


Extracellular Vesicles Isolated from Plasma of Multiple Myeloma Patients Treated with Daratumumab Express CD38, PD-L1, and the Complement Inhibitory Proteins CD55 and CD59
 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Population and Sample Collection
2.2. Antibodies and Reagents
2.3. EV Isolation and Iodixanol Density Gradient Separation
2.3.1. Peripheral Blood (PB) Platelet Free Plasma (PB PFP)
2.3.2. Bone Marrow (BM) Aspirate Platelet Free Plasma (BM PFP)
2.3.3. Iodixanol Density Gradient Centrifugation
2.4. Flow Cytometric Analysis
2.4.1. EV Detection and Counting
2.4.2. EV-Bead Conjugated Flow Cytometry
2.5. SDS-Polyacrylamide Gel Electrophoresis (SDS-PAGE) and Western Blot Analysis
2.6. Transmission Electron Microscopy and Nanoparticle Tracking Analysis
2.7. Mass spectrometry
Data Analysis
2.8. Statistical Analysis
3. Results
3.1. Patient Characteristics
3.2. Expression of EV Markers on MM PB and BM EVs
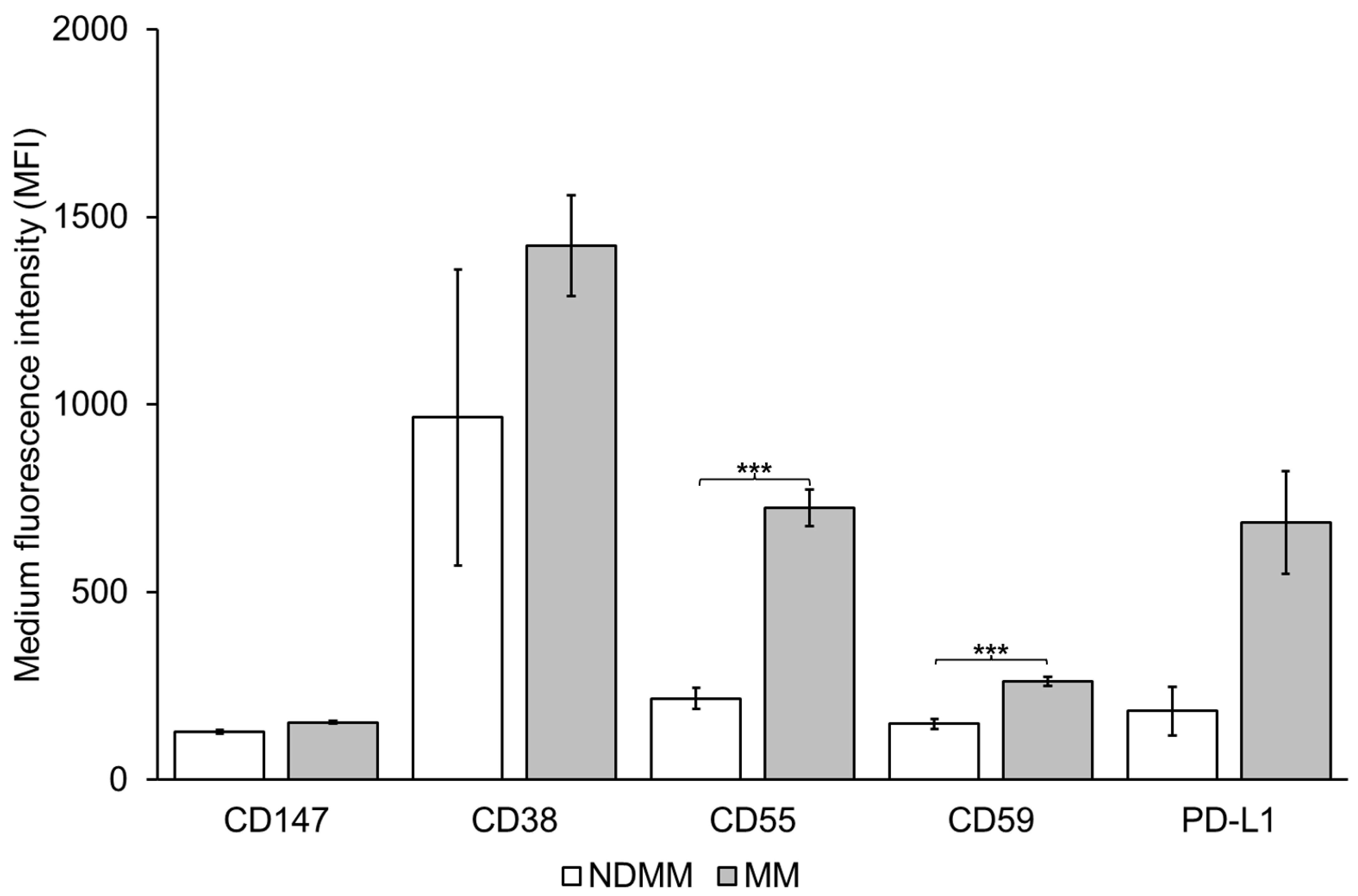
3.3. Expression of CD38, CD55, CD59 and PD-L1 on MM PB EVs
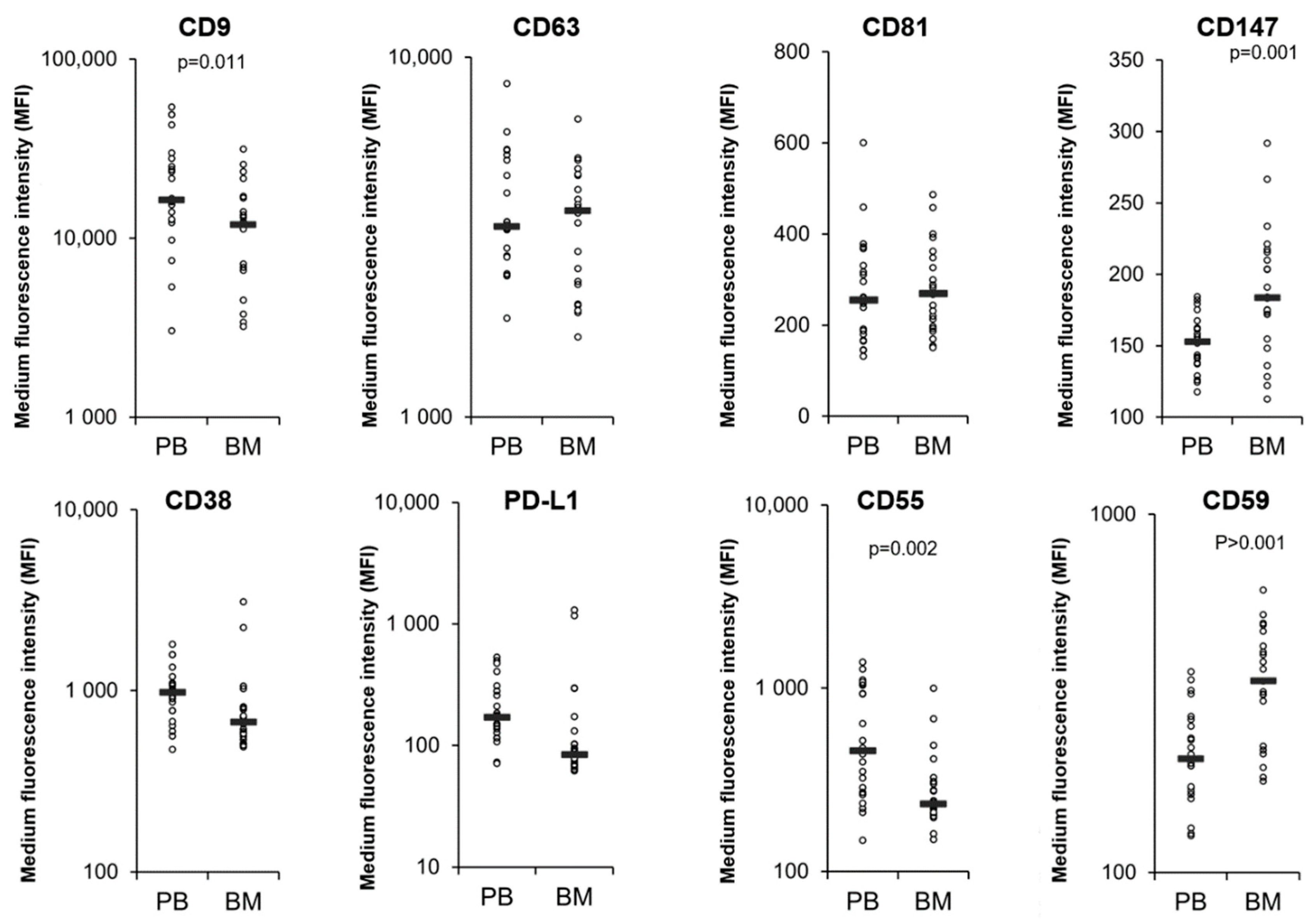
3.4. Comparison of MM PB and BM EVs
3.5. DARA Is Present in MM PB EVs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kyle, R.A.; Rajkumar, S.V. Multiple myeloma. Blood 2008, 111, 2962–2972. [Google Scholar] [CrossRef]
- Moreau, P.; Richardson, P.G.; Cavo, M.; Orlowski, R.Z.; San Miguel, J.F.; Palumbo, A.; Harousseau, J.L. Proteasome inhibitors in multiple myeloma: 10 Years later. Blood 2012, 120, 947–959. [Google Scholar] [CrossRef]
- Quach, H.; Kalff, A.; Spencer, A. Lenalidomide in multiple myeloma: Current status and future potential. Am. J. Hematol. 2012, 87, 1089–1095. [Google Scholar] [CrossRef]
- Lokhorst, H.M.; Plesner, T.; Laubach, J.P.; Nahi, H.; Gimsing, P.; Hansson, M.; Minnema, M.C.; Lassen, U.; Krejcik, J.; Palumbo, A.; et al. Targeting CD38 with Daratumumab Monotherapy in Multiple Myeloma. N. Engl. J. Med. 2015, 373, 1207–1219. [Google Scholar] [CrossRef]
- de Weers, M.; Tai, Y.T.; van der Veer, M.S.; Bakker, J.M.; Vink, T.; Jacobs, D.C.; Oomen, L.A.; Peipp, M.; Valerius, T.; Slootstra, J.W.; et al. Daratumumab, a novel therapeutic human CD38 monoclonal antibody, induces killing of multiple myeloma and other hematological tumors. J. Immunol. 2011, 186, 1840–1848. [Google Scholar] [CrossRef]
- Krejcik, J.; Casneuf, T.; Nijhof, I.S.; Verbist, B.; Bald, J.; Plesner, T.; Syed, K.; Liu, K.; van de Donk, N.W.; Weiss, B.M.; et al. Daratumumab depletes CD38+ immune regulatory cells, promotes T-cell expansion, and skews T-cell repertoire in multiple myeloma. Blood 2016, 128, 384–394. [Google Scholar] [CrossRef]
- Marlein, C.R.; Piddock, R.E.; Mistry, J.J.; Zaitseva, L.; Hellmich, C.; Horton, R.H.; Zhou, Z.; Auger, M.J.; Bowles, K.M.; Rushworth, S.A. CD38-Driven Mitochondrial Trafficking Promotes Bioenergetic Plasticity in Multiple Myeloma. Cancer Res. 2019, 79, 2285–2297. [Google Scholar] [CrossRef]
- Ghose, J.; Viola, D.; Terrazas, C.; Caserta, E.; Troadec, E.; Khalife, J.; Gunes, E.G.; Sanchez, J.; McDonald, T.; Marcucci, G.; et al. Daratumumab induces CD38 internalization and impairs myeloma cell adhesion. Oncoimmunology 2018, 7, e1486948. [Google Scholar] [CrossRef]
- Chatterjee, S.; Daenthanasanmak, A.; Chakraborty, P.; Wyatt, M.W.; Dhar, P.; Selvam, S.P.; Fu, J.; Zhang, J.; Nguyen, H.; Kang, I.; et al. CD38-NAD+ Axis Regulates Immunotherapeutic Anti-Tumor T Cell Response. Cell Metab. 2018, 27, 85–100.e8. [Google Scholar] [CrossRef]
- Martin, T.G.; Corzo, K.; Chiron, M.; Velde, H.V.; Abbadessa, G.; Campana, F.; Solanki, M.; Meng, R.; Lee, H.; Wiederschain, D.; et al. Therapeutic Opportunities with Pharmacological Inhibition of CD38 with Isatuximab. Cells 2019, 8, 1522. [Google Scholar] [CrossRef]
- Nijhof, I.S.; Casneuf, T.; van Velzen, J.; van Kessel, B.; Axel, A.E.; Syed, K.; Groen, R.W.; van Duin, M.; Sonneveld, P.; Minnema, M.C.; et al. CD38 expression and complement inhibitors affect response and resistance to daratumumab therapy in myeloma. Blood 2016, 128, 959–970. [Google Scholar] [CrossRef]
- Overdijk, M.B.; Verploegen, S.; Bögels, M.; van Egmond, M.; Lammerts van Bueren, J.J.; Mutis, T.; Groen, R.W.; Breij, E.; Martens, A.C.; Bleeker, W.K.; et al. Antibody-mediated phagocytosis contributes to the anti-tumor activity of the therapeutic antibody daratumumab in lymphoma and multiple myeloma. MAbs 2015, 7, 311–321. [Google Scholar] [CrossRef]
- Malavasi, F.; Faini, A.C.; Morandi, F.; Castella, B.; Incarnato, D.; Oliviero, S.; Horenstein, A.L.; Massaia, M.; van de Donk, N.; Richardson, P.G. Molecular dynamics of targeting CD38 in multiple myeloma. Br. J. Haematol. 2021, 193, 581–591. [Google Scholar] [CrossRef]
- Sanchez, L.; Dardac, A.; Madduri, D.; Richard, S.; Richter, J. B-cell maturation antigen (BCMA) in multiple myeloma: The new frontier of targeted therapies. Ther. Adv. Hematol. 2021, 12, 2040620721989585. [Google Scholar] [CrossRef]
- Gandhi, U.H.; Cornell, R.F.; Lakshman, A.; Gahvari, Z.J.; McGehee, E.; Jagosky, M.H.; Gupta, R.; Varnado, W.; Fiala, M.A.; Chhabra, S.; et al. Outcomes of patients with multiple myeloma refractory to CD38-targeted monoclonal antibody therapy. Leukemia 2019, 33, 2266–2275. [Google Scholar] [CrossRef]
- Szabo, A.G.; Thorsen, J.; Iversen, K.F.; Levring, M.B.; Preiss, B.; Helleberg, C.; Breinholt, M.F.; Hermansen, E.; Gjerdrum, L.M.R.; Bønløkke, S.T.; et al. The clinical course and life expectancy of patients with multiple myeloma who discontinue their first daratumumab-containing line of therapy. Am. J. Hematol. 2021, 138, 3779–3781. [Google Scholar] [CrossRef]
- Nijhof, I.S.; Groen, R.W.; Lokhorst, H.M.; van Kessel, B.; Bloem, A.C.; van Velzen, J.; de Jong-Korlaar, R.; Yuan, H.; Noort, W.A.; Klein, S.K.; et al. Upregulation of CD38 expression on multiple myeloma cells by all-trans retinoic acid improves the efficacy of daratumumab. Leukemia 2015, 29, 2039–2049. [Google Scholar] [CrossRef]
- Frerichs, K.A.; Minnema, M.C.; Levin, M.D.; Broijl, A.; Bos, G.M.J.; Kersten, M.J.; Mutis, T.; Verkleij, C.P.M.; Nijhof, I.S.; Maas-Bosman, P.W.C.; et al. Efficacy and safety of daratumumab combined with all-trans retinoic acid in relapsed/refractory multiple myeloma. Blood Adv. 2021, 5, 5128–5139. [Google Scholar] [CrossRef]
- Plesner, T.; van de Donk, N.; Richardson, P.G. Controversy in the Use of CD38 Antibody for Treatment of Myeloma: Is High CD38 Expression Good or Bad? Cells 2020, 9, 378. [Google Scholar] [CrossRef]
- Tkach, M.; Théry, C. Communication by Extracellular Vesicles: Where We Are and Where We Need to Go. Cell 2016, 164, 1226–1232. [Google Scholar] [CrossRef]
- Skotland, T.; Sandvig, K.; Llorente, A. Lipids in exosomes: Current knowledge and the way forward. Prog. Lipid Res. 2017, 66, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Thery, C.; Ostrowski, M.; Segura, E. Membrane vesicles as conveyors of immune responses. Nat. Rev. Immunol. 2009, 9, 581–593. [Google Scholar] [CrossRef] [PubMed]
- Nishida-Aoki, N.; Tominaga, N.; Takeshita, F.; Sonoda, H.; Yoshioka, Y.; Ochiya, T. Disruption of Circulating Extracellular Vesicles as a Novel Therapeutic Strategy against Cancer Metastasis. Mol. Ther. 2017, 25, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.G.; Grizzle, W.E. Exosomes and cancer: A newly described pathway of immune suppression. Clin. Cancer Res. 2011, 17, 959–964. [Google Scholar] [CrossRef] [PubMed]
- Maia, J.; Caja, S.; Strano Moraes, M.C.; Couto, N.; Costa-Silva, B. Exosome-Based Cell-Cell Communication in the Tumor Microenvironment. Front. Cell Dev. Biol. 2018, 6, 18. [Google Scholar] [CrossRef]
- Théry, C. Exosomes: Secreted vesicles and intercellular communications. F1000 Biol. Rep. 2011, 3, 15. [Google Scholar] [CrossRef]
- Ciravolo, V.; Huber, V.; Ghedini, G.C.; Venturelli, E.; Bianchi, F.; Campiglio, M.; Morelli, D.; Villa, A.; Della Mina, P.; Menard, S.; et al. Potential role of HER2-overexpressing exosomes in countering trastuzumab-based therapy. J. Cell. Physiol. 2012, 227, 658–667. [Google Scholar] [CrossRef]
- Donin, N.; Jurianz, K.; Ziporen, L.; Schultz, S.; Kirschfink, M.; Fishelson, Z. Complement resistance of human carcinoma cells depends on membrane regulatory proteins, protein kinases and sialic acid. Clin. Exp. Immunol. 2003, 131, 254–263. [Google Scholar] [CrossRef]
- Gelderman, K.A.; Tomlinson, S.; Ross, G.D.; Gorter, A. Complement function in mAb-mediated cancer immunotherapy. Trends Immunol. 2004, 25, 158–164. [Google Scholar] [CrossRef]
- Rosenblatt, J.; Avigan, D. Targeting the PD-1/PD-L1 axis in multiple myeloma: A dream or a reality? Blood 2017, 129, 275–279. [Google Scholar] [CrossRef]
- Chen, G.; Huang, A.C.; Zhang, W.; Zhang, G.; Wu, M.; Xu, W.; Yu, Z.; Yang, J.; Wang, B.; Sun, H.; et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature 2018, 560, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.; Tamada, K.; et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat. Med. 2002, 8, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, M.; Tamura, H.; Sunakawa, M.; Kondo-Onodera, A.; Okuyama, N.; Hamada, Y.; Moriya, K.; Choi, I.; Tamada, K.; Inokuchi, K. Myeloma Drug Resistance Induced by Binding of Myeloma B7-H1 (PD-L1) to PD-1. Cancer Immunol. Res. 2016, 4, 779–788. [Google Scholar] [CrossRef] [PubMed]
- Tamura, H.; Ishibashi, M.; Yamashita, T.; Tanosaki, S.; Okuyama, N.; Kondo, A.; Hyodo, H.; Shinya, E.; Takahashi, H.; Dong, H.; et al. Marrow stromal cells induce B7-H1 expression on myeloma cells, generating aggressive characteristics in multiple myeloma. Leukemia 2013, 27, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Ayala-Mar, S.; Donoso-Quezada, J.; González-Valdez, J. Clinical Implications of Exosomal PD-L1 in Cancer Immunotherapy. J. Immunol. Res. 2021, 2021, 8839978. [Google Scholar] [CrossRef]
- Verkleij, C.P.M.; Jhatakia, A.; Broekmans, M.E.C.; Frerichs, K.A.; Zweegman, S.; Mutis, T.; Bezman, N.A.; van de Donk, N. Preclinical Rationale for Targeting the PD-1/PD-L1 Axis in Combination with a CD38 Antibody in Multiple Myeloma and Other CD38-Positive Malignancies. Cancers 2020, 12, 3713. [Google Scholar] [CrossRef]
- Rajkumar, S.V.; Dimopoulos, M.A.; Palumbo, A.; Blade, J.; Merlini, G.; Mateos, M.-V.; Kumar, S.; Hillengass, J.; Kastritis, E.; Richardson, P.; et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014, 15, e538–e548. [Google Scholar] [CrossRef]
- Harris, P.A.; Taylor, R.; Minor, B.L.; Elliott, V.; Fernandez, M.; O’Neal, L.; McLeod, L.; Delacqua, G.; Delacqua, F.; Kirby, J.; et al. The REDCap consortium: Building an international community of software platform partners. J. Biomed. Inform. 2019, 95, 103208. [Google Scholar] [CrossRef]
- Harris, P.A.; Taylor, R.; Thielke, R.; Payne, J.; Gonzalez, N.; Conde, J.G. Research electronic data capture (REDCap)—A metadata-driven methodology and workflow process for providing translational research informatics support. J. Biomed. Inform. 2009, 42, 377–381. [Google Scholar] [CrossRef]
- Livshits, M.A.; Khomyakova, E.; Evtushenko, E.G.; Lazarev, V.N.; Kulemin, N.A.; Semina, S.E.; Generozov, E.V.; Govorun, V.M. Isolation of exosomes by differential centrifugation: Theoretical analysis of a commonly used protocol. Sci. Rep. 2015, 5, 17319. [Google Scholar] [CrossRef]
- Brennan, K.; Martin, K.; FitzGerald, S.P.; O’Sullivan, J.; Wu, Y.; Blanco, A.; Richardson, C.; Mc Gee, M.M. A comparison of methods for the isolation and separation of extracellular vesicles from protein and lipid particles in human serum. Sci. Rep. 2020, 10, 1039. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Brennan, K.; Fernández, A.B.; Mc Gee, M.M. Cyclophilin A regulates secretion of tumour-derived extracellular vesicles. Transl. Oncol. 2021, 14, 101112. [Google Scholar] [CrossRef] [PubMed]
- Meier, F.; Beck, S.; Grassl, N.; Lubeck, M.; Park, M.A.; Raether, O.; Mann, M. Parallel Accumulation-Serial Fragmentation (PASEF): Multiplying Sequencing Speed and Sensitivity by Synchronized Scans in a Trapped Ion Mobility Device. J. Proteome Res. 2015, 14, 5378–5387. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, S.V.; Harousseau, J.L.; Durie, B.; Anderson, K.C.; Dimopoulos, M.; Kyle, R.; Blade, J.; Richardson, P.; Orlowski, R.; Siegel, D.; et al. Consensus recommendations for the uniform reporting of clinical trials: Report of the International Myeloma Workshop Consensus Panel 1. Blood 2011, 117, 4691–4695. [Google Scholar] [CrossRef]
- Palumbo, A.; Avet-Loiseau, H.; Oliva, S.; Lokhorst, H.M.; Goldschmidt, H.; Rosinol, L.; Richardson, P.; Caltagirone, S.; Lahuerta, J.J.; Facon, T.; et al. Revised International Staging System for Multiple Myeloma: A Report From International Myeloma Working Group. J. Clin. Oncol. 2015, 33, 2863–2869. [Google Scholar] [CrossRef]
- Arendt, B.K.; Walters, D.K.; Wu, X.; Tschumper, R.C.; Huddleston, P.M.; Henderson, K.J.; Dispenzieri, A.; Jelinek, D.F. Increased expression of extracellular matrix metalloproteinase inducer (CD147) in multiple myeloma: Role in regulation of myeloma cell proliferation. Leukemia 2012, 26, 2286–2296. [Google Scholar] [CrossRef][Green Version]
- Łacina, P.; Butrym, A.; Frontkiewicz, D.; Mazur, G.; Bogunia-Kubik, K. Soluble CD147 (BSG) as a Prognostic Marker in Multiple Myeloma. Curr. Issues Mol. Biol. 2022, 44, 350–359. [Google Scholar] [CrossRef]
- De Luca, L.; Laurenzana, I.; Trino, S.; Lamorte, D.; Caivano, A.; Musto, P. An update on extracellular vesicles in multiple myeloma: A focus on their role in cell-to-cell cross-talk and as potential liquid biopsy biomarkers. Expert Rev. Mol. Diagn. 2019, 19, 249–258. [Google Scholar] [CrossRef]
- Aung, T.; Chapuy, B.; Vogel, D.; Wenzel, D.; Oppermann, M.; Lahmann, M.; Weinhage, T.; Menck, K.; Hupfeld, T.; Koch, R.; et al. Exosomal evasion of humoral immunotherapy in aggressive B-cell lymphoma modulated by ATP-binding cassette transporter A3. Proc. Natl. Acad. Sci. USA 2011, 108, 15336–15341. [Google Scholar] [CrossRef]
- Shah, R.; Patel, T.; Freedman, J.E. Circulating Extracellular Vesicles in Human Disease. N. Engl. J. Med. 2018, 379, 958–966. [Google Scholar] [CrossRef]
- Poggio, M.; Hu, T.; Pai, C.C.; Chu, B.; Belair, C.D.; Chang, A.; Montabana, E.; Lang, U.E.; Fu, Q.; Fong, L.; et al. Suppression of Exosomal PD-L1 Induces Systemic Anti-tumor Immunity and Memory. Cell 2019, 177, 414–427. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Li, C.W.; Chan, L.C.; Wei, Y.; Hsu, J.M.; Xia, W.; Cha, J.H.; Hou, J.; Hsu, J.L.; Sun, L.; et al. Exosomal PD-L1 harbors active defense function to suppress T cell killing of breast cancer cells and promote tumor growth. Cell Res. 2018, 28, 862–864. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo, E.; Vorholt, D.; Blakemore, S.; Sackey, B.; Nolte, J.L.; Barbarino, V.; Schmitz, J.; Nickel, N.; Bachurski, D.; Lobastova, L.; et al. Extracellular vesicles and PD-L1 suppress macrophages, inducing therapy resistance in TP53-deficient B-cell malignancies. Blood 2022, 139, 3617–3629. [Google Scholar] [CrossRef] [PubMed]
- Parsa, A.T.; Waldron, J.S.; Panner, A.; Crane, C.A.; Parney, I.F.; Barry, J.J.; Cachola, K.E.; Murray, J.C.; Tihan, T.; Jensen, M.C.; et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat. Med. 2007, 13, 84–88. [Google Scholar] [CrossRef]
- Lastwika, K.J.; Wilson, W., 3rd; Li, Q.K.; Norris, J.; Xu, H.; Ghazarian, S.R.; Kitagawa, H.; Kawabata, S.; Taube, J.M.; Yao, S.; et al. Control of PD-L1 Expression by Oncogenic Activation of the AKT-mTOR Pathway in Non-Small Cell Lung Cancer. Cancer Res. 2016, 76, 227–238. [Google Scholar] [CrossRef]
- Akbay, E.A.; Koyama, S.; Carretero, J.; Altabef, A.; Tchaicha, J.H.; Christensen, C.L.; Mikse, O.R.; Cherniack, A.D.; Beauchamp, E.M.; Pugh, T.J.; et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov. 2013, 3, 1355–1363. [Google Scholar] [CrossRef]
- Casey, S.C.; Tong, L.; Li, Y.; Do, R.; Walz, S.; Fitzgerald, K.N.; Gouw, A.M.; Baylot, V.; Gütgemann, I.; Eilers, M.; et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science 2016, 352, 227–231. [Google Scholar] [CrossRef]
- Dorand, R.D.; Nthale, J.; Myers, J.T.; Barkauskas, D.S.; Avril, S.; Chirieleison, S.M.; Pareek, T.K.; Abbott, D.W.; Stearns, D.S.; Letterio, J.J.; et al. Cdk5 disruption attenuates tumor PD-L1 expression and promotes antitumor immunity. Science 2016, 353, 399–403. [Google Scholar] [CrossRef]
- Ribas, A.; Hu-Lieskovan, S. What does PD-L1 positive or negative mean? J. Exp. Med. 2016, 213, 2835–2840. [Google Scholar] [CrossRef]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef]
- Herbst, R.S.; Soria, J.C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef] [PubMed]
- McDermott, D.F.; Sosman, J.A.; Sznol, M.; Massard, C.; Gordon, M.S.; Hamid, O.; Powderly, J.D.; Infante, J.R.; Fassò, M.; Wang, Y.V.; et al. Atezolizumab, an Anti-Programmed Death-Ligand 1 Antibody, in Metastatic Renal Cell Carcinoma: Long-Term Safety, Clinical Activity, and Immune Correlates From a Phase Ia Study. J. Clin. Oncol. 2016, 34, 833–842. [Google Scholar] [CrossRef] [PubMed]
- van de Donk, N.W.; Janmaat, M.L.; Mutis, T.; Lammerts van Bueren, J.J.; Ahmadi, T.; Sasser, A.K.; Lokhorst, H.M.; Parren, P.W. Monoclonal antibodies targeting CD38 in hematological malignancies and beyond. Immunol. Rev. 2016, 270, 95–112. [Google Scholar] [CrossRef] [PubMed]
- You, T.; Hu, W.; Ge, X.; Shen, J.; Qin, X. Application of a novel inhibitor of human CD59 for the enhancement of complement-dependent cytolysis on cancer cells. Cell. Mol. Immunol. 2011, 8, 157–163. [Google Scholar] [CrossRef]
- Xiong, L.; Edwards, C.K., 3rd; Zhou, L. The biological function and clinical utilization of CD147 in human diseases: A review of the current scientific literature. Int. J. Mol. Sci. 2014, 15, 17411. [Google Scholar] [CrossRef]
- Tang, Y.; Nakada, M.T.; Kesavan, P.; McCabe, F.; Millar, H.; Rafferty, P.; Bugelski, P.; Yan, L. Extracellular matrix metalloproteinase inducer stimulates tumor angiogenesis by elevating vascular endothelial cell growth factor and matrix metalloproteinases. Cancer Res. 2005, 65, 3193–3199. [Google Scholar] [CrossRef]
- Zhu, D.; Wang, Z.; Zhao, J.J.; Calimeri, T.; Meng, J.; Hideshima, T.; Fulciniti, M.; Kang, Y.; Ficarro, S.B.; Tai, Y.T.; et al. The Cyclophilin A-CD147 complex promotes the proliferation and homing of multiple myeloma cells. Nat. Med. 2015, 21, 572–580. [Google Scholar] [CrossRef]
- Amit-Cohen, B.C.; Rahat, M.M.; Rahat, M.A. Tumor cell-macrophage interactions increase angiogenesis through secretion of EMMPRIN. Front. Physiol. 2013, 4, 178. [Google Scholar] [CrossRef]
- Panchabhai, S.; Kelemen, K.; Ahmann, G.; Sebastian, S.; Mantei, J.; Fonseca, R. Tumor-associated macrophages and extracellular matrix metalloproteinase inducer in prognosis of multiple myeloma. Leukemia 2016, 30, 951–954. [Google Scholar] [CrossRef]
- Panchabhai, S.; Schlam, I.; Sebastian, S.; Fonseca, R. PKM2 and other key regulators of Warburg effect positively correlate with CD147 (EMMPRIN) gene expression and predict survival in multiple myeloma. Leukemia 2017, 31, 991–994. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient Characteristics | Responders n = 19 | Non-Responders n = 38 |
|---|---|---|
| Age *; years; median (range) | 61 (49–83) | 66 (47–84) |
| Sex; n (%) | ||
| Female | 7 (37) | 20 (53) |
| Male | 12 (63) | 18 (47) |
| Immunoglobulin subtype *; n (%) | ||
| IgG | 12 (63) | 25 (66) |
| IgA | 1 (5) | 3 (8) |
| Light-chain only | 6 (32) | 9 (23.5) |
| Non secretory | 0 (0) | 1 (2.5) |
| ISS *; n(%) | ||
| I | 5 (26) | 13 (34) |
| II | 8 (42) | 8 (21) |
| III | 4 (21) | 5 (13) |
| Unknown | 2 (11) | 12 (32) |
| ECOG performance status *; n (%) | ||
| 0 | 9 (47) | 20 (53) |
| 1 | 7 (37) | 6 (16) |
| 2 | 0 (0) | 0 (0) |
| 3 | 0 (0) | 2 (5) |
| Unknown | 3 (16) | 10 (26) |
| Fluorescence in situ hybridization §; n (%) | ||
| High-risk | 1 (5) | 9 (24) |
| Standard-risk | 15 (79) | 19 (50) |
| Unknown | 3 (16) | 10 (26) |
| Number of prior lines of therapy; median (range) | 2 (1–5) | 4 (0–17) |
| DARA-Specific Sequences only Detected in DARA Treated MM Patient EVs | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| MM Patient | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
| Light chain | SNWPPTFGQGTKVEIKRTVAAPSVFIFPPSDEQLKSGTASVVCLLNNFYPR | SLEPEDFAVY | SNWPPTFGQGTKVEIKRTVAAPSVFIFPPSDEQLKSGTASVVCLLNNFYPR | SNWPPTFGQGTKVEIKRTVAAPSVFIFPPSDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSK | ||||||
| Heavy chain | STSGGT | SGGTAA | GPSVFPLAPSSKSTSGGTAALGCLVK | GGTAAL | GPSVFPLAPSSKSTSGGTAALGCLVK | SCDKTHTCPPCPAPELLGGPSVFLFPPKPKDTLMISR | TKGPSVF | |||
| KAKGQP | SGVHTF | ALTSGV | STKGPSVFPLA | |||||||
| KPSNTK | TYICNVNHK | KGPSVF | ||||||||
| VEPKSCDK | SCDKTHTCPPCPAPELLGGPSVFLFPPKPKDTLMISR | HKPSNTKVD | ||||||||
| PSVFLFP | EEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYK | |||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brennan, K.; Iversen, K.F.; Blanco-Fernández, A.; Lund, T.; Plesner, T.; Mc Gee, M.M. Extracellular Vesicles Isolated from Plasma of Multiple Myeloma Patients Treated with Daratumumab Express CD38, PD-L1, and the Complement Inhibitory Proteins CD55 and CD59. Cells 2022, 11, 3365. https://doi.org/10.3390/cells11213365
Brennan K, Iversen KF, Blanco-Fernández A, Lund T, Plesner T, Mc Gee MM. Extracellular Vesicles Isolated from Plasma of Multiple Myeloma Patients Treated with Daratumumab Express CD38, PD-L1, and the Complement Inhibitory Proteins CD55 and CD59. Cells. 2022; 11(21):3365. https://doi.org/10.3390/cells11213365
Chicago/Turabian StyleBrennan, Kieran, Katrine F. Iversen, Alfonso Blanco-Fernández, Thomas Lund, Torben Plesner, and Margaret M. Mc Gee. 2022. "Extracellular Vesicles Isolated from Plasma of Multiple Myeloma Patients Treated with Daratumumab Express CD38, PD-L1, and the Complement Inhibitory Proteins CD55 and CD59" Cells 11, no. 21: 3365. https://doi.org/10.3390/cells11213365
APA StyleBrennan, K., Iversen, K. F., Blanco-Fernández, A., Lund, T., Plesner, T., & Mc Gee, M. M. (2022). Extracellular Vesicles Isolated from Plasma of Multiple Myeloma Patients Treated with Daratumumab Express CD38, PD-L1, and the Complement Inhibitory Proteins CD55 and CD59. Cells, 11(21), 3365. https://doi.org/10.3390/cells11213365