
Combination Treatment of Retinoic Acid Plus Focal Adhesion Kinase Inhibitor Prevents Tumor Growth and Breast Cancer Cell Metastasis

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
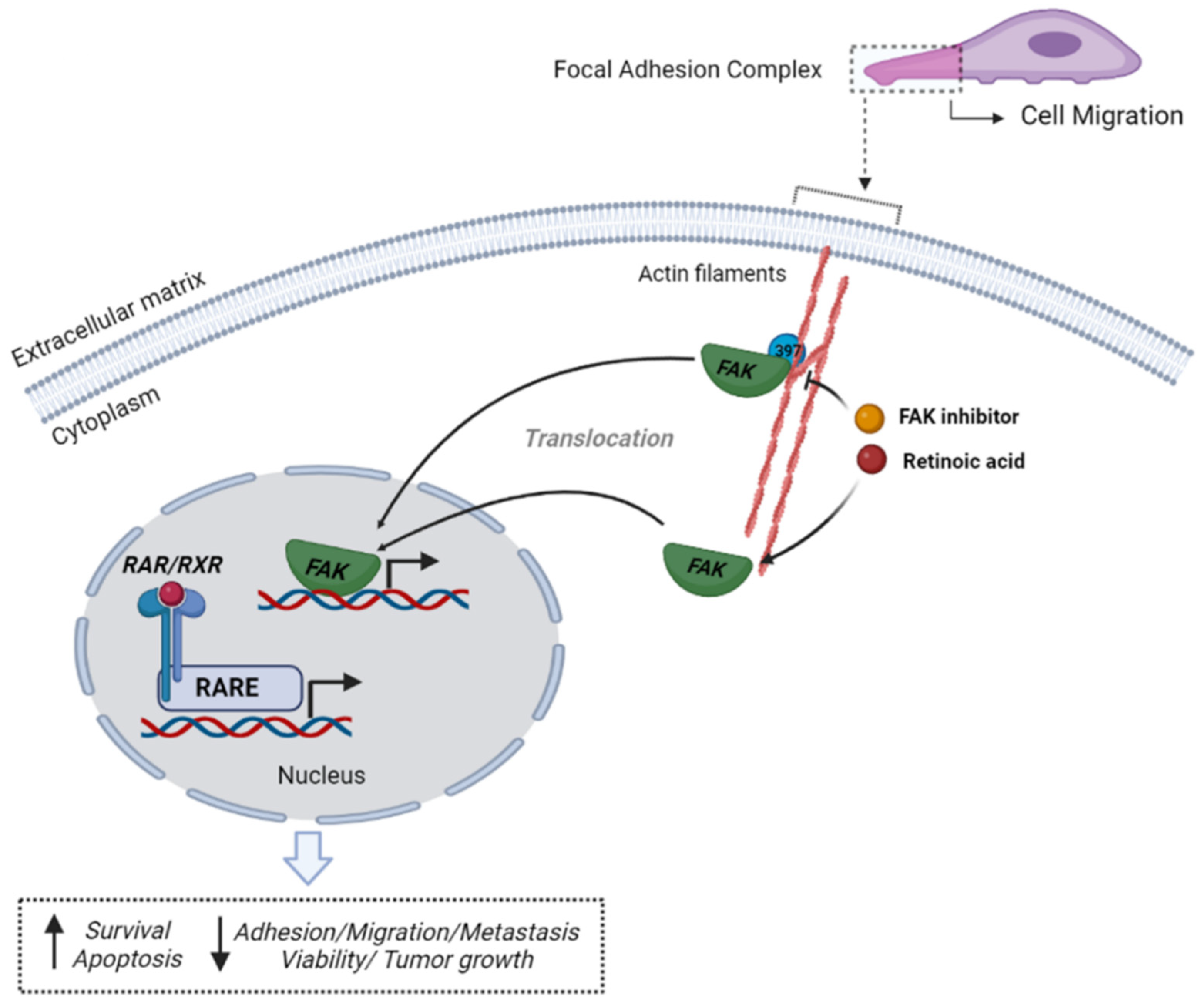
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Treatments
2.2. Bioinformatics
2.2.1. Basal Gene Expression Study in Eight Cell Lines
2.2.2. Gene-Expression Study in RA and Control BC Cell Lines
2.3. Reverse Transcription—Quantitative PCR
2.4. Immunoblotting
2.5. Cell Adhesion Assay
2.6. Wound Healing Assay
2.7. Cell Viability
2.8. Analysis of Drug Interactions
2.9. Cell Immunofluorescence
2.10. Animals
- (1)
- CONTROL (indicated as CON): animals inoculated with LM3 cells received an empty subcutaneous silastic pellet.
- (2)
- RA: animals inoculated with LM3 cells received a subcutaneous silastic pellet containing RA (10 mg).
- (3)
- FAKi: animals inoculated with LM3 cells (pretreated with FAKi) and received an empty subcutaneous silastic pellet.
- (4)
- RA plus FAKi (indicated as RA+FAKi): animals inoculated with LM3 cells (pretreated with FAKi) and received a subcutaneous silastic pellet containing RA (10 mg).
2.10.1. Orthotopic Tumor Growth
2.10.2. Experimental Lung Metastasis Assay
2.11. Histological Analysis
2.12. Statistical Analysis
3. Results
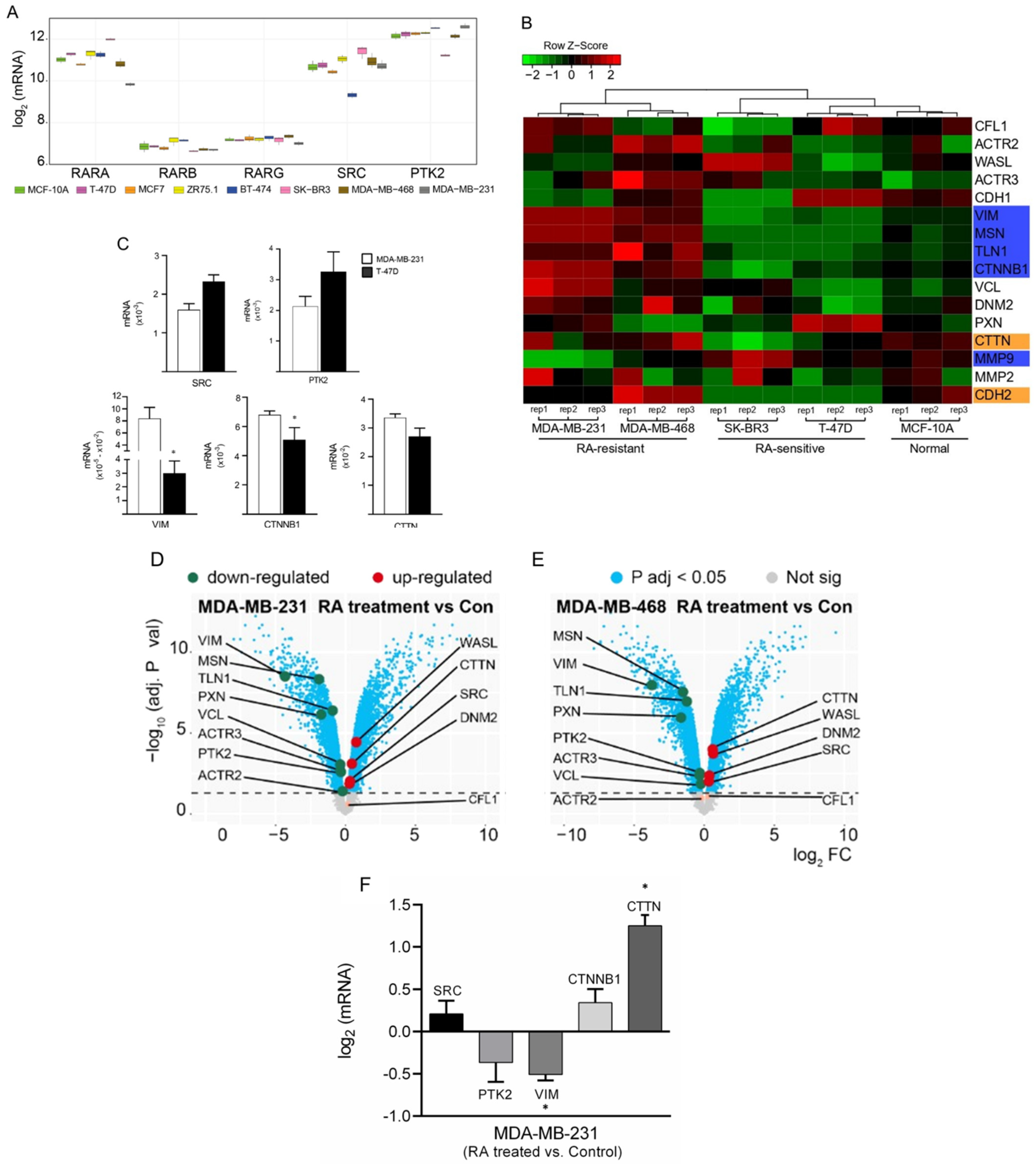
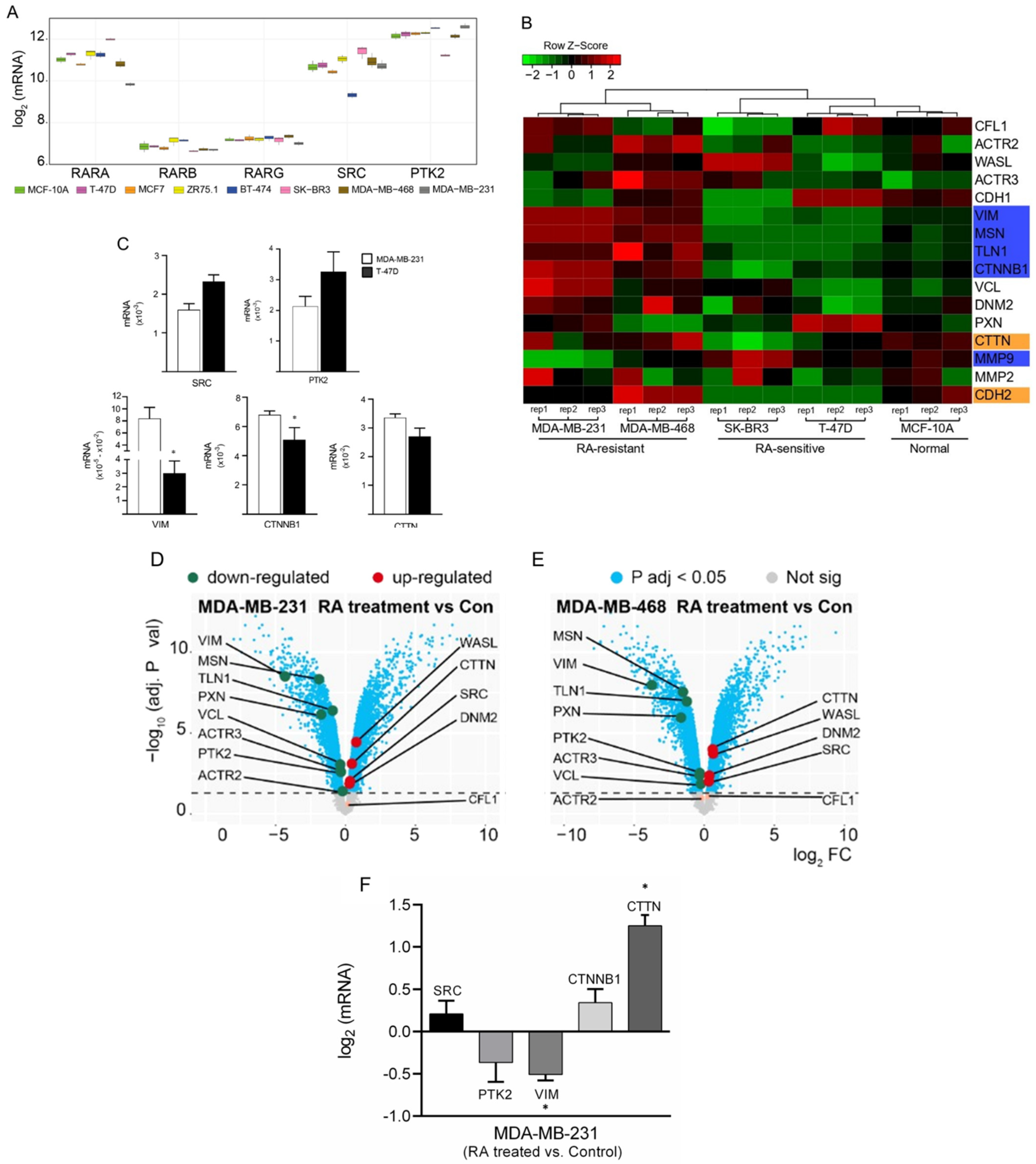
3.1. Differential Gene Expression Analysis of Cancer-Related Genes among BC Cells
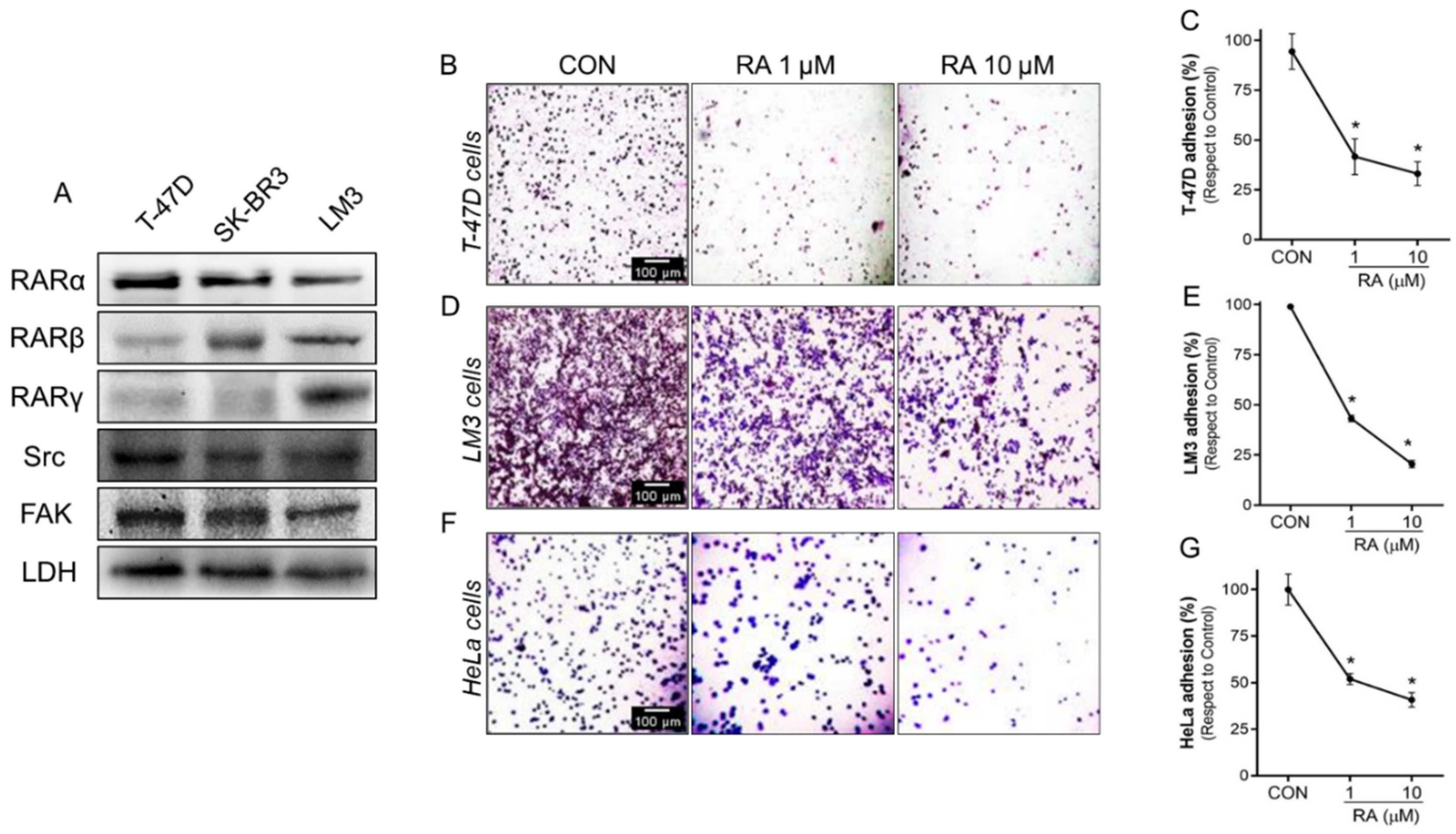
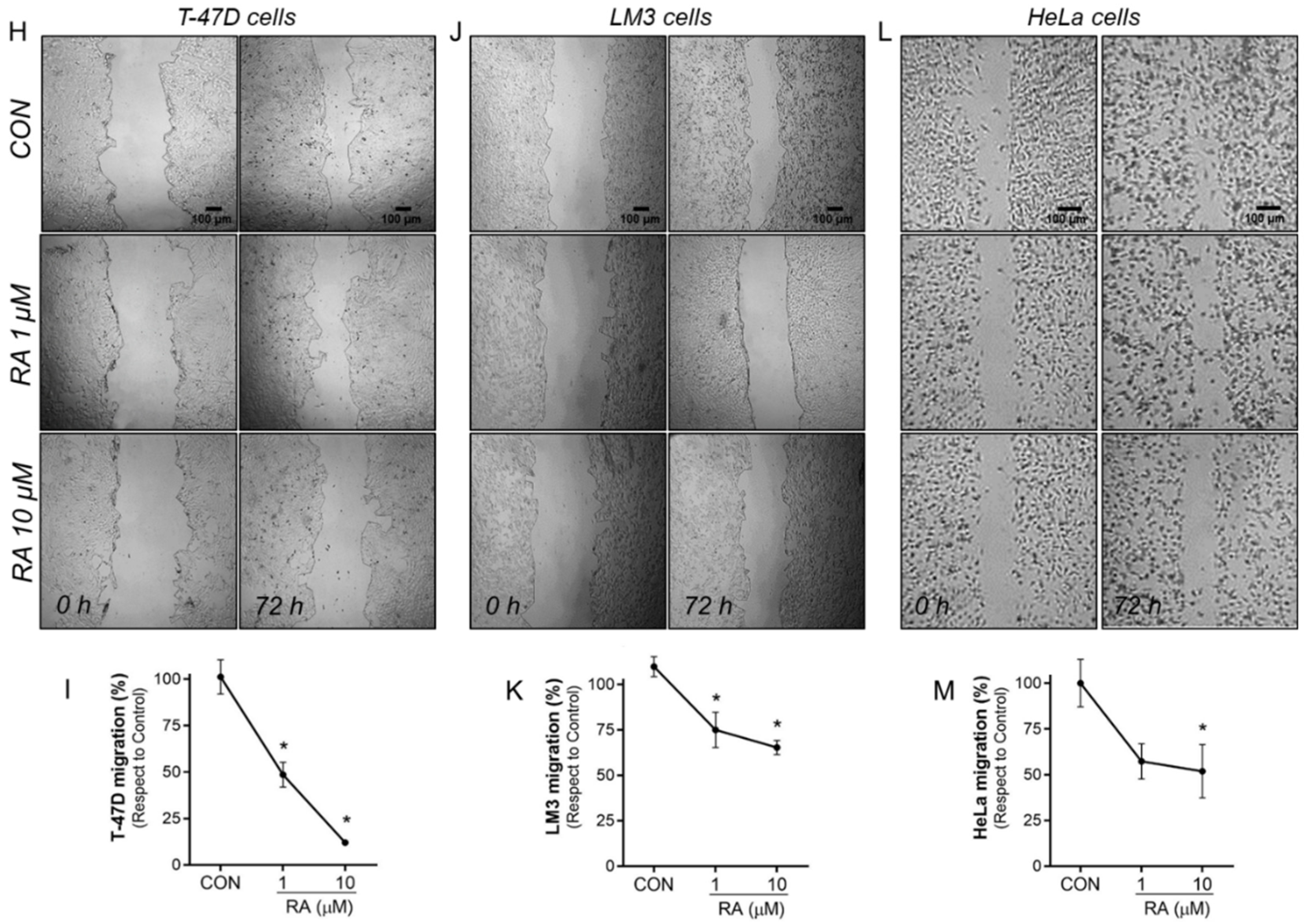
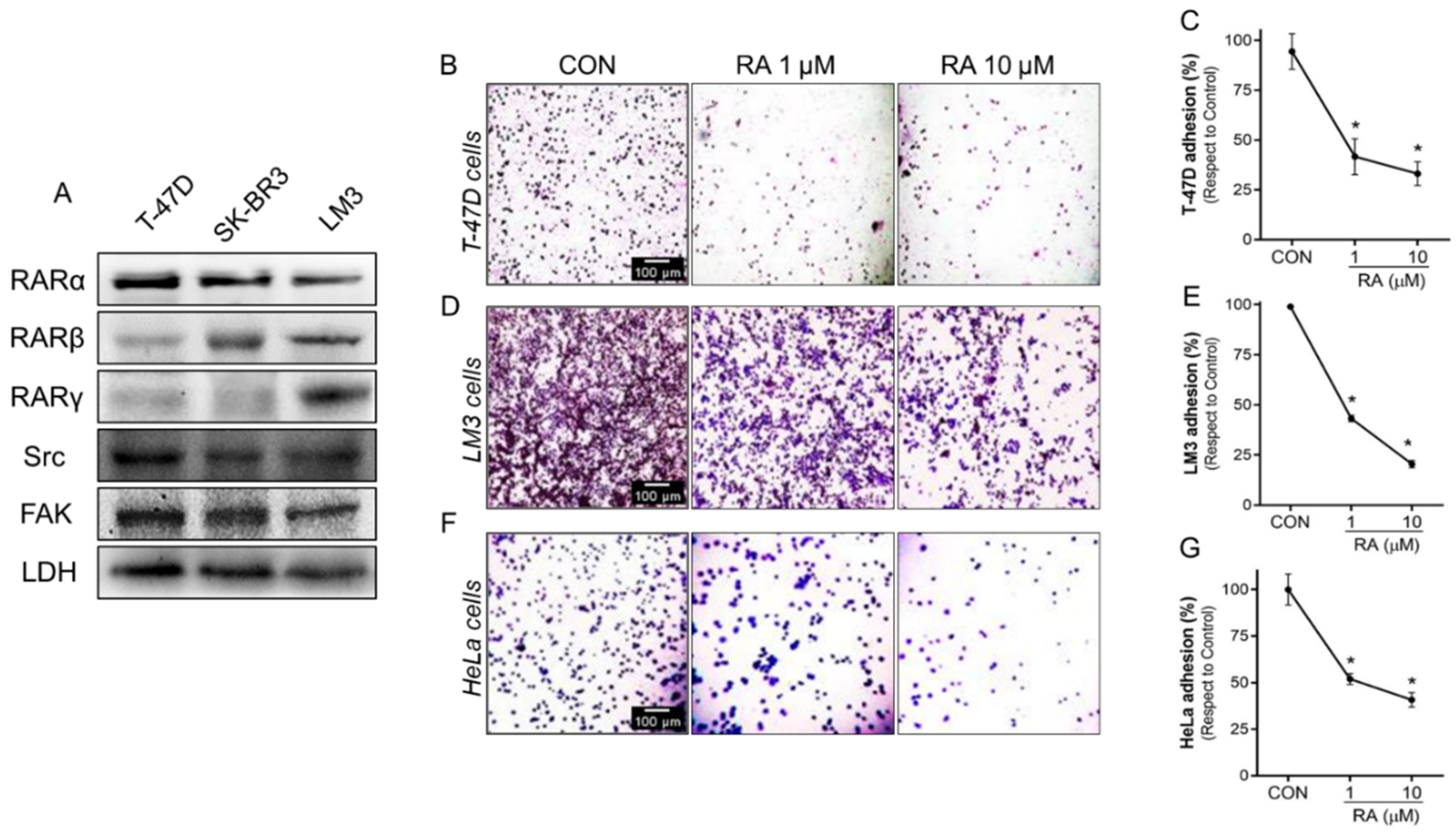
3.2. RA Reduces Cell Adhesion and Migration in Tumoral Cells
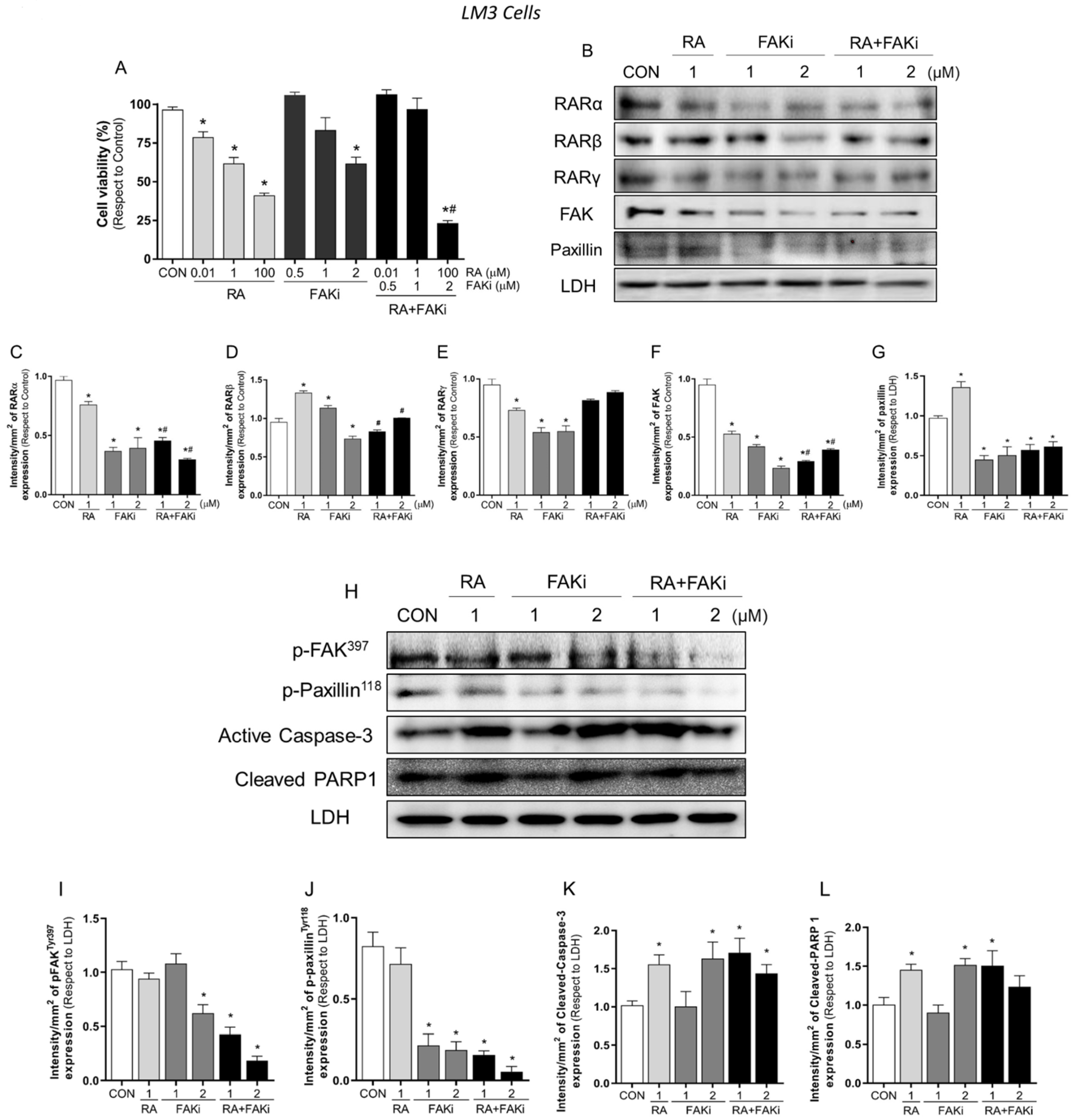
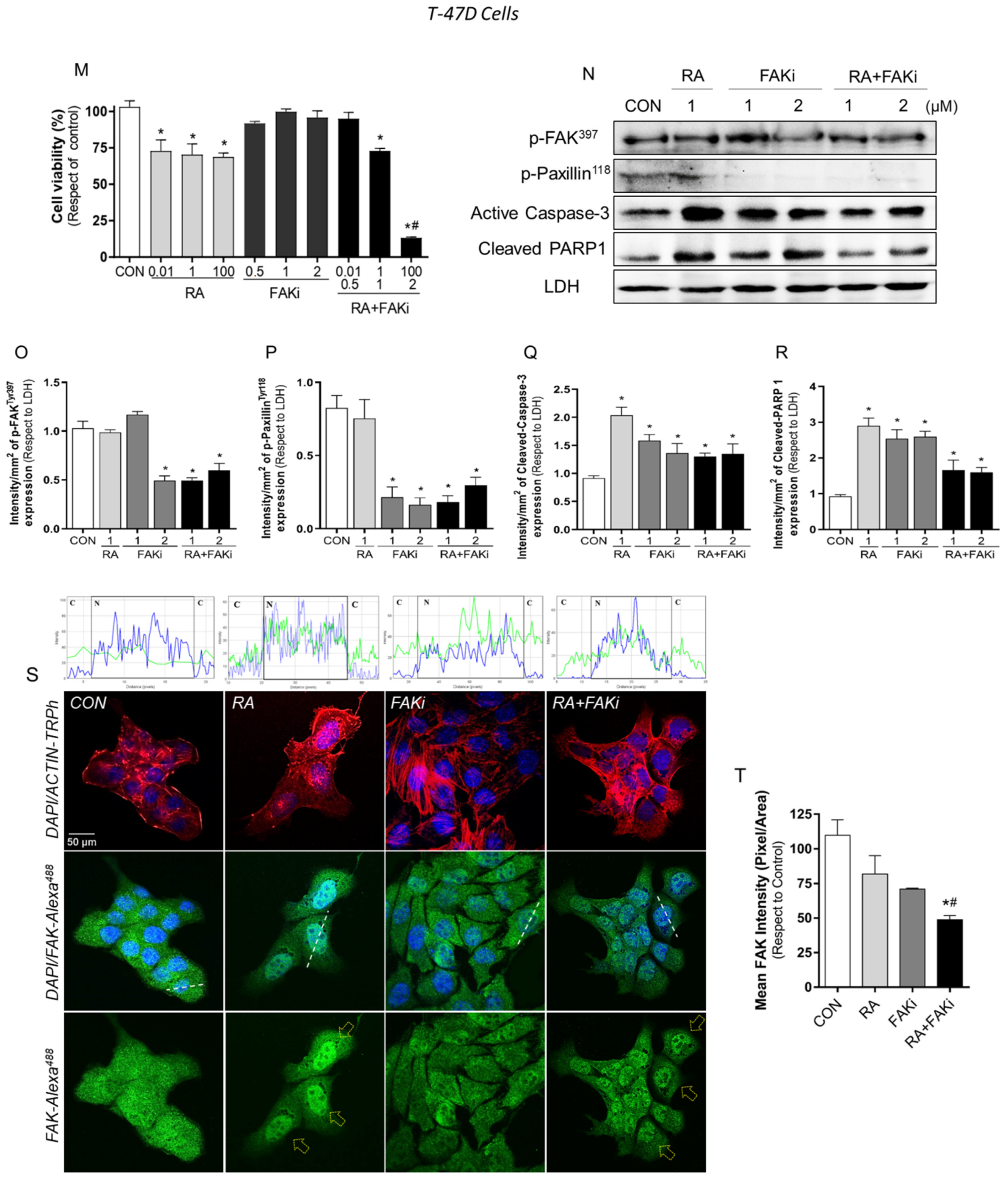
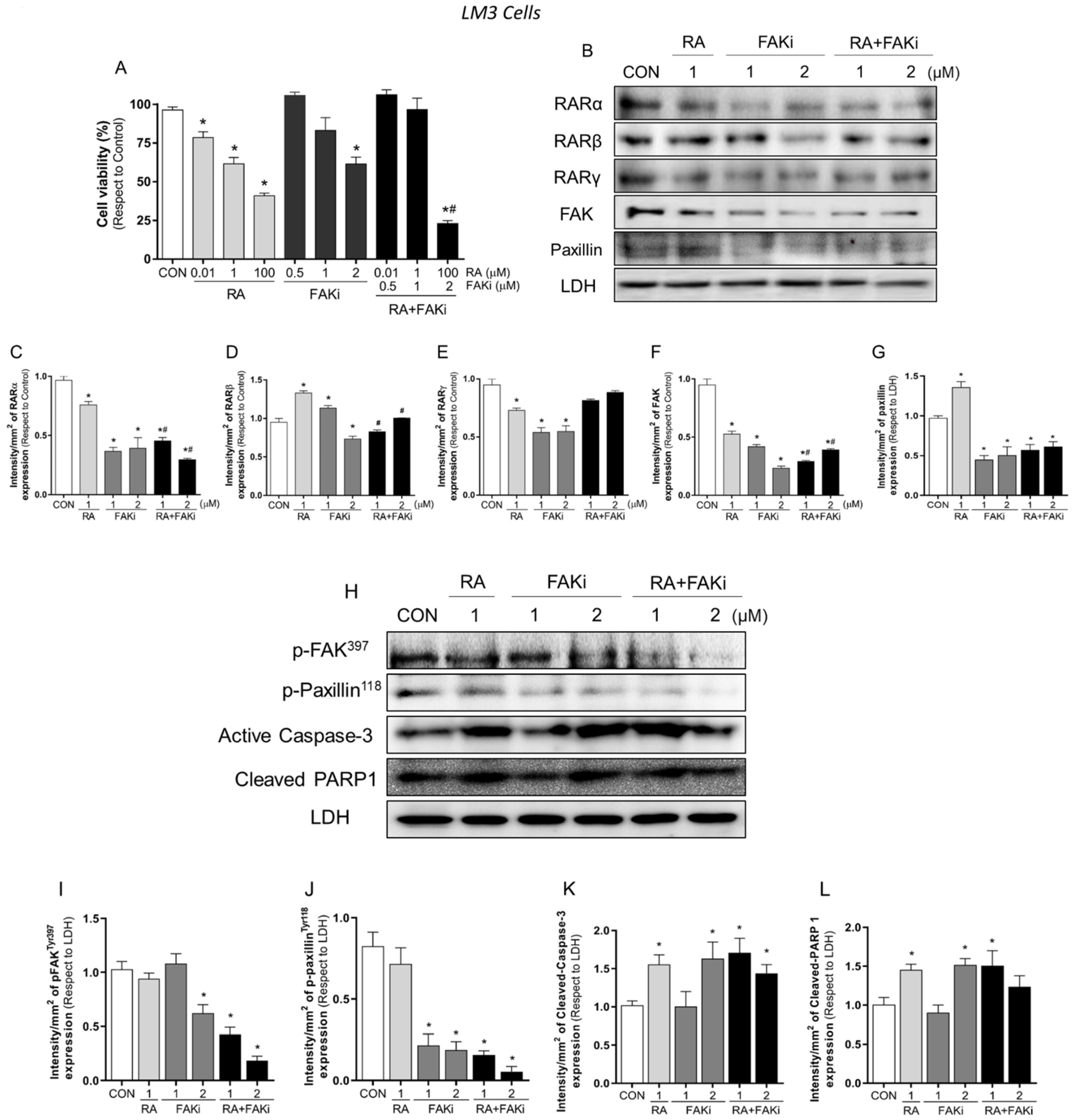
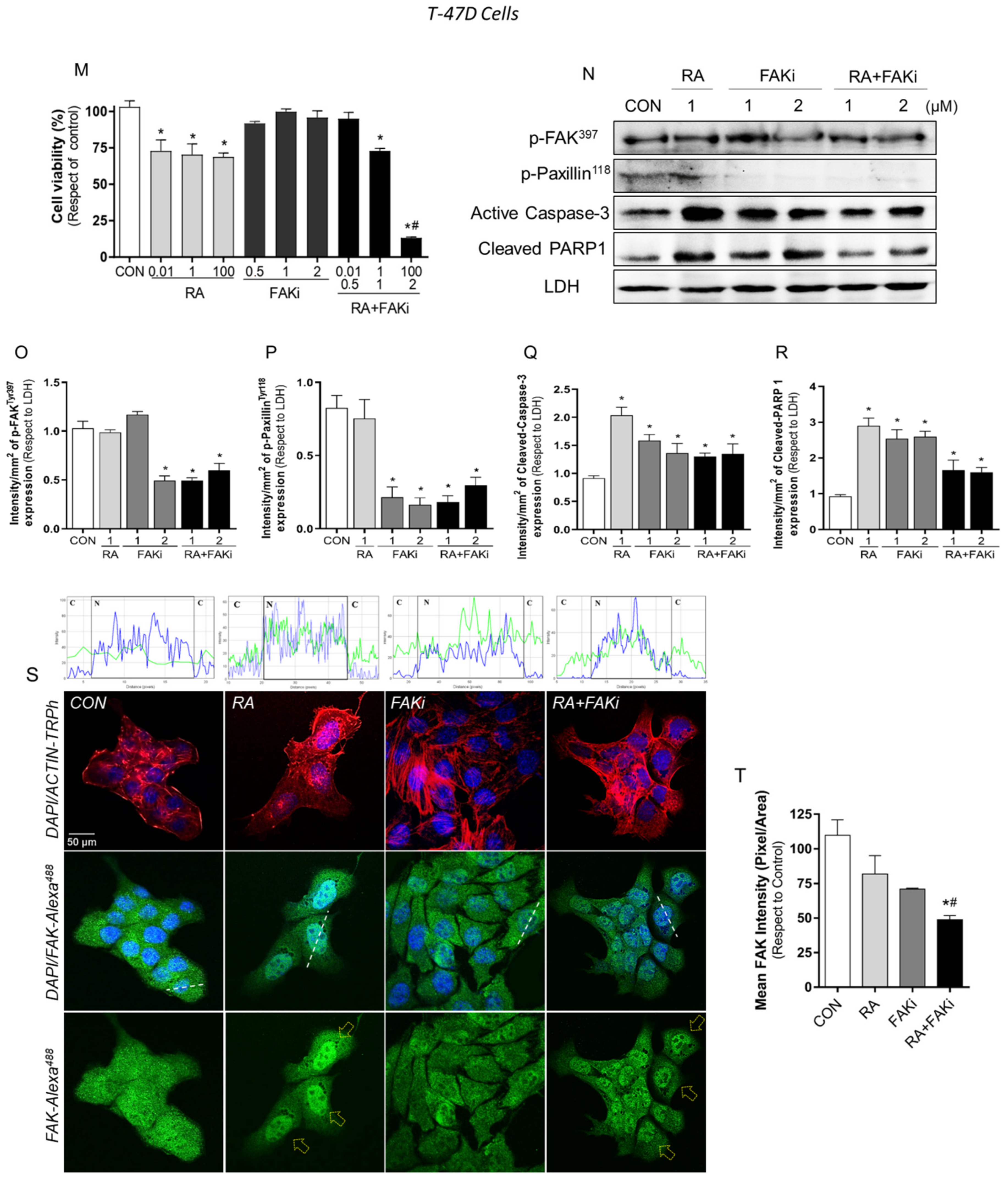
3.3. Treatment with RA Plus FAKi Reduces RARα, FAK, and Paxillin Expression Decreasing LM3 Cells Viability via Caspasa-3 and PARP1 Cleavage
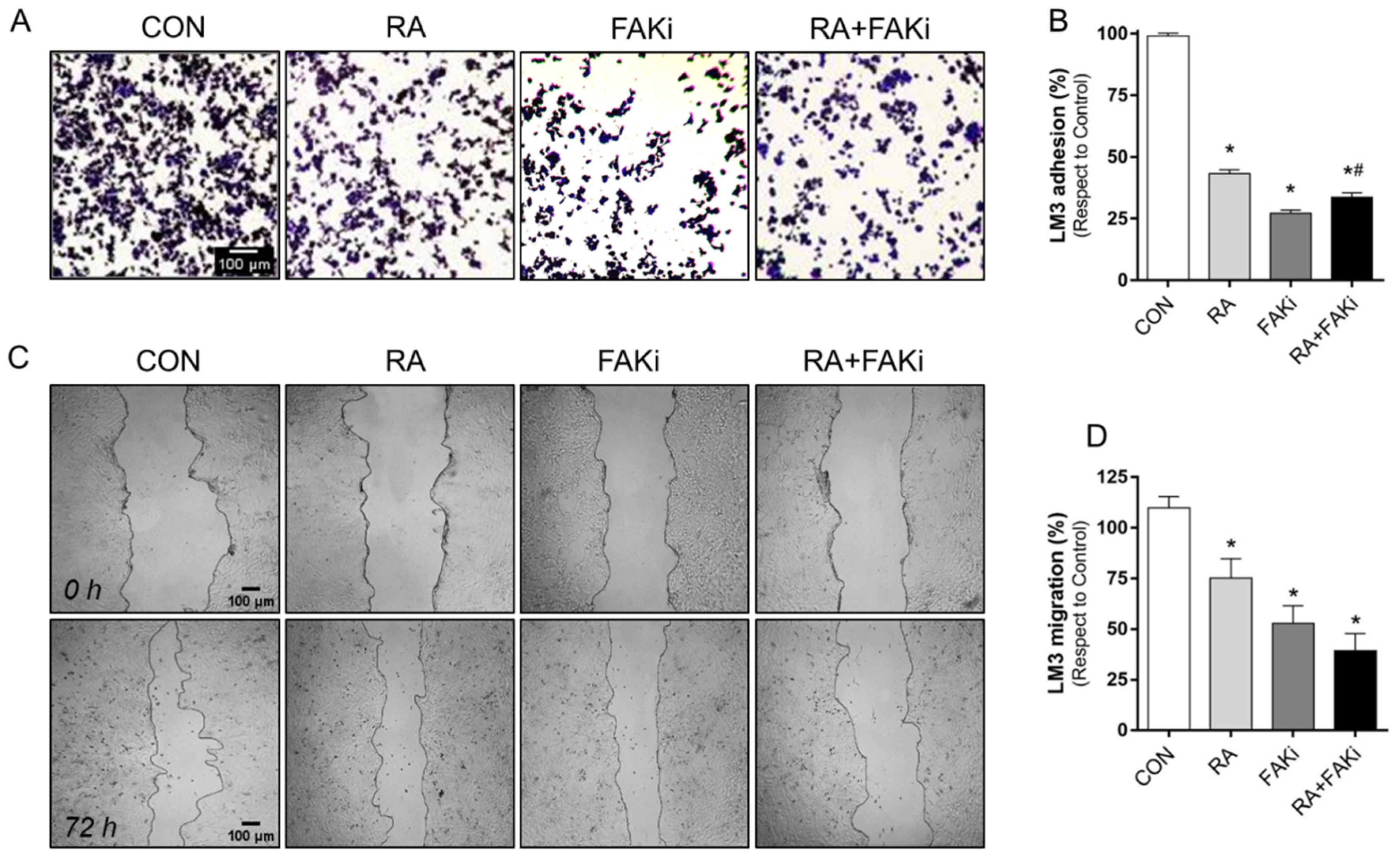
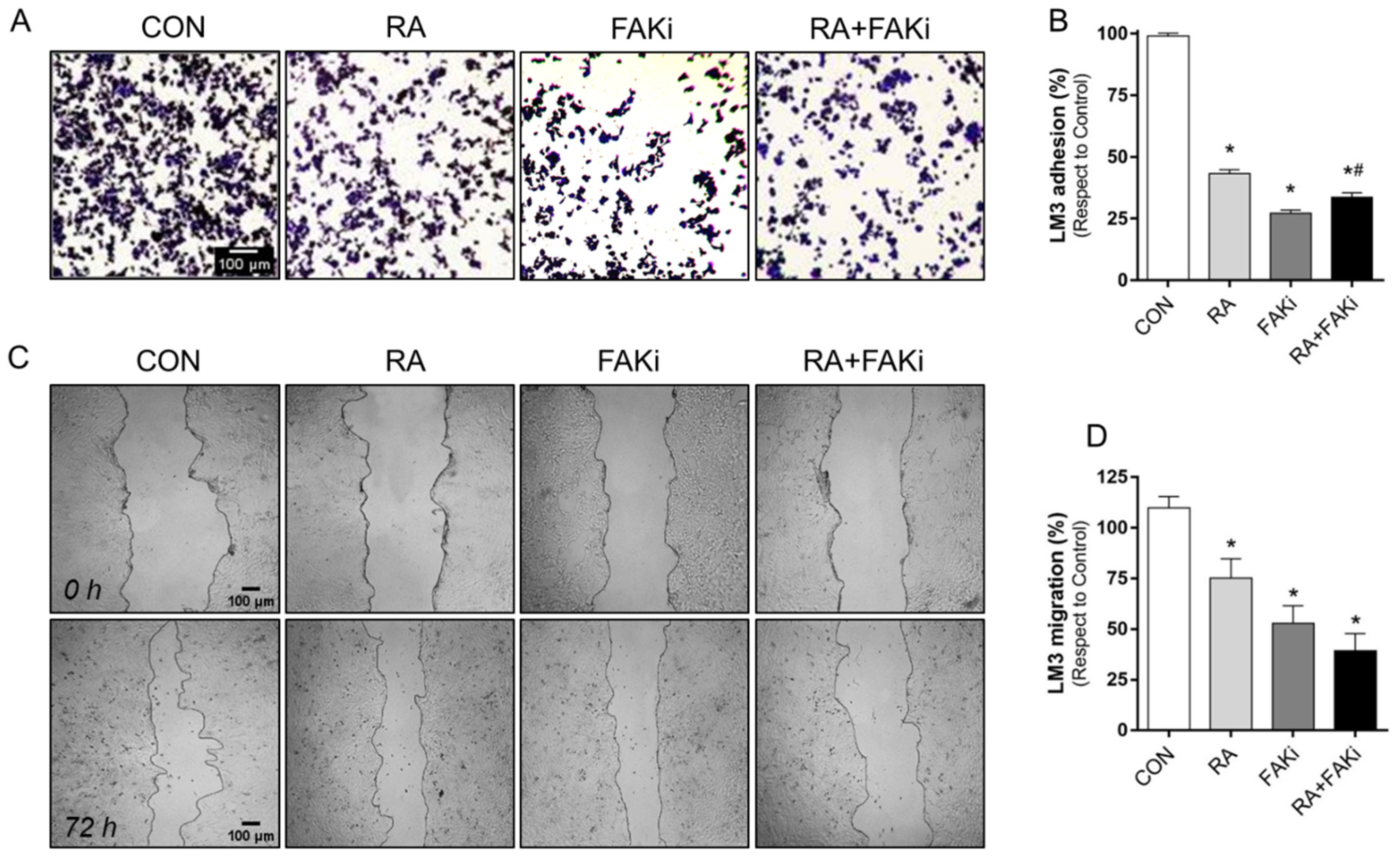
3.4. RA and FAKi Combination Improve LM3 Adhesion and Migration Inhibition
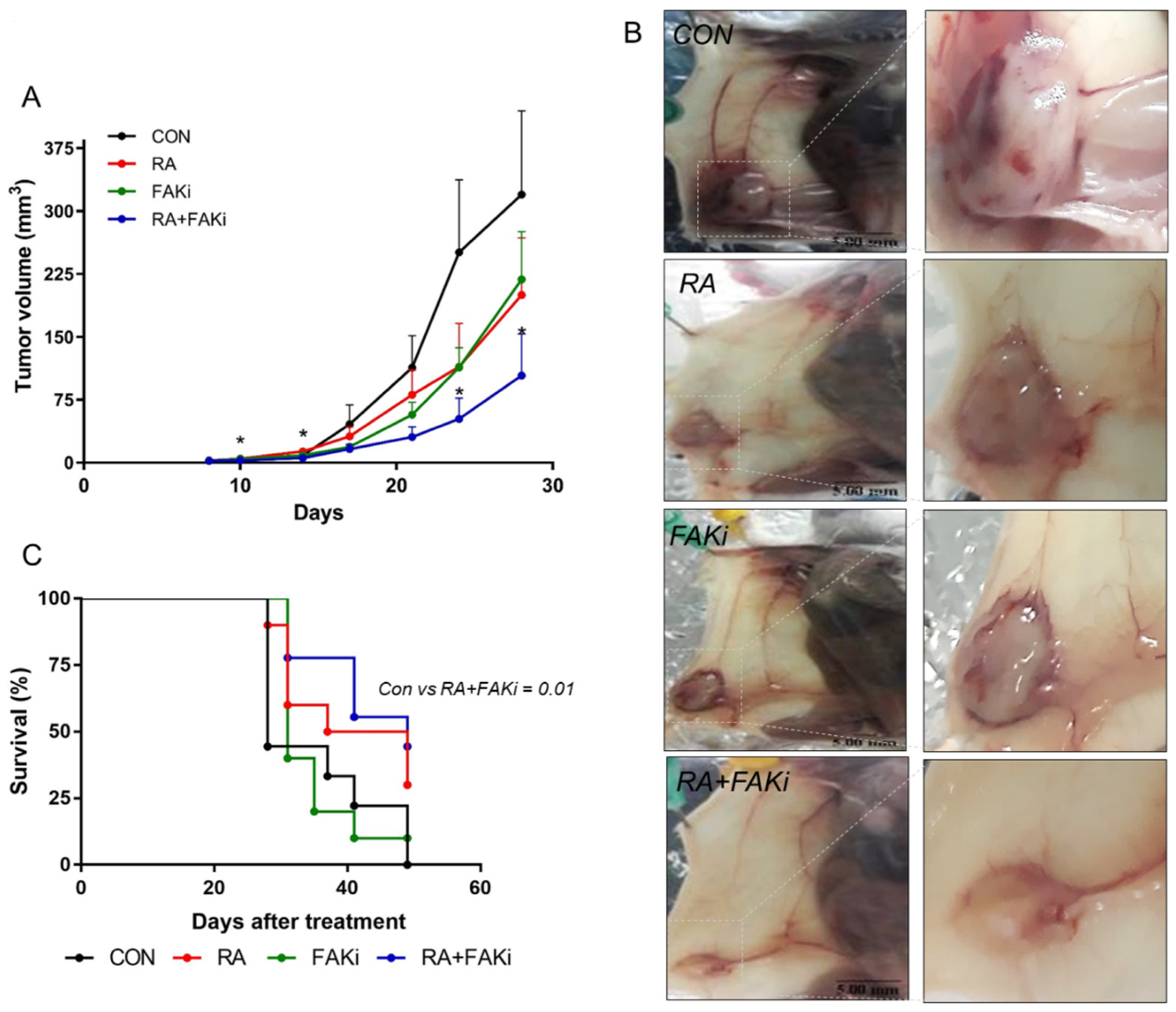
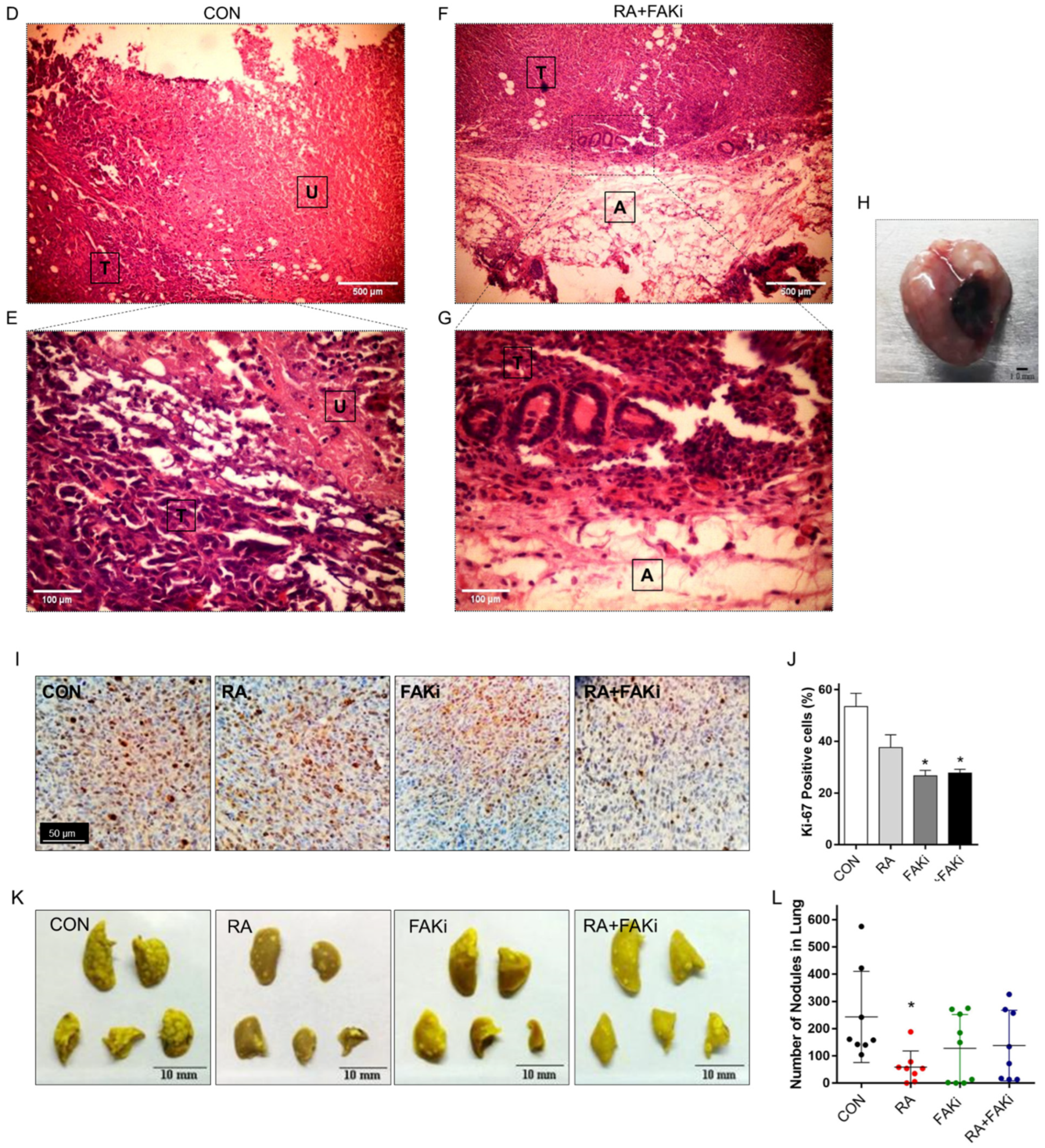
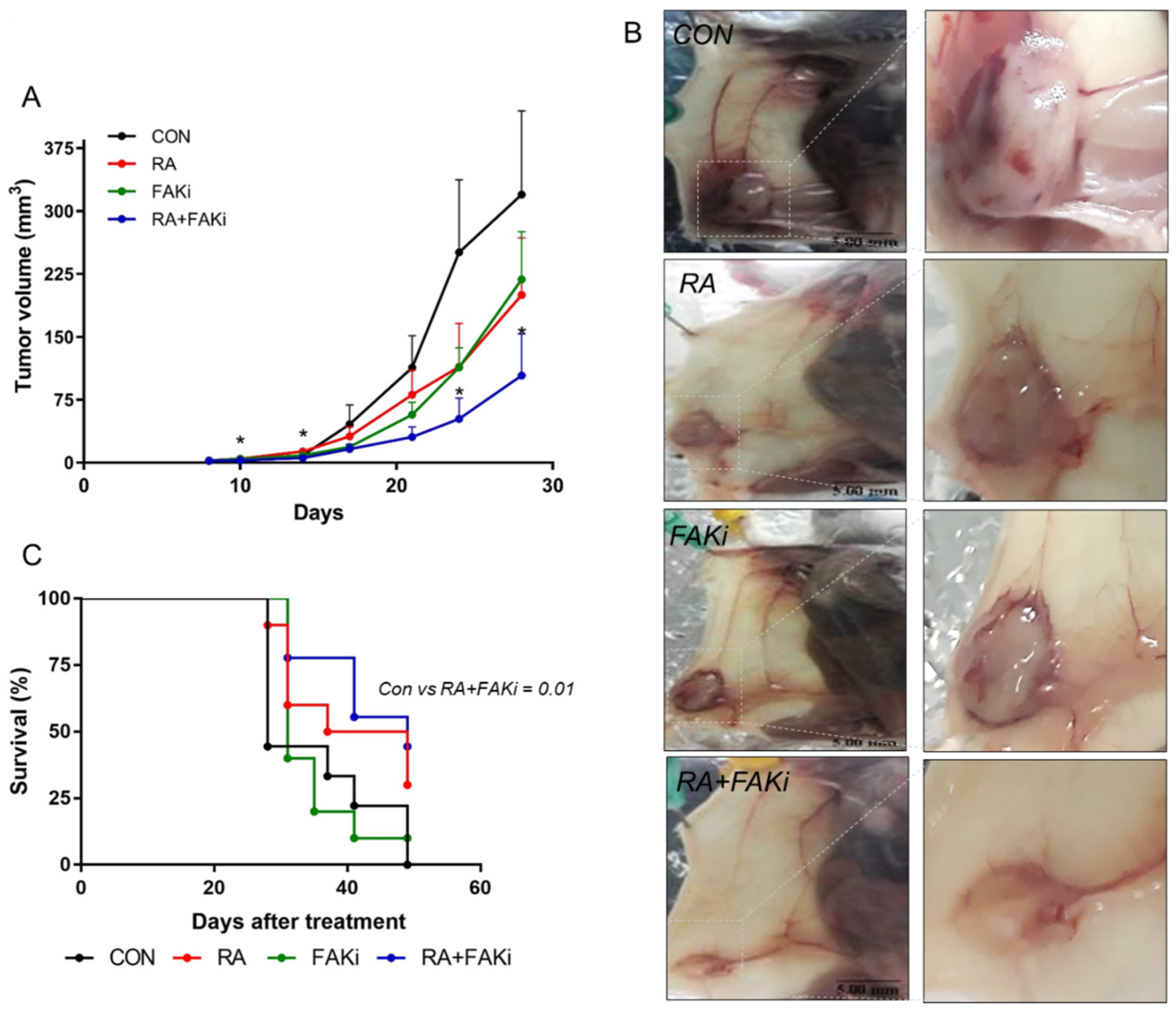
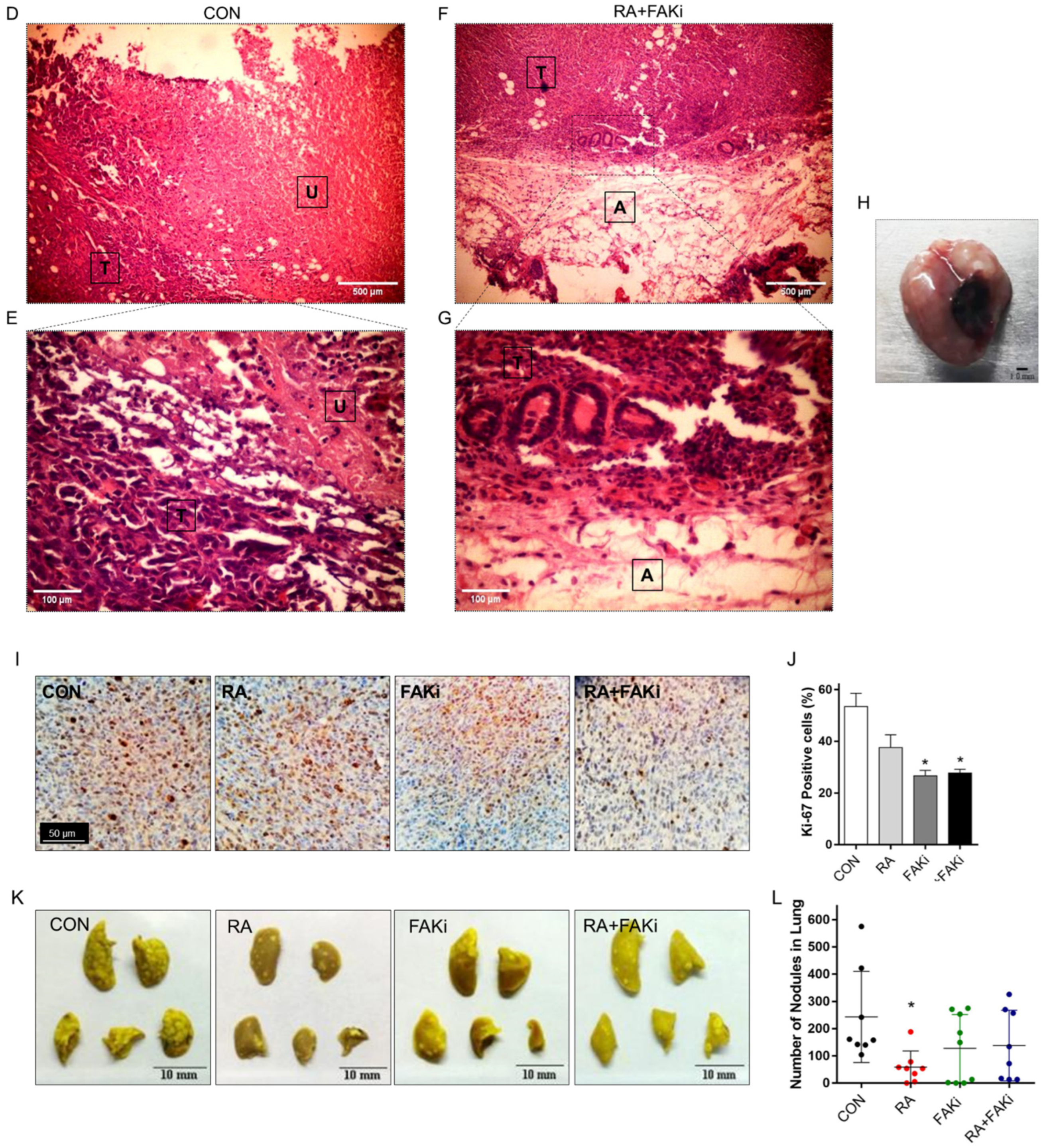
3.5. RA Plus FAKi Reduces Tumor Growth and Metastasis, Increasing Mice Survival
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schenk, T.; Stengel, S.; Zelent, A. Unlocking the potential of retinoic acid in anticancer therapy. Br. J. Cancer 2014, 111, 2039–2045. [Google Scholar] [CrossRef]
- Dobrotkova, V.; Chlapek, P.; Mazanek, P.; Sterba, J.; Veselska, R. Traffic lights for retinoids in oncology: Molecular markers of retinoid resistance and sensitivity and their use in the management of cancer differentiation therapy. BMC Cancer 2018, 18, 1059. [Google Scholar] [CrossRef]
- Lalevee, S.; Anno, Y.N.; Chatagnon, A.; Samarut, E.; Poch, O.; Laudet, V.; Benoit, G.; Lecompte, O.; Rochette-Egly, C. Genome-wide in silico identification of new conserved and functional retinoic acid receptor response elements (direct repeats separated by 5 bp). J. Biol. Chem. 2011, 286, 33322–33334. [Google Scholar] [CrossRef]
- Kumar, S.; Cunningham, T.J.; Duester, G. Nuclear receptor corepressors Ncor1 and Ncor2 (Smrt) are required for retinoic acid-dependent repression of Fgf8 during somitogenesis. Dev. Biol. 2016, 418, 204–215. [Google Scholar] [CrossRef]
- Chambon, P. A decade of molecular biology of retinoic acid receptors. FASEB J. 1996, 10, 940–954. [Google Scholar] [CrossRef]
- Al Tanoury, Z.; Piskunov, A.; Rochette-Egly, C. Vitamin A and retinoid signaling: Genomic and nongenomic effects. J. Lipid Res. 2013, 54, 1761–1775. [Google Scholar] [CrossRef]
- Giuli, M.V.; Hanieh, P.N.; Giuliani, E.; Rinaldi, F.; Marianecci, C.; Screpanti, I.; Checquolo, S.; Carafa, M. Current Trends in ATRA Delivery for Cancer Therapy. Pharmaceutics 2020, 12, 707. [Google Scholar] [CrossRef]
- Lo-Coco, F.; Avvisati, G.; Vignetti, M.; Thiede, C.; Orlando, S.M.; Iacobelli, S.; Ferrara, F.; Fazi, P.; Cicconi, L.; Di Bona, E.; et al. Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N. Engl. J. Med. 2013, 369, 111–121. [Google Scholar] [CrossRef]
- Uray, I.P.; Dmitrovsky, E.; Brown, P.H. Retinoids and rexinoids in cancer prevention: From laboratory to clinic. Semin. Oncol. 2016, 43, 49–64. [Google Scholar] [CrossRef]
- Bushue, N.; Wan, Y.J. Retinoid pathway and cancer therapeutics. Adv. Drug Deliv. Rev. 2010, 62, 1285–1298. [Google Scholar] [CrossRef] [Green Version]
- Garattini, E.; Bolis, M.; Garattini, S.K.; Fratelli, M.; Centritto, F.; Paroni, G.; Gianni, M.; Zanetti, A.; Pagani, A.; Fisher, J.N.; et al. Retinoids and breast cancer: From basic studies to the clinic and back again. Cancer Treat. Rev. 2014, 40, 739–749. [Google Scholar] [CrossRef]
- Costantini, L.; Molinari, R.; Farinon, B.; Merendino, N. Retinoic Acids in the Treatment of Most Lethal Solid Cancers. J. Clin. Med. 2020, 9, 360. [Google Scholar] [CrossRef]
- Gupta, G.P.; Massague, J. Cancer metastasis: Building a framework. Cell 2006, 127, 679–695. [Google Scholar] [CrossRef]
- Singletary, S.E.; Connolly, J.L. Breast cancer staging: Working with the sixth edition of the AJCC Cancer Staging Manual. CA Cancer J. Clin. 2006, 56, 37–47; quiz 50–31. [Google Scholar] [CrossRef]
- Nagano, M.; Hoshino, D.; Koshikawa, N.; Akizawa, T.; Seiki, M. Turnover of focal adhesions and cancer cell migration. Int. J. Cell Biol. 2012, 2012, 310616. [Google Scholar] [CrossRef]
- Shortrede, J.E.; Uzair, I.D.; Neira, F.J.; Flamini, M.I.; Sanchez, A.M. Paxillin, a novel controller in the signaling of estrogen to FAK/N-WASP/Arp2/3 complex in breast cancer cells. Mol. Cell. Endocrinol. 2016, 430, 56–67. [Google Scholar] [CrossRef]
- Zhao, X.; Guan, J.L. Focal adhesion kinase and its signaling pathways in cell migration and angiogenesis. Adv. Drug Deliv. Rev. 2011, 63, 610–615. [Google Scholar] [CrossRef]
- Agochiya, M.; Brunton, V.G.; Owens, D.W.; Parkinson, E.K.; Paraskeva, C.; Keith, W.N.; Frame, M.C. Increased dosage and amplification of the focal adhesion kinase gene in human cancer cells. Oncogene 1999, 18, 5646–5653. [Google Scholar] [CrossRef] [PubMed]
- Cance, W.G.; Harris, J.E.; Iacocca, M.V.; Roche, E.; Yang, X.; Chang, J.; Simkins, S.; Xu, L. Immunohistochemical analyses of focal adhesion kinase expression in benign and malignant human breast and colon tissues: Correlation with preinvasive and invasive phenotypes. Clin. Cancer Res. 2000, 6, 2417–2423. [Google Scholar] [PubMed]
- Miyazaki, T.; Kato, H.; Nakajima, M.; Sohda, M.; Fukai, Y.; Masuda, N.; Manda, R.; Fukuchi, M.; Tsukada, K.; Kuwano, H. FAK overexpression is correlated with tumour invasiveness and lymph node metastasis in oesophageal squamous cell carcinoma. Br. J. Cancer 2003, 89, 140–145. [Google Scholar] [CrossRef] [Green Version]
- Lv, P.C.; Jiang, A.Q.; Zhang, W.M.; Zhu, H.L. FAK inhibitors in Cancer, a patent review. Expert Opin. Ther. Pat. 2018, 28, 139–145. [Google Scholar] [CrossRef]
- Schultze, A.; Fiedler, W. Therapeutic potential and limitations of new FAK inhibitors in the treatment of cancer. Expert Opin. Investig. Drugs 2010, 19, 777–788. [Google Scholar] [CrossRef]
- Mohanty, A.; Pharaon, R.R.; Nam, A.; Salgia, S.; Kulkarni, P.; Massarelli, E. FAK-targeted and combination therapies for the treatment of cancer: An overview of phase I and II clinical trials. Expert Opin. Investig. Drugs 2020, 29, 399–409. [Google Scholar] [CrossRef]
- Mousson, A.; Sick, E.; Carl, P.; Dujardin, D.; De Mey, J.; Ronde, P. Targeting Focal Adhesion Kinase Using Inhibitors of Protein-Protein Interactions. Cancers 2018, 10, 278. [Google Scholar] [CrossRef]
- Berardi, D.E.; Ariza Bareno, L.; Amigo, N.; Canonero, L.; Pelagatti, M.L.N.; Motter, A.N.; Taruselli, M.A.; Diaz Bessone, M.I.; Cirigliano, S.M.; Edelstein, A.; et al. All-trans retinoic acid and protein kinase C alpha/beta1 inhibitor combined treatment targets cancer stem cells and impairs breast tumor progression. Sci. Rep. 2021, 11, 6044. [Google Scholar] [CrossRef]
- Flamini, M.I.; Gauna, G.V.; Sottile, M.L.; Nadin, B.S.; Sanchez, A.M.; Vargas-Roig, L.M. Retinoic acid reduces migration of human breast cancer cells: Role of retinoic acid receptor beta. J. Cell. Mol. Med. 2014, 18, 1113–1123. [Google Scholar] [CrossRef]
- Sanchez, A.M.; Shortrede, J.E.; Vargas-Roig, L.M.; Flamini, M.I. Retinoic acid induces nuclear FAK translocation and reduces breast cancer cell adhesion through Moesin, FAK, and Paxillin. Mol. Cell. Endocrinol. 2016, 430, 1–11. [Google Scholar] [CrossRef]
- Vanderhoeven, F.; Redondo, A.L.; Martinez, A.L.; Vargas-Roig, L.M.; Sanchez, A.M.; Flamini, M.I. Synergistic antitumor activity by combining trastuzumab with retinoic acid in HER2 positive human breast cancer cells. Oncotarget 2018, 9, 26527–26542. [Google Scholar] [CrossRef]
- Urtreger, A.; Ladeda, V.; Puricelli, L.; Rivelli, A.; Vidal, M.; Delustig, E.; Joffe, E. Modulation of fibronectin expression and proteolytic activity associated with the invasive and metastatic phenotype in two new murine mammary tumor cell lines. Int. J. Oncol. 1997, 11, 489–496. [Google Scholar] [CrossRef]
- Davis, S.; Meltzer, P.S. GEOquery: A bridge between the Gene Expression Omnibus (GEO) and BioConductor. Bioinformatics 2007, 23, 1846–1847. [Google Scholar] [CrossRef] [Green Version]
- Johnson, W.E.; Li, C.; Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 2007, 8, 118–127. [Google Scholar] [CrossRef]
- Leek, J.T.; Johnson, W.E.; Parker, H.S.; Jaffe, A.E.; Storey, J.D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 2012, 28, 882–883. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Untergasser, A.; Ruijter, J.M.; Benes, V.; van den Hoff, M.J.B. Web-based LinRegPCR: Application for the visualization and analysis of (RT)-qPCR amplification and melting data. BMC Bioinform. 2021, 22, 1–18. [Google Scholar] [CrossRef]
- Chou, T.C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharm. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef]
- Chou, T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef]
- Kelley, J.B.; Paschal, B.M. Fluorescence-based quantification of nucleocytoplasmic transport. Methods 2019, 157, 106–114. [Google Scholar] [CrossRef]
- Zou, D.; Yoon, H.S.; Anjomshoaa, A.; Perez, D.; Fukuzawa, R.; Guilford, P.; Humar, B. Increased levels of active c-Src distinguish invasive from in situ lobular lesions. Breast Cancer Res. 2009, 11, R45. [Google Scholar] [CrossRef]
- Elsberger, B.; Fullerton, R.; Zino, S.; Jordan, F.; Mitchell, T.J.; Brunton, V.G.; Mallon, E.A.; Shiels, P.G.; Edwards, J. Breast cancer patients’ clinical outcome measures are associated with Src kinase family member expression. Br. J. Cancer 2010, 103, 899–909. [Google Scholar] [CrossRef]
- Lark, A.L.; Livasy, C.A.; Dressler, L.; Moore, D.T.; Millikan, R.C.; Geradts, J.; Iacocca, M.; Cowan, D.; Little, D.; Craven, R.J.; et al. High focal adhesion kinase expression in invasive breast carcinomas is associated with an aggressive phenotype. Mod. Pathol. 2005, 18, 1289–1294. [Google Scholar] [CrossRef] [Green Version]
- Centritto, F.; Paroni, G.; Bolis, M.; Garattini, S.K.; Kurosaki, M.; Barzago, M.M.; Zanetti, A.; Fisher, J.N.; Scott, M.F.; Pattini, L.; et al. Cellular and molecular determinants of all-trans retinoic acid sensitivity in breast cancer: Luminal phenotype and RARalpha expression. EMBO Mol. Med. 2015, 7, 950–972. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Mira-y-Lopez, R.; Nakajo, S.; Nakaya, K.; Jing, Y. Expression of estrogen receptor alpha, retinoic acid receptor alpha and cellular retinoic acid binding protein II genes is coordinately regulated in human breast cancer cells. Oncogene 2005, 24, 4362–4369. [Google Scholar] [CrossRef]
- Schneider, S.M.; Offterdinger, M.; Huber, H.; Grunt, T.W. Activation of retinoic acid receptor alpha is sufficient for full induction of retinoid responses in SK-BR-3 and T47D human breast cancer cells. Cancer Res. 2000, 60, 5479–5487. [Google Scholar]
- Sun, S.Y.; Wan, H.; Yue, P.; Hong, W.K.; Lotan, R. Evidence that retinoic acid receptor beta induction by retinoids is important for tumor cell growth inhibition. J. Biol. Chem. 2000, 275, 17149–17153. [Google Scholar] [CrossRef]
- Xu, Q.; Jitkaew, S.; Choksi, S.; Kadigamuwa, C.; Qu, J.; Choe, M.; Jang, J.; Liu, C.; Liu, Z.G. The cytoplasmic nuclear receptor RARgamma controls RIP1 initiated cell death when cIAP activity is inhibited. Nat. Commun. 2017, 8, 425. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Jian, Z.Y.; Shen, X.F.; Wei, X.M.; Yu, G.Z.; Zeng, X.T. Promoter Methylation of the Retinoic Acid Receptor Beta2 (RARbeta2) Is Associated with Increased Risk of Breast Cancer: A PRISMA Compliant Meta-Analysis. PLoS ONE 2015, 10, e0140329. [Google Scholar] [CrossRef]
- Yari, K.; Rahimi, Z. Promoter Methylation Status of the Retinoic Acid Receptor-Beta 2 Gene in Breast Cancer Patients: A Case Control Study and Systematic Review. Breast Care 2019, 14, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Guan, J.L. Signal transduction by focal adhesion kinase in cancer. Cancer Metastasis Rev. 2009, 28, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Coyle, K.M.; Maxwell, S.; Thomas, M.L.; Marcato, P. Profiling of the transcriptional response to all-trans retinoic acid in breast cancer cells reveals RARE-independent mechanisms of gene expression. Sci. Rep. 2017, 7, 16684. [Google Scholar] [CrossRef] [PubMed]
- Zanetti, A.; Affatato, R.; Centritto, F.; Fratelli, M.; Kurosaki, M.; Barzago, M.M.; Bolis, M.; Terao, M.; Garattini, E.; Paroni, G. All-trans-retinoic Acid Modulates the Plasticity and Inhibits the Motility of Breast Cancer Cells: Role of NOTCH1 and Transforming Growth Factor (TGFbeta). J. Biol. Chem. 2015, 290, 17690–17709. [Google Scholar] [CrossRef]
- Cui, J.; Gong, M.; He, Y.; Li, Q.; He, T.; Bi, Y. All-trans retinoic acid inhibits proliferation, migration, invasion and induces differentiation of hepa1-6 cells through reversing EMT in vitro. Int. J. Oncol. 2016, 48, 349–357. [Google Scholar] [CrossRef] [Green Version]
- Woo, Y.J.; Jang, K.L. All-trans retinoic acid activates E-cadherin expression via promoter hypomethylation in the human colon carcinoma HCT116 cells. Biochem. Biophys. Res. Commun. 2012, 425, 944–949. [Google Scholar] [CrossRef]
- Zuo, L.; Yang, X.; Lu, M.; Hu, R.; Zhu, H.; Zhang, S.; Zhou, Q.; Chen, F.; Gui, S.; Wang, Y. All-Trans Retinoic Acid Inhibits Human Colorectal Cancer Cells RKO Migration via Downregulating Myosin Light Chain Kinase Expression through MAPK Signaling Pathway. Nutr. Cancer 2016, 68, 1225–1233. [Google Scholar] [CrossRef]
- Chauhan, A.; Khan, T. Focal adhesion kinase-An emerging viable target in cancer and development of focal adhesion kinase inhibitors. Chem. Biol. Drug Des. 2021, 97, 774–794. [Google Scholar] [CrossRef]
- Hao, H.; Naomoto, Y.; Bao, X.; Watanabe, N.; Sakurama, K.; Noma, K.; Motoki, T.; Tomono, Y.; Fukazawa, T.; Shirakawa, Y.; et al. Focal adhesion kinase as potential target for cancer therapy (Review). Oncol. Rep. 2009, 22, 973–979. [Google Scholar]
- Lu, Y.; Sun, H. Progress in the Development of Small Molecular Inhibitors of Focal Adhesion Kinase (FAK). J. Med. Chem. 2020, 63, 14382–14403. [Google Scholar] [CrossRef]
- Parsons, J.T.; Slack-Davis, J.; Tilghman, R.; Roberts, W.G. Focal adhesion kinase: Targeting adhesion signaling pathways for therapeutic intervention. Clin. Cancer Res. 2008, 14, 627–632. [Google Scholar] [CrossRef]
- Van Heusden, J.; Wouters, W.; Ramaekers, F.C.; Krekels, M.D.; Dillen, L.; Borgers, M.; Smets, G. All-trans-retinoic acid metabolites significantly inhibit the proliferation of MCF-7 human breast cancer cells in vitro. Br. J. Cancer 1998, 77, 26–32. [Google Scholar] [CrossRef]
- Golubovskaya, V.M.; Figel, S.; Ho, B.T.; Johnson, C.P.; Yemma, M.; Huang, G.; Zheng, M.; Nyberg, C.; Magis, A.; Ostrov, D.A.; et al. A small molecule focal adhesion kinase (FAK) inhibitor, targeting Y397 site: 1-(2-hydroxyethyl)-3, 5, 7-triaza-1-azoniatricyclo [3.3.1.1(3,7)] decane; bromide effectively inhibits FAK autophosphorylation activity and decreases cancer cell viability, clonogenicity and tumor growth in vivo. Carcinogenesis 2012, 33, 1004–1013. [Google Scholar] [CrossRef]
- Walsh, C.; Tanjoni, I.; Uryu, S.; Tomar, A.; Nam, J.O.; Luo, H.; Phillips, A.; Patel, N.; Kwok, C.; McMahon, G.; et al. Oral delivery of PND-1186 FAK inhibitor decreases tumor growth and spontaneous breast to lung metastasis in pre-clinical models. Cancer Biol. 2010, 9, 778–790. [Google Scholar] [CrossRef]
- Toma, S.; Isnardi, L.; Raffo, P.; Dastoli, G.; De Francisci, E.; Riccardi, L.; Palumbo, R.; Bollag, W. Effects of all-trans-retinoic acid and 13-cis-retinoic acid on breast-cancer cell lines: Growth inhibition and apoptosis induction. Int. J. Cancer 1997, 70, 619–627. [Google Scholar] [CrossRef]
- Abdolahi, M.; Shokri, F.; Hosseini, M.; Shadanian, M.; Saboor-Yaraghi, A.A. The combined effects of all-trans-retinoic acid and docosahexaenoic acid on the induction of apoptosis in human breast cancer MCF-7 cells. J. Cancer Res. Ther. 2016, 12, 204–208. [Google Scholar] [CrossRef]
- Koay, D.C.; Zerillo, C.; Narayan, M.; Harris, L.N.; DiGiovanna, M.P. Anti-tumor effects of retinoids combined with trastuzumab or tamoxifen in breast cancer cells: Induction of apoptosis by retinoid/trastuzumab combinations. Breast Cancer Res. 2010, 12, R62. [Google Scholar] [CrossRef]
- Hong, T.K.; Lee-Kim, Y.C. Effects of retinoic acid isomers on apoptosis and enzymatic antioxidant system in human breast cancer cells. Nutr. Res. Pract. 2009, 3, 77–83. [Google Scholar] [CrossRef]
- Brigger, D.; Schlafli, A.M.; Garattini, E.; Tschan, M.P. Activation of RARalpha induces autophagy in SKBR3 breast cancer cells and depletion of key autophagy genes enhances ATRA toxicity. Cell Death Dis. 2015, 6, e1861. [Google Scholar] [CrossRef] [PubMed]
- Golubovskaya, V.M.; Gross, S.; Kaur, A.S.; Wilson, R.I.; Xu, L.H.; Yang, X.H.; Cance, W.G. Simultaneous inhibition of focal adhesion kinase and SRC enhances detachment and apoptosis in colon cancer cell lines. Mol. Cancer Res. MCR 2003, 1, 755–764. [Google Scholar]
- Golubovskaya, V.M.; Virnig, C.; Cance, W.G. TAE226-induced apoptosis in breast cancer cells with overexpressed Src or EGFR. Mol. Carcinog. 2008, 47, 222–234. [Google Scholar] [CrossRef]
- Tanjoni, I.; Walsh, C.; Uryu, S.; Tomar, A.; Nam, J.O.; Mielgo, A.; Lim, S.T.; Liang, C.; Koenig, M.; Sun, C.; et al. PND-1186 FAK inhibitor selectively promotes tumor cell apoptosis in three-dimensional environments. Cancer Biol. Ther. 2010, 9, 764–777. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; He, D.H.; Zajac-Kaye, M.; Hochwald, S.N. A small molecule FAK kinase inhibitor, GSK2256098, inhibits growth and survival of pancreatic ductal adenocarcinoma cells. Cell Cycle 2014, 13, 3143–3149. [Google Scholar] [CrossRef]
- Dutta, A.; Sen, T.; Banerji, A.; Das, S.; Chatterjee, A. Studies on Multifunctional Effect of All-Trans Retinoic Acid (ATRA) on Matrix Metalloproteinase-2 (MMP-2) and Its Regulatory Molecules in Human Breast Cancer Cells (MCF-7). J. Oncol. 2009, 2009, 627840. [Google Scholar] [CrossRef]
- Dutta, A.; Sen, T.; Chatterjee, A. All-trans retinoic acid (ATRA) downregulates MMP-9 by modulating its regulatory molecules. Cell Adhes. Migr. 2010, 4, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Lietha, D.; Cai, X.; Ceccarelli, D.F.; Li, Y.; Schaller, M.D.; Eck, M.J. Structural basis for the autoinhibition of focal adhesion kinase. Cell 2007, 129, 1177–1187. [Google Scholar] [CrossRef]
- Lim, S.T.; Chen, X.L.; Lim, Y.; Hanson, D.A.; Vo, T.T.; Howerton, K.; Larocque, N.; Fisher, S.J.; Schlaepfer, D.D.; Ilic, D. Nuclear FAK promotes cell proliferation and survival through FERM-enhanced p53 degradation. Mol. Cell 2008, 29, 9–22. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Yi, Q.; Tang, L. The roles of nuclear focal adhesion kinase (FAK) on Cancer: A focused review. J. Exp. Clin. Cancer Res. 2019, 38, 250. [Google Scholar] [CrossRef]
- Berardi, D.E.; Bessone, M.I.; Motter, A.; Bal de Kier Joffe, E.D.; Urtreger, A.J.; Todaro, L.B. Involvement of protein kinase C alpha and delta activities on the induction of the retinoic acid system in mammary cancer cells. Mol. Carcinog. 2015, 54, 1110–1121. [Google Scholar] [CrossRef]
- Huang, S.; Chen, Y.; Liang, Z.M.; Li, N.N.; Liu, Y.; Zhu, Y.; Liao, D.; Zhou, X.Z.; Lu, K.P.; Yao, Y.; et al. Targeting Pin1 by All-Trans Retinoic Acid (ATRA) Overcomes Tamoxifen Resistance in Breast Cancer via Multifactorial Mechanisms. Front. Cell Dev. Biol. 2019, 7, 322. [Google Scholar] [CrossRef]
- Wei, S.; Kozono, S.; Kats, L.; Nechama, M.; Li, W.; Guarnerio, J.; Luo, M.; You, M.H.; Yao, Y.; Kondo, A.; et al. Active Pin1 is a key target of all-trans retinoic acid in acute promyelocytic leukemia and breast cancer. Nat. Med. 2015, 21, 457–466. [Google Scholar] [CrossRef]
- Gudas, L.J.; Wagner, J.A. Retinoids regulate stem cell differentiation. J. Cell. Physiol. 2011, 226, 322–330. [Google Scholar] [CrossRef]
- Kolev, V.N.; Tam, W.F.; Wright, Q.G.; McDermott, S.P.; Vidal, C.M.; Shapiro, I.M.; Xu, Q.; Wicha, M.S.; Pachter, J.A.; Weaver, D.T. Inhibition of FAK kinase activity preferentially targets cancer stem cells. Oncotarget 2017, 8, 51733–51747. [Google Scholar] [CrossRef]
- Luo, M.; Fan, H.; Nagy, T.; Wei, H.; Wang, C.; Liu, S.; Wicha, M.S.; Guan, J.L. Mammary epithelial-specific ablation of the focal adhesion kinase suppresses mammary tumorigenesis by affecting mammary cancer stem/progenitor cells. Cancer Res. 2009, 69, 466–474. [Google Scholar] [CrossRef]
- Chansri, N.; Kawakami, S.; Yamashita, F.; Hashida, M. Inhibition of liver metastasis by all-trans retinoic acid incorporated into O/W emulsions in mice. Int. J. Pharm. 2006, 321, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Xian, M.; Xiang, S.; Xiang, D.; Shao, X.; Wang, J.; Cao, J.; Yang, X.; Yang, B.; Ying, M.; et al. All-Trans Retinoic Acid Prevents Osteosarcoma Metastasis by Inhibiting M2 Polarization of Tumor-Associated Macrophages. Cancer Immunol. Res. 2017, 5, 547–559. [Google Scholar] [CrossRef]
- Grace, V.M.B.; Saranya, S.; Wilson, D.D. Protective role of All Trans Retinoic Acid on B16F10 melanoma cell line metastasis in C57BL/6 mice by enhancing RAR- beta protein and homeostasis maintenance. J. Histotechnol. 2021, 44, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Bessone, M.I.D.; Berardi, D.E.; Cirigliano, S.M.; Delbart, D.I.; Peters, M.G.; Todaro, L.B.; Urtreger, A.J. Protein Kinase C Alpha (PKCalpha) overexpression leads to a better response to retinoid acid therapy through Retinoic Acid Receptor Beta (RARbeta) activation in mammary cancer cells. J. Cancer Res. Clin. Oncol. 2020, 146, 3241–3253. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castro-Guijarro, A.C.; Vanderhoeven, F.; Mondaca, J.M.; Redondo, A.L.; Zoppino, F.C.M.; Fernandez-Muñoz, J.M.; Sanchez, A.M.; Flamini, M.I. Combination Treatment of Retinoic Acid Plus Focal Adhesion Kinase Inhibitor Prevents Tumor Growth and Breast Cancer Cell Metastasis. Cells 2022, 11, 2988. https://doi.org/10.3390/cells11192988
Castro-Guijarro AC, Vanderhoeven F, Mondaca JM, Redondo AL, Zoppino FCM, Fernandez-Muñoz JM, Sanchez AM, Flamini MI. Combination Treatment of Retinoic Acid Plus Focal Adhesion Kinase Inhibitor Prevents Tumor Growth and Breast Cancer Cell Metastasis. Cells. 2022; 11(19):2988. https://doi.org/10.3390/cells11192988
Chicago/Turabian StyleCastro-Guijarro, Ana Carla, Fiorella Vanderhoeven, Joselina Magali Mondaca, Analía Lourdes Redondo, Felipe Carlos Martin Zoppino, Juan Manuel Fernandez-Muñoz, Angel Matias Sanchez, and Marina Inés Flamini. 2022. "Combination Treatment of Retinoic Acid Plus Focal Adhesion Kinase Inhibitor Prevents Tumor Growth and Breast Cancer Cell Metastasis" Cells 11, no. 19: 2988. https://doi.org/10.3390/cells11192988
APA StyleCastro-Guijarro, A. C., Vanderhoeven, F., Mondaca, J. M., Redondo, A. L., Zoppino, F. C. M., Fernandez-Muñoz, J. M., Sanchez, A. M., & Flamini, M. I. (2022). Combination Treatment of Retinoic Acid Plus Focal Adhesion Kinase Inhibitor Prevents Tumor Growth and Breast Cancer Cell Metastasis. Cells, 11(19), 2988. https://doi.org/10.3390/cells11192988