
Stage-Dependent Activity and Pro-Chondrogenic Function of PI3K/AKT during Cartilage Neogenesis from Mesenchymal Stromal Cells

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Isolation and Expansion of MSCs and ACs
2.2. Cartilage Neogenesis from MSCs and ACs
2.3. Quantitative Gene Expression Analysis
2.4. Western Blotting
2.5. ALP Enzyme Activity
2.6. GAG and DNA Quantification
2.7. Histology
2.8. Statistical Analyses
3. Results
3.1. Inverse Regulation of PI3K/AKT Activation in MSC versus AC Chondrogenic Cultures
3.2. Elevated AKT Activation Is Independent of TGFβ and Cell-Autonomous BMP and WNT Activity
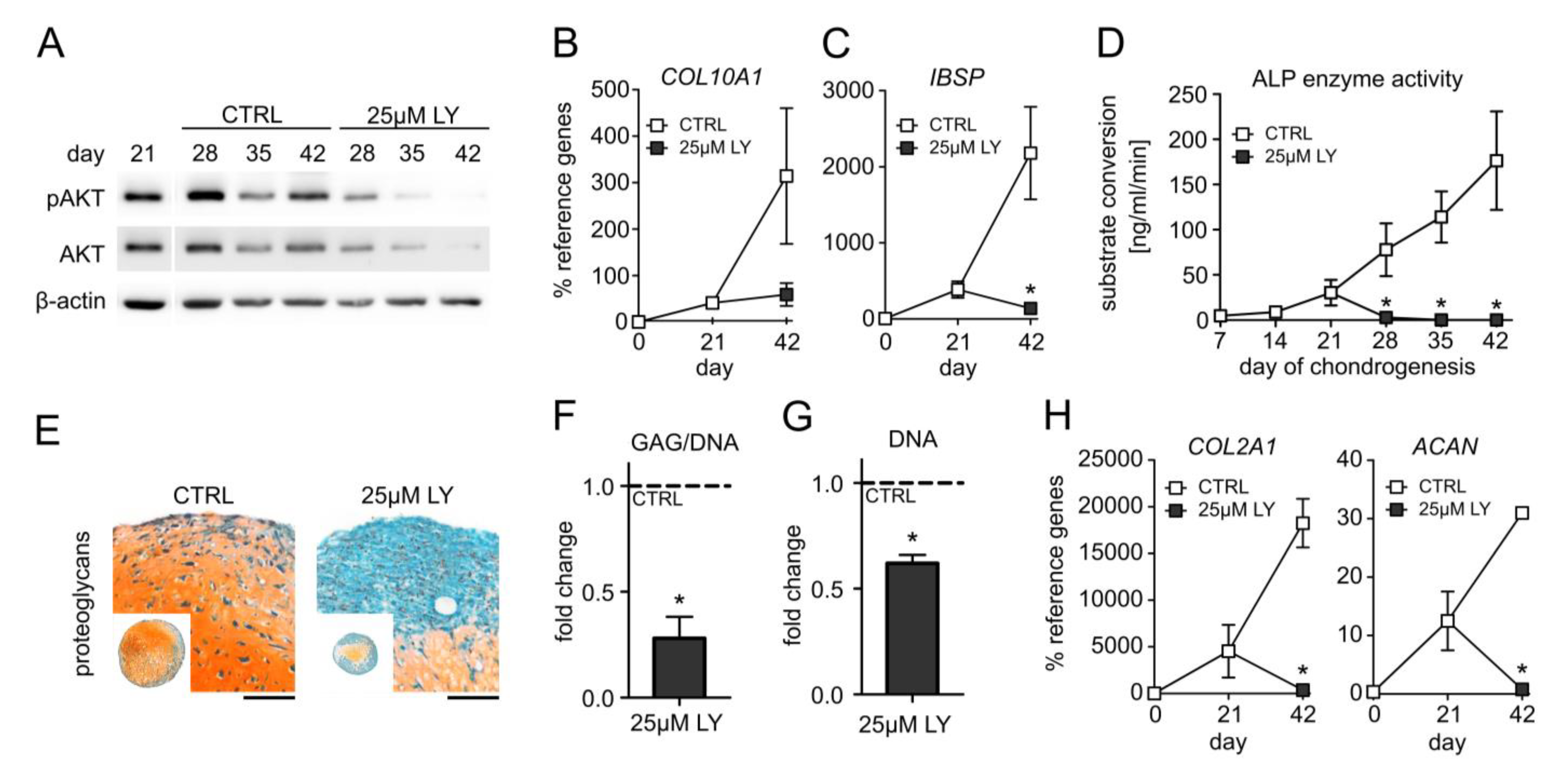
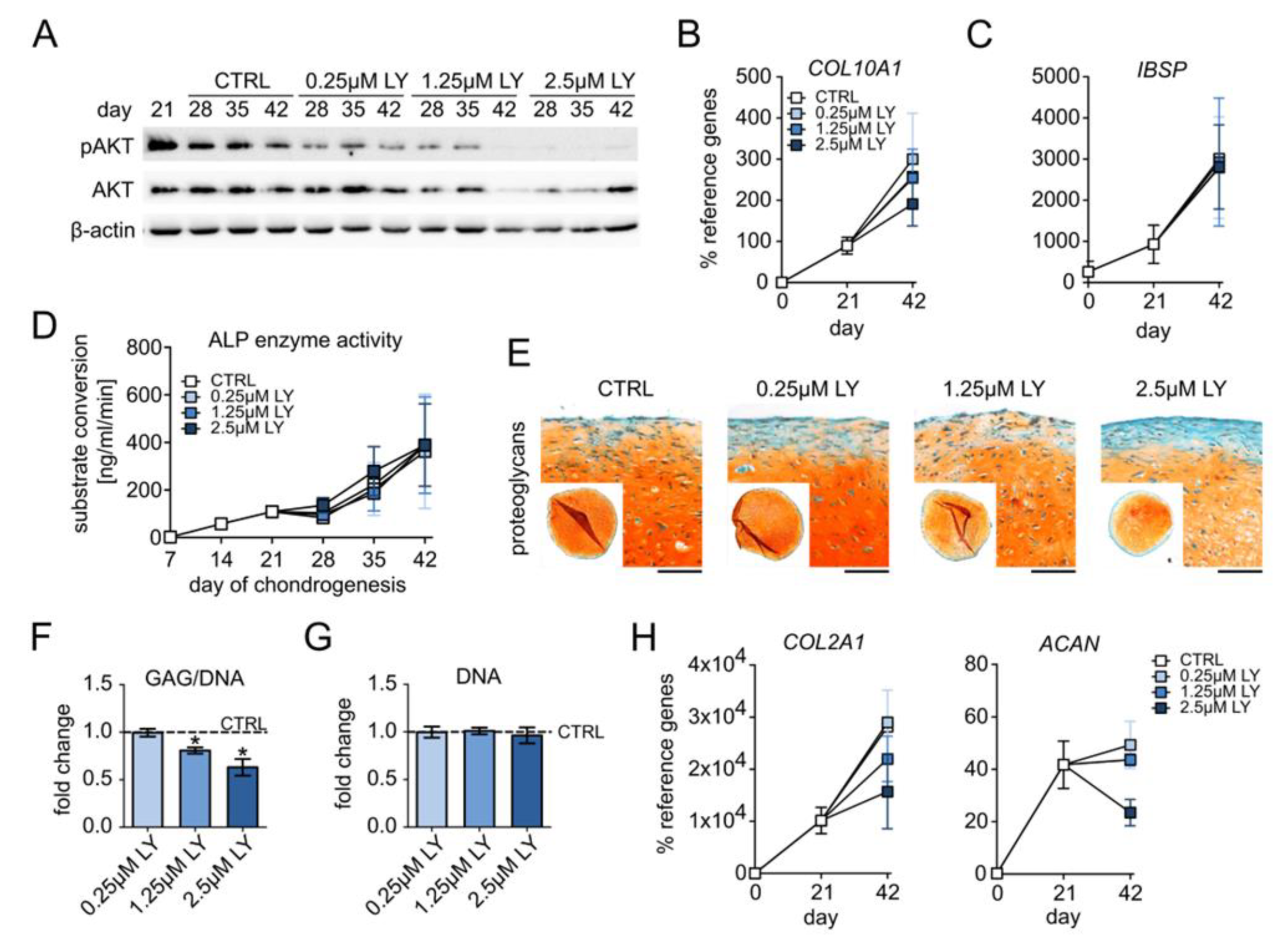
3.3. Elevated PI3K/AKT Activation Is Essential for High Proteoglycan Deposition
3.4. PI3K/AKT Activity Is Important for TGFβ Signaling and High SOX9 Protein Levels
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yoo, J.U.; Barthel, T.S.; Nishimura, K.; Solchaga, L.; Caplan, A.I.; Goldberg, V.M.; Johnstone, B. The chondrogenic potential of human bone-marrow-derived mesenchymal progenitor cells. J. Bone Jt. Surg. Am. 1998, 80, 1745–1757. [Google Scholar] [CrossRef] [PubMed]
- Winter, A.; Breit, S.; Parsch, D.; Benz, K.; Steck, E.; Hauner, H.; Weber, R.M.; Ewerbeck, V.; Richter, W. Cartilage-like gene expression in differentiated human stem cell spheroids: A comparison of bone marrow-derived and adipose tissue-derived stromal cells. Arthritis Rheum. 2003, 48, 418–429. [Google Scholar] [CrossRef] [PubMed]
- Diederichs, S.; Tonnier, V.; Marz, M.; Dreher, S.I.; Geisbusch, A.; Richter, W. Regulation of WNT5A and WNT11 during MSC in vitro chondrogenesis: WNT inhibition lowers BMP and hedgehog activity, and reduces hypertrophy. Cell. Mol. Life Sci. 2019, 76, 3875–3889. [Google Scholar] [CrossRef] [PubMed]
- Pelttari, K.; Winter, A.; Steck, E.; Goetzke, K.; Hennig, T.; Ochs, B.G.; Aigner, T.; Richter, W. Premature induction of hypertrophy during in vitro chondrogenesis of human mesenchymal stem cells correlates with calcification and vascular invasion after ectopic transplantation in SCID mice. Arthritis Rheum. 2006, 54, 3254–3266. [Google Scholar] [CrossRef]
- Fischer, J.; Dickhut, A.; Rickert, M.; Richter, W. Human articular chondrocytes secrete parathyroid hormone-related protein and inhibit hypertrophy of mesenchymal stem cells in coculture during chondrogenesis. Arthritis Rheum. 2010, 62, 2696–2706. [Google Scholar] [CrossRef]
- Leijten, J.C.; Emons, J.; Sticht, C.; van Gool, S.; Decker, E.; Uitterlinden, A.; Rappold, G.; Hofman, A.; Rivadeneira, F.; Scherjon, S.; et al. Gremlin 1, frizzled-related protein, and Dkk-1 are key regulators of human articular cartilage homeostasis. Arthritis Rheum. 2012, 64, 3302–3312. [Google Scholar] [CrossRef]
- Dexheimer, V.; Gabler, J.; Bomans, K.; Sims, T.; Omlor, G.; Richter, W. Differential expression of TGF-β superfamily members and role of Smad1/5/9-signalling in chondral versus endochondral chondrocyte differentiation. Sci. Rep. 2016, 6, 36655. [Google Scholar] [CrossRef]
- Dreher, S.I.; Fischer, J.; Walker, T.; Diederichs, S.; Richter, W. Significance of MEF2C and RUNX3 Regulation for Endochondral Differentiation of Human Mesenchymal Progenitor Cells. Front. Cell. Dev. Biol. 2020, 8, 81. [Google Scholar] [CrossRef]
- Franco, R.A.G.; McKenna, E.; Robey, P.G.; Shajib, M.S.; Crawford, R.W.; Doran, M.R.; Futrega, K. Inhibition of BMP signaling with LDN 193189 can influence bone marrow stromal cell fate but does not prevent hypertrophy during chondrogenesis. Stem Cell Rep. 2022, 17, 616–632. [Google Scholar] [CrossRef]
- Fischer, J.; Ortel, M.; Hagmann, S.; Hoeflich, A.; Richter, W. Role of PTHrP(1-34) Pulse Frequency Versus Pulse Duration to Enhance Mesenchymal Stromal Cell Chondrogenesis. J. Cell. Physiol. 2016, 231, 2673–2681. [Google Scholar] [CrossRef]
- Beier, F.; Loeser, R.F. Biology and pathology of Rho GTPase, PI-3 kinase-Akt, and MAP kinase signaling pathways in chondrocytes. J. Cell. Biochem. 2010, 110, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [PubMed]
- Steele-Perkins, G.; Turner, J.; Edman, J.C.; Hari, J.; Pierce, S.B.; Stover, C.; Rutter, W.J.; Roth, R.A. Expression and characterization of a functional human insulin-like growth factor I receptor. J. Biol. Chem. 1988, 263, 11486–11492. [Google Scholar] [CrossRef]
- Denley, A.; Cosgrove, L.J.; Booker, G.W.; Wallace, J.C.; Forbes, B.E. Molecular interactions of the IGF system. Cytokine Growth Factor Rev. 2005, 16, 421–439. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cheng, Z.; Elalieh, H.Z.; Nakamura, E.; Nguyen, M.T.; Mackem, S.; Clemens, T.L.; Bikle, D.D.; Chang, W. IGF-1R signaling in chondrocytes modulates growth plate development by interacting with the PTHrP/Ihh pathway. J. Bone Miner. Res. 2011, 26, 1437–1446. [Google Scholar] [CrossRef]
- Ulici, V.; Hoenselaar, K.D.; Agoston, H.; McErlain, D.D.; Umoh, J.; Chakrabarti, S.; Holdsworth, D.W.; Beier, F. The role of Akt1 in terminal stages of endochondral bone formation: Angiogenesis and ossification. Bone 2009, 45, 1133–1145. [Google Scholar] [CrossRef]
- Peng, X.D.; Xu, P.Z.; Chen, M.L.; Hahn-Windgassen, A.; Skeen, J.; Jacobs, J.; Sundararajan, D.; Chen, W.S.; Crawford, S.E.; Coleman, K.G.; et al. Dwarfism, impaired skin development, skeletal muscle atrophy, delayed bone development, and impeded adipogenesis in mice lacking Akt1 and Akt2. Genes Dev. 2003, 17, 1352–1365. [Google Scholar] [CrossRef]
- Louvi, A.; Accili, D.; Efstratiadis, A. Growth-promoting interaction of IGF-II with the insulin receptor during mouse embryonic development. Dev. Biol. 1997, 189, 33–48. [Google Scholar] [CrossRef]
- Heilig, J.; Paulsson, M.; Zaucke, F. Insulin-like growth factor 1 receptor (IGF1R) signaling regulates osterix expression and cartilage matrix mineralization during endochondral ossification. Bone 2016, 83, 48–57. [Google Scholar] [CrossRef]
- Hunziker, E.B.; Wagner, J.; Zapf, J. Differential effects of insulin-like growth factor I and growth hormone on developmental stages of rat growth plate chondrocytes in vivo. J. Clin. Investig. 1994, 93, 1078–1086. [Google Scholar] [CrossRef]
- Luyten, F.P.; Hascall, V.C.; Nissley, S.P.; Morales, T.I.; Reddi, A.H. Insulin-like growth factors maintain steady-state metabolism of proteoglycans in bovine articular cartilage explants. Arch. Biochem. Biophys. 1988, 267, 416–425. [Google Scholar] [CrossRef]
- Osborn, K.D.; Trippel, S.B.; Mankin, H.J. Growth factor stimulation of adult articular cartilage. J. Orthop. Res. 1989, 7, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Yaeger, P.C.; Masi, T.L.; de Ortiz, J.L.; Binette, F.; Tubo, R.; McPherson, J.M. Synergistic action of transforming growth factor-beta and insulin-like growth factor-I induces expression of type II collagen and aggrecan genes in adult human articular chondrocytes. Exp. Cell Res. 1997, 237, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Chubinskaya, S.; Hakimiyan, A.A.; Rappoport, L.; Yanke, A.; Rueger, D.C.; Cole, B.J. Response of human chondrocytes prepared for autologous implantation to growth factors. J. Knee Surg. 2008, 21, 192–199. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Maor, G.; Hochberg, Z.; Silbermann, M. Insulin-like growth factor I accelerates proliferation and differentiation of cartilage progenitor cells in cultures of neonatal mandibular condyles. Acta Endocrinol. 1993, 128, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Cigan, A.D.; Nims, R.J.; Albro, M.B.; Esau, J.D.; Dreyer, M.P.; Vunjak-Novakovic, G.; Hung, C.T.; Ateshian, G.A. Insulin, ascorbate, and glucose have a much greater influence than transferrin and selenous acid on the in vitro growth of engineered cartilage in chondrogenic media. Tissue Eng. Part. A 2013, 19, 1941–1948. [Google Scholar] [CrossRef]
- Starkman, B.G.; Cravero, J.D.; Delcarlo, M.; Loeser, R.F. IGF-I stimulation of proteoglycan synthesis by chondrocytes requires activation of the PI 3-kinase pathway but not ERK MAPK. Biochem. J. 2005, 389, 723–729. [Google Scholar] [CrossRef]
- Ahmed, N.; Iu, J.; Brown, C.E.; Taylor, D.W.; Kandel, R.A. Serum- and growth-factor-free three-dimensional culture system supports cartilage tissue formation by promoting collagen synthesis via Sox9-Col2a1 interaction. Tissue Eng. Part. A 2014, 20, 2224–2233. [Google Scholar] [CrossRef]
- Cheng, C.C.; Uchiyama, Y.; Hiyama, A.; Gajghate, S.; Shapiro, I.M.; Risbud, M.V. PI3K/AKT regulates aggrecan gene expression by modulating Sox9 expression and activity in nucleus pulposus cells of the intervertebral disc. J. Cell. Physiol. 2009, 221, 668–676. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Park, J.I.; Loeser, R.F. Oxidative stress inhibits insulin-like growth factor-I induction of chondrocyte proteoglycan synthesis through differential regulation of phosphatidylinositol 3-Kinase-Akt and MEK-ERK MAPK signaling pathways. J. Biol. Chem. 2009, 284, 31972–31981. [Google Scholar] [CrossRef]
- Fischer, J.; Knoch, N.; Sims, T.; Rosshirt, N.; Richter, W. Time-dependent contribution of BMP, FGF, IGF, and HH signaling to the proliferation of mesenchymal stroma cells during chondrogenesis. J. Cell. Physiol. 2018, 233, 8962–8970. [Google Scholar] [CrossRef] [PubMed]
- Dexheimer, V.; Frank, S.; Richter, W. Proliferation as a Requirement for In Vitro Chondrogenesis of Human Mesenchymal Stem Cells. Stem Cells Dev. 2012, 21, 2160–2169. [Google Scholar] [CrossRef] [PubMed]
- Cooper, K.L.; Oh, S.; Sung, Y.; Dasari, R.R.; Kirschner, M.W.; Tabin, C.J. Multiple phases of chondrocyte enlargement underlie differences in skeletal proportions. Nature 2013, 495, 375–378. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhou, J.; Bondy, C.A. Igf1 promotes longitudinal bone growth by insulin-like actions augmenting chondrocyte hypertrophy. FASEB J. 1999, 13, 1985–1990. [Google Scholar] [CrossRef]
- Liu, J.P.; Baker, J.; Perkins, A.S.; Robertson, E.J.; Efstratiadis, A. Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r). Cell 1993, 75, 59–72. [Google Scholar] [CrossRef]
- Wang, Y.; Nishida, S.; Sakata, T.; Elalieh, H.Z.; Chang, W.; Halloran, B.P.; Doty, S.B.; Bikle, D.D. Insulin-like growth factor-I is essential for embryonic bone development. Endocrinology 2006, 147, 4753–4761. [Google Scholar] [CrossRef]
- Baker, J.; Liu, J.P.; Robertson, E.J.; Efstratiadis, A. Role of insulin-like growth factors in embryonic and postnatal growth. Cell 1993, 75, 73–82. [Google Scholar] [CrossRef]
- Zhang, F.; He, Q.; Tsang, W.P.; Garvey, W.T.; Chan, W.Y.; Wan, C. Insulin exerts direct, IGF-1 independent actions in growth plate chondrocytes. Bone Res. 2014, 2, 14012. [Google Scholar] [CrossRef]
- Mushtaq, T.; Bijman, P.; Ahmed, S.F.; Farquharson, C. Insulin-like growth factor-I augments chondrocyte hypertrophy and reverses glucocorticoid-mediated growth retardation in fetal mice metatarsal cultures. Endocrinology 2004, 145, 2478–2486. [Google Scholar] [CrossRef]
- Abbaspour, A.; Takata, S.; Matsui, Y.; Katoh, S.; Takahashi, M.; Yasui, N. Continuous infusion of insulin-like growth factor-I into the epiphysis of the tibia. Int. Orthop. 2008, 32, 395–402. [Google Scholar] [CrossRef]
- Phornphutkul, C.; Wu, K.Y.; Auyeung, V.; Chen, Q.; Gruppuso, P.A. mTOR signaling contributes to chondrocyte differentiation. Dev. Dyn. 2008, 237, 702–712. [Google Scholar] [CrossRef] [PubMed]
- Phornphutkul, C.; Wu, K.Y.; Gruppuso, P.A. The role of insulin in chondrogenesis. Mol. Cell. Endocrinol. 2006, 249, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Leach, R.M., Jr.; Richards, M.P.; Praul, C.A.; Ford, B.C.; McMurtry, J.P. Investigation of the insulin-like growth factor system in the avian epiphyseal growth plate. Domest. Anim. Endocrinol. 2007, 33, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Choukair, D.; Hügel, U.; Sander, A.; Uhlmann, L.; Tönshoff, B. Inhibition of IGF-I-related intracellular signaling pathways by proinflammatory cytokines in growth plate chondrocytes. Pediatr. Res. 2014, 76, 245–251. [Google Scholar] [CrossRef]
- Ciarmatori, S.; Kiepe, D.; Haarmann, A.; Huegel, U.; Tönshoff, B. Signaling mechanisms leading to regulation of proliferation and differentiation of the mesenchymal chondrogenic cell line RCJ3.1C5.18 in response to IGF-I. J. Mol. Endocrinol. 2007, 38, 493–508. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, X.; Li, H.; Chen, C.; Hu, B.; Niu, X.; Li, Q.; Zhao, B.; Xie, Z.; Wang, Y. Exosomes/tricalcium phosphate combination scaffolds can enhance bone regeneration by activating the PI3K/Akt signaling pathway. Stem Cell. Res. Ther. 2016, 7, 136. [Google Scholar] [CrossRef]
- Baker, N.; Sohn, J.; Tuan, R.S. Promotion of human mesenchymal stem cell osteogenesis by PI3-kinase/Akt signaling, and the influence of caveolin-1/cholesterol homeostasis. Stem Cell. Res. Ther. 2015, 6, 238. [Google Scholar] [CrossRef]
- Xia, P.; Gu, R.; Zhang, W.; Shao, L.; Li, F.; Wu, C.; Sun, Y. MicroRNA-200c promotes osteogenic differentiation of human bone mesenchymal stem cells through activating the AKT/β-Catenin signaling pathway via downregulating Myd88. J. Cell. Physiol. 2019, 234, 22675–22686. [Google Scholar] [CrossRef]
- Ye, C.; Zhang, W.; Hang, K.; Chen, M.; Hou, W.; Chen, J.; Chen, X.; Chen, E.; Tang, L.; Lu, J.; et al. Extracellular IL-37 promotes osteogenic differentiation of human bone marrow mesenchymal stem cells via activation of the PI3K/AKT signaling pathway. Cell Death Dis. 2019, 10, 753. [Google Scholar] [CrossRef]
- Farndale, R.W.; Buttle, D.J.; Barrett, A.J. Improved quantitation and discrimination of sulphated glycosaminoglycans by use of dimethylmethylene blue. Biochim. Biophys. Acta 1986, 883, 173–177. [Google Scholar] [CrossRef]
- Mueller, M.B.; Fischer, M.; Zellner, J.; Berner, A.; Dienstknecht, T.; Prantl, L.; Kujat, R.; Nerlich, M.; Tuan, R.S.; Angele, P. Hypertrophy in mesenchymal stem cell chondrogenesis: Effect of TGF-beta isoforms and chondrogenic conditioning. Cells Tissues Organs 2010, 192, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Narcisi, R.; Cleary, M.A.; Brama, P.A.; Hoogduijn, M.J.; Tüysüz, N.; ten Berge, D.; van Osch, G.J. Long-term expansion, enhanced chondrogenic potential, and suppression of endochondral ossification of adult human MSCs via WNT signaling modulation. Stem Cell Rep. 2015, 4, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Kita, K.; Kimura, T.; Nakamura, N.; Yoshikawa, H.; Nakano, T. PI3K/Akt signaling as a key regulatory pathway for chondrocyte terminal differentiation. Genes Cells 2008, 13, 839–850. [Google Scholar] [CrossRef] [PubMed]
- Ikegami, D.; Akiyama, H.; Suzuki, A.; Nakamura, T.; Nakano, T.; Yoshikawa, H.; Tsumaki, N. Sox9 sustains chondrocyte survival and hypertrophy in part through Pik3ca-Akt pathways. Development 2011, 138, 1507–1519. [Google Scholar] [CrossRef]
- Fukai, A.; Kawamura, N.; Saito, T.; Oshima, Y.; Ikeda, T.; Kugimiya, F.; Higashikawa, A.; Yano, F.; Ogata, N.; Nakamura, K.; et al. Akt1 in murine chondrocytes controls cartilage calcification during endochondral ossification under physiologic and pathologic conditions. Arthritis Rheum. 2010, 62, 826–836. [Google Scholar] [CrossRef]
- Ulici, V.; Hoenselaar, K.D.; Gillespie, J.R.; Beier, F. The PI3K pathway regulates endochondral bone growth through control of hypertrophic chondrocyte differentiation. BMC Dev. Biol. 2008, 8, 40. [Google Scholar] [CrossRef]
- Luo, K. Signaling Cross Talk between TGF-β/Smad and Other Signaling Pathways. Cold Spring Harb. Perspect. Biol. 2017, 9, a022137. [Google Scholar] [CrossRef]
- Danielpour, D.; Song, K. Cross-talk between IGF-I and TGF-beta signaling pathways. Cytokine Growth Factor Rev. 2006, 17, 59–74. [Google Scholar] [CrossRef]
- Bakin, A.V.; Tomlinson, A.K.; Bhowmick, N.A.; Moses, H.L.; Arteaga, C.L. Phosphatidylinositol 3-kinase function is required for transforming growth factor beta-mediated epithelial to mesenchymal transition and cell migration. J. Biol. Chem. 2000, 275, 36803–36810. [Google Scholar] [CrossRef]
- Thien, A.; Prentzell, M.T.; Holzwarth, B.; Kläsener, K.; Kuper, I.; Boehlke, C.; Sonntag, A.G.; Ruf, S.; Maerz, L.; Nitschke, R.; et al. TSC1 activates TGF-β-Smad2/3 signaling in growth arrest and epithelial-to-mesenchymal transition. Dev. Cell 2015, 32, 617–630. [Google Scholar] [CrossRef]
- Tsukazaki, T.; Chiang, T.A.; Davison, A.F.; Attisano, L.; Wrana, J.L. SARA, a FYVE domain protein that recruits Smad2 to the TGFbeta receptor. Cell 1998, 95, 779–791. [Google Scholar] [CrossRef]
- Fernandez-Cancio, M.; Esteban, C.; Carrascosa, A.; Toran, N.; Andaluz, P.; Audi, L. IGF-I and not IGF-II expression is regulated by glucocorticoids in human fetal epiphyseal chondrocytes. Growth Horm. IGF Res. 2008, 18, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Steinert, A.F.; Palmer, G.D.; Pilapil, C.; Nöth, U.; Evans, C.H.; Ghivizzani, S.C. Enhanced in vitro chondrogenesis of primary mesenchymal stem cells by combined gene transfer. Tissue Eng. Part. A 2009, 15, 1127–1139. [Google Scholar] [CrossRef] [PubMed]
- Frisch, J.; Venkatesan, J.K.; Rey-Rico, A.; Schmitt, G.; Madry, H.; Cucchiarini, M. Influence of insulin-like growth factor I overexpression via recombinant adeno-associated vector gene transfer upon the biological activities and differentiation potential of human bone marrow-derived mesenchymal stem cells. Stem Cell. Res. Ther. 2014, 5, 103. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Chun, H.; Baek, J.; Sadik, J.E.; Shirazyan, A.; Razavi, P.; Lopez, N.; Lyons, K.M. The TGFβ type I receptor TGFβRI functions as an inhibitor of BMP signaling in cartilage. Proc. Natl. Acad. Sci. USA 2019, 116, 15570–15579. [Google Scholar] [CrossRef]
- Gharbi, S.I.; Zvelebil, M.J.; Shuttleworth, S.J.; Hancox, T.; Saghir, N.; Timms, J.F.; Waterfield, M.D. Exploring the specificity of the PI3K family inhibitor LY294002. Biochem. J. 2007, 404, 15–21. [Google Scholar] [CrossRef]
- Bain, J.; Plater, L.; Elliott, M.; Shpiro, N.; Hastie, C.J.; McLauchlan, H.; Klevernic, I.; Arthur, J.S.; Alessi, D.R.; Cohen, P. The selectivity of protein kinase inhibitors: A further update. Biochem. J. 2007, 408, 297–315. [Google Scholar] [CrossRef]
- Davies, S.P.; Reddy, H.; Caivano, M.; Cohen, P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J. 2000, 351, 95–105. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klampfleuthner, F.A.M.; Lotz, B.; Renkawitz, T.; Richter, W.; Diederichs, S. Stage-Dependent Activity and Pro-Chondrogenic Function of PI3K/AKT during Cartilage Neogenesis from Mesenchymal Stromal Cells. Cells 2022, 11, 2965. https://doi.org/10.3390/cells11192965
Klampfleuthner FAM, Lotz B, Renkawitz T, Richter W, Diederichs S. Stage-Dependent Activity and Pro-Chondrogenic Function of PI3K/AKT during Cartilage Neogenesis from Mesenchymal Stromal Cells. Cells. 2022; 11(19):2965. https://doi.org/10.3390/cells11192965
Chicago/Turabian StyleKlampfleuthner, Felicia A. M., Benedict Lotz, Tobias Renkawitz, Wiltrud Richter, and Solvig Diederichs. 2022. "Stage-Dependent Activity and Pro-Chondrogenic Function of PI3K/AKT during Cartilage Neogenesis from Mesenchymal Stromal Cells" Cells 11, no. 19: 2965. https://doi.org/10.3390/cells11192965
APA StyleKlampfleuthner, F. A. M., Lotz, B., Renkawitz, T., Richter, W., & Diederichs, S. (2022). Stage-Dependent Activity and Pro-Chondrogenic Function of PI3K/AKT during Cartilage Neogenesis from Mesenchymal Stromal Cells. Cells, 11(19), 2965. https://doi.org/10.3390/cells11192965