Next, a tetracycline-inducible SpCas9 (iCas9) expression cassette and an EGFP-targeting gRNA were sequentially introduced into these cells to generate the EGFP/iCas9/gRNA-EGFP reporter cell line (Figure 1D). Upon doxycycline (Dox) induction of SpCas9 and subsequent cleavage of the EGFP coding sequence in the H2AX locus, the EGFP signal loss could be quantified and serve as an indicator of SpCas9 activity (Figure 1D and Supplementary Figure S2C). In the absence of Dox induction, the two compounds (100 μM) had no effects on EGFP expression in the reporter cells (Supplementary Figure S2D). When we examined cell survival using CCK-8 assays, we found no cytotoxicity in cells cultured with C12 (100 μM) while C4 appeared slightly cytotoxic (Supplementary Figure S2E). Upon induction, more EGFP+ cells could be observed in cells that were also treated with the two compounds, indicating the ability of the two compounds to inhibit SpCas9 activity in cellulo. We estimated the efficiency of SpCas9 inhibition at 44.0% and 17.1%, respectively, in this assay (Figure 1E). Overall, our data showed that the two candidate compounds could inhibit the activity of SpCas9 in vitro and in cellulo.
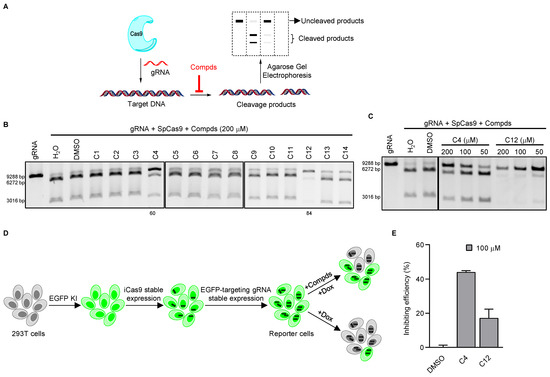
Figure 1.
Identification of small-molecule inhibitors of SpCas9 through unbiased screens. (A) In vitro Cas9 cleavage assays were carried out in the presence of different compounds. Cleavage products were then resolved by agarose gel electrophoresis. (B,C) SpCas9–gRNA (58.8 nM) mixtures containing various compounds at the indicated concentrations were incubated with a linearized plasmid (3.48 nM) for in vitro DNA cleavage assays. The number indicates the compound’s inhibition rate. (D,E) To monitor the cleavage activity of SpCas9 in live cells, we generated reporter 293T cells where EGFP was knocked into the H2AX locus, then tetracycline-inducible Cas9 (iCas9) and an EGFP-targeting gRNA were stably expressed in the cells. The reporter cells were induced with doxycycline (2 μg/mL) in the presence of various compounds (100 μM) and examined for EGFP+ cells by flow cytometry after 72 h (D). Results were quantified and plotted in (E). Error bars represent SD from two biological repeats.
3.2. Exploring the Structure–Activity Relationships of the Candidate Inhibitors
Since the inhibitory activity of C4 and C12 on SpCas9 is not strong, to enhance the activity of these two compounds, we next examined groups of structural analogs of the two compounds in cleavage assays (Supplementary Figure S3A,B). We found that multiple C4 analogs could effectively inhibit the activity of SpCas9 at a 200 μM concentration (Figure 2A). When the activities were correlated with structures (Supplementary Figure S3A), we noticed that the triazole ring in C4 analogs needed to be directly connected to the benzene ring, as in the case of C4a vs. C4b and C4v. Additionally, the inhibitory activity was lost if the chlorine group in the left benzene ring was substituted with a nitro group as in the case of C4a vs. C4m. For C12 derivatives, only C12a and C12b exhibited strong inhibitory activities at 200 μM concentration (Figure 2A). The remaining analogs either lost the activity or only weakly inhibited SpCas9 (e.g., C12c and C12d) in the assays. When the R1 position was substituted with phenyl, such as C12 to C12d, the compounds were able to retain some inhibitory activity (Figure 2D and Supplementary Figure S3B), suggesting that the benzene ring at the R1 position was critical to the activity of the C12 series of compounds.
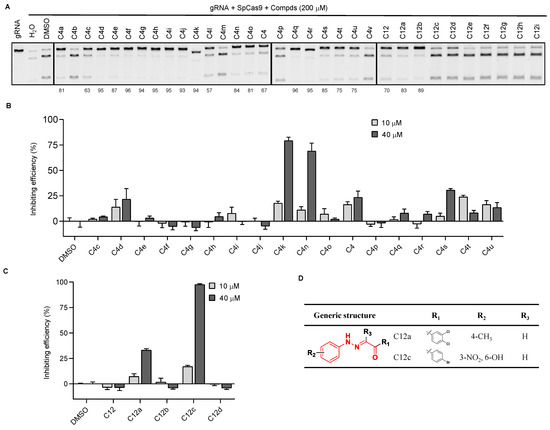
Figure 2.
Analysis of structure–activity relationships using analogs of the SpCas9 inhibitors we identified. (A) Similar in vitro DNA cleavage assays were carried out as described above using the three groups of inhibitor analogs. The number indicates the compound’s inhibition rate. (B,C) Similar in cellulo assessment of the inhibitory activities of the compound analogs was carried out as described above using the reporter 293T cells. Error bars represent SD from three biological repeats. (D) The chemical structure of C12a and C12c.
We next used the reporter cell line to evaluate the activities of the candidate small-molecule analogs in live cells at much lower concentrations (Figure 2B,C). Of the analogs tested, C4k, C4n, and C12c showed ~50% SpCas9 inhibition at 40 μM, and C12a showed ~33% inhibition. Of these four compounds, C12a and C12c did not negatively affect EGFP expression in uninduced reporter 293T cells (Supplementary Figure S3C) and became the focus of our more in-depth studies.
3.3. Compound Optimization Based on Intracellular Activities
A search of the SPECS website (https://www.specs.net/, assessed on 28 April 2019) using the parent chain of C12a and C12c (Figure 2D) yielded 800+ hits. Because the benzene ring in the R1 position is important for the inhibiting activity of the C12 series compounds, we preferentially selected compounds with phenyl at R1 position. In addition, in consideration of the diversity of compound structures, we artificially selected some compounds with carbon rings or heterocycles at R1 position. Finally, we selected 37 hits from the list (hitherto referred to as the SP series) and determined their in vitro and in cellulo activities as described above (Supplementary Figure S4 and Supplementary Table S1). At 200 μM concentration, multiple SP series compounds could block in vitro SpCas9 cleavage by >50% (Figure 3A). When certain SP compounds were further tested in our CRISPR reporter cells, SP9, SP12, and SP32 showed >50% inhibition at 40 μM concentration; however, these compounds exhibited less than 50% inhibition at 10 μM concentration (Supplementary Figure S5A). The inhibition rate of SP5, SP6, SP8, SP11, SP19, SP28, SP33, SP34, and SP36 were less than 50% at 40 μM concentration (Supplementary Figure S5A). SP2, SP14, SP15, SP24, and SP29 showed 50% inhibition at 10 μM concentration (Supplementary Figure S5A), although four of these compounds (except for SP2) could reduce EGFP expression (albeit slightly) in uninduced cells at 40 μM (Supplementary Figure S5B). Next, we tested the inhibitory activities of these five compounds at even lower concentrations. Among the five, SP15 and SP24 could both inhibit SpCas9 cleavage in cells to a similar extent at 4 and 8 μM concentrations, but only SP24 retained its level of inhibitory activity at 2 μM concentration (Figure 3B). While SP2 appeared not as effective as SP24, its activity changed little from 40 to 2 μM, likely a reflection of the extensive structural differences that exist between the two inhibitors. We calculated the EC50 of SP2 and SP24 to be 5.07 μM and 0.57 μM, respectively, through titration experiments using the reporter cells (Figure 3C,D). Neither compound appeared to affect SpCas9 expression at the concentrations tested, supporting a direct inhibition of the SpCas9–gRNA complex (Figure 3E). Importantly, in vitro LbCas12a-mediated cleavage assays found no inhibition of LbCas12a by either compound at 100 μM concentration (Supplementary Figure S6), indicating that their activities were specific for SpCas9.
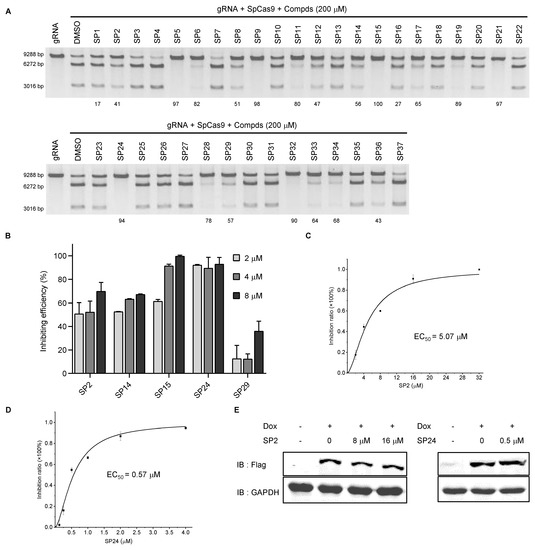
Figure 3.
Analysis of in vitro and in cellulo activities of SP series compounds. (A,B) The SP series compounds were examined in vitro DNA cleavage assays (A) and reporter cells (B) as above. The number indicates the compound’s inhibition rate. (C,D) Reporter cells were cultured in Dox (2 μg/mL) plus DMSO or SP2/SP24 at the indicated concentrations for 72 h before flow cytometry analysis. SP2 and SP24 EC50 were calculated to be 5.07 μM (C) and 0.57 μM (D), respectively. Error bars represent SD from three biological repeats. (E) Flag-tagged SpCas9 protein expression was determined by western blotting with GAPDH serving as the loading control.
3.4. SP2 and SP24 Can Improve the Specificity of SpCas9 in Cells
While the EGFP cDNA had been inserted into the endogenous H2AX locus, we wanted to determine whether SP2 and SP24 could effectively inhibit SpCas9-mediated cleavage of bona fide endogenous loci. To this end, we first established iCas9-expressing U2OS cells. We then selected four gRNAs that target different sites in three endogenous gene loci and stably expressed these guides in the iCas9 U2OS cells. The cells were subsequently cultured with or without Dox before being collected for DNA extraction and deep sequencing (Figure 4A). Cleavage at all four sites could be detected even in the absence of Dox, an indication of basal SpCas9 expression caused by the leakiness of the tet-on promoter [42,43,44] (Figure 4B–E). As expected, the mutation frequency (indel, %) was low for both on- and off-target cleavage without Dox induction, although considerable variation existed between the four target sites, where EMX1-1 had the highest on- as well as off-targeting levels (Figure 4B). In the absence of inhibitors, Dox-induced Cas9 expression (DMSO alone samples) led to varying degrees of increases in on-site cleavage, from ~3 fold for VEGFA-3 (Figure 4D) to ~10 fold for EMX1-2 (Figure 4E). With inhibitor treatment, the effect on SpCas9 cleavage was also site-dependent. For ZSCAN2 and VEGFA-3, inhibition was minimal with either compound for both on- and off-site editing, with the exception of SP2 on ZSCAN2 off-targeting that showed small but appreciable inhibition (Figure 4C). For EMX1-1, decreases in the average editing efficiency at on-target sites were not significant, but with 2.5 μM SP2 off-target editing decreased from 35.7% to 21.7% (Figure 4B). For EMX1-2, on-site editing decreased from ~23% to ~18% with both compounds, and 2.5 μM SP2 was able to reduce SpCas9 off-target editing by 63.3% (Figure 4E). At all four tested sites, the ratio of on-target to off-target cleavage activity was higher in the uninduced group (-Dox) than in the DMSO-treated induced group (Figure 4B-E), consistent with the idea that SpCas9 may preferentially cleave target sites at low concentrations [45,46,47]. Following Dox induction (+Dox), the cleavage activity ratio of on-target versus off-target sites was consistently higher in the inhibitor treatment groups than the corresponding DMSO control groups (Figure 4B–E), indicating improved specificity due to the presence of SP2 and SP24. Our results thus far indicate that both SP2 and SP24 can inhibit the cleavage activity of SpCas9 in cells, and the extent of their inhibition is highly dependent on the specific genomic sequence context. Although we have proven that SP2 and SP24 can improve the specificity of SpCas9 in U2OS cells, it is necessary to verify the inhibitory activity of compounds on SpCas9 at more endogenous loci in different cell types.
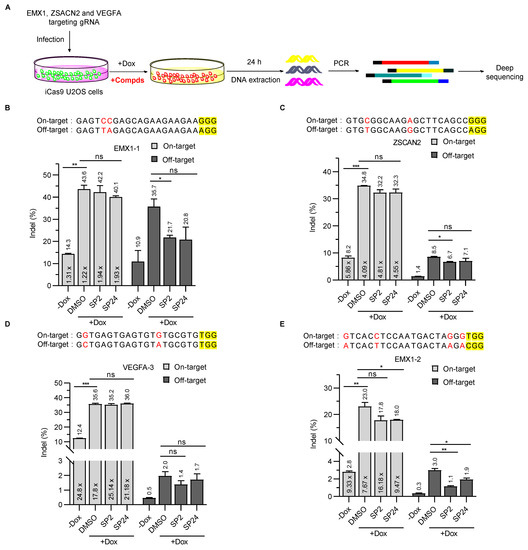
Figure 4.
SP2 and SP24 can improve the specificity of SpCas9 in cells. (A) To investigate the ability of SP2 and SP24 to inhibit SpCas9 in cells, we established iCas9-expressing U2OS cell lines that also stably expressed gRNAs targeting different endogenous sites. The cells were then cultured with or without Dox and SP2 (2.5 μM)/SP24 (1 μM for VEGFA-3 locus, and 0.5 μM for others) for 24 h before genomic DNA extraction and deep sequencing of on- and off-target sites. (B,E) The percentage of indels at on- and off-target sites for gRNAs targeting EMX1-1 (B), ZSCAN2 (C), VEGFA (D), and EMX1-2 (E) were calculated and plotted as shown. On-target and off-target sites editing efficiency of SpCas9 with SP2 and SP24 treatment at EMX1-1 (B), ZSCAN2 (C), VEGFA (D), EMX1-2 (E) locus. Data are from two independent biological replicates. The multiple value in the panel columns was the ratio of on-target vs. off-target indels that were calculated for each condition and target site. PAM sequences are highlighted in yellow. Mismatched bases in on- and off-target sites are marked in red. Error bars indicate mean ± SD. The p values were determined by two-tailed unpaired t-test, ns means no significance, (*) p < 0.05, (**) p < 0.01, and (***) p < 0.001.
3.5. Examining the Inhibitory Mechanisms of SP2 and SP24
To elucidate the mechanisms underlying the inhibitory activities of SP2 and SP24, we carried out competitive fluorescence polarization (FP) assays, where interaction of the SpCas9–gRNA complex with the fluorescently labeled target DNA could enhance fluorescence polarization signals. As expected, adding unlabeled target DNA (PAM-DNA) to the reaction decreased FP signals (Figure 5A,B). The addition of SP2 at 50 μM did not lead to changes in FP signals of dSpCas9 (nuclease dead SpCas9)-gRNA and DNA (Figure 5B), slightly affect (<5%) FP signals of SpCas9-gRNA, and DNA (Figure 5A), indicating that SP2 may not interfere with the binding of target DNA to the SpCas9–gRNA or dSpCas9–gRNA complex and that SP2 might directly act on SpCas9. In comparison, addition of SP24 led to a significant reduction in FP signals (Figure 5A,B). Compared with DMSO, the addition of SP2 and SP24 did not affect the fluorescence polarization value of fluorescence-labeled DNA (Supplementary Figure S7). Our data suggest that SP24 might function by blocking the interaction between the SpCas9–gRNA complex and the target DNA. To further probe the binding between the compounds and SpCas9, we purified the EGFP-tagged SpCas9 from E. coil BL21 (DE3) and carried out microscale thermophoresis assays (MST) (Supplementary Figure S8). We calculated the EC50 of SP2 for SpCas9 and SpCas9-gRNA to be 44.23 ± 35.40 μM and 5.63 ± 3.65 μM, respectively (Figure 5C,D). And the EC50 of SP24 to SpCas9 and SpCas9–gRNA complex was 14.31 ± 6.9 μM and 7.24 ± 3.16 μM, respectively (Figure 5E,F). These data support a direct interaction between the SpCas9–gRNA complex and the two compounds. Next, we used the SYBYL 2.1.1 Surflex-dock module [40,41] to examine the interaction sites between the SpCas9–gRNA complex (PDB code 4ZT9) and the compounds. Based on molecular docking analysis, SP2 interacts with Tyr72 and Lys65 of SpCas9. In this model, SP2 could form four hydrogen bonds with the gRNA in the SpCas9–gRNA complex, namely cytosine at position 18, guanine at position 19, guanine at position 21, and adenine at position 51 (Supplementary Figure S9). This finding helps explain our observation that the affinity between SP2 and the SpCas9–gRNA complex appeared higher than SP2 and SpCas9. Analysis of SP24 also indicated that the nitro group of SP24 could form hydrogen bonds with the NH2 group of Lys735. The NH2 group of C67 in the gRNA could also interact with the nitro group of SP24 by hydrogen bonding. And the oxygen atom of the methoxy group in the benzene ring of SP24 could form a hydrogen bond with the hydrogen atom of the peptide bond between Asp54 and Ser55 (Figure 5G). These three hydrogen bonding interactions could lead to the inhibition of SpCas9 by the compounds and helps explain why the inhibitory activity of SP24 appeared greater than that of SP16 and that SP15 was better than SP1 (Figure 3A and Supplementary Figure S4). In addition, the groups between the two benzene rings of SP24 could also form hydrogen bonds with Asp54, Gln1101, and Lys1200 in SpCas9 (Figure 5G). Taken together, our data not only shed light on how SpCas9 activity may be inhibited by small molecules in cellulo and in vitro, but also help lay the groundwork for further compound modifications and optimization to facilitate the implementation of CRISPR–Cas systems in clinical settings.
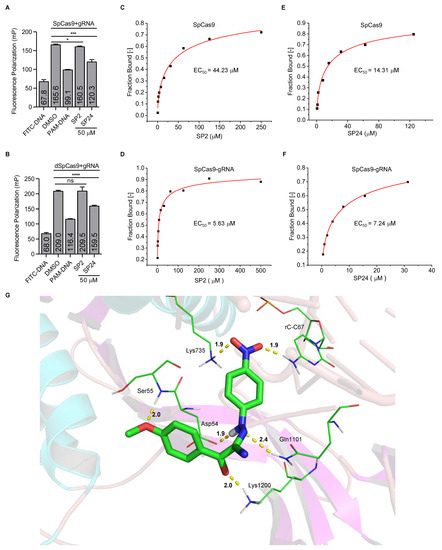
Figure 5.
Investigating the inhibition mechanisms of the identified compounds. (A,B) Each compound (50 nM) was added to a SpCas9-gRNA (1:1) (A) or dSpCas9-gRNA (1:1) (B) mixture before addition of the FITC-labeled target DNA (25 nM). Fluorescence polarization was assessed by a multilabel reader within 10 min. FITC-DNA, FITC labeled double-stranded target DNA. PAM-DNA, unlabeled double-stranded target DNA (1.25 μM). Error bars represent mean ± SD (n = 3). The p values were determined by two-tailed unpaired t-test, ns means no significance, (*) p < 0.05, (***) p < 0.001, and (****) p < 0.0001. (C–F) For MST assays, the compounds were titrated with SpCas9 or SpCas9-gRNA as indicated. The concentration of SpCas9–EGFP fusion protein was 200 nM. The mixture was kept at room temperature for 10 min. Then the sample was loaded into the NanoTemper standard capillaries. The EC50 of SP2 to SpCas9 and SpCas9–gRNA RNP complex is 44.23 ± 35.40 μM (C) and 5.63 ± 3.65 μM (D), respectively. The EC50 of SP24 to SpCas9 and SpCas9–gRNA complex is 14.31 ± 6.9 μM (E) and 7.24 ± 3.16 μM (F), respectively. (G) We propose that SP24 inserts into the active site of SpCas9 (PDB: 4ZT9). The ribbons represent SpCas9 or gRNA. The green rod in the middle represents compound SP24. Amino acid residues or bases that interact with the compound are represented by green lines. The yellow dashed lines represent the hydrogen bond interaction between the compound and the SpCas9–gRNA complex. Carbon, nitrogen, oxygen, and hydrogen atoms are shown in green, blue, red, and white, respectively.
4. Discussion
In this study, we identified several small-molecule inhibitors of SpCas9, with SP24 showing the highest inhibitory activity in cellulo. Our results indicate that SP24 could inhibit the cleavage activity of SpCas9, both in genomically integrated reporter sequences and at bona fide endogenous loci. However, whether compounds such as SP24 can effectively inhibit Cas9 nucleases in vivo remains to be addressed. Therefore, examining the efficacy and safety of compounds such as SP24 in animal models will be critical to our understanding of how promising inhibitory compounds as well as CRISPR–Cas systems function and interact with each other in vivo. We observed that the inhibition rates of the two compounds in 293T reporter cells were higher than those in U2OS cells. Because the rate of GFP degradation will affect the evaluation of inhibition rates, slower GFP degradation may lead to a higher calculated inhibition rate. Therefore, more rapid and sensitive reporter cells are needed to evaluate the inhibitory activity of compounds on SpCas9 in cellulo.
While in-depth studies of inhibitor-Cas9/gRNA interactions can only be possible with high-resolution structures of the complex obtained through crystallography or other means, clues on how chemical compounds such as SP2 and SP24 can achieve effective inhibition can already be gleaned from in vitro assays such as FP. For instance, our FP assays showed that SP24 could affect the interaction between the SpCas9–gRNA complex and its target DNA. Further investigation is warranted where residues predicted by molecular docking analysis may be mutated to better pinpoint the residues that are crucial to SP24 action. In addition, we found that SP2 and SP24 significantly inhibit the cleavage activity of SpCas9 at off-target sites while minimally affecting on-target binding, resulting in improved specificity of SpCas9 in U2OS cells. Given the varied inhibitory effects of the compounds on different endogenous target sites, more work is also needed to investigate whether factors such as specific genomic sequences, cell type, and chromatin structure may be determining factors as well. We observed that SP2 and SP24 have little effect on the activity of SpCas9 at endogenous on-target sites, which may depend on the low affinity between the compound and SpCas9. It is necessary to optimize the structure of these compounds and improve the affinity between the compound and SpCas9, to improve the inhibitory activity of these compounds.
The structure of SP24 is different from previously reported small-molecule inhibitors [33,48], and the binding pocket is likely to be different as well. In fact, our data suggest that SpCas9 selectivity could be improved by the compounds. More potent and selective inhibitors of SpCas9 should be attainable based on existing structural information of SpCas9 [49]. We surmise that a cocktail of small-molecule compounds with different structures may be more effective at inhibiting the Cas9 nuclease. In a recent study, SpCas9 variants that could selectively cleave target sites were engineered using a crystal-structure-guided design [49], but the variant is less active in mammalian cells than the wild-type SpCas9 [50]. The mutation sites of the variants are concentrated in the region where SpCas9 interacts with DNA distal from the protospacer adjacent motifs (PAM). Other mutations can improve SpCas9 fidelity by weakening the interaction between SpCas9 and targeted DNA [51,52] or weakening the intermolecular interaction between the domains of SpCas9 [53]. It is possible that mutations in other regions of SpCas9 can also improve SpCas9 its selectivity. Small-molecule inhibitors such as SP2 and SP24 should aid in the identification of such residues/regions on the Cas nucleases. Such endeavors will enable finer control of CRISPR–Cas-mediated cleavage, both its specificity and active time window, in order to reduce the risks of genomic instability and unwanted mutations. In addition, CRISPR–Cas-mediated gene drive has great potential in improving human health, but an uncontrolled gene drive will bring unpredictable risks. Inhibitors of Cas proteins can change the intensity of the gene drive, which is a powerful safety tool [22,54]. Furthermore, routine medical procedures such as blood transfusions, childbirth, and dental surgery may have higher risk of bacterial infection [55,56]. Phage therapy as a means of fighting bacterial infection [55], these small-molecule inhibitors of Cas protein can inhibit bacterial Cas activity and increase the efficacy of bacteriophage therapy [15]. Finally, the inhibitor of Cas proteins can reduce the cytotoxicity caused by the continuous activation of the Cas protein [57].
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/cells11223574/s1, Figures S1 and S2: Related to Figure 1, Identification of small-molecule inhibitors of SpCas9 through unbiased screens; Figure S3: Related to Figure 2. Analysis of structure–activity relationships using analogs of the SpCas9 inhibitors we identified; Figure S4: Related to Figure 3. The chemical structure of SP series compounds; Figure S5: Related to Figure 3. Analysis of SP series compound in cellulo activities; Figure S6: In vitro LbCas12a cleavage assays were carried out in the presence of different SP series compounds; Figure S7: Fluorescence polarization to detect the interaction between the target DNA and SP2 (or SP24); Figure S8: Prokaryotic expression of SpCas9, dSpCas9, and SpCas9-EGFP; Figure S9: The proposed binding model of SP2 into the active site of SpCas9 (PDB: 4ZT9); Table S1: ID number of SP series compounds in SPECS website; Table S2: List of amplicon sequence data used in this study; Table S3: List of oligos sequence used in this study.
Author Contributions
Conceptualization, Z.S., P.L. and J.W.; methodology, Y.Y., D.L., F.W., B.C. and F.L.; software, G.W. and Y.Y.; validation, Y.Y., D.L. and F.W.; formal analysis, Y.Y., D.L. and G.W.; investigation, Y.Y., D.L., P.L. and Z.S.; resources, Y.Y., D.L., F.W., B.C., F.L., G.W., Y.R., J.W., P.L. and Z.S.; data curation, Y.Y., D.L., F.W. and G.W.; writing—original draft preparation, Y.Y. and P.L.; writing—review and editing, P.L. and Z.S.; visualization, Y.Y. and D.L.; supervision, Z.S., P.L. and J.W.; project administration, Y.Y., D.L., F.W., B.C., F.L., G.W., Y.R., J.W., P.L. and Z.S.; funding acquisition, Y.R., J.W., P.L. and Z.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China, funding numbers 31930058 and 91640119, National Key R&D Program of China, funding numbers 2018YFA0107003.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All raw data of this paper are available upon request. The high-throughput sequencing data from this study have been deposited into the NCBI Sequence Read Archive database (SRA; https://www.ncbi.nlm.nih.gov/sra, assessed on 11 April 2022) under Accession Number NCBI: PRJNA825343 and listed in Supplementary Table S2.
Acknowledgments
We thank Jiaqi Liu and Yuxi Chen for helpful advice on protein expression and purification. Additionally, we are grateful for the suggestions made by anonymous reviewers.
Conflicts of Interest
The authors declare no conflict of interest.
Appendix A. Compound Synthesis and Characterization
Appendix A.1. Chemistry
Common reagents and solvents were obtained from commercial suppliers and used without further purification. The compounds were obtained by column chromatography using 200–300 mesh silica gel. The thin-layer chromatography (TLC) with pre-coated glass plates (silica gel 60 GF 254, Qingdao Haiyang Chemical, China) was used to monitor the reaction process. 1H and 13C spectra were obtained on Varian Mercury-Plus 400 or 600 MHz spectrometers (Bruker, Karlsruhe, Germany). Chemical shifts (δ) were expressed in parts per million (ppm) while the singlet (0 ppm) for tetramethylsilane (TMS). The abbreviations were used to describe peak signal, which s, d, t, q, and m refer to singlet, doublet, triplet, quadruplet, and multiplet, respectively. Coupling constants (J) were quoted in HZ. High-resolution mass spectrometry (HRMS) experiments were performed using mass spectrometer with ESI as the ionization.
Appendix A.2. Synthesis of C4 Series Compounds
The synthesis of C4 series compounds and their detailed characterization will be described elsewhere (Y.Y., manuscript in preparation).
Appendix A.3. Synthesis of C9 and C12 Series Compounds
The C9 and C12 series compounds were synthesized using the following steps (Scheme A1).

Scheme A1.
Synthesis pathway of C12 series compounds.

General Procedure for the Preparation of C12 Series Compounds
Ethyl acetoacetate (10 mmol) was added into a flask, and then added an aqueous solution of KOH (3.5 g, 100 mL water), and the mixture was stirred at room temperature for 24 h. Once cooled to 0 °C and slowly added concentrated HCl (4.5 mL), the ice water (15 mL) was then added to the mixture. p-nitroaniline(10 mmol) was added to another flask, and then added water (10 mL) and concentrated HCl (3 mL). Once cooled to 0 °C, the mixture was then stirred for 30 min at 0 °C. NaNO2 (10 mmol) aqueous solution was added slowly, the temperature should not exceed 5 °C. The mixture was stirred for 30 min to obtain diazonium salt. The freshly prepared diazonium salt was added dropwise to the first flask, and the solution was kept at 0–5 °C. The solution became turbid, and CH3COONa aqueous solution (8.2 g, 100 mL ice water) was added dropwise. The mixture was stirred at 0–5 °C for 2 h. The reaction was close to complete, and a yellow solid was obtained by suction filtration.
C9: 1-(2-(4-nitrophenyl) hydrazono)butanone
Orange solid; Yield: 76%; mp: 190.3–191.7 °C; 1H NMR (600 MHz, DMSO-d6) δ 8.18 (d, J = 9.2 Hz, 2H), 7.43 (s, 1H), 7.29 (d, J = 9.0 Hz, 2H), 2.84 (q, J = 6.8 Hz, 2H), 1.04 (t, J = 7.3 Hz, 3H). 13C NMR (151 MHz, DMSO-d6) δ 236.86, 187.08, 177.91, 175.53, 163.48, 66.88, 45.73. MS (ESI) m/z: 222.0873 [M+H]+.
C12: 1-(2-(4-nitrophenyl) hydrazono)-4-bromo-acetophenone
Yellow solid; Yield: 77%; mp: 223.3–224 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.01 (s, 1H), 8.19 (d, J = 7.4 Hz, 2H), 7.91 (d, J = 8.5 Hz, 2H), 7.77 (d, J = 4.7 Hz, 2H), 7.74 (s, 1H), 7.20 (d, J = 9.1 Hz, 2H). 13C NMR (151 MHz, DMSO-d6) δ 188.72, 149.23, 141.27, 137.95, 136.02, 131.98, 131.81, 126.96, 126.42, 113.80. MS (ESI) m/z: 369.9792 [M+Na]+.
C12a: 1-(2-(4-nitrophenyl) hydrazono)-3,4-dichlor-acetophenone
Yellow solid; Yield: 69%; mp: 216.3–218°C; 1H NMR (400 MHz, DMSO-d6) δ 12.08 (s, 1H), 8.20 (d, J = 9.2 Hz, 2H), 8.13 (s, 1H), 7.95 (d, J = 10.2 Hz, 1H), 7.82 (d, J = 8.3 Hz, 1H), 7.76 (s, 1H), 7.20 (d, J = 9.1 Hz, 2H). MS (ESI) m/z: 335.9957 [M-H]+.
C12b: 1-(4-bromophenyl)-2-(2-(4-chlorophenyl)hydrazono)ethanone
Yellow solid; Yield: 65%; mp: 235.2–237.2 °C; 1H NMR (400 MHz, DMSO-d6) δ 11.57 (s, 1H), 7.88 (d, J = 8.4 Hz, 2H), 7.71 (d, J = 8.5 Hz, 2H), 7.65 (s, 1H), 7.33 (d, J = 8.8 Hz, 2H), 7.08 (d, J = 8.8 Hz, 2H). 13C NMR (151 MHz, DMSO-d6) δ 188.66, 142.44, 136.57, 134.53, 131.87, 131.63, 129.75, 126.41, 125.74, 115.65. MS (ESI) m/z: 358.9592 [M+Na]+.
C12c: 1-(4-bromophenyl)-2-(2-(2-hydroxy-5-nitrophenyl)hydrazono)ethanone
Yellow solid; Yield: 76%; mp: 246.6–247.2 °C; 1H NMR (600 MHz, DMSO-d6) δ 11.66 (s, 1H), 11.33 (s, 1H), 8.11 (s, 1H), 7.97 (s, 1H), 7.92 (d, J = 8.1 Hz, 1H), 7.76 (t, J = 9.9 Hz,2H), 7.01 (d, J = 8.8 Hz, 3H). 13C NMR (151 MHz, DMSO-d6) δ 188.96, 151.23, 140.77, 136.64, 136.31, 132.39, 131.87, 131.53, 126.52, 118.57, 115.17, 108.89. MS (ESI) m/z: 385.9750 [M+Na]+.
C12d: 1-(2,4-difluorophenyl)-2-(2-(4-nitrophenyl)hydrazono)-2-(1H-1,2,4-triazol-1-yl)ethanone
Yellow solid; Yield: 73%; 1H NMR (600 MHz, DMSO-d6) δ 9.36 (s, 1H), 8.53 (d, J = 7.7 Hz, 2H), 8.44 (s, 1H), 8.29 (s, 1H), 8.04 (d, J = 7.6 Hz, 2H), 7.54 (s,1H), 7.36–7.29 (m, 1H), 7.26 (d, J = 9.8 Hz, 1H). 13C NMR (151 MHz, DMSO-d6) δ 182.56, 152.75, 147.82, 146.80, 146.78, 142.32, 132.97, 129.31, 128.43, 125.71, 115.03, 111.90, 111.76, 104.74. MS (ESI) m/z: 373.0844 [M+H]+.
C12e: 2-(2-(2-hydroxy-5-nitrophenyl)hydrazono)-1-phenylbutane-1,3-dione
Yellow solid; Yield: 70%; 1H NMR (600 MHz, DMSO-d6) δ 12.18 (s, 1H), 7.90 (s, 3H), 7.71 (d, J = 6.2 Hz, 1H), 7.68 (s, 1H), 7.53 (d, J = 17.8 Hz, 2H), 7.08 (d, J = 7.7 Hz, 1H), 2.54 (d, J = 9.9 Hz, 3H). 13C NMR (151 MHz, DMSO-d6) δ 198.06, 191.82, 152.48, 140.57, 138.29, 134.29, 132.62, 130.60, 130.37, 128.23, 121.68, 115.90, 110.22, 30.71. MS (ESI) m/z: 350.0744 [M+Na]+.
C12f: 2-(2-(3-chlorophenyl)hydrazono)-1-(2,4-difluorophenyl)-2-(1H-1,2,4-triazol-1-yl)ethanone
Brown solid; Yield: 60%; 1H NMR (600 MHz, DMSO-d6) δ 11.31 (s, 1H), 8.99 (s, 1H), 8.46 (s, 1H), 7.84 (s, 1H), 7.52 (s, 1H), 7.43–7.27 (m, 2H), 7.19 (s, 1H), 7.12 (d, J = 23.3 Hz, 2H). 13C NMR (151 MHz, DMSO-d6) δ 182.85, 153.12, 147.06, 144.02, 134.25, 133.17, 131.58, 128.15, 123.59, 122.99, 115.19, 113.97, 112.01, 104.90. MS (ESI) m/z: 236.0613 [M+H]+.
C12g: 2-(2-(3-nitrophenyl)hydrazono)-1-phenylbutane-1,3-dione
Yellow solid; Yield: 67%; 1H NMR (600 MHz, DMSO-d6) δ 11.76 (s, 1H), 8.12 (s, 1H), 7.93–7.76 (m, 4H), 7.71 (s, 1H), 7.64–7.52 (m, 3H), 2.54 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ 196.11, 148.90, 145.64, 140.90, 135.87, 134.83, 131.08, 129.48, 121.21, 116.94, 109.44. MS (ESI) m/z: 334.0792 [M+Na]+.
C12h: 2-(2-(3-chlorophenyl) hydrazono)-5,5,5-trifluoro-1-phenylpentane-1,3-dione
Gray solid; Yield: 75%; 1H NMR (600 MHz, DMSO-d6) δ 12.34 (s, 1H), 7.77 (d, J = 7.8 Hz, 2H), 7.51–7.29 (m, 5H), 7.23 (d, J = 7.9 Hz, 1H), 2.42 (s, 2H). 13C NMR (151 MHz, DMSO-d6) δ 188.74, 153.31, 149.01, 144.06, 143.48, 134.50, 131.51, 130.42, 129.52, 127.96, 119.29, 118.70, 21.66. MS (ESI) m/z: 391.0430 [M+Na]+.
C12i: 1-(2-(2-methoxy-5-nitrophenyl)hydrazono)propan-2-one
Yellow solid; Yield: 79%; 1H NMR (600 MHz, DMSO-d6) δ 8.08–8.06 (m, 1H), 8.05 (s, 2H), 7.82 (s, 1H), 7.59 (d, J = 8.8 Hz, 1H), 2.38 (s, 3H), 2.36 (s, 3H). 13C NMR (151 MHz, DMSO-d6) δ 202.15, 152.67, 145.46, 144.96, 131.45, 128.36, 117.90, 108.56, 29.46, 22.71. MS (ESI) m/z: 224.0687 [M+Na]+.
References
- Makarova, K.S.; Wolf, Y.I.; Alkhnbashi, O.S.; Costa, F.; Shah, S.A.; Saunders, S.J.; Barrangou, R.; Brouns, S.J.; Charpentier, E.; Haft, D.H.; et al. An updated evolutionary classification of CRISPR-Cas systems. Nat. Rev. Microbiol. 2015, 13, 722–736. [Google Scholar] [CrossRef] [PubMed]
- Deveau, H.; Garneau, J.E.; Moineau, S. CRISPR/Cas system and its role in phage-bacteria interactions. Annu. Rev. Microbiol. 2010, 64, 475–493. [Google Scholar] [CrossRef] [PubMed]
- Marraffini, L.A.; Sontheimer, E.J. CRISPR interference: RNA-directed adaptive immunity in bacteria and archaea. Nat. Rev. Genet. 2010, 11, 181–190. [Google Scholar] [CrossRef]
- Koonin, E.V.; Makarova, K.S. CRISPR-Cas: Evolution of an RNA-based adaptive immunity system in prokaryotes. RNA Biol. 2013, 10, 679–686. [Google Scholar] [CrossRef]
- Makarova, K.S.; Wolf, Y.I.; Koonin, E.V. The basic building blocks and evolution of CRISPR-CAS systems. Biochem. Soc. Trans. 2013, 41, 1392–1400. [Google Scholar] [CrossRef] [PubMed]
- Barrangou, R.; Marraffini, L.A. CRISPR-Cas systems: Prokaryotes upgrade to adaptive immunity. Mol. Cell 2014, 54, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Jore, M.M.; Brouns, S.J.; van der Oost, J. RNA in defense: CRISPRs protect prokaryotes against mobile genetic elements. Cold Spring Harb Perspect. Biol. 2012, 4, a003657. [Google Scholar] [CrossRef]
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D.A.; Horvath, P. CRISPR provides acquired resistance against viruses in prokaryotes. Science 2007, 315, 1709–1712. [Google Scholar] [CrossRef]
- Marraffini, L.A.; Sontheimer, E.J. CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. Science 2008, 322, 1843–1845. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Sternberg, S.H.; LaFrance, B.; Kaplan, M.; Doudna, J.A. Conformational control of DNA target cleavage by CRISPR-Cas9. Nature 2015, 527, 110–113. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, T.; Ou, J.; Huang, J.; Liang, P. Homology-based repair induced by CRISPR-Cas nucleases in mammalian embryo genome editing. Protein Cell 2022, 13, 316–335. [Google Scholar] [CrossRef]
- Gangopadhyay, S.A.; Cox, K.J.; Manna, D.; Lim, D.; Maji, B.; Zhou, Q.; Choudhary, A. Precision Control of CRISPR-Cas9 Using Small Molecules and Light. Biochemistry 2019, 58, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.; Merkel, O.M. Immunogenicity of Cas9 Protein. J. Pharm. Sci. 2020, 109, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Enache, O.M.; Rendo, V.; Abdusamad, M.; Lam, D.; Davison, D.; Pal, S.; Currimjee, N.; Hess, J.; Pantel, S.; Nag, A.; et al. Cas9 activates the p53 pathway and selects for p53-inactivating mutations. Nat. Genet. 2020, 52, 662–668. [Google Scholar] [CrossRef]
- Dolgin, E. The kill-switch for CRISPR that could make gene-editing safer. Nature 2020, 577, 308–310. [Google Scholar] [CrossRef] [PubMed]
- Wiegand, T.; Karambelkar, S.; Bondy-Denomy, J.; Wiedenheft, B. Structures and Strategies of Anti-CRISPR-Mediated Immune Suppression. Annu. Rev. Microbiol. 2020, 74, 21–37. [Google Scholar] [CrossRef]
- Yin, Y.; Yang, B.; Entwistle, S. Bioinformatics Identification of Anti-CRISPR Loci by Using Homology, Guilt-by-Association, and CRISPR Self-Targeting Spacer Approaches. mSystems 2019, 4, e00455-19. [Google Scholar] [CrossRef]
- Bondy-Denomy, J.; Pawluk, A.; Maxwell, K.L.; Davidson, A.R. Bacteriophage genes that inactivate the CRISPR/Cas bacterial immune system. Nature 2013, 493, 429–432. [Google Scholar] [CrossRef] [PubMed]
- Marino, N.D.; Pinilla-Redondo, R.; Csorgo, B.; Bondy-Denomy, J. Anti-CRISPR protein applications: Natural brakes for CRISPR-Cas technologies. Nat. Methods 2020, 17, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.J.; Hinner, M.J. Getting across the cell membrane: An overview for small molecules, peptides, and proteins. Methods Mol. Biol. 2015, 1266, 29–53. [Google Scholar] [CrossRef] [PubMed]
- Nihongaki, Y.; Yamamoto, S.; Kawano, F.; Suzuki, H.; Sato, M. CRISPR-Cas9-based photoactivatable transcription system. Chem. Biol. 2015, 22, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Polstein, L.R.; Gersbach, C.A. A light-inducible CRISPR-Cas9 system for control of endogenous gene activation. Nat. Chem. Biol. 2015, 11, 198–200. [Google Scholar] [CrossRef]
- Hemphill, J.; Borchardt, E.K.; Brown, K.; Asokan, A.; Deiters, A. Optical Control of CRISPR/Cas9 Gene Editing. J. Am. Chem. Soc. 2015, 137, 5642–5645. [Google Scholar] [CrossRef]
- Zhang, Y.; Ling, X.; Su, X.; Zhang, S.; Wang, J.; Zhang, P.; Feng, W.; Zhu, Y.Y.; Liu, T.; Tang, X. Optical Control of a CRISPR/Cas9 System for Gene Editing by Using Photolabile crRNA. Angew. Chem. 2020, 59, 20895–20899. [Google Scholar] [CrossRef]
- Zetsche, B.; Volz, S.E.; Zhang, F. A split-Cas9 architecture for inducible genome editing and transcription modulation. Nat. Biotechnol. 2015, 33, 139–142. [Google Scholar] [CrossRef]
- Yu, Y.; Wu, X.; Guan, N.; Shao, J.; Li, H.; Chen, Y.; Ping, Y.; Li, D.; Ye, H. Engineering a far-red light-activated split-Cas9 system for remote-controlled genome editing of internal organs and tumors. Sci. Adv. 2020, 6, eabb1777. [Google Scholar] [CrossRef]
- Wright, A.V.; Sternberg, S.H.; Taylor, D.W.; Staahl, B.T.; Bardales, J.A.; Kornfeld, J.E.; Doudna, J.A. Rational design of a split-Cas9 enzyme complex. Proc. Natl. Acad. Sci. USA 2015, 112, 2984–2989. [Google Scholar] [CrossRef]
- Nunez, J.K.; Harrington, L.B.; Doudna, J.A. Chemical and Biophysical Modulation of Cas9 for Tunable Genome Engineering. ACS Chem. Biol. 2016, 11, 681–688. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, C.; Zhang, J.; Lee, J.H.; Jiao, J.; Cheng, D.; Liu, L.; Kim, H.W.; Tao, Y.; Li, M. Spatiotemporal control of CRISPR/Cas9 gene editing. Signal. Transduct. Target Ther. 2021, 6, 238. [Google Scholar] [CrossRef] [PubMed]
- Maji, B.; Gangopadhyay, S.A.; Lee, M.; Shi, M.; Wu, P.; Heler, R.; Mok, B.; Lim, D.; Siriwardena, S.U.; Paul, B.; et al. A High-Throughput Platform to Identify Small-Molecule Inhibitors of CRISPR-Cas9. Cell 2019, 177, 1067–1079.e19. [Google Scholar] [CrossRef] [PubMed]
- Acharya, S.; Mishra, A.; Paul, D.; Ansari, A.H.; Azhar, M.; Kumar, M.; Rauthan, R.; Sharma, N.; Aich, M.; Sinha, D.; et al. Francisella novicida Cas9 interrogates genomic DNA with very high specificity and can be used for mammalian genome editing. Proc. Natl. Acad. Sci. USA 2019, 116, 20959–20968. [Google Scholar] [CrossRef]
- Chen, Y.; Zhi, S.; Liang, P.; Zheng, Q.; Liu, M.; Zhao, Q.; Ren, J.; Cui, J.; Huang, J.; Liu, Y.; et al. Single AAV-Mediated CRISPR-SaCas9 Inhibits HSV-1 Replication by Editing ICP4 in Trigeminal Ganglion Neurons. Mol. Ther. Methods Clin. Dev. 2020, 18, 33–43. [Google Scholar] [CrossRef]
- Nakade, S.; Tsubota, T.; Sakane, Y.; Kume, S.; Sakamoto, N.; Obara, M.; Daimon, T.; Sezutsu, H.; Yamamoto, T.; Sakuma, T.; et al. Microhomology-mediated end-joining-dependent integration of donor DNA in cells and animals using TALENs and CRISPR/Cas9. Nat. Commun. 2014, 5, 5560. [Google Scholar] [CrossRef]
- Sakuma, T.; Nakade, S.; Sakane, Y.; Suzuki, K.T.; Yamamoto, T. MMEJ-assisted gene knock-in using TALENs and CRISPR-Cas9 with the PITCh systems. Nat. Protoc. 2016, 11, 118–133. [Google Scholar] [CrossRef]
- Clement, K.; Rees, H.; Canver, M.C.; Gehrke, J.M.; Farouni, R.; Hsu, J.Y.; Cole, M.A.; Liu, D.R.; Joung, J.K.; Bauer, D.E.; et al. CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol. 2019, 37, 224–226. [Google Scholar] [CrossRef]
- Wienken, C.J.; Baaske, P.; Rothbauer, U.; Braun, D.; Duhr, S. Protein-binding assays in biological liquids using microscale thermophoresis. Nat. Commun. 2010, 1, 100. [Google Scholar] [CrossRef]
- Jain, A.N. Surflex-Dock 2.1: Robust performance from ligand energetic modeling, ring flexibility, and knowledge-based search. J. Comput. Aided Mol. Des. 2007, 21, 281–306. [Google Scholar] [CrossRef]
- Jain, A.N. Surflex: Fully automatic flexible molecular docking using a molecular similarity-based search engine. J. Med. Chem. 2003, 46, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Dow, L.E.; Fisher, J.; O'Rourke, K.P.; Muley, A.; Kastenhuber, E.R.; Livshits, G.; Tschaharganeh, D.F.; Socci, N.D.; Lowe, S.W. Inducible in vivo genome editing with CRISPR-Cas9. Nat. Biotechnol. 2015, 33, 390–394. [Google Scholar] [CrossRef] [PubMed]
- Das, A.T.; Tenenbaum, L.; Berkhout, B. Tet-On Systems For Doxycycline-inducible Gene Expression. Curr. Gene Ther. 2016, 16, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Gossen, M.; Freundlieb, S.; Bender, G.; Muller, G.; Hillen, W.; Bujard, H. Transcriptional activation by tetracyclines in mammalian cells. Science 1995, 268, 1766–1769. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.D.; Scott, D.A.; Weinstein, J.A.; Ran, F.A.; Konermann, S.; Agarwala, V.; Li, Y.; Fine, E.J.; Wu, X.; Shalem, O.; et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013, 31, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Foden, J.A.; Khayter, C.; Maeder, M.L.; Reyon, D.; Joung, J.K.; Sander, J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013, 31, 822–826. [Google Scholar] [CrossRef]
- Cao, J.; Wu, L.; Zhang, S.M.; Lu, M.; Cheung, W.K.; Cai, W.; Gale, M.; Xu, Q.; Yan, Q. An easy and efficient inducible CRISPR/Cas9 platform with improved specificity for multiple gene targeting. Nucleic Acids Res. 2016, 44, e149. [Google Scholar] [CrossRef]
- Lee, S.W.; Tran, K.T.; Vazquez-Uribe, R.; Gotfredsen, C.H.; Clausen, M.H.; Mendez, B.L.; Montoya, G.; Bach, A.; Sommer, M.O.A. Identification and Optimization of Novel Small-Molecule Cas9 Inhibitors by Cell-Based High-Throughput Screening. J. Med. Chem. 2022, 65, 3266–3305. [Google Scholar] [CrossRef]
- Bravo, J.P.K.; Liu, M.S.; Hibshman, G.N.; Dangerfield, T.L.; Jung, K.; McCool, R.S.; Johnson, K.A.; Taylor, D.W. Structural basis for mismatch surveillance by CRISPR-Cas9. Nature 2022, 603, 343–347. [Google Scholar] [CrossRef]
- Kulcsár, P.I.; Tálas, A.; Welker, E. SuperFi-Cas9 exhibits extremely high fidelity but reduced activity in mammalian cells. bioRxiv 2022. [Google Scholar] [CrossRef]
- Kleinstiver, B.P.; Pattanayak, V.; Prew, M.S.; Tsai, S.Q.; Nguyen, N.T.; Zheng, Z.; Joung, J.K. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature 2016, 529, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Slaymaker, I.M.; Gao, L.; Zetsche, B.; Scott, D.A.; Yan, W.X.; Zhang, F. Rationally engineered Cas9 nucleases with improved specificity. Science 2016, 351, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.S.; Dagdas, Y.S.; Kleinstiver, B.P.; Welch, M.M.; Sousa, A.A.; Harrington, L.B.; Sternberg, S.H.; Joung, J.K.; Yildiz, A.; Doudna, J.A. Enhanced proofreading governs CRISPR-Cas9 targeting accuracy. Nature 2017, 550, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Basgall, E.M.; Goetting, S.C.; Goeckel, M.E.; Giersch, R.M.; Roggenkamp, E.; Schrock, M.N.; Halloran, M.; Finnigan, G.C. Gene drive inhibition by the anti-CRISPR proteins AcrIIA2 and AcrIIA4 in Saccharomyces cerevisiae. Microbiology 2018, 164, 464–474. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, R.; Pires, D.P.; Costa, A.R.; Azeredo, J. Phage Therapy: Going Temperate? Trends Microbiol. 2019, 27, 368–378. [Google Scholar] [CrossRef]
- Cappare, P.; D'Ambrosio, R.; De Cunto, R.; Darvizeh, A.; Nagni, M.; Gherlone, E. The Usage of an Air Purifier Device with HEPA 14 Filter during Dental Procedures in COVID-19 Pandemic: A Randomized Clinical Trial. Int. J. Environ. Res. Public Health 2022, 19, 5139. [Google Scholar] [CrossRef]
- Li, C.; Psatha, N.; Gil, S.; Wang, H.; Papayannopoulou, T.; Lieber, A. HDAd5/35(++) Adenovirus Vector Expressing Anti-CRISPR Peptides Decreases CRISPR/Cas9 Toxicity in Human Hematopoietic Stem Cells. Mol. Ther. Methods Clin. Dev. 2018, 9, 390–401. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).