

Endothelial-to-Mesenchymal Transition in Atherosclerosis: Friend or Foe?

 , , ,
, , ,
Abstract
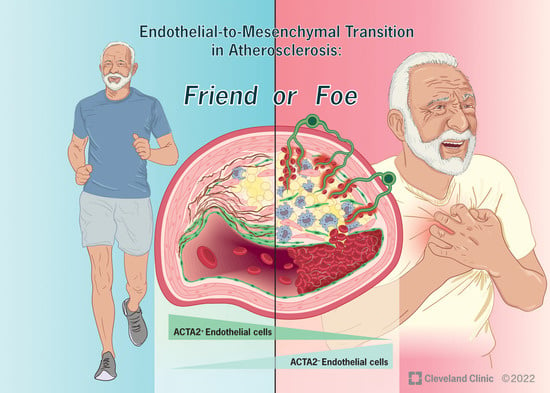
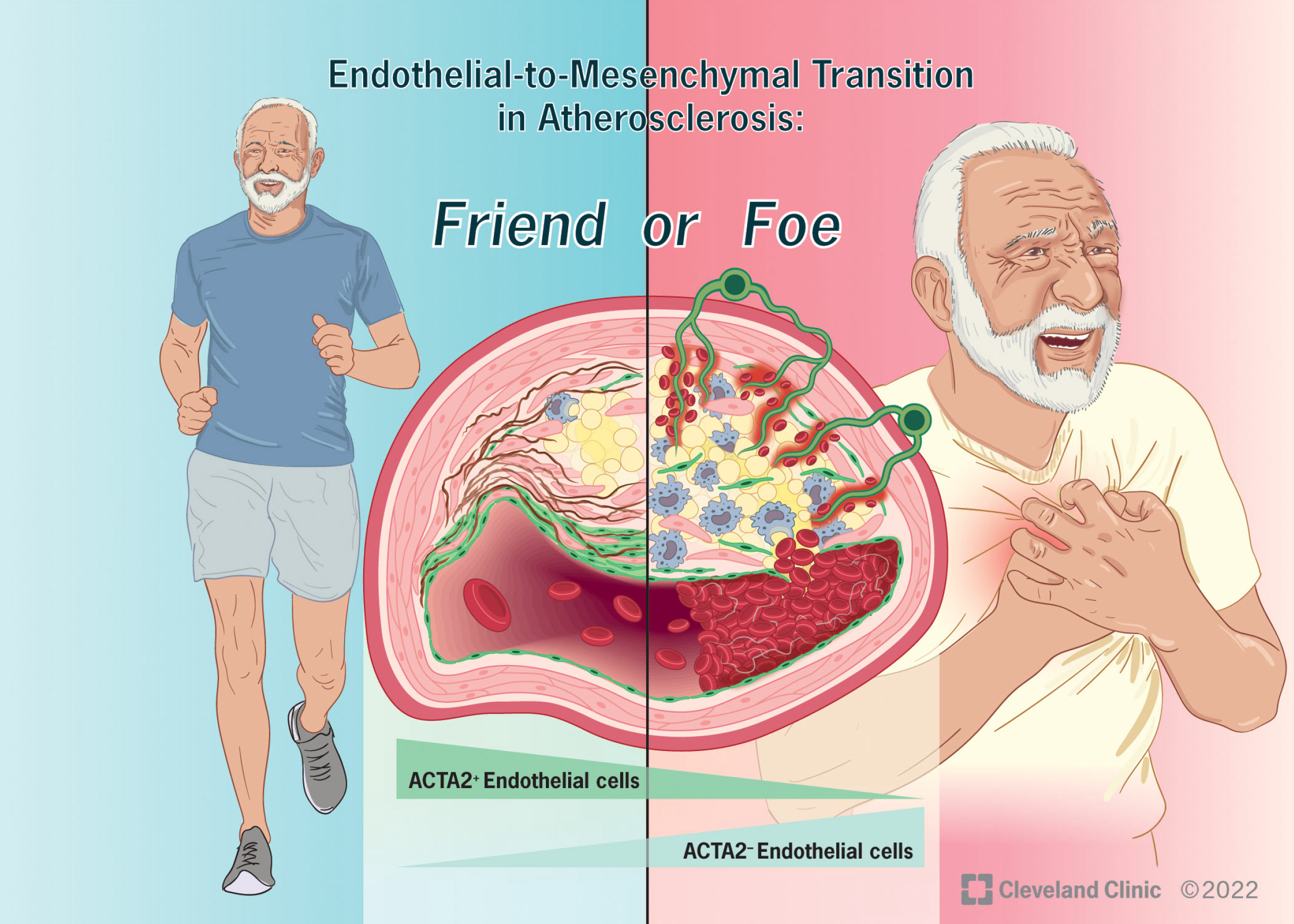{kind=link}
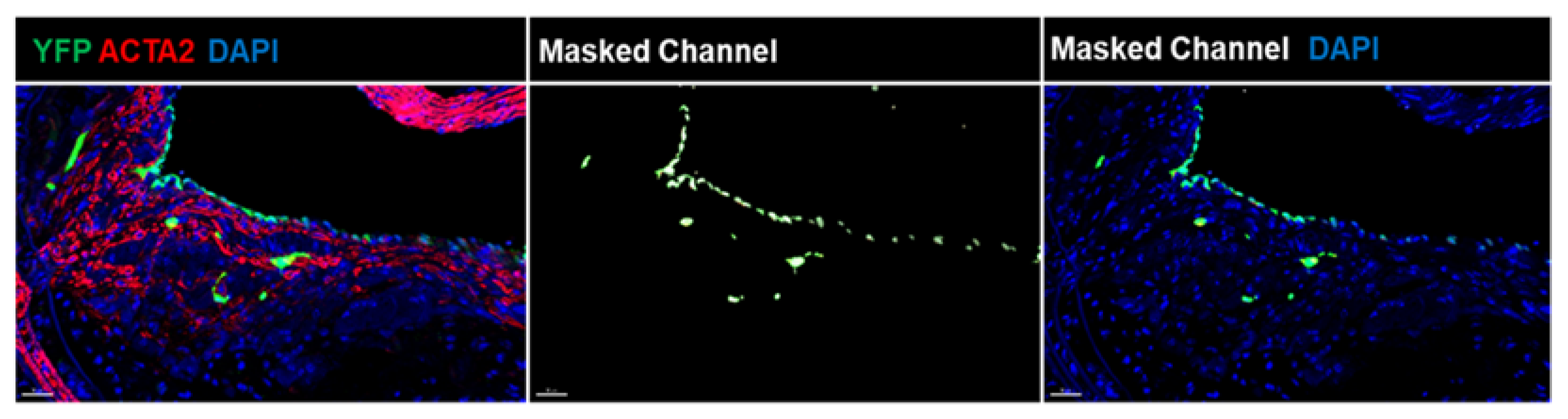{kind=link}
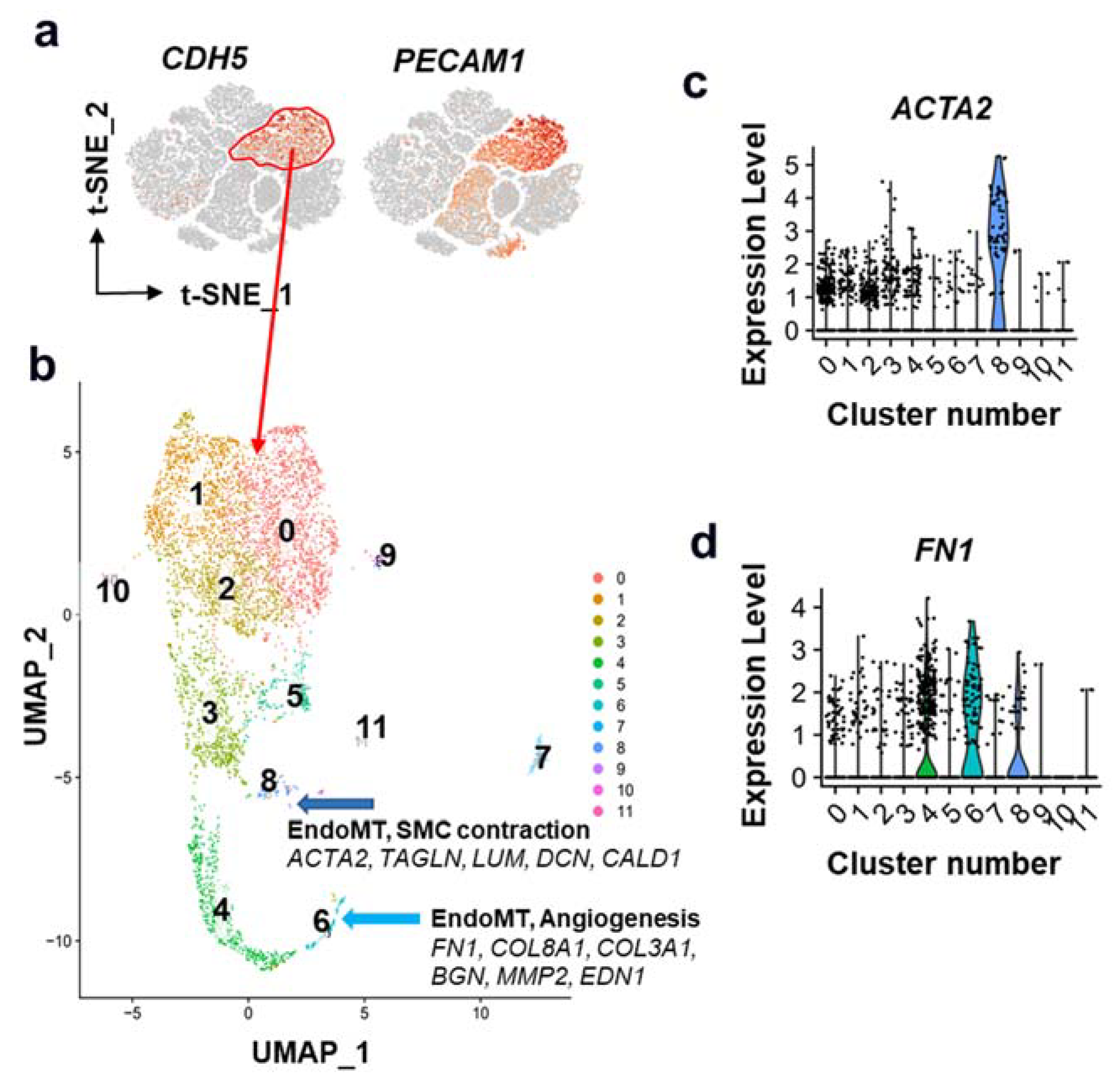{kind=link}
1. Introduction
2. Potential Detrimental Role of EndoMT in Atherosclerosis
3. Potential Athero-Protective Role of EndoMT
4. Multiple Sub-Phenotypes of EndoMT in Atherosclerosis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Markwald, R.R.; Krook, J.M.; Kitten, G.T.; Runyan, R.B. Endocardial cushion tissue development: Structural analyses on the attachment of extracellular matrix to migrating mesenchymal cell surfaces. Scanning Electron Microsc. 1981, Pt 2, 261–274. [Google Scholar]
- Runyan, R.B.; Markwald, R.R. Invasion of mesenchyme into three-dimensional collagen gels: A regional and temporal analysis of interaction in embryonic heart tissue. Dev. Biol. 1983, 95, 108–114. [Google Scholar] [CrossRef]
- Pardali, E.; Sanchez-Duffhues, G.; Gomez-Puerto, M.C.; Ten Dijke, P. TGF-β-Induced Endothelial-Mesenchymal Transition in Fibrotic Diseases. Int. J. Mol. Sci. 2017, 18, 2157. [Google Scholar] [CrossRef]
- Maleszewska, M.; Moonen, J.-R.A.J.; Huijkman, N.; van de Sluis, B.; Krenning, G.; Harmsen, M.C. IL-1β and TGFβ2 synergistically induce endothelial to mesenchymal transition in an NFκB-dependent manner. Immunobiology 2013, 218, 443–454. [Google Scholar] [CrossRef]
- Huang, M.; Yang, F.; Zhang, D.; Lin, M.; Duan, H.; El-Mayta, R.; Zhang, L.; Qin, L.; Shewale, S.V.; Pei, L.; et al. Endothelial plasticity drives aberrant vascularization and impedes cardiac repair after myocardial infarction. Nat. Cardiovasc. Res. 2022, 1, 372–388. [Google Scholar] [CrossRef]
- Liebner, S.; Cattelino, A.; Gallini, R.; Rudini, N.; Iurlaro, M.; Piccolo, S.; Dejana, E. β-Catenin is required for endothelial-mesenchymal transformation during heart cushion development in the mouse. J. Cell Biol. 2004, 166, 359–367. [Google Scholar] [CrossRef]
- Widyantoro, B.; Emoto, N.; Nakayama, K.; Anggrahini, D.W.; Adiarto, S.; Iwasa, N.; Yagi, K.; Miyagawa, K.; Rikitake, Y.; Suzuki, T.; et al. Endothelial Cell–Derived Endothelin-1 Promotes Cardiac Fibrosis in Diabetic Hearts Through Stimulation of Endothelial-to-Mesenchymal Transition. Circulation 2010, 121, 2407–2418. [Google Scholar] [CrossRef]
- Peng, Q.; Shan, D.; Cui, K.; Li, K.; Zhu, B.; Wu, H.; Wang, B.; Wong, S.; Norton, V.; Dong, Y.; et al. The Role of Endothelial-to-Mesenchymal Transition in Cardiovascular Disease. Cells 2022, 11, 1834. [Google Scholar] [CrossRef]
- Newby, A. Fibrous cap formation or destruction—The critical importance of vascular smooth muscle cell proliferation, migration and matrix formation. Cardiovasc. Res. 1999, 41, 345–360. [Google Scholar] [CrossRef]
- Finn, A.V.; Nakano, M.; Narula, J.; Kolodgie, F.D.; Virmani, R. Concept of vulnerable/unstable plaque. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1282–1292. [Google Scholar] [CrossRef]
- Evrard, S.M.; Lecce, L.; Michelis, K.C.; Nomura-Kitabayashi, A.; Pandey, G.; Purushothaman, K.-R.; d’Escamard, V.; Li, J.R.; Hadri, L.; Fujitani, K.; et al. Endothelial to mesenchymal transition is common in atherosclerotic lesions and is associated with plaque instability. Nat. Commun. 2016, 7, 11853. [Google Scholar] [CrossRef]
- Chen, P.-Y.; Qin, L.; Barnes, C.; Charisse, K.; Yi, T.; Zhang, X.; Ali, R.; Medina, P.P.; Yu, J.; Slack, F.J.; et al. FGF regulates TGF-beta signaling and endothelial-to-mesenchymal transition via control of let-7 miRNA expression. Cell Rep. 2012, 2, 1684–1696. [Google Scholar] [CrossRef]
- Chen, P.-Y.; Qin, L.; Li, G.; Wang, Z.; Dahlman, J.E.; Malagon-Lopez, J.; Gujja, S.; Cilfone, N.A.; Kauffman, K.J.; Sun, L.; et al. Endothelial TGF-β signalling drives vascular inflammation and atherosclerosis. Nat. Metab. 2019, 1, 912–926. [Google Scholar] [CrossRef]
- Cooley, B.C.; Nevado, J.; Mellad, J.; Yang, D.; St Hilaire, C.; Negro, A.; Fang, F.; Chen, G.; San, H.; Walts, A.D.; et al. TGF-β signaling mediates endothelial-to-mesenchymal transition (EndMT) during vein graft remodeling. Sci. Transl. Med. 2014, 6, 227ra34. [Google Scholar] [CrossRef]
- Chen, P.-Y.; Qin, L.; Baeyens, N.; Li, G.; Afolabi, T.; Budatha, M.; Tellides, G.; Schwartz, M.A.; Simons, M. Endothelial-to-mesenchymal transition drives atherosclerosis progression. J. Clin. Investig. 2015, 125, 4514–4528. [Google Scholar] [CrossRef]
- Shin, J.; Tkachenko, S.; Chaklader, M.; Pletz, C.; Singh, K.; Bulut, G.B.; Han, Y.; Mitchell, K.; Baylis, R.A.; Kuzmin, A.A.; et al. Endothelial OCT4 is atheroprotective by preventing metabolic and phenotypic dysfunction. Cardiovasc. Res. 2022, 118, 2458–2477. [Google Scholar] [CrossRef]
- Alencar, G.F.; Owsiany, K.M.; Karnewar, S.; Sukhavasi, K.; Mocci, G.; Nguyen, A.; Williams, C.M.; Shamsuzzaman, S.; Mokry, M.; Henderson, C.A.; et al. The Stem Cell Pluripotency Genes Klf4 and Oct4 Regulate Complex SMC Phenotypic Changes Critical in Late-Stage Atherosclerotic Lesion Pathogenesis. Circulation 2020, 142, 2045–2059. [Google Scholar] [CrossRef]
- Gomez, D.; Shankman, L.S.; Nguyen, A.T.; Owens, G.K. Detection of histone modifications at specific gene loci in single cells in histological sections. Nat. Methods 2013, 10, 171–177. [Google Scholar] [CrossRef]
- Newman, A.A.C.; Serbulea, V.; Baylis, R.A.; Shankman, L.S.; Bradley, X.; Alencar, G.F.; Owsiany, K.; Deaton, R.A.; Karnewar, S.; Shamsuzzaman, S.; et al. Multiple cell types contribute to the atherosclerotic lesion fibrous cap by PDGFRβ and bioenergetic mechanisms. Nat. Metab. 2021, 3, 166–181. [Google Scholar] [CrossRef]
- Mehta, V.; Pang, K.-L.; Givens, C.S.; Chen, Z.; Huang, J.; Sweet, D.T.; Jo, H.; Reader, J.S.; Tzima, E. Mechanical forces regulate endothelial-to-mesenchymal transition and atherosclerosis via an Alk5-Shc mechanotransduction pathway. Sci. Adv. 2021, 7, eabg5060. [Google Scholar] [CrossRef]
- Liang, G.; Wang, S.; Shao, J.; Jin, Y.-J.; Xu, L.; Yan, Y.; Günther, S.; Wang, L.; Offermanns, S. Tenascin-X Mediates Flow-Induced Suppression of EndMT and Atherosclerosis. Circ. Res. 2022, 130, 1647–1659. [Google Scholar] [CrossRef]
- Chen, L.; Shang, C.; Wang, B.; Wang, G.; Jin, Z.; Yao, F.; Yue, Z.; Bai, L.; Wang, R.; Zhao, S.; et al. HDAC3 inhibitor suppresses endothelial-to-mesenchymal transition via modulating inflammatory response in atherosclerosis. Biochem. Pharmacol. 2021, 192, 114716. [Google Scholar] [CrossRef]
- Liu, S.; Xu, D.; Li, M.; Zhang, Y.; Li, Q.; Li, T.; Ren, L. Icariin attenuates endothelial-mesenchymal transition via H19/miR-148b-3p/ELF5 in ox-LDL-stimulated HUVECs. Mol. Ther. Nucleic Acids 2021, 23, 464–475. [Google Scholar] [CrossRef]
- Lai, B.; Li, Z.; He, M.; Wang, Y.; Chen, L.; Zhang, J.; Yang, Y.; Shyy, J.Y.-J. Atheroprone flow enhances the endothelial-to-mesenchymal transition. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1293–H1303. [Google Scholar] [CrossRef]
- Huang, Q.; Gan, Y.; Yu, Z.; Wu, H.; Zhong, Z. Endothelial to Mesenchymal Transition: An Insight in Atherosclerosis. Front. Cardiovasc. Med. 2021, 8, 734550. [Google Scholar] [CrossRef]
- Virmani, R.; Kolodgie, F.D.; Burke, A.P.; Farb, A.; Schwartz, S.M. Lessons From Sudden Coronary Death. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1262–1275. [Google Scholar] [CrossRef]
- Davies, M.J.; Richardson, P.D.; Woolf, N.; Katz, D.R.; Mann, J. Risk of thrombosis in human atherosclerotic plaques: Role of extracellular lipid, macrophage, and smooth muscle cell content. Heart 1993, 69, 377–381. [Google Scholar] [CrossRef]
- Newman, A.A.C.; Baylis, R.A.; Hess, D.L.; Griffith, S.D.; Shankman, L.S.; Cherepanova, O.A.; Owens, G.K. Irradiation abolishes smooth muscle investment into vascular lesions in specific vascular beds. JCI Insight 2018, 3, e121017. [Google Scholar] [CrossRef]
- Karnewar, S.; Baylis, R.; Bradley, X.; Karnewar, V.; Alencar, G.; Owens, G. Regression of Advanced Atherosclerotic Lesions in Response to Long Term Chow Diet Feeding is associated With Increased IL1b-dependent EndoMT. Atheroscler. Thromb. Vacular Biol. 2020, 40, AMP132. [Google Scholar]
- Wirka, R.C.; Wagh, D.; Paik, D.T.; Pjanic, M.; Nguyen, T.; Miller, C.L.; Kundu, R.; Nagao, M.; Coller, J.; Koyano, T.K.; et al. Atheroprotective roles of smooth muscle cell phenotypic modulation and the TCF21 disease gene as revealed by single-cell analysis. Nat. Med. 2019, 25, 1280–1289. [Google Scholar] [CrossRef]
- Dobnikar, L.; Taylor, A.L.; Chappell, J.; Oldach, P.; Harman, J.L.; Oerton, E.; Dzierzak, E.; Bennett, M.R.; Spivakov, M.; Jørgensen, H.F. Disease-relevant transcriptional signatures identified in individual smooth muscle cells from healthy mouse vessels. Nat. Commun. 2018, 9, 4567. [Google Scholar] [CrossRef]
- Pan, H.; Xue, C.; Auerbach, B.J.; Fan, J.; Bashore, A.C.; Cui, J.; Yang, D.Y.; Trignano, S.B.; Liu, W.; Shi, J.; et al. Single-Cell Genomics Reveals a Novel Cell State During Smooth Muscle Cell Phenotypic Switching and Potential Therapeutic Targets for Atherosclerosis in Mouse and Human. Circulation 2020, 142, 2060–2075. [Google Scholar] [CrossRef]
- Orr, A.W.; Sanders, J.M.; Bevard, M.; Coleman, E.; Sarembock, I.J.; Schwartz, M.A. The subendothelial extracellular matrix modulates NF-κB activation by flow. J. Cell Biol. 2005, 169, 191–202. [Google Scholar] [CrossRef]
- Rohwedder, I.; Montanez, E.; Beckmann, K.; Bengtsson, E.; Dunér, P.; Nilsson, J.; Soehnlein, O.; Fässler, R. Plasma fibronectin deficiency impedes atherosclerosis progression and fibrous cap formation. EMBO Mol. Med. 2012, 4, 564–576. [Google Scholar] [CrossRef]
- Al-Yafeai, Z.; Yurdagul, A.; Peretik, J.M.; Alfaidi, M.; Murphy, P.A.; Orr, A.W. Endothelial FN (Fibronectin) Deposition by α5β1 Integrins Drives Atherogenic Inflammation. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 2601–2614. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gole, S.; Tkachenko, S.; Masannat, T.; Baylis, R.A.; Cherepanova, O.A. Endothelial-to-Mesenchymal Transition in Atherosclerosis: Friend or Foe? Cells 2022, 11, 2946. https://doi.org/10.3390/cells11192946
Gole S, Tkachenko S, Masannat T, Baylis RA, Cherepanova OA. Endothelial-to-Mesenchymal Transition in Atherosclerosis: Friend or Foe? Cells. 2022; 11(19):2946. https://doi.org/10.3390/cells11192946
Chicago/Turabian StyleGole, Sarin, Svyatoslav Tkachenko, Tarek Masannat, Richard A. Baylis, and Olga A. Cherepanova. 2022. "Endothelial-to-Mesenchymal Transition in Atherosclerosis: Friend or Foe?" Cells 11, no. 19: 2946. https://doi.org/10.3390/cells11192946
APA StyleGole, S., Tkachenko, S., Masannat, T., Baylis, R. A., & Cherepanova, O. A. (2022). Endothelial-to-Mesenchymal Transition in Atherosclerosis: Friend or Foe? Cells, 11(19), 2946. https://doi.org/10.3390/cells11192946