
A Ca2+-Mediated Switch of Epiplakin from a Diffuse to Keratin-Bound State Affects Keratin Dynamics


 and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Immunofluorescence Microscopy
2.3. Co-Sedimentation Assay
2.4. Preparation of Protein Lysates from Cultured Cells
2.5. SDS-PAGE
2.6. Western Blot
2.7. Cloning of cDNAs Encoding Short EPPK1
2.8. Generation of Stable Cell Lines
2.9. Generation of CRISPR/Cas9 Knock-in and EPPK1−/− Cell Lines
2.10. EPPK1−/−
2.11. Knock-in
2.12. Live Cell Imaging
2.13. Time-Lapse Microscopy of Cells during Fixation
2.14. Time-Lapse Microscopy of Cells during Drug Treatment
2.15. UV-A Irradiation
2.16. Monitoring of the Keratin Flow
2.17. Laminin-Rich Coating
2.18. Intensity Correlation Analysis
3. Results
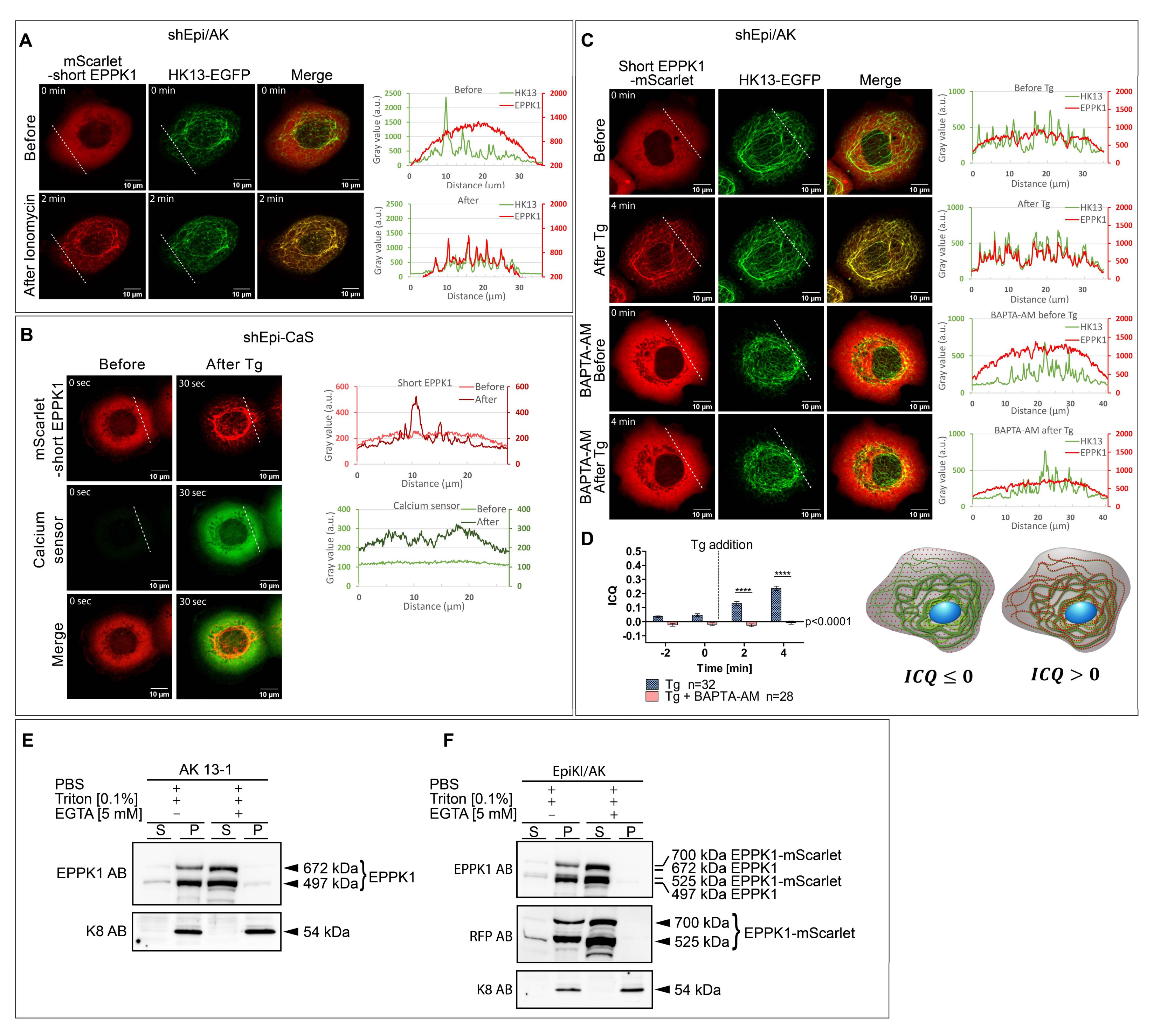
3.1. EPPK1 Is Diffusely Distributed throughout the Cytoplasm of Vital Cells under Standard Cell Culture Conditions but Rapidly Re-Localizes to Keratin Filaments during Fixation
3.2. EPPK1 Re-Localizes to Keratin Filaments during Stress in a Reversible Manner
3.3. EPPK1 Re-Localization to Keratin Filaments Is Ca2+-Dependent
3.4. EPPK1 Association with Keratin Filaments Slows down Keratin Dynamics
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jacob, J.T.; Coulombe, P.A.; Kwan, R.; Omary, M.B. Types I and II Keratin Intermediate Filaments. Cold Spring Harb. Perspect. Biol. 2018, 10, a018275. [Google Scholar] [CrossRef]
- Toivola, D.M.; Boor, P.; Alam, C.; Strnad, P. Keratins in health and disease. Curr. Opin. Cell Biol. 2015, 32, 73–81. [Google Scholar] [CrossRef]
- Yoon, S.; Leube, R.E. Keratin intermediate filaments: Intermediaries of epithelial cell migration. Essays Biochem. 2019, 63, 521–533. [Google Scholar] [CrossRef]
- Windoffer, R.; Beil, M.; Magin, T.M.; Leube, R.E. Cytoskeleton in motion: The dynamics of keratin intermediate filaments in epithelia. J. Cell Biol. 2011, 194, 669–678. [Google Scholar] [CrossRef]
- Sawant, M.S.; Leube, R.E. Consequences of Keratin Phosphorylation for Cytoskeletal Organization and Epithelial Functions. Int. Rev. Cell Mol. Biol. 2017, 330, 171–225. [Google Scholar] [CrossRef]
- Moch, M.; Windoffer, R.; Schwarz, N.; Pohl, R.; Omenzetter, A.; Schnakenberg, U.; Herb, F.; Chaisaowong, K.; Merhof, D.; Ramms, L.; et al. Effects of Plectin Depletion on Keratin Network Dynamics and Organization. PLoS ONE 2016, 11, e0149106. [Google Scholar] [CrossRef] [PubMed]
- Prechova, M.; Adamova, Z.; Schweizer, A.L.; Maninova, M.; Bauer, A.; Kah, D.; Meier-Menches, S.M.; Wiche, G.; Fabry, B.; Gregor, M. Plectin-mediated cytoskeletal crosstalk controls cell tension and cohesion in epithelial sheets. J. Cell Biol. 2022, 221, e202105146. [Google Scholar] [CrossRef] [PubMed]
- Sonnenberg, A.; Liem, R.K. Plakins in development and disease. Exp. Cell Res. 2007, 313, 2189–2203. [Google Scholar] [CrossRef] [PubMed]
- Leung, C.L.; Green, K.J.; Liem, R.K. Plakins: A family of versatile cytolinker proteins. Trends Cell Biol. 2002, 12, 37–45. [Google Scholar] [CrossRef]
- Fogl, C.; Mohammed, F.; Al-Jassar, C.; Jeeves, M.; Knowles, T.J.; Rodriguez-Zamora, P.; White, S.A.; Odintsova, E.; Overduin, M.; Chidgey, M. Mechanism of intermediate filament recognition by plakin repeat domains revealed by envoplakin targeting of vimentin. Nat. Commun. 2016, 7, 10827. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, F.; Trieber, C.; Overduin, M.; Chidgey, M. Molecular mechanism of intermediate filament recognition by plakin proteins. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2020, 1867, 118801. [Google Scholar] [CrossRef] [PubMed]
- Spazierer, D.; Fuchs, P.; Pröll, V.; Janda, L.; Oehler, S.; Fischer, I.; Hauptmann, R.; Wiche, G. Epiplakin gene analysis in mouse reveals a single exon encoding a 725 kDa protein with expression restricted to epithelial tissues. J. Biol. Chem. 2003, 278, 31657–31666. [Google Scholar] [CrossRef]
- Ishikawa, K.; Furuhashi, M.; Sasaki, T.; Kudoh, J.; Tsuchisaka, A.; Hashimoto, T.; Sasaki, T.; Yoshioka, H.; Eshima, N.; Matsuda-Hirose, H.; et al. Intragenic copy number variation within human epiplakin 1 (EPPK1) generates variation of molecular size of epiplakin. J. Dermatol. Sci. 2018, 91, 228–231. [Google Scholar] [CrossRef] [PubMed]
- Ueo, D.; Furuhashi, M.; Sasaki, T.; Kudoh, J.; Parry, D.A.D.; Winter, D.J.; Sasaki, T.; Hashimoto, T.; Tsuruta, D.; Fujiwara, S. Intragenic copy number variation in mouse epiplakin 1 (Eppk1) and the conservation of the repeat structures in the lower vertebrates. J. Dermatol. Sci. 2021, 103, 186–189. [Google Scholar] [CrossRef]
- Fujiwara, S.; Takeo, N.; Otani, Y.; Parry, D.A.; Kunimatsu, M.; Lu, R.; Sasaki, M.; Matsuo, N.; Khaleduzzaman, M.; Yoshioka, H. Epiplakin, a novel member of the Plakin family originally identified as a 450-kDa human epidermal autoantigen. Structure and tissue localization. J. Biol. Chem. 2001, 276, 13340–13347. [Google Scholar] [CrossRef]
- Jang, S.I.; Kalinin, A.; Takahashi, K.; Marekov, L.N.; Steinert, P.M. Characterization of human epiplakin: RNAi-mediated epiplakin depletion leads to the disruption of keratin and vimentin IF networks. J. Cell Sci. 2005, 118, 781–793. [Google Scholar] [CrossRef]
- Spazierer, D.; Raberger, J.; Gross, K.; Fuchs, P.; Wiche, G. Stress-induced recruitment of epiplakin to keratin networks increases their resistance to hyperphosphorylation-induced disruption. J. Cell Sci. 2008, 121, 825–833. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Szabo, S.; Wogenstein, K.L.; Osterreicher, C.H.; Guldiken, N.; Chen, Y.; Doler, C.; Wiche, G.; Boor, P.; Haybaeck, J.; Strnad, P.; et al. Epiplakin attenuates experimental mouse liver injury by chaperoning keratin reorganization. J. Hepatol. 2015, 62, 1357–1366. [Google Scholar] [CrossRef]
- Wang, W.; Sumiyoshi, H.; Yoshioka, H.; Fujiwara, S. Interactions between epiplakin and intermediate filaments. J. Dermatol. 2006, 33, 518–527. [Google Scholar] [CrossRef]
- Spazierer, D.; Fuchs, P.; Reipert, S.; Fischer, I.; Schmuth, M.; Lassmann, H.; Wiche, G. Epiplakin is dispensable for skin barrier function and for integrity of keratin network cytoarchitecture in simple and stratified epithelia. Mol. Cell Biol. 2006, 26, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Goto, M.; Sumiyoshi, H.; Sakai, T.; Fassler, R.; Ohashi, S.; Adachi, E.; Yoshioka, H.; Fujiwara, S. Elimination of epiplakin by gene targeting results in acceleration of keratinocyte migration in mice. Mol. Cell Biol. 2006, 26, 548–558. [Google Scholar] [CrossRef] [PubMed]
- Wögenstein, K.L.; Szabo, S.; Lunova, M.; Wiche, G.; Haybaeck, J.; Strnad, P.; Boor, P.; Wagner, M.; Fuchs, P. Epiplakin deficiency aggravates murine caerulein-induced acute pancreatitis and favors the formation of acinar keratin granules. PLoS ONE 2014, 9, e108323. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef]
- Koch, B.; Nijmeijer, B.; Kueblbeck, M.; Cai, Y.; Walther, N.; Ellenberg, J. Generation and validation of homozygous fluorescent knock-in cells using CRISPR–Cas9 genome editing. Nat. Protoc. 2018, 13, 1465–1487. [Google Scholar] [CrossRef]
- Gruber, F.; Oskolkova, O.; Leitner, A.; Mildner, M.; Mlitz, V.; Lengauer, B.; Kadl, A.; Mrass, P.; Kronke, G.; Binder, B.R.; et al. Photooxidation generates biologically active phospholipids that induce heme oxygenase-1 in skin cells. J. Biol. Chem. 2007, 282, 16934–16941. [Google Scholar] [CrossRef]
- Pora, A.; Yoon, S.; Dreissen, G.; Hoffmann, B.; Merkel, R.; Windoffer, R.; Leube, R.E. Regulation of keratin network dynamics by the mechanical properties of the environment in migrating cells. Sci. Rep. 2020, 10, 4574. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.-K.; Wang, M.-J.J. Image thresholding by minimizing the measures of fuzzines. Pattern Recognit. 1995, 28, 41–51. [Google Scholar] [CrossRef]
- Li, Q.; Lau, A.; Morris, T.J.; Guo, L.; Fordyce, C.B.; Stanley, E.F. A Syntaxin 1, Gαo, and N-Type Calcium Channel Complex at a Presynaptic Nerve Terminal: Analysis by Quantitative Immunocolocalization. J. Neurosci. 2004, 24, 4070–4081. [Google Scholar] [CrossRef]
- Windoffer, R.; Leube, R.E. Detection of cytokeratin dynamics by time-lapse fluorescence microscopy in living cells. J. Cell Sci. 1999, 112 Pt 24, 4521–4534. [Google Scholar] [CrossRef]
- Strnad, P.; Windoffer, R.; Leube, R.E. Induction of rapid and reversible cytokeratin filament network remodeling by inhibition of tyrosine phosphatases. J. Cell Sci. 2002, 115, 4133–4148. [Google Scholar] [CrossRef] [PubMed]
- Werner, N.S.; Windoffer, R.; Strnad, P.; Grund, C.; Leube, R.E.; Magin, T.M. Epidermolysis bullosa simplex-type mutations alter the dynamics of the keratin cytoskeleton and reveal a contribution of actin to the transport of keratin subunits. Mol. Biol. Cell 2004, 15, 990–1002. [Google Scholar] [CrossRef]
- Subramaniam, T.; Fauzi, M.B.; Lokanathan, Y.; Law, J.X. The Role of Calcium in Wound Healing. Int. J. Mol. Sci. 2021, 22, 6486. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J. The Inositol Trisphosphate/Calcium Signaling Pathway in Health and Disease. Physiol. Rev. 2016, 96, 1261–1296. [Google Scholar] [CrossRef]
- Wales, P.; Schuberth, C.E.; Aufschnaiter, R.; Fels, J.; García-Aguilar, I.; Janning, A.; Dlugos, C.P.; Schäfer-Herte, M.; Klingner, C.; Wälte, M.; et al. Calcium-mediated actin reset (CaAR) mediates acute cell adaptations. eLife 2016, 5, e19850. [Google Scholar] [CrossRef] [PubMed]
- Ridge, K.M.; Linz, L.; Flitney, F.W.; Kuczmarski, E.R.; Chou, Y.H.; Omary, M.B.; Sznajder, J.I.; Goldman, R.D. Keratin 8 phosphorylation by protein kinase C delta regulates shear stress-mediated disassembly of keratin intermediate filaments in alveolar epithelial cells. J. Biol. Chem. 2005, 280, 30400–30405. [Google Scholar] [CrossRef] [PubMed]
- Sivaramakrishnan, S.; Schneider, J.L.; Sitikov, A.; Goldman, R.D.; Ridge, K.M. Shear stress induced reorganization of the keratin intermediate filament network requires phosphorylation by protein kinase C zeta. Mol. Biol. Cell 2009, 20, 2755–2765. [Google Scholar] [CrossRef]
- Fuchs, P.; Drexler, C.; Ratajczyk, S.; Eckhart, L. Comparative genomics reveals evolutionary loss of epiplakin in cetaceans. Sci. Rep. 2022, 12, 1112. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Abbreviation | Origin of Cell Line | Special Features |
|---|---|---|
| AK 13-1 | A 431 | HK13-EGFP |
| shEpi/AK | AK 13-1 | HK13-EGFP mScarlet-short EPPK1 |
| EpiKI/AK | AK 13-1 | HK13-EGFP mScarlet-tagged endogenous EPPK1 |
| EpiKO/AK 2C4 | AK 13-1 | HK13-EGFP EPPK1−/− |
| EpiKO/AK 3F2 | AK 13-1 | HK13-EGFP EPPK1−/− |
| shEpi-KO/AK 2C4 | EpiKO/AK 2C4 | HK13-EGFP EPPK1−/− mScarlet-short EPPK1 |
| shEpi-KO/AK 3F2 | EpiKO/AK 3F2 | HK13-EGFP EPPK1−/− mScarlet-short EPPK1 |
| shEpi-CaS | A-431 | mScarlet-short EPPK1-mScarlet Ca2+ sensor |
| shEpi/PK | PK 18-5 | HK18-YFP mCherry-short EPPK1 |
| shEpi/MCF7 | MCF7 | K14-EYFP mScarlet-short EPPK1 |
| Antibody | Vendor/Source | Reference Number | Target | Species | Dilution |
|---|---|---|---|---|---|
| Troma 1 | Developmental Studies Hybridoma Bank, University of Iowa | - | Keratin 8 | Rat | IB: 1:1000 |
| Anti RFP | chromotek | 6g6 | mScarlet | Mouse | IB: 1:1000 |
| EPPK1#66 | ThermoFisher | PA5-66869 | Human EPPK1 | rabbit | IB: 1:200 |
| EPPK1#24 | Abcam | ab247172 | Human EPPK1 | rabbit | IF: 1:50 |
| Antibody | Vendor | Species | Dilution |
|---|---|---|---|
| HRPO anti mouse IgGs | Jackson ImmunoResearch Labs | Goat | IB: 1:20,000 |
| HRPO anti rat IgGs | Jackson ImmunoResearch Labs | Goat | IB: 1:20,000 |
| HRPO anti rabbit IgGs | Vector Laboratories | Goat | IB: 1:20,000 |
| Alexa Fluor 647 anti rabbit | Jackson ImmunoResearch Labs | Donkey | IF: 1:500 |
| Substance | Vendor | Reference Number | Concentration Used |
|---|---|---|---|
| Thapsigargin | Sigma Aldrich | T9033 | 200 nM |
| Hydrogen peroxide | Sigma Aldrich | H1009 | 2.5 mM |
| Ionomycin | Sigma Aldrich | I0634 | 165 nM |
| ATP | Sigma Aldrich | 11140965001 | 50 µM |
| Bradykinin-acetate | Sigma Aldrich | B3259 | 500 nM |
| BAPTA/AM | Sigma Aldrich | 196419 | 20 µM |
| Urea | Sigma Aldrich | U5378 | 300 nM |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ratajczyk, S.; Drexler, C.; Windoffer, R.; Leube, R.E.; Fuchs, P. A Ca2+-Mediated Switch of Epiplakin from a Diffuse to Keratin-Bound State Affects Keratin Dynamics. Cells 2022, 11, 3077. https://doi.org/10.3390/cells11193077
Ratajczyk S, Drexler C, Windoffer R, Leube RE, Fuchs P. A Ca2+-Mediated Switch of Epiplakin from a Diffuse to Keratin-Bound State Affects Keratin Dynamics. Cells. 2022; 11(19):3077. https://doi.org/10.3390/cells11193077
Chicago/Turabian StyleRatajczyk, Sonia, Corinne Drexler, Reinhard Windoffer, Rudolf E. Leube, and Peter Fuchs. 2022. "A Ca2+-Mediated Switch of Epiplakin from a Diffuse to Keratin-Bound State Affects Keratin Dynamics" Cells 11, no. 19: 3077. https://doi.org/10.3390/cells11193077
APA StyleRatajczyk, S., Drexler, C., Windoffer, R., Leube, R. E., & Fuchs, P. (2022). A Ca2+-Mediated Switch of Epiplakin from a Diffuse to Keratin-Bound State Affects Keratin Dynamics. Cells, 11(19), 3077. https://doi.org/10.3390/cells11193077