
A New Model of Acute Exacerbation of Experimental Pulmonary Fibrosis in Mice
 and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethical Approval
2.2. Animals
2.3. Experimental Design
2.4. Lung Sampling for Analyses
2.5. Microscopy Analyses
2.6. Biochemical and Molecular Biology Analyses
2.7. Statistics
3. Results
3.1. Acute Injury by Last Double-Dose Bleomycin Induces a Rapid Clinical and Respiratory Deterioration
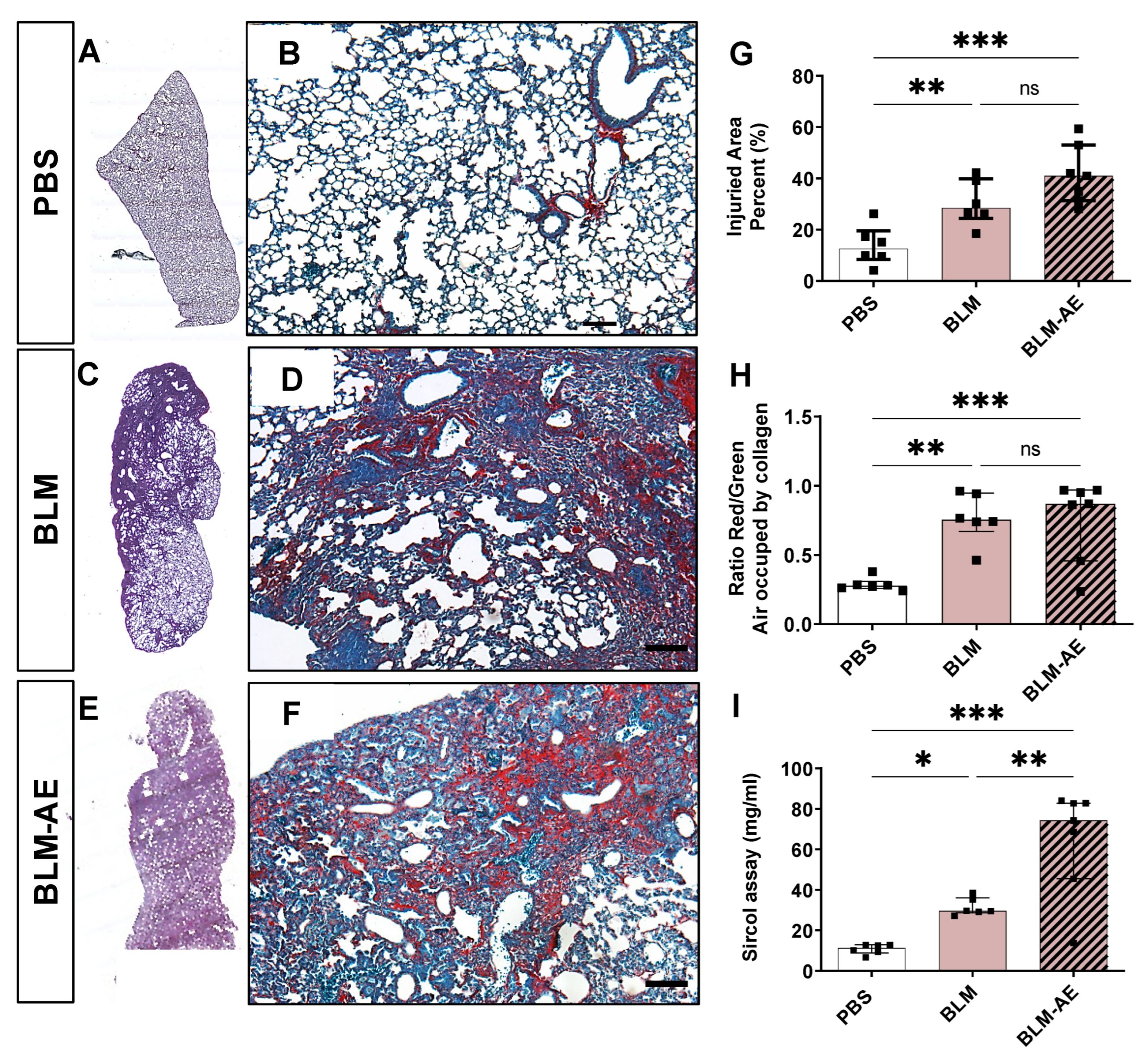
3.2. Acute Injury by Last Double-Dose Bleomycin Instillation Increases Lung Remodeling and Worsens Fibrosis
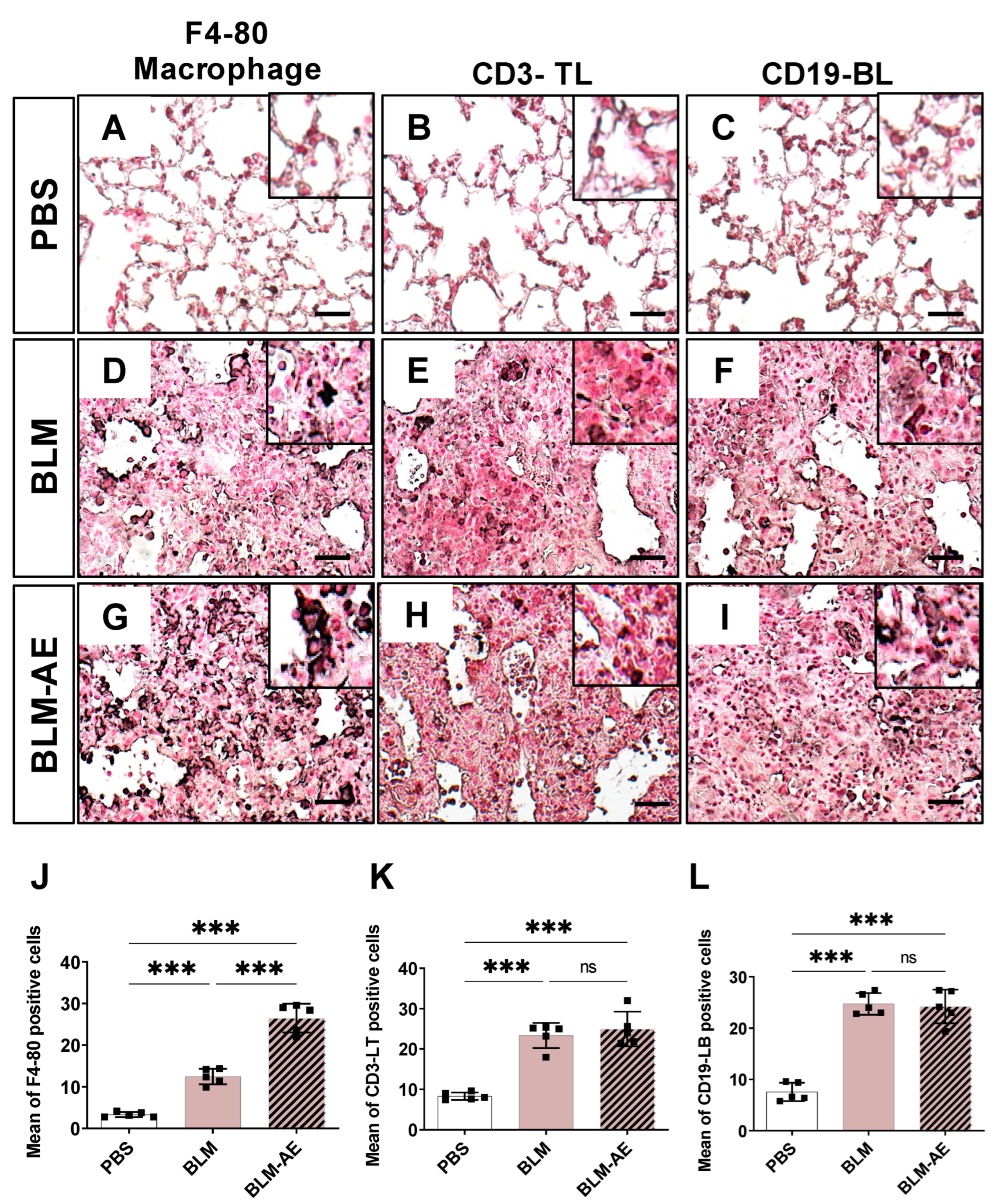
3.3. Lung Inflammation Is Observed in the Acute Exacerbation Model of Pulmonary Fibrosis
3.4. HIF-1a Is Associated with the Worsening Effect of Acute Injury on Lung Fibrosis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Selman, M.; Pardo, A. Idiopathic pulmonary fibrosis: An epithelial/fibroblastic cross-talk disorder. Respir. Res. 2002, 3, 3. [Google Scholar] [CrossRef]
- Cottin, V.; Spagnolo, P.; Bonniaud, P.; Nolin, M.; Dalon, F.; Kirchgässler, K.U.; Kamath, T.V.; Van Ganse, E.; Belhassen, M. Mortality and Respiratory-Related Hospitalizations in Idiopathic Pulmonary Fibrosis Not Treated With Antifibrotics. Front. Med. 2021, 8, 802989. [Google Scholar] [CrossRef] [PubMed]
- Collard, H.R.; Ryerson, C.J.; Corte, T.J.; Jenkins, G.; Kondoh, Y.; Lederer, D.J.; Lee, J.S.; Maher, T.M.; Wells, A.U.; Antoniou, K.M.; et al. Acute Exacerbation of Idiopathic Pulmonary Fibrosis. An International Working Group Report. Am. J. Respir. Crit. Care Med. 2016, 194, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Maher, T.M.; Whyte, M.K.B.; Hoyles, R.K.; Parfrey, H.; Ochiai, Y.; Mathieson, N.; Turnbull, A.; Williamson, N.; Bennett, B.M. Development of a Consensus Statement for the Definition, Diagnosis, and Treatment of Acute Exacerbations of Idiopathic Pulmonary Fibrosis Using the Delphi Technique. Adv. Ther. 2015, 32, 929–943. [Google Scholar] [CrossRef] [PubMed]
- Churg, A.; Wright, J.L.; Tazelaar, H.D. Acute exacerbations of fibrotic interstitial lung disease. Histopathology 2011, 58, 525–530. [Google Scholar] [CrossRef]
- Borzone, G.; Moreno, R.; Urrea, R.; Meneses, M.; Oyarzún, M.; Lisboa, C. Bleomycin-induced chronic lung damage does not resemble human idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2001, 163, 1648–1653. [Google Scholar] [CrossRef] [PubMed]
- Chung, M.P.; Monick, M.M.; Hamzeh, N.Y.; Butler, N.S.; Powers, L.S.; Hunninghake, G.W. Role of repeated lung injury and genetic background in bleomycin-induced fibrosis. Am. J. Respir. Cell Mol. Biol. 2003, 29 Pt 1, 375–380. [Google Scholar] [CrossRef]
- Degryse, A.L.; Lawson, W.E. Progress toward improving animal models for idiopathic pulmonary fibrosis. Am. J. Med. Sci. 2011, 341, 444–449. [Google Scholar] [CrossRef] [PubMed]
- Limjunyawong, N.; Mitzner, W.; Horton, M.R. A mouse model of chronic idiopathic pulmonary fibrosis. Physiol. Rep. 2014, 2, e00249. [Google Scholar] [CrossRef] [PubMed]
- Degryse, A.L.; Tanjore, H.; Xu, X.C.; Polosukhin, V.V.; Jones, B.R.; McMahon, F.B.; Gleaves, L.A.; Blackwell, T.S.; Lawson, W.E. Repetitive intratracheal bleomycin models several features of idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010, 299, L442–L452. [Google Scholar] [CrossRef]
- Redente, E.F.; Black, B.P.; Backos, D.S.; Bahadur, A.N.; Humphries, S.M.; Lynch, D.A.; Tuder, R.M.; Zemans, R.L.; Riches, D.W.H. Persistent, Progressive Pulmonary Fibrosis and Epithelial Remodeling in Mice. Am. J. Respir. Cell Mol. Biol. 2021, 64, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Ashley, S.L.; Jegal, Y.; Moore, T.A.; van Dyk, L.F.; Laouar, Y.; Moore, B.B. γ-Herpes virus-68, but not Pseudomonas aeruginosa or influenza A (H1N1), exacerbates established murine lung fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L219–L230. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Qiu, H.; Zhao, M.M.; Chen, S.S.; Wu, Q.; Zhou, N.Y.; Lu, L.-Q.; Song, J.-C.; Tang, D.-L.; Weng, D.; et al. IL-17A contributes to HSV1 infection-induced acute lung injury in a mouse model of pulmonary fibrosis. J. Cell. Mol. Med. 2019, 23, 908–919. [Google Scholar] [CrossRef] [PubMed]
- McMillan, T.R.; Moore, B.B.; Weinberg, J.B.; Vannella, K.M.; Fields, W.B.; Christensen, P.J.; van Dyk, L.F.; Toews, G.B. Exacerbation of established pulmonary fibrosis in a murine model by gammaherpesvirus. Am. J. Respir. Crit. Care Med. 2008, 177, 771–780. [Google Scholar] [CrossRef]
- Wei, Y.R.; Qiu, H.; Wu, Q.; Du, Y.K.; Yin, Z.F.; Chen, S.S.; Jin, Y.-P.; Zhao, M.-M.; Wang, C.; Weng, D.; et al. Establishment of the mouse model of acute exacerbation of idiopathic pulmonary fibrosis. Exp. Lung Res. 2016, 42, 75–86. [Google Scholar] [CrossRef]
- Haine, L.; Bravais, J.; Yegen, C.H.; Bernaudin, J.F.; Marchant, D.; Planès, C.; Voituron, N.; Boncoeur, E. Sleep Apnea in Idiopathic Pulmonary Fibrosis: A Molecular Investigation in an Experimental Model of Fibrosis and Intermittent Hypoxia. Life 2021, 11, 973. [Google Scholar] [CrossRef] [PubMed]
- Verheijden, K.A.T.; Henricks, P.A.J.; Redegeld, F.A.; Garssen, J.; Folkerts, G. Measurement of Airway Function Using Invasive and Non-Invasive Methods in Mild and Severe Models for Allergic Airway Inflammation in Mice. Front. Pharmacol. 2014, 5, 190. Available online: https://www.frontiersin.org/article/10.3389/fphar.2014.00190 (accessed on 4 May 2022). [CrossRef]
- Gille, T.; Didier, M.; Rotenberg, C.; Delbrel, E.; Marchant, D.; Sutton, A.; Dard, N.; Haine, L.; Voituron, N.; Bernaudin, J.-F.; et al. Intermittent Hypoxia Increases the Severity of Bleomycin-Induced Lung Injury in Mice. Oxidative Med. Cell. Longev. 2018, 2018, 1240192. [Google Scholar] [CrossRef]
- Sesé, L.; Nunes, H.; Cottin, V.; Sanyal, S.; Didier, M.; Carton, Z.; Israel-Biet, D.; Crestani, B.; Cadranel, J.; Wallaert, B.; et al. Role of atmospheric pollution on the natural history of idiopathic pulmonary fibrosis. Thorax 2018, 73, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Johannson, K.A.; Vittinghoff, E.; Lee, K.; Balmes, J.R.; Ji, W.; Kaplan, G.G.; Kim, D.S.; Collard, H.R. Acute exacerbation of idiopathic pulmonary fibrosis associated with air pollution exposure. Eur. Respir. J. 2014, 43, 1124–1131. [Google Scholar] [CrossRef] [PubMed]
- Biondini, D.; Balestro, E.; Sverzellati, N.; Cocconcelli, E.; Bernardinello, N.; Ryerson, C.J.; Spagnolo, P. Acute exacerbations of idiopathic pulmonary fibrosis (AE-IPF): An overview of current and future therapeutic strategies. Expert Rev. Respir. Med. 2020, 14, 405–414. [Google Scholar] [CrossRef]
- Moore, B.B.; Hogaboam, C.M. Murine models of pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 294, L152–L160. [Google Scholar] [CrossRef]
- Kimura, T.; Nojiri, T.; Hosoda, H.; Shintani, Y.; Inoue, M.; Miyazato, M.; Okumura, M.; Kangawa, K. Exacerbation of bleomycin-induced injury by lipopolysaccharide in mice: Establishment of a mouse model for acute exacerbation of interstitial lung diseases. Eur. J. Cardiothorac. Surg. 2015, 48, e85–e91. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Remy-Jardin, M.; Myers, J.L.; Richeldi, L.; Ryerson, C.J.; Lederer, D.J.; Behr, J.; Cottin, V.; Danoff, S.K.; Morell, F.; et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2018, 198, e44–e68. [Google Scholar] [CrossRef] [PubMed]
- King, T.E.; Pardo, A.; Selman, M. Idiopathic pulmonary fibrosis. Lancet 2011, 378, 1949–1961. [Google Scholar] [CrossRef]
- Selman, M.; King, T.E.; Pardo, A. American Thoracic Society, European Respiratory Society, American College of Chest Physicians. Idiopathic pulmonary fibrosis: Prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Ann. Intern. Med. 2001, 134, 136–151. [Google Scholar] [CrossRef]
- Álvarez, D.; Cárdenes, N.; Sellarés, J.; Bueno, M.; Corey, C.; Hanumanthu, V.S.; Peng, Y.; D’Cunha, H.; Sembrat, J.; Nouraie, M.; et al. IPF lung fibroblasts have a senescent phenotype. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 313, L1164–L1173. [Google Scholar]
- Disayabutr, S.; Kim, E.K.; Cha, S.I.; Green, G.; Naikawadi, R.P.; Jones, K.D.; Golden, J.A.; Schroeder, A.; Matthay, M.A.; Kukreja, J.; et al. miR-34 miRNAs Regulate Cellular Senescence in Type II Alveolar Epithelial Cells of Patients with Idiopathic Pulmonary Fibrosis. PLoS ONE 2016, 11, e0158367. [Google Scholar] [CrossRef]
- Wu, Q.; Zhang, K.J.; Jiang, S.M.; Fu, L.; Shi, Y.; Tan, R.B.; Cui, J.; Zhou, Y. p53: A Key Protein That Regulates Pulmonary Fibrosis. Oxidative Med. Cell. Longev. 2020, 2020, e6635794. [Google Scholar] [CrossRef]
- Konigsberg, I.R.; Borie, R.; Walts, A.D.; Cardwell, J.; Rojas, M.; Metzger, F.; Hauck, S.M.; Fingerlin, T.E.; Yang, I.V.; Schwartz, D.A. Molecular Signatures of Idiopathic Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2021, 65, 430–441. [Google Scholar] [CrossRef]
- Chen, S.S.; Yin, Z.F.; Chen, T.; Qiu, H.; Wei, Y.R.; Du, S.S.; Jin, Y.-P.; Zhao, M.-M.; Wu, Q.; Weng, D.; et al. Development of a non-infectious rat model of acute exacerbation of idiopathic pulmonary fibrosis. J. Thorac. Dis. 2017, 9, 96–105. [Google Scholar] [CrossRef]
- Marchioni, A.; Tonelli, R.; Ball, L.; Fantini, R.; Castaniere, I.; Cerri, S.; Luppi, F.; Malerba, M.; Pelosi, P.; Clini, E. Acute exacerbation of idiopathic pulmonary fibrosis: Lessons learned from acute respiratory distress syndrome? Crit. Care 2018, 22, 80. [Google Scholar] [CrossRef] [PubMed]
- Schupp, J.C.; Binder, H.; Jäger, B.; Cillis, G.; Zissel, G.; Müller-Quernheim, J.; Prasse, A. Macrophage activation in acute exacerbation of idiopathic pulmonary fibrosis. PLoS ONE 2015, 10, e0116775. [Google Scholar] [CrossRef] [PubMed]
- Papiris, S.A.; Tomos, I.P.; Karakatsani, A.; Spathis, A.; Korbila, I.; Analitis, A.; Kolilekas, L.; Kagouridis, K.; Loukides, S.; Karakitsos, P.; et al. High levels of IL-6 and IL-8 characterize early-on idiopathic pulmonary fibrosis acute exacerbations. Cytokine 2018, 102, 168–172. [Google Scholar] [CrossRef]
- Mellor, H.R.; Harris, A.L. The role of the hypoxia-inducible BH3-only proteins BNIP3 and BNIP3L in cancer. Cancer Metastasis Rev. 2007, 26, 553–566. [Google Scholar] [CrossRef] [PubMed]
- Ueno, M.; Maeno, T.; Nomura, M.; Aoyagi-Ikeda, K.; Matsui, H.; Hara, K.; Tanaka, T.; Iso, T.; Suga, T.; Kurabayashi, M. Hypoxia-inducible factor-1α mediates TGF-β-induced PAI-1 production in alveolar macrophages in pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 300, L740–L752. [Google Scholar] [CrossRef] [PubMed]
- Chao, J.; Wood, J.G.; Gonzalez, N.C. Alveolar macrophages initiate the systemic microvascular inflammatory response to alveolar hypoxia. Respir. Physiol. Neurobiol. 2011, 178, 439–448. [Google Scholar] [CrossRef]
- Uzunhan, Y.; Bernard, O.; Marchant, D.; Dard, N.; Vanneaux, V.; Larghero, J.; Gille, T.; Clerici, C.; Valeyre, M.; Nunes, H.; et al. Mesenchymal stem cells protect from hypoxia-induced alveolar epithelial-mesenchymal transition. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 310, L439–L451. [Google Scholar] [CrossRef] [PubMed]
- Burman, A.; Kropski, J.A.; Calvi, C.L.; Serezani, A.P.; Pascoalino, B.D.; Han, W.; Sherrill, T.; Gleaves, L.; Lawson, W.E.; Young, L.R.; et al. Localized hypoxia links ER stress to lung fibrosis through induction of C/EBP homologous protein. JCI Insight 2018, 3, e99543. [Google Scholar] [CrossRef]
- Delbrel, E.; Soumare, A.; Naguez, A.; Label, R.; Bernard, O.; Bruhat, A.; Fafournoux, P.; Tremblais, G.; Marchant, D.; Gille, T.; et al. HIF-1α triggers ER stress and CHOP-mediated apoptosis in alveolar epithelial cells, a key event in pulmonary fibrosis. Sci. Rep. 2018, 8, 17939. [Google Scholar] [CrossRef]
- Delbrel, E.; Uzunhan, Y.; Soumare, A.; Gille, T.; Marchant, D.; Planès, C.; Boncoeur, E. ER Stress is Involved in Epithelial-To-Mesenchymal Transition of Alveolar Epithelial Cells Exposed to a Hypoxic Microenvironment. Int. J. Mol. Sci. 2019, 20, 1299. [Google Scholar] [CrossRef] [PubMed]
- Tzouvelekis, A.; Harokopos, V.; Paparountas, T.; Oikonomou, N.; Chatziioannou, A.; Vilaras, G.; Tsiambas, E.; Karameris, A.; Bouros, D.; Aidinis, V. Comparative expression profiling in pulmonary fibrosis suggests a role of hypoxia-inducible factor-1alpha in disease pathogenesis. Am. J. Respir. Crit. Care Med. 2007, 176, 1108–1119. [Google Scholar] [CrossRef] [PubMed]
- Suresh, M.V.; Balijepalli, S.; Zhang, B.; Singh, V.V.; Swamy, S.; Panicker, S.; Dolgachev, V.A.; Subramanian, C.; Ramakrishnan, S.K.; Thomas, B.; et al. Hypoxia-Inducible Factor (HIF)-1α Promotes Inflammation and Injury Following Aspiration-Induced Lung Injury in Mice. Shock 2019, 52, 612–621. [Google Scholar] [CrossRef] [PubMed]
- Finnerty, J.P.; Ponnuswamy, A.; Dutta, P.; Abdelaziz, A.; Kamil, H. Efficacy of antifibrotic drugs, nintedanib and pirfenidone, in treatment of progressive pulmonary fibrosis in both idiopathic pulmonary fibrosis (IPF) and non-IPF: A systematic review and meta-analysis. BMC Pulm. Med. 2021, 21, 411. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | References | Host | Antibody type | Dilution |
|---|---|---|---|---|
| F4-80 | D2S9R, Cell Signaling Technology | Rabbit | Monoclonal | 1:250 |
| CD3 | D4V8L, Cell Signaling Technology | Rabbit | Monoclonal | 1:200 |
| CD19 | D4V4L, Cell Signaling Technology | Rabbit | Monoclonal | 1:800 |
| HIF-1α | NB100-479, Novus Biological | Rabbit | Polyclonal | 1:750 |
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| BNIP3 | 5′-TTT-GGG-ATC-TAC-ATT-GGA-AGG-C-3′ | 5′-GTG-CAG-ACA-CCC-AAG-GAT-CA-3′ |
| SERPINE1 | 5′-GCA-CAA-CCC-GAC-AGA-GAC-AA-3′ | 5′-ATG-AAG-GCG-TCT-CTT-CCC-AC-3′ |
| 18S | 5′-GTA-AGT-GCG-GGC-CAT-AAG-CTT-3′ | 5′-AGT-CAA-GTT-CGA-CCG-TCT-TCT-CA-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yegen, C.-H.; Haine, L.; Da Costa Ferreira, K.; Marchant, D.; Bernaudin, J.-F.; Planès, C.; Voituron, N.; Boncoeur, E. A New Model of Acute Exacerbation of Experimental Pulmonary Fibrosis in Mice. Cells 2022, 11, 3379. https://doi.org/10.3390/cells11213379
Yegen C-H, Haine L, Da Costa Ferreira K, Marchant D, Bernaudin J-F, Planès C, Voituron N, Boncoeur E. A New Model of Acute Exacerbation of Experimental Pulmonary Fibrosis in Mice. Cells. 2022; 11(21):3379. https://doi.org/10.3390/cells11213379
Chicago/Turabian StyleYegen, Céline-Hivda, Liasmine Haine, Kevin Da Costa Ferreira, Dominique Marchant, Jean-Francois Bernaudin, Carole Planès, Nicolas Voituron, and Emilie Boncoeur. 2022. "A New Model of Acute Exacerbation of Experimental Pulmonary Fibrosis in Mice" Cells 11, no. 21: 3379. https://doi.org/10.3390/cells11213379
APA StyleYegen, C.-H., Haine, L., Da Costa Ferreira, K., Marchant, D., Bernaudin, J.-F., Planès, C., Voituron, N., & Boncoeur, E. (2022). A New Model of Acute Exacerbation of Experimental Pulmonary Fibrosis in Mice. Cells, 11(21), 3379. https://doi.org/10.3390/cells11213379