

Implications of a Neuronal Receptor Family, Metabotropic Glutamate Receptors, in Cancer Development and Progression
 , , , ,
, , , ,
Abstract
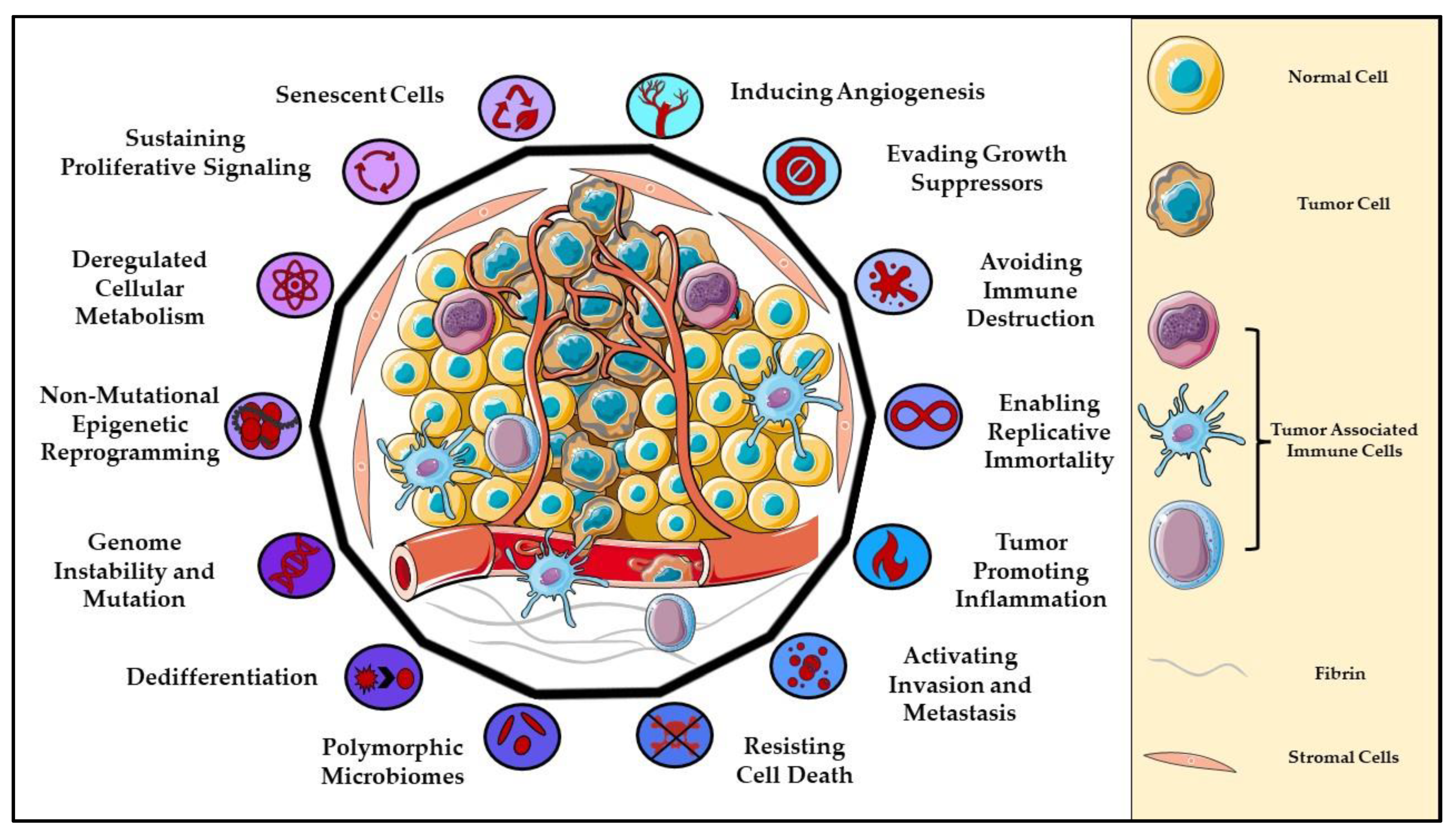
1. Introduction to Cancer
2. Metabotropic Glutamate Receptor Biology
3. Involvement of mGluR1 Signaling in Melanomagenesis and Progression
4. The Role of Metabotropic Glutamate Receptors in Various Cancers
4.1. Breast Cancer
4.2. Colorectal Cancer
4.3. Glioma
4.4. Kidney Cancer
4.5. Melanoma
4.6. Oral Squamous Cell Carcinoma
4.7. Osteosarcoma
4.8. Pancreatic Cancer
4.9. Prostate Cancer
4.10. mGluRs in T-Cell Biology and T-Cell Cancers

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancers | GRMs | Oncogene/Tumor Suppressor | Reference |
|---|---|---|---|
| Breast Cancer | GRM1, GRM8 | Oncogene | [105,106,107] |
| GRM4 | Tumor Suppressor | [108] | |
| Colorectal Carcinoma | GRM4 | Oncogene | [100] |
| Glioma | GRM1, GRM2, GRM3 | Oncogene | [88,91,93,119] |
| Kidney Cancer | GRM1 | Oncogene | [89,138] |
| GRM3, GRM4, GRM5 | Function Unknown | [124] | |
| Melanoma | GRM1, GRM3, GRM5 | Oncogene | [38,125,126,128] |
| Oral Squamous Cell Carcinoma | GRM5 | Oncogene | [98] |
| Osteosarcoma | GRM4 GRM5 | Tumor Suppressor Oncogene | [131,132] |
| Pancreatic Cancer | GRM1 | Function Unknown | [133,136] |
| Prostate Cancer | GRM1 | Oncogene | [97] |
| T-Cell Cancers | GRM1-GRM8 | Function Unknown | [86,140,141] |
5. Altered Glutamate Metabolism in Cancers
6. Discussion and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Slipicevic, A.; Herlyn, M. KIT in melanoma: Many shades of gray. J. Investig. Dermatol. 2015, 135, 337–338. [Google Scholar] [CrossRef] [PubMed]
- Dahl, C.; Abildgaard, C.; Riber-Hansen, R.; Steiniche, T.; Lade-Keller, J.; Guldberg, P. KIT is a frequent target for epigenetic silencing in cutaneous melanoma. J. Investig. Dermatol. 2015, 135, 516–524. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Shi, Q.; Wang, W. Double agents: Genes with both oncogenic and tumor-suppressor functions. Oncogenesis 2018, 7, 25. [Google Scholar] [CrossRef]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337.e310. [Google Scholar] [CrossRef]
- Vogelstein, B.; Kinzler, K.W. Cancer genes and the pathways they control. Nat. Med. 2004, 10, 789–799. [Google Scholar] [CrossRef]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Klotz-Weigand, L.; Enz, R. Metabotropic Glutamate Receptors at Ribbon Synapses in the Retina and Cochlea. Cells 2022, 11, 1097. [Google Scholar] [CrossRef] [PubMed]
- Lämmermann, T.; Kastenmüller, W. Concepts of GPCR-controlled navigation in the immune system. Immunol. Rev. 2019, 289, 205–231. [Google Scholar] [CrossRef]
- Basith, S.; Cui, M.; Macalino, S.J.Y.; Park, J.; Clavio, N.A.B.; Kang, S.; Choi, S. Exploring G Protein-Coupled Receptors (GPCRs) Ligand Space via Cheminformatics Approaches: Impact on Rational Drug Design. Front. Pharmacol. 2018, 9, 128. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Yang, D.; Wu, M.; Guo, Y.; Guo, W.; Zhong, L.; Cai, X.; Dai, A.; Jang, W.; Shakhnovich, E.I.; et al. Common activation mechanism of class A GPCRs. eLife 2019, 8, e50279. [Google Scholar] [CrossRef] [PubMed]
- Tuteja, N. Signaling through G protein coupled receptors. Plant Signal. Behav. 2009, 4, 942–947. [Google Scholar] [CrossRef] [PubMed]
- Kobilka, B.K. G protein coupled receptor structure and activation. Biochim. Biophys. Acta 2007, 1768, 794–807. [Google Scholar] [CrossRef]
- Cook, J.V.; Eidne, K.A. An intramolecular disulfide bond between conserved extracellular cysteines in the gonadotropin-releasing hormone receptor is essential for binding and activation. Endocrinology 1997, 138, 2800–2806. [Google Scholar] [CrossRef][Green Version]
- Ritter, S.L.; Hall, R.A. Fine-tuning of GPCR activity by receptor-interacting proteins. Nat. Rev. Mol. Cell. Biol. 2009, 10, 819–830. [Google Scholar] [CrossRef]
- Niswender, C.M.; Conn, P.J. Metabotropic glutamate receptors: Physiology, pharmacology, and disease. Annu. Rev. Pharmacol. Toxicol 2010, 50, 295–322. [Google Scholar] [CrossRef]
- Cao, J.; Huang, S.; Qian, J.; Huang, J.; Jin, L.; Su, Z.; Yang, J.; Liu, J. Evolution of the class C GPCR Venus flytrap modules involved positive selected functional divergence. BMC Evol. Biol. 2009, 9, 67. [Google Scholar] [CrossRef] [PubMed]
- Teh, J.L.; Chen, S. Glutamatergic signaling in cellular transformation. Pigment Cell Melanoma Res. 2012, 25, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Whorton, M.R.; MacKinnon, R. X-ray structure of the mammalian GIRK2-βγ G-protein complex. Nature 2013, 498, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Kano, H.; Toyama, Y.; Imai, S.; Iwahashi, Y.; Mase, Y.; Yokogawa, M.; Osawa, M.; Shimada, I. Structural mechanism underlying G protein family-specific regulation of G protein-gated inwardly rectifying potassium channel. Nat. Commun. 2019, 10, 2008. [Google Scholar] [CrossRef]
- Zamponi, G.W.; Currie, K.P. Regulation of Ca(V)2 calcium channels by G protein coupled receptors. Biochim. Biophys. Acta 2013, 1828, 1629–1643. [Google Scholar] [CrossRef]
- Herlitze, S.; Garcia, D.E.; Mackie, K.; Hille, B.; Scheuer, T.; Catterall, W.A. Modulation of Ca2+ channels by G-protein beta gamma subunits. Nature 1996, 380, 258–262. [Google Scholar] [CrossRef]
- De Waard, M.; Liu, H.; Walker, D.; Scott, V.E.; Gurnett, C.A.; Campbell, K.P. Direct binding of G-protein betagamma complex to voltage-dependent calcium channels. Nature 1997, 385, 446–450. [Google Scholar] [CrossRef]
- Eddy, K.; Chen, S. Glutamatergic Signaling a Therapeutic Vulnerability in Melanoma. Cancers 2021, 13, 3874. [Google Scholar] [CrossRef]
- Marciel, M.P.; Khadka, V.S.; Deng, Y.; Kilicaslan, P.; Pham, A.; Bertino, P.; Lee, K.; Chen, S.; Glibetic, N.; Hoffmann, F.W.; et al. Selenoprotein K deficiency inhibits melanoma by reducing calcium flux required for tumor growth and metastasis. Oncotarget 2018, 9, 13407–13422. [Google Scholar] [CrossRef]
- Lee, J.; Munguba, H.; Gutzeit, V.A.; Singh, D.R.; Kristt, M.; Dittman, J.S.; Levitz, J. Defining the Homo- and Heterodimerization Propensities of Metabotropic Glutamate Receptors. Cell Rep. 2020, 31, 107891. [Google Scholar] [CrossRef]
- Maksymetz, J.; Moran, S.P.; Conn, P.J. Targeting metabotropic glutamate receptors for novel treatments of schizophrenia. Mol. Brain 2017, 10, 15. [Google Scholar] [CrossRef] [PubMed]
- Kryszkowski, W.; Boczek, T. The G Protein-Coupled Glutamate Receptors as Novel Molecular Targets in Schizophrenia Treatment-A Narrative Review. J. Clin. Med. 2021, 10, 1475. [Google Scholar] [CrossRef] [PubMed]
- Orgován, Z.; Ferenczy, G.G.; Keserű, G.M. Allosteric Molecular Switches in Metabotropic Glutamate Receptors. ChemMedChem 2021, 16, 81–93. [Google Scholar] [CrossRef]
- Llinas Del Torrent, C.; Pérez-Benito, L.; Tresadern, G. Computational Drug Design Applied to the Study of Metabotropic Glutamate Receptors. Molecules 2019, 24, 1098. [Google Scholar] [CrossRef]
- Schöneberg, T.; Liebscher, I. Mutations in G Protein-Coupled Receptors: Mechanisms, Pathophysiology and Potential Therapeutic Approaches. Pharmacol. Rev. 2021, 73, 89–119. [Google Scholar] [CrossRef]
- Luo, J.; Sun, P.; Siwko, S.; Liu, M.; Xiao, J. The role of GPCRs in bone diseases and dysfunctions. Bone Res. 2019, 7, 19. [Google Scholar] [CrossRef]
- Wang, J.; Gareri, C.; Rockman, H.A. G-Protein-Coupled Receptors in Heart Disease. Circ. Res. 2018, 123, 716–735. [Google Scholar] [CrossRef]
- Pollock, P.M.; Cohen-Solal, K.; Sood, R.; Namkoong, J.; Martino, J.J.; Koganti, A.; Zhu, H.; Robbins, C.; Makalowska, I.; Shin, S.S.; et al. Melanoma mouse model implicates metabotropic glutamate signaling in melanocytic neoplasia. Nat. Genet. 2003, 34, 108–112. [Google Scholar] [CrossRef]
- Namkoong, J.; Shin, S.S.; Lee, H.J.; Marín, Y.E.; Wall, B.A.; Goydos, J.S.; Chen, S. Metabotropic glutamate receptor 1 and glutamate signaling in human melanoma. Cancer Res. 2007, 67, 2298–2305. [Google Scholar] [CrossRef]
- Chen, S.; Teicher, L.C.; Kazim, D.; Pollack, R.E.; Wise, L.S. Commitment of mouse fibroblasts to adipocyte differentiation by DNA transfection. Science 1989, 244, 582–585. [Google Scholar] [CrossRef]
- Colón-Teicher, L.; Wise, L.S.; Martino, J.J.; Baskin, L.; Sakoulas, G.; Pollack, R.E.; Chen, S. Genomic sequences capable of committing mouse and rat fibroblasts to adipogenesis. Nucleic Acids Res. 1993, 21, 2223–2228. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhu, H.; Wetzel, W.J.; Philbert, M.A. Spontaneous melanocytosis in transgenic mice. J. Investig. Dermatol. 1996, 106, 1145–1151. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Reuhl, K.; Zhang, X.; Botha, R.; Ryan, K.; Wei, J.; Chen, S. Development of heritable melanoma in transgenic mice. J. Investig. Dermatol. 1998, 110, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Funusaka, Y.; Harada, T.; Aiba, A.; Nishigori, C. Expression of metabotropic glutamate receptor 1 and phosphorylated extracellular signal-regulated kinase 1/2 proteins in human melanocytic lesions. Pigm. Cell Res. 2006, 19, 256. [Google Scholar]
- Wangari-Talbot, J.; Wall, B.A.; Goydos, J.S.; Chen, S. Functional effects of GRM1 suppression in human melanoma cells. Mol. Cancer Res. 2012, 10, 1440–1450. [Google Scholar] [CrossRef]
- Chong, J.A.; Tapia-Ramírez, J.; Kim, S.; Toledo-Aral, J.J.; Zheng, Y.; Boutros, M.C.; Altshuller, Y.M.; Frohman, M.A.; Kraner, S.D.; Mandel, G. REST: A mammalian silencer protein that restricts sodium channel gene expression to neurons. Cell 1995, 80, 949–957. [Google Scholar] [CrossRef]
- Schoenherr, C.J.; Anderson, D.J. The neuron-restrictive silencer factor (NRSF): A coordinate repressor of multiple neuron-specific genes. Science 1995, 267, 1360–1363. [Google Scholar] [CrossRef]
- Crepaldi, L.; Lackner, C.; Corti, C.; Ferraguti, F. Transcriptional activators and repressors for the neuron-specific expression of a metabotropic glutamate receptor. J. Biol. Chem. 2007, 282, 17877–17889. [Google Scholar] [CrossRef]
- Lee, H.J.; Wall, B.A.; Wangari-Talbot, J.; Chen, S. Regulation of mGluR1 expression in human melanocytes and melanoma cells. Biochim. Biophys. Acta 2012, 1819, 1123–1131. [Google Scholar] [CrossRef]
- Shin, S.S.; Namkoong, J.; Wall, B.A.; Gleason, R.; Lee, H.J.; Chen, S. Oncogenic activities of metabotropic glutamate receptor 1 (Grm1) in melanocyte transformation. Pigment Cell Melanoma Res. 2008, 21, 368–378. [Google Scholar] [CrossRef]
- Ohtani, Y.; Harada, T.; Funasaka, Y.; Nakao, K.; Takahara, C.; Abdel-Daim, M.; Sakai, N.; Saito, N.; Nishigori, C.; Aiba, A. Metabotropic glutamate receptor subtype-1 is essential for in vivo growth of melanoma. Oncogene 2008, 27, 7162–7170. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.S.; Wall, B.A.; Goydos, J.S.; Chen, S. AKT2 is a downstream target of metabotropic glutamate receptor 1 (Grm1). Pigment Cell Melanoma Res. 2010, 23, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Marín, Y.E.; Namkoong, J.; Cohen-Solal, K.; Shin, S.S.; Martino, J.J.; Oka, M.; Chen, S. Stimulation of oncogenic metabotropic glutamate receptor 1 in melanoma cells activates ERK1/2 via PKCepsilon. Cell Signal. 2006, 18, 1279–1286. [Google Scholar] [CrossRef] [PubMed]
- Marín, Y.E.; Namkoong, J.; Shin, S.S.; Raines, J.; Degenhardt, K.; White, E.; Chen, S. Grm5 expression is not required for the oncogenic role of Grm1 in melanocytes. Neuropharmacology 2005, 49 (Suppl. S1), 70–79. [Google Scholar] [CrossRef] [PubMed]
- Teh, J.L.; Shah, R.; Shin, S.S.; Wen, Y.; Mehnert, J.M.; Goydos, J.; Chen, S. Metabotropic glutamate receptor 1 mediates melanocyte transformation via transactivation of insulin-like growth factor 1 receptor. Pigment Cell Melanoma Res. 2014, 27, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.C.; Sierra, J.; Yu, L.J.; Cerchio, R., Jr.; Wall, B.A.; Goydos, J.; Chen, S. Activation of Grm1 expression by mutated BRaf (V600E) in vitro and in vivo. Oncotarget 2018, 9, 5861–5875. [Google Scholar] [CrossRef]
- Wen, Y.; Li, J.; Koo, J.; Shin, S.S.; Lin, Y.; Jeong, B.S.; Mehnert, J.M.; Chen, S.; Cohen-Sola, K.A.; Goydos, J.S. Activation of the glutamate receptor GRM1 enhances angiogenic signaling to drive melanoma progression. Cancer Res. 2014, 74, 2499–2509. [Google Scholar] [CrossRef]
- Massoumi, R.; Kuphal, S.; Hellerbrand, C.; Haas, B.; Wild, P.; Spruss, T.; Pfeifer, A.; Fässler, R.; Bosserhoff, A.K. Down-regulation of CYLD expression by Snail promotes tumor progression in malignant melanoma. J. Exp. Med. 2009, 206, 221–232. [Google Scholar] [CrossRef]
- De Jel, M.M.; Schott, M.; Lamm, S.; Neuhuber, W.; Kuphal, S.; Bosserhoff, A.K. Loss of CYLD accelerates melanoma development and progression in the Tg(Grm1) melanoma mouse model. Oncogenesis 2019, 8, 56. [Google Scholar] [CrossRef]
- Sun, S.C. CYLD: A tumor suppressor deubiquitinase regulating NF-kappaB activation and diverse biological processes. Cell Death Differ. 2010, 17, 25–34. [Google Scholar] [CrossRef]
- Marin, Y.E.; Wall, B.A.; Wang, S.; Namkoong, J.; Martino, J.J.; Suh, J.; Lee, H.J.; Rabson, A.B.; Yang, C.S.; Chen, S.; et al. Curcumin downregulates the constitutive activity of NF-kappaB and induces apoptosis in novel mouse melanoma cells. Melanoma Res. 2007, 17, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Schott, M.; Kappelmann-Fenzl, M.; Fischer, S.; Fernandez-Barrena, M.G.; Pineda-Lucena, A.; Ávila, M.A.; Kuphal, S.; Bosserhoff, A.K. Impact of CYLD on chromatin structure and histone methylation in malignant melanoma. Int. J. Mol. Med. 2022, 49, 66. [Google Scholar] [CrossRef] [PubMed]
- Cronin, J.C.; Watkins-Chow, D.E.; Incao, A.; Hasskamp, J.H.; Schönewolf, N.; Aoude, L.G.; Hayward, N.K.; Bastian, B.C.; Dummer, R.; Loftus, S.K.; et al. SOX10 ablation arrests cell cycle, induces senescence, and suppresses melanomagenesis. Cancer Res. 2013, 73, 5709–5718. [Google Scholar] [CrossRef] [PubMed]
- Tudrej, K.B.; Czepielewska, E.; Kozłowska-Wojciechowska, M. SOX10-MITF pathway activity in melanoma cells. Arch. Med. Sci. 2017, 13, 1493–1503. [Google Scholar] [CrossRef] [PubMed]
- Eddy, K.; Shah, R.; Chen, S. Decoding Melanoma Development and Progression: Identification of Therapeutic Vulnerabilities. Front. Oncol. 2021, 10, 626129. [Google Scholar] [CrossRef] [PubMed]
- Yip, D.; Le, M.N.; Chan, J.L.; Lee, J.H.; Mehnert, J.A.; Yudd, A.; Kempf, J.; Shih, W.J.; Chen, S.; Goydos, J.S. A phase 0 trial of riluzole in patients with resectable stage III and IV melanoma. Clin. Cancer Res. 2009, 15, 3896–3902. [Google Scholar] [CrossRef]
- Mehnert, J.M.; Silk, A.W.; Lee, J.H.; Dudek, L.; Jeong, B.S.; Li, J.; Schenkel, J.M.; Sadimin, E.; Kane, M.; Lin, H.; et al. A phase II trial of riluzole, an antagonist of metabotropic glutamate receptor 1 (GRM1) signaling, in patients with advanced melanoma. Pigment Cell Melanoma Res. 2018, 31, 534–540. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.; Singh, S.J.; Eddy, K.; Filipp, F.V.; Chen, S. Concurrent Targeting of Glutaminolysis and Metabotropic Glutamate Receptor 1 (GRM1) Reduces Glutamate Bioavailability in GRM1(+) Melanoma. Cancer Res. 2019, 79, 1799–1809. [Google Scholar] [CrossRef] [PubMed]
- Wall, B.A.; Wangari-Talbot, J.; Shin, S.S.; Schiff, D.; Sierra, J.; Yu, L.J.; Khan, A.; Haffty, B.; Goydos, J.S.; Chen, S. Disruption of GRM1-mediated signalling using riluzole results in DNA damage in melanoma cells. Pigment Cell Melanoma Res. 2014, 27, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.S.; Jeong, B.S.; Wall, B.A.; Li, J.; Shan, N.L.; Wen, Y.; Goydos, J.S.; Chen, S. Participation of xCT in melanoma cell proliferation in vitro and tumorigenesis in vivo. Oncogenesis 2018, 7, 86. [Google Scholar] [CrossRef]
- Cerchio, R., Jr.; Marinaro, C.; Foo, T.K.; Xia, B.; Chen, S. Nonhomologous end-joining repair is likely involved in the repair of double-stranded DNA breaks induced by riluzole in melanoma cells. Melanoma Res. 2020, 30, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Caumont, A.S.; Octave, J.N.; Hermans, E. Specific regulation of rat glial cell line-derived neurotrophic factor gene expression by riluzole in C6 glioma cells. J. Neurochem. 2006, 97, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Yohay, K.; Tyler, B.; Weaver, K.D.; Pardo, A.C.; Gincel, D.; Blakeley, J.; Brem, H.; Rothstein, J.D. Efficacy of local polymer-based and systemic delivery of the anti-glutamatergic agents riluzole and memantine in rat glioma models. J. Neurosurg. 2014, 120, 854–863. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Tsuji, S.; Nakamura, S.; Egashira, Y.; Shimazawa, M.; Nakayama, N.; Yano, H.; Iwama, T.; Hara, H. Riluzole enhances the antitumor effects of temozolomide via suppression of MGMT expression in glioblastoma. J. Neurosurg. 2020, 134, 701–710. [Google Scholar] [CrossRef] [PubMed]
- Raghubir, M.; Azeem, S.M.; Hasnat, R.; Rahman, C.N.; Wong, L.; Yan, S.; Huang, Y.Q.; Zhagui, R.; Blyufer, A.; Tariq, I.; et al. Riluzole-induced apoptosis in osteosarcoma is mediated through Yes-associated protein upon phosphorylation by c-Abl Kinase. Sci. Rep. 2021, 11, 20974. [Google Scholar] [CrossRef] [PubMed]
- Coppedè, F.; Migliore, L. DNA damage in neurodegenerative diseases. Mutat. Res. 2015, 776, 84–97. [Google Scholar] [CrossRef]
- Vallée, A.; Vallée, J.N.; Guillevin, R.; Lecarpentier, Y. Riluzole: A therapeutic strategy in Alzheimer’s disease by targeting the WNT/β-catenin pathway. Aging 2020, 12, 3095–3113. [Google Scholar] [CrossRef]
- Kok, J.R.; Palminha, N.M.; Dos Santos Souza, C.; El-Khamisy, S.F.; Ferraiuolo, L. DNA damage as a mechanism of neurodegeneration in ALS and a contributor to astrocyte toxicity. Cell. Mol. Life Sci. 2021, 78, 5707–5729. [Google Scholar] [CrossRef]
- Van Kan, H.J.; Groeneveld, G.J.; Kalmijn, S.; Spieksma, M.; van den Berg, L.H.; Guchelaar, H.J. Association between CYP1A2 activity and riluzole clearance in patients with amyotrophic lateral sclerosis. Br. J. Clin. Pharmacol. 2005, 59, 310–313. [Google Scholar] [CrossRef]
- Kan, Z.; Jaiswal, B.S.; Stinson, J.; Janakiraman, V.; Bhatt, D.; Stern, H.M.; Yue, P.; Haverty, P.M.; Bourgon, R.; Zheng, J.; et al. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature 2010, 466, 869–873. [Google Scholar] [CrossRef] [PubMed]
- Esseltine, J.L.; Willard, M.D.; Wulur, I.H.; Lajiness, M.E.; Barber, T.D.; Ferguson, S.S. Somatic mutations in GRM1 in cancer alter metabotropic glutamate receptor 1 intracellular localization and signaling. Mol. Pharmacol. 2013, 83, 770–780. [Google Scholar] [CrossRef] [PubMed]
- Sjöblom, T.; Jones, S.; Wood, L.D.; Parsons, D.W.; Lin, J.; Barber, T.D.; Mandelker, D.; Leary, R.J.; Ptak, J.; Silliman, N.; et al. The consensus coding sequences of human breast and colorectal cancers. Science 2006, 314, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Durinck, S.; Ho, C.; Wang, N.J.; Liao, W.; Jakkula, L.R.; Collisson, E.A.; Pons, J.; Chan, S.W.; Lam, E.T.; Chu, C.; et al. Temporal dissection of tumorigenesis in primary cancers. Cancer Discov. 2011, 1, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Stransky, N.; Egloff, A.M.; Tward, A.D.; Kostic, A.D.; Cibulskis, K.; Sivachenko, A.; Kryukov, G.V.; Lawrence, M.S.; Sougnez, C.; McKenna, A.; et al. The mutational landscape of head and neck squamous cell carcinoma. Science 2011, 333, 1157–1160. [Google Scholar] [CrossRef]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef]
- Stepulak, A.; Luksch, H.; Gebhardt, C.; Uckermann, O.; Marzahn, J.; Sifringer, M.; Rzeski, W.; Staufner, C.; Brocke, K.S.; Turski, L.; et al. Expression of glutamate receptor subunits in human cancers. Histochem. Cell Biol. 2009, 132, 435–445. [Google Scholar] [CrossRef]
- Brocke, K.S.; Staufner, C.; Luksch, H.; Geiger, K.D.; Stepulak, A.; Marzahn, J.; Schackert, G.; Temme, A.; Ikonomidou, C. Glutamate receptors in pediatric tumors of the central nervous system. Cancer Biol. Ther. 2010, 9, 455–468. [Google Scholar] [CrossRef]
- Zhang, C.; Yuan, X.R.; Li, H.Y.; Zhao, Z.J.; Liao, Y.W.; Wang, X.Y.; Su, J.; Sang, S.S.; Liu, Q. Anti-cancer effect of metabotropic glutamate receptor 1 inhibition in human glioma U87 cells: Involvement of PI3K/Akt/mTOR pathway. Cell Physiol. Biochem. 2015, 35, 419–432. [Google Scholar] [CrossRef]
- Martino, J.J.; Wall, B.A.; Mastrantoni, E.; Wilimczyk, B.J.; La Cava, S.N.; Degenhardt, K.; White, E.; Chen, S. Metabotropic glutamate receptor 1 (Grm1) is an oncogene in epithelial cells. Oncogene 2013, 32, 4366–4376. [Google Scholar] [CrossRef]
- Kalariti, N.; Lembessis, P.; Papageorgiou, E.; Pissimissis, N.; Koutsilieris, M. Regulation of the mGluR5, EAAT1 and GS expression by glucocorticoids in MG-63 osteoblast-like osteosarcoma cells. J. Musculoskelet. Neuronal Interact. 2007, 7, 113–118. [Google Scholar]
- D’Onofrio, M.; Arcella, A.; Bruno, V.; Ngomba, R.T.; Battaglia, G.; Lombari, V.; Ragona, G.; Calogero, A.; Nicoletti, F. Pharmacological blockade of mGlu2/3 metabotropic glutamate receptors reduces cell proliferation in cultured human glioma cells. J. Neurochem. 2003, 84, 1288–1295. [Google Scholar] [CrossRef] [PubMed]
- Prickett, T.D.; Samuels, Y. Molecular pathways: Dysregulated glutamatergic signaling pathways in cancer. Clin. Cancer Res. 2012, 18, 4240–4246. [Google Scholar] [CrossRef] [PubMed]
- Arcella, A.; Carpinelli, G.; Battaglia, G.; D’Onofrio, M.; Santoro, F.; Ngomba, R.T.; Bruno, V.; Casolini, P.; Giangaspero, F.; Nicoletti, F. Pharmacological blockade of group II metabotropic glutamate receptors reduces the growth of glioma cells in vivo. Neuro-Oncology 2005, 7, 236–245. [Google Scholar] [CrossRef] [PubMed]
- Ferrigno, A.; Berardo, C.; Di Pasqua, L.G.; Siciliano, V.; Richelmi, P.; Vairetti, M. Localization and role of metabotropic glutamate receptors subtype 5 in the gastrointestinal tract. World J. Gastroenterol. 2017, 23, 4500–4507. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Network. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef] [PubMed]
- Frati, C.; Marchese, C.; Fisichella, G.; Copani, A.; Nasca, M.R.; Storto, M.; Nicoletti, F. Expression of functional mGlu5 metabotropic glutamate receptors in human melanocytes. J. Cell Physiol. 2000, 183, 364–372. [Google Scholar] [CrossRef]
- Koochekpour, S.; Majumdar, S.; Azabdaftari, G.; Attwood, K.; Scioneaux, R.; Subramani, D.; Manhardt, C.; Lorusso, G.D.; Willard, S.S.; Thompson, H.; et al. Serum Glutamate Levels Correlate with Gleason Score and Glutamate Blockade Decreases Proliferation, Migration, and Invasion and Induces Apoptosis in Prostate Cancer Cells. Clin. Cancer Res. 2012, 18, 5888–5901. [Google Scholar] [CrossRef]
- Park, S.-Y.; Lee, S.-A.; Han, I.-H.; Yoo, B.-C.; Lee, S.-H.; Park, J.-Y.; Cha, I.-H.; Kim, J.; Choi, S.-W. Clinical significance of metabotropic glutamate receptor 5 expression in oral squamous cell carcinoma. Oncol. Rep. 2007, 17, 81–87. [Google Scholar] [CrossRef][Green Version]
- Pissimissis, N.; Papageorgiou, E.; Lembessis, P.; Armakolas, A.; Koutsilieris, M. The Glutamatergic System Expression in Human PC-3 and LNCaP Prostate Cancer Cells. Anticancer. Res. 2009, 29, 371. [Google Scholar]
- Chang, H.J.; Yoo, B.C.; Lim, S.-B.; Jeong, S.-Y.; Kim, W.H.; Park, J.-G. Metabotropic Glutamate Receptor 4 Expression in Colorectal Carcinoma and Its Prognostic Significance. Clin. Cancer Res. 2005, 11, 3288–3295. [Google Scholar] [CrossRef]
- Teh, J.L.; Shah, R.; La Cava, S.; Dolfi, S.C.; Mehta, M.S.; Kongara, S.; Price, S.; Ganesan, S.; Reuhl, K.R.; Hirshfield, K.M.; et al. Metabotropic glutamate receptor 1 disrupts mammary acinar architecture and initiates malignant transformation of mammary epithelial cells. Breast Cancer Res. Treat. 2015, 151, 57–73. [Google Scholar] [CrossRef]
- Bastiaansen, A.E.M.; Timmermans, A.M.; Smid, M.; van Deurzen, C.H.M.; Hulsenboom, E.S.P.; Prager-van der Smissen, W.J.C.; Foekens, R.; Trapman-Jansen, A.; Sillevis Smitt, P.A.E.; Luider, T.M.; et al. Metabotropic glutamate receptor 1 is associated with unfavorable prognosis in ER-negative and triple-negative breast cancer. Sci. Rep. 2020, 10, 22292. [Google Scholar] [CrossRef]
- Mehta, M.S.; Dolfi, S.C.; Bronfenbrener, R.; Bilal, E.; Chen, C.; Moore, D.; Lin, Y.; Rahim, H.; Aisner, S.; Kersellius, R.D.; et al. Metabotropic glutamate receptor 1 expression and its polymorphic variants associate with breast cancer phenotypes. PLoS ONE 2013, 8, e69851. [Google Scholar] [CrossRef]
- Stires, H.; Heckler, M.M.; Fu, X.; Li, Z.; Grasso, C.S.; Quist, M.J.; Lewis, J.A.; Klimach, U.; Zwart, A.; Mahajan, A.; et al. Integrated molecular analysis of Tamoxifen-resistant invasive lobular breast cancer cells identifies MAPK and GRM/mGluR signaling as therapeutic vulnerabilities. Mol. Cell. Endocrinol. 2018, 471, 105–117. [Google Scholar] [CrossRef]
- Speyer, C.L.; Hachem, A.H.; Assi, A.A.; Johnson, J.S.; DeVries, J.A.; Gorski, D.H. Metabotropic glutamate receptor-1 as a novel target for the antiangiogenic treatment of breast cancer. PLoS ONE 2014, 9, e88830. [Google Scholar] [CrossRef]
- Sexton, R.E.; Hachem, A.H.; Assi, A.A.; Bukhsh, M.A.; Gorski, D.H.; Speyer, C.L. Metabotropic glutamate receptor-1 regulates inflammation in triple negative breast cancer. Sci. Rep. 2018, 8, 16008. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Xie, S.; Yuan, S.; Zhang, Y.; Bai, Y.; Chu, L.; Wu, Z.; Guo, N.; Wang, Q.; Zhang, J. Metabotropic Glutamate Receptor 8 Is Regulated by miR-33a-5p and Functions as an Oncogene in Breast Cancer. J. Oncol. 2021, 2021, 8002087. [Google Scholar] [CrossRef]
- Xiao, B.; Chen, D.; Zhou, Q.; Hang, J.; Zhang, W.; Kuang, Z.; Sun, Z.; Li, L. Glutamate metabotropic receptor 4 (GRM4) inhibits cell proliferation, migration and invasion in breast cancer and is regulated by miR-328-3p and miR-370-3p. BMC Cancer 2019, 19, 891. [Google Scholar] [CrossRef] [PubMed]
- Yoo, B.C.; Jeon, E.; Hong, S.H.; Shin, Y.K.; Chang, H.J.; Park, J.G. Metabotropic glutamate receptor 4-mediated 5-Fluorouracil resistance in a human colon cancer cell line. Clin. Cancer Res. 2004, 10, 4176–4184. [Google Scholar] [CrossRef]
- Peters, G.J.; van Groeningen, C.J.; Laurensse, E.J.; Pinedo, H.M. A comparison of 5-fluorouracil metabolism in human colorectal cancer and colon mucosa. Cancer 1991, 68, 1903–1909. [Google Scholar] [CrossRef]
- Wang, F.S.; Aschele, C.; Sobrero, A.; Chang, Y.M.; Bertino, J.R. Decreased folylpolyglutamate synthetase expression: A novel mechanism of fluorouracil resistance. Cancer Res. 1993, 53, 3677–3680. [Google Scholar] [PubMed]
- Beck, A.; Etienne, M.C.; Chéradame, S.; Fischel, J.L.; Formento, P.; Renée, N.; Milano, G. A role for dihydropyrimidine dehydrogenase and thymidylate synthase in tumour sensitivity to fluorouracil. Eur. J. Cancer 1994, 30a, 1517–1522. [Google Scholar] [CrossRef]
- Peters, G.J.; Backus, H.H.; Freemantle, S.; van Triest, B.; Codacci-Pisanelli, G.; van der Wilt, C.L.; Smid, K.; Lunec, J.; Calvert, A.H.; Marsh, S.; et al. Induction of thymidylate synthase as a 5-fluorouracil resistance mechanism. Biochim. Biophys. Acta 2002, 1587, 194–205. [Google Scholar] [CrossRef]
- Bapat, A.R.; Zarow, C.; Danenberg, P.V. Human leukemic cells resistant to 5-fluoro-2′-deoxyuridine contain a thymidylate synthetase with lower affinity for nucleotides. J. Biol. Chem. 1983, 258, 4130–4136. [Google Scholar] [CrossRef]
- Berger, S.H.; Barbour, K.W.; Berger, F.G. A naturally occurring variation in thymidylate synthase structure is associated with a reduced response to 5-fluoro-2′-deoxyuridine in a human colon tumor cell line. Mol. Pharmacol. 1988, 34, 480–484. [Google Scholar] [PubMed]
- Spears, C.P.; Gustavsson, B.G.; Berne, M.; Frösing, R.; Bernstein, L.; Hayes, A.A. Mechanisms of innate resistance to thymidylate synthase inhibition after 5-fluorouracil. Cancer Res. 1988, 48, 5894–5900. [Google Scholar]
- Adamson, D.C.; Rasheed, B.A.; McLendon, R.E.; Bigner, D.D. Central nervous system. Cancer Biomark. 2010, 9, 193–210. [Google Scholar] [CrossRef] [PubMed]
- Jessen, K.R. Glial cells. Int. J. Biochem. Cell Biol. 2004, 36, 1861–1867. [Google Scholar] [CrossRef]
- Khan, A.J.; LaCava, S.; Mehta, M.; Schiff, D.; Thandoni, A.; Jhawar, S.; Danish, S.; Haffty, B.G.; Chen, S. The glutamate release inhibitor riluzole increases DNA damage and enhances cytotoxicity in human glioma cells, in vitro and in vivo. Oncotarget 2019, 10, 2824–2834. [Google Scholar] [CrossRef]
- Willard, S.S.; Koochekpour, S. Glutamate signaling in benign and malignant disorders: Current status, future perspectives, and therapeutic implications. Int. J. Biol. Sci. 2013, 9, 728–742. [Google Scholar] [CrossRef]
- Medikonda, R.; Choi, J.; Pant, A.; Saleh, L.; Routkevitch, D.; Tong, L.; Belcaid, Z.; Kim, Y.H.; Jackson, C.M.; Jackson, C.; et al. Synergy between glutamate modulation and anti-programmed cell death protein 1 immunotherapy for glioblastoma. J. Neurosurg. 2022, 136, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Carbone, M.; Duty, S.; Rattray, M. Riluzole elevates GLT-1 activity and levels in striatal astrocytes. Neurochem. Int. 2012, 60, 31–38. [Google Scholar] [CrossRef]
- Dall’Igna, O.P.; Bobermin, L.D.; Souza, D.O.; Quincozes-Santos, A. Riluzole increases glutamate uptake by cultured C6 astroglial cells. Int. J. Dev. Neurosci. 2013, 31, 482–486. [Google Scholar] [CrossRef]
- Huang, C.Y.; Hsueh, Y.M.; Chen, L.C.; Cheng, W.C.; Yu, C.C.; Chen, W.J.; Lu, T.L.; Lan, K.J.; Lee, C.H.; Huang, S.P.; et al. Clinical significance of glutamate metabotropic receptors in renal cell carcinoma risk and survival. Cancer Med. 2018, 7, 6104–6111. [Google Scholar] [CrossRef] [PubMed]
- Prickett, T.D.; Wei, X.; Cardenas-Navia, I.; Teer, J.K.; Lin, J.C.; Walia, V.; Gartner, J.; Jiang, J.; Cherukuri, P.F.; Molinolo, A.; et al. Exon capture analysis of G protein-coupled receptors identifies activating mutations in GRM3 in melanoma. Nature Genet. 2011, 43, 1119–1126. [Google Scholar] [CrossRef]
- Neto, A.; Ceol, C.J. Melanoma-associated GRM3 variants dysregulate melanosome trafficking and cAMP signaling. Pigment Cell Melanoma Res. 2018, 31, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.G.; Leapman, R.D.; Zhang, G.; Lai, B.; Valencia, J.C.; Cardarelli, C.O.; Vieira, W.D.; Hearing, V.J.; Gottesman, M.M. Influence of melanosome dynamics on melanoma drug sensitivity. J. Natl. Cancer Inst. 2009, 101, 1259–1271. [Google Scholar] [CrossRef]
- Choi, K.Y.; Chang, K.; Pickel, J.M.; Badger, J.D., 2nd; Roche, K.W. Expression of the metabotropic glutamate receptor 5 (mGluR5) induces melanoma in transgenic mice. Proc. Natl. Acad. Sci. USA 2011, 108, 15219–15224. [Google Scholar] [CrossRef] [PubMed]
- Markopoulos, A.K. Current aspects on oral squamous cell carcinoma. Open Dent. J. 2012, 6, 126–130. [Google Scholar] [CrossRef]
- Rigi-Ladiz, M.A.; Baranzehi, T.; Hassanpour, B.; Ashraf, M.J.; Farhad-Mollashahi, L.; Kordi-Tamandani, D.M. DNA methylation and expression status of glutamate receptor genes in patients with oral squamous cell carcinoma. Meta Gene 2019, 20, 100555. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, N.; Wei, X.; Chen, B.; Zhang, Y.; Zhao, Y.; Hu, X.; Hou, S. GRM4 inhibits the proliferation, migration, and invasion of human osteosarcoma cells through interaction with CBX4. Biosci. Biotechnol. Biochem. 2020, 84, 279–289. [Google Scholar] [CrossRef]
- Liao, S.; Ruiz, Y.; Gulzar, H.; Yelskaya, Z.; Ait Taouit, L.; Houssou, M.; Jaikaran, T.; Schvarts, Y.; Kozlitina, K.; Basu-Roy, U.; et al. Osteosarcoma cell proliferation and survival requires mGluR5 receptor activity and is blocked by Riluzole. PLoS ONE 2017, 12, e0171256. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Li, Y.; Wei, Y.; Xu, Y.; Wang, R.; Fu, Z.; Zheng, S.; Zhou, Q.; Zhou, Y.; Chen, R.; et al. Anticancer effect of HOTTIP regulates human pancreatic cancer via the metabotropic glutamate receptor 1 pathway. Oncol. Lett. 2018, 16, 1937–1942. [Google Scholar] [CrossRef]
- Javed, Z.; Ahmed Shah, F.; Rajabi, S.; Raza, Q.; Iqbal, Z.; Ullah, M.; Ahmad, T.; Salehi, B.; Sharifi-Rad, M.; Pezzani, R.; et al. LncRNAs as Potential Therapeutic Targets in Thyroid Cancer. Asian Pac. J. Cancer Prev. 2020, 21, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Chen, L.; Li, C.; Huang, R.; Guo, M.; Ning, S.; Ji, J.; Guo, X.; Lou, G.; Jia, X.; et al. The multifaceted roles of long noncoding RNAs in pancreatic cancer: An update on what we know. Cancer Cell Int. 2020, 20, 41. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Xiao, L.; Yu, H. Expression levels of long non-coding RNA HOXA distal transcript antisense RNA and metabotropic glutamate receptor 1 in pancreatic carcinoma, and their prognostic values. Oncol. Lett. 2018, 15, 9464–9470. [Google Scholar] [CrossRef] [PubMed]
- Koochekpour, S. Glutamate, a metabolic biomarker of aggressiveness and a potential therapeutic target for prostate cancer. Asian J. Androl. 2013, 15, 212–213. [Google Scholar] [CrossRef]
- Yu, L.J.; Wall, B.A.; Wangari-Talbot, J.; Chen, S. Metabotropic glutamate receptors in cancer. Neuropharmacology 2017, 115, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.; Shourideh, M.; Koochekpour, S. Identification of Novel GRM1 Mutations and Single Nucleotide Polymorphisms in Prostate Cancer Cell Lines and Tissues. PLoS ONE 2014, 9, e103204. [Google Scholar] [CrossRef]
- Ganor, Y.; Levite, M. The neurotransmitter glutamate and human T cells: Glutamate receptors and glutamate-induced direct and potent effects on normal human T cells, cancerous human leukemia and lymphoma T cells, and autoimmune human T cells. J. Neural Transm. 2014, 121, 983–1006. [Google Scholar] [CrossRef]
- Ganor, Y.; Levite, M. Glutamate in the Immune System: Glutamate Receptors in Immune Cells, Potent Effects, Endogenous Production and Involvement in Disease. In Nerve-Driven Immunity: Neurotransmitters and Neuropeptides in the Immune System; Levite, M., Ed.; Springer: Vienna, Austria, 2012; pp. 121–161. [Google Scholar]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar] [CrossRef] [PubMed]
- Bott, A.J.; Maimouni, S.; Zong, W.X. The Pleiotropic Effects of Glutamine Metabolism in Cancer. Cancers 2019, 11, 770. [Google Scholar] [CrossRef]
- Deberardinis, R.J.; Sayed, N.; Ditsworth, D.; Thompson, C.B. Brick by brick: Metabolism and tumor cell growth. Curr. Opin. Genet. Dev. 2008, 18, 54–61. [Google Scholar] [CrossRef]
- Michalak, K.P.; Maćkowska-Kędziora, A.; Sobolewski, B.; Woźniak, P. Key Roles of Glutamine Pathways in Reprogramming the Cancer Metabolism. Oxid. Med. Cell Longev. 2015, 2015, 964321. [Google Scholar] [CrossRef]
- Christa, L.; Simon, M.T.; Flinois, J.P.; Gebhardt, R.; Brechot, C.; Lasserre, C. Overexpression of glutamine synthetase in human primary liver cancer. Gastroenterology 1994, 106, 1312–1320. [Google Scholar] [CrossRef]
- Osada, T.; Sakamoto, M.; Nagawa, H.; Yamamoto, J.; Matsuno, Y.; Iwamatsu, A.; Muto, T.; Hirohashi, S. Acquisition of glutamine synthetase expression in human hepatocarcinogenesis: Relation to disease recurrence and possible regulation by ubiquitin-dependent proteolysis. Cancer 1999, 85, 819–831. [Google Scholar] [CrossRef]
- Sappington, D.R.; Siegel, E.R.; Hiatt, G.; Desai, A.; Penney, R.B.; Jamshidi-Parsian, A.; Griffin, R.J.; Boysen, G. Glutamine drives glutathione synthesis and contributes to radiation sensitivity of A549 and H460 lung cancer cell lines. Biochim. Biophys. Acta 2016, 1860, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Venneti, S.; Nagrath, D. Glutaminolysis: A Hallmark of Cancer Metabolism. Annu. Rev. Biomed. Eng. 2017, 19, 163–194. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Alesi, G.N.; Kang, S. Glutaminolysis as a target for cancer therapy. Oncogene 2016, 35, 3619–3625. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.B.; Erickson, J.W.; Fuji, R.; Ramachandran, S.; Gao, P.; Dinavahi, R.; Wilson, K.F.; Ambrosio, A.L.; Dias, S.M.; Dang, C.V.; et al. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell 2010, 18, 207–219. [Google Scholar] [CrossRef]
- Van den Heuvel, A.P.; Jing, J.; Wooster, R.F.; Bachman, K.E. Analysis of glutamine dependency in non-small cell lung cancer: GLS1 splice variant GAC is essential for cancer cell growth. Cancer Biol. Ther. 2012, 13, 1185–1194. [Google Scholar] [CrossRef]
- Seltzer, M.J.; Bennett, B.D.; Joshi, A.D.; Gao, P.; Thomas, A.G.; Ferraris, D.V.; Tsukamoto, T.; Rojas, C.J.; Slusher, B.S.; Rabinowitz, J.D.; et al. Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer Res. 2010, 70, 8981–8987. [Google Scholar] [CrossRef]
- Zhang, J.; Mao, S.; Guo, Y.; Wu, Y.; Yao, X.; Huang, Y. Inhibition of GLS suppresses proliferation and promotes apoptosis in prostate cancer. Biosci. Rep. 2019, 39, BSR20181826. [Google Scholar] [CrossRef]
- Xiang, Y.; Stine, Z.E.; Xia, J.; Lu, Y.; O’Connor, R.S.; Altman, B.J.; Hsieh, A.L.; Gouw, A.M.; Thomas, A.G.; Gao, P.; et al. Targeted inhibition of tumor-specific glutaminase diminishes cell-autonomous tumorigenesis. J. Clin. Investig. 2015, 125, 2293–2306. [Google Scholar] [CrossRef]
- Masamha, C.P.; LaFontaine, P. Molecular targeting of glutaminase sensitizes ovarian cancer cells to chemotherapy. J. Cell Biochem. 2018, 119, 6136–6145. [Google Scholar] [CrossRef]
- Shen, Y.A.; Chen, C.L.; Huang, Y.H.; Evans, E.E.; Cheng, C.C.; Chuang, Y.J.; Zhang, C.; Le, A. Inhibition of glutaminolysis in combination with other therapies to improve cancer treatment. Curr. Opin. Chem. Biol. 2021, 62, 64–81. [Google Scholar] [CrossRef]
- Ren, L.; Ruiz-Rodado, V.; Dowdy, T.; Huang, S.; Issaq, S.H.; Beck, J.; Wang, H.; Tran Hoang, C.; Lita, A.; Larion, M.; et al. Glutaminase-1 (GLS1) inhibition limits metastatic progression in osteosarcoma. Cancer Metab. 2020, 8, 4. [Google Scholar] [CrossRef] [PubMed]
- Momcilovic, M.; Bailey, S.T.; Lee, J.T.; Fishbein, M.C.; Magyar, C.; Braas, D.; Graeber, T.; Jackson, N.J.; Czernin, J.; Emberley, E.; et al. Targeted Inhibition of EGFR and Glutaminase Induces Metabolic Crisis in EGFR Mutant Lung Cancer. Cell Rep. 2017, 18, 601–610. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Jin, J.; Bao, X.; Zhan, W.H.; Han, T.Y.; Gan, M.; Zhang, C.; Wang, J. Inhibition of mitochondrial glutaminase activity reverses acquired erlotinib resistance in non-small cell lung cancer. Oncotarget 2016, 7, 610–621. [Google Scholar] [CrossRef]
- Liu, P.; Ge, M.; Hu, J.; Li, X.; Che, L.; Sun, K.; Cheng, L.; Huang, Y.; Pilo, M.G.; Cigliano, A.; et al. A functional mammalian target of rapamycin complex 1 signaling is indispensable for c-Myc-driven hepatocarcinogenesis. Hepatology 2017, 66, 167–181. [Google Scholar] [CrossRef] [PubMed]
- Csibi, A.; Lee, G.; Yoon, S.O.; Tong, H.; Ilter, D.; Elia, I.; Fendt, S.M.; Roberts, T.M.; Blenis, J. The mTORC1/S6K1 pathway regulates glutamine metabolism through the eIF4B-dependent control of c-Myc translation. Curr. Biol. CB 2014, 24, 2274–2280. [Google Scholar] [CrossRef]
- Howell, J.J.; Ricoult, S.J.H.; Ben-Sahra, I.; Manning, B.D. A growing role for mTOR in promoting anabolic metabolism. Biochem. Soc. Trans. 2013, 41, 906–912. [Google Scholar] [CrossRef]
- Tong, Y.; Guo, D.; Lin, S.-H.; Liang, J.; Yang, D.; Ma, C.; Shao, F.; Li, M.; Yu, Q.; Jiang, Y.; et al. SUCLA2-coupled regulation of GLS succinylation and activity counteracts oxidative stress in tumor cells. Mol. Cell 2021, 81, 2303–2316.e2308. [Google Scholar] [CrossRef]
- Greene, K.S.; Lukey, M.J.; Wang, X.; Blank, B.; Druso, J.E.; Lin, M.J.; Stalnecker, C.A.; Zhang, C.; Negrón Abril, Y.; Erickson, J.W.; et al. SIRT5 stabilizes mitochondrial glutaminase and supports breast cancer tumorigenesis. Proc. Natl. Acad. Sci. USA 2019, 116, 26625–26632. [Google Scholar] [CrossRef]
- Shah, R.; Chen, S. Metabolic Signaling Cascades Prompted by Glutaminolysis in Cancer. Cancers 2020, 12, 2624. [Google Scholar] [CrossRef]
- Jin, H.; Wang, S.; Zaal, E.A.; Wang, C.; Wu, H.; Bosma, A.; Jochems, F.; Isima, N.; Jin, G.; Lieftink, C.; et al. A powerful drug combination strategy targeting glutamine addiction for the treatment of human liver cancer. eLife 2020, 9, e56749. [Google Scholar] [CrossRef]
- Wang, L.; Zhu, L.; Wu, K.; Chen, Y.; Lee, D.Y.; Gucek, M.; Sack, M.N. Mitochondrial General Control of Amino Acid Synthesis 5 Like 1 Regulates Glutaminolysis, Mammalian Target of Rapamycin Complex 1 Activity, and Murine Liver Regeneration. Hepatology 2020, 71, 643–657. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Tchernyshyov, I.; Chang, T.C.; Lee, Y.S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458, 762–765. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.-J.; Chen, H.-Y.; Zeng, Z.; Deng, S.; Zhu, S.; Ye, Z.; He, C.; Liu, M.-L.; Huang, K.; Zhong, J.-X.; et al. Nutrient Stress-Dysregulated Antisense lncRNA GLS-AS Impairs GLS-Mediated Metabolism and Represses Pancreatic Cancer Progression. Cancer Res. 2019, 79, 1398–1412. [Google Scholar] [CrossRef] [PubMed]
- Lampa, M.; Arlt, H.; He, T.; Ospina, B.; Reeves, J.; Zhang, B.; Murtie, J.; Deng, G.; Barberis, C.; Hoffmann, D.; et al. Glutaminase is essential for the growth of triple-negative breast cancer cells with a deregulated glutamine metabolism pathway and its suppression synergizes with mTOR inhibition. PLoS ONE 2017, 12, e0185092. [Google Scholar] [CrossRef] [PubMed]
- Busca, R.; Bertolotto, C.; Ortonne, J.P.; Ballotti, R. Inhibition of the phosphatidylinositol 3-kinase/p70(S6)-kinase pathway induces B16 melanoma cell differentiation. J. Biol. Chem. 1996, 271, 31824–31830. [Google Scholar] [CrossRef] [PubMed]
- Speyer, C.L.; Smith, J.S.; Banda, M.; DeVries, J.A.; Mekani, T.; Gorski, D.H. Metabotropic glutamate receptor-1: A potential therapeutic target for the treatment of breast cancer. Breast Cancer Res. Treat. 2012, 132, 565–573. [Google Scholar] [CrossRef]
- Speyer, C.L.; Nassar, M.A.; Hachem, A.H.; Bukhsh, M.A.; Jafry, W.S.; Khansa, R.M.; Gorski, D.H. Riluzole mediates anti-tumor properties in breast cancer cells independent of metabotropic glutamate receptor-1. Breast Cancer Res. Treat. 2016, 157, 217–228. [Google Scholar] [CrossRef]
- Dolfi, S.C.; Medina, D.J.; Kareddula, A.; Paratala, B.; Rose, A.; Dhami, J.; Chen, S.; Ganesan, S.; Mackay, G.; Vazquez, A.; et al. Riluzole exerts distinct antitumor effects from a metabotropic glutamate receptor 1-specific inhibitor on breast cancer cells. Oncotarget 2017, 8, 44639–44653. [Google Scholar] [CrossRef]
- Isola, A.L.; Eddy, K.; Zembrzuski, K.; Goydos, J.S.; Chen, S. Exosomes released by metabotropic glutamate receptor 1 (GRM1) expressing melanoma cells increase cell migration and invasiveness. Oncotarget 2018, 9, 1187–1199. [Google Scholar] [CrossRef]
- Wall, B.A.; Yu, L.J.; Khan, A.; Haffty, B.; Goydos, J.S.; Chen, S. Riluzole is a radio-sensitizing agent in an in vivo model of brain metastasis derived from GRM1 expressing human melanoma cells. Pigment Cell Melanoma Res. 2015, 28, 105–109. [Google Scholar] [CrossRef]
- Le, M.N.; Chan, J.L.; Rosenberg, S.A.; Nabatian, A.S.; Merrigan, K.T.; Cohen-Solal, K.A.; Goydos, J.S. The glutamate release inhibitor Riluzole decreases migration, invasion, and proliferation of melanoma cells. J. Investig. Dermatol. 2010, 130, 2240–2249. [Google Scholar] [CrossRef] [PubMed]
- Datta, A.; Kim, H.; McGee, L.; Johnson, A.E.; Talwar, S.; Marugan, J.; Southall, N.; Hu, X.; Lal, M.; Mondal, D.; et al. High-throughput screening identified selective inhibitors of exosome biogenesis and secretion: A drug repurposing strategy for advanced cancer. Sci. Rep. 2018, 8, 8161. [Google Scholar] [CrossRef] [PubMed]
- Ridky, T.W.; Chow, J.M.; Wong, D.J.; Khavari, P.A. Invasive three-dimensional organotypic neoplasia from multiple normal human epithelia. Nat. Med. 2010, 16, 1450–1455. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eddy, K.; Eddin, M.N.; Fateeva, A.; Pompili, S.V.B.; Shah, R.; Doshi, S.; Chen, S. Implications of a Neuronal Receptor Family, Metabotropic Glutamate Receptors, in Cancer Development and Progression. Cells 2022, 11, 2857. https://doi.org/10.3390/cells11182857
Eddy K, Eddin MN, Fateeva A, Pompili SVB, Shah R, Doshi S, Chen S. Implications of a Neuronal Receptor Family, Metabotropic Glutamate Receptors, in Cancer Development and Progression. Cells. 2022; 11(18):2857. https://doi.org/10.3390/cells11182857
Chicago/Turabian StyleEddy, Kevinn, Mohamad Naser Eddin, Anna Fateeva, Stefano Vito Boccadamo Pompili, Raj Shah, Saurav Doshi, and Suzie Chen. 2022. "Implications of a Neuronal Receptor Family, Metabotropic Glutamate Receptors, in Cancer Development and Progression" Cells 11, no. 18: 2857. https://doi.org/10.3390/cells11182857
APA StyleEddy, K., Eddin, M. N., Fateeva, A., Pompili, S. V. B., Shah, R., Doshi, S., & Chen, S. (2022). Implications of a Neuronal Receptor Family, Metabotropic Glutamate Receptors, in Cancer Development and Progression. Cells, 11(18), 2857. https://doi.org/10.3390/cells11182857