
Wrangling Actin Assemblies: Actin Ring Dynamics during Cell Wound Repair


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
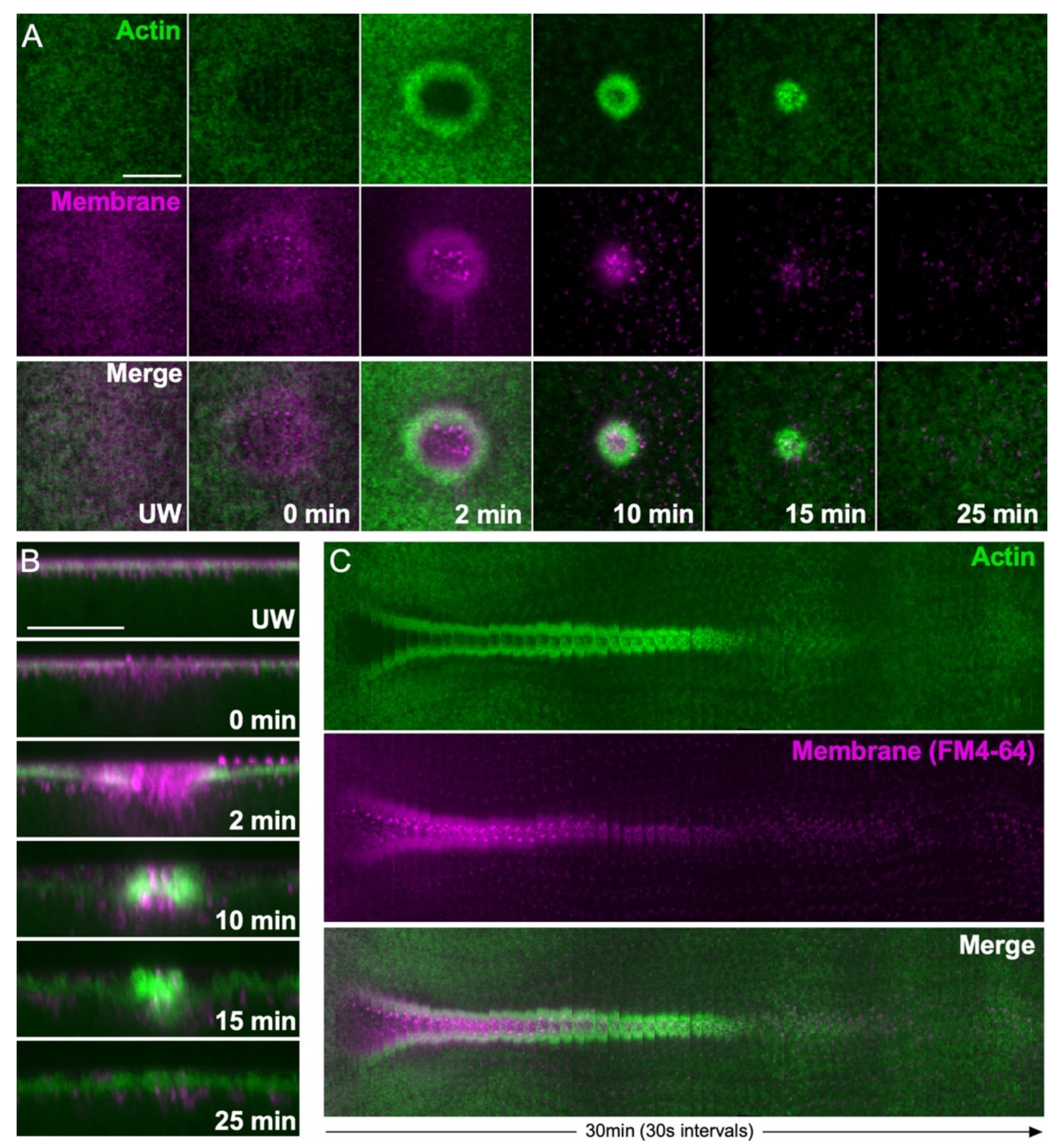
2. Actin Ring Formation
2.1. Actin Stabilization at Wounds
2.2. Actin Recruitment to Wounds
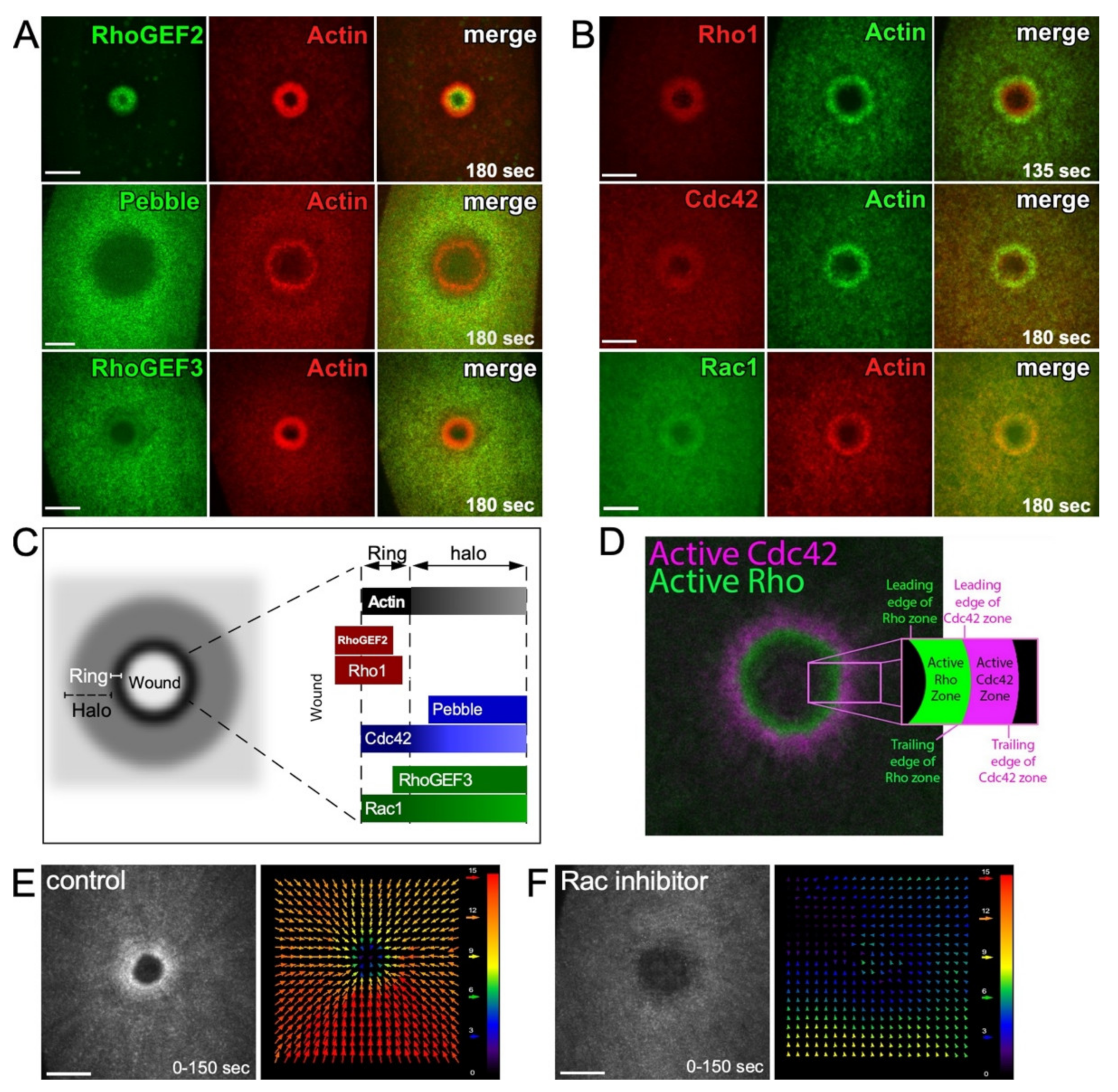
2.3. Actin Organization at Wounds: Rho Family GTPases
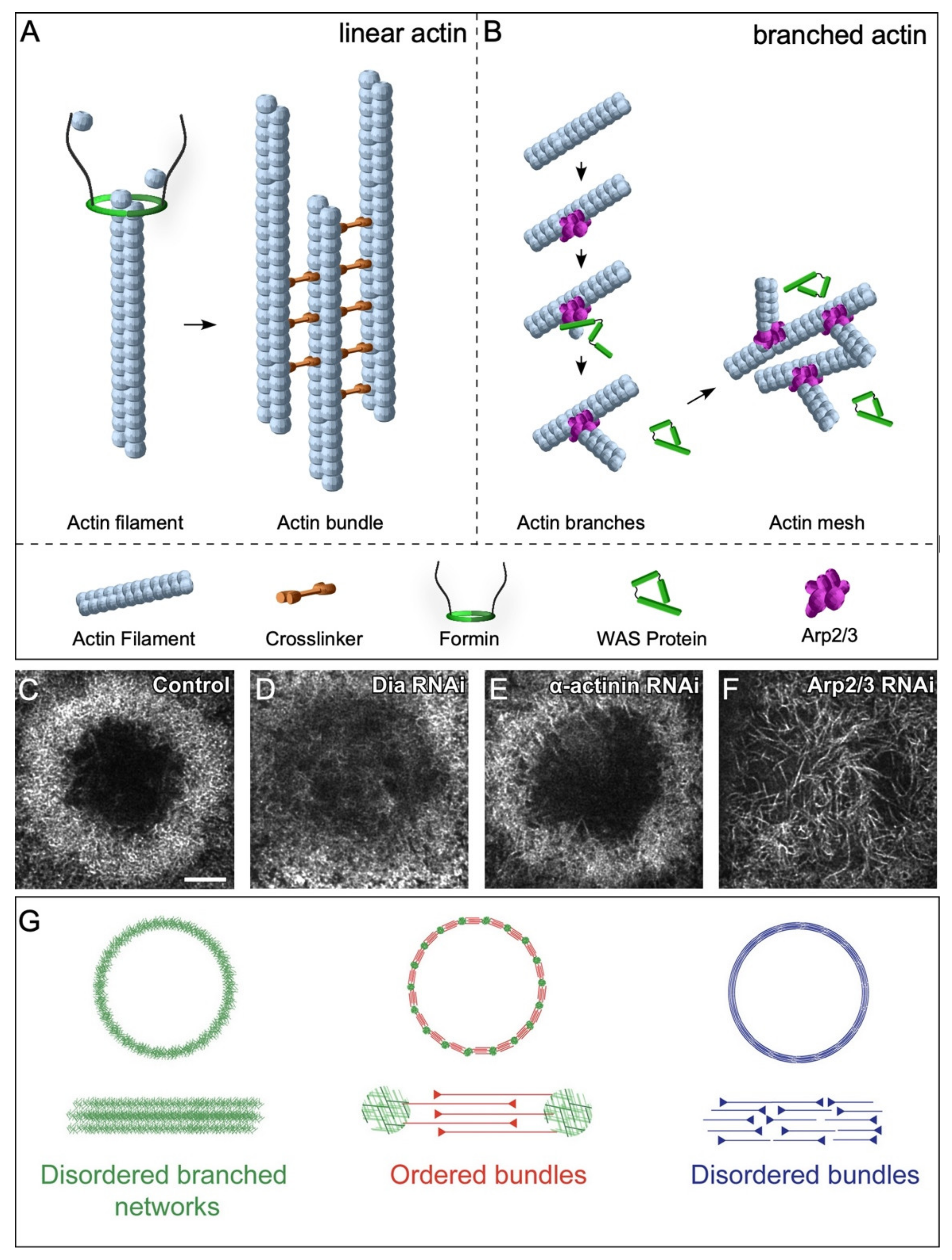
2.3.1. Linear Actin
2.3.2. Branched Actin
2.4. Actin Organization at Wounds: Other Factors
2.5. Actin Architectures in Cell Wound Repair
3. Actin Ring Translocation
3.1. Diversity in Translocation Mechanisms
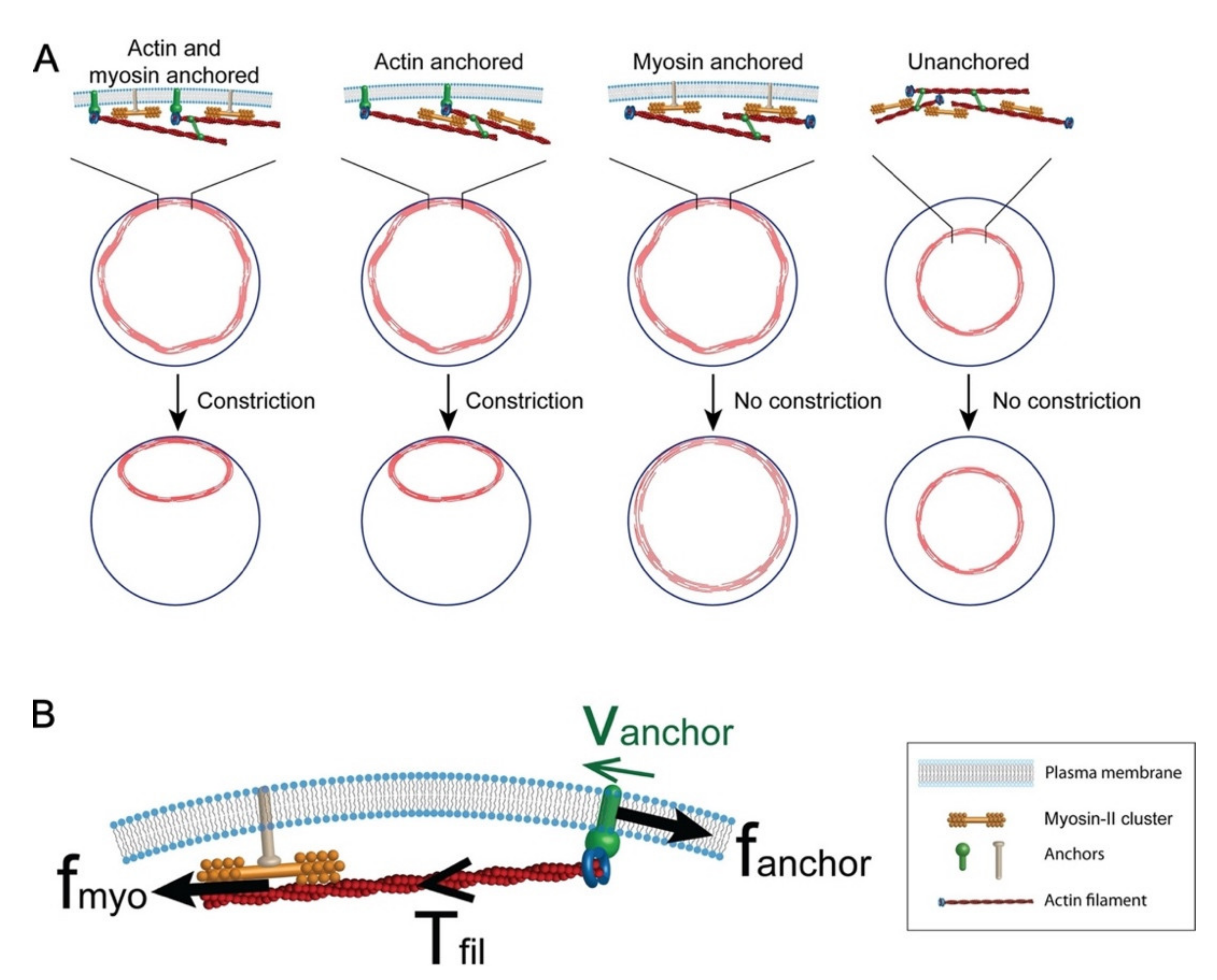
3.1.1. Actomyosin Ring Contraction
3.1.2. Actin Treadmilling
3.1.3. F-Actin Buckling Model
3.2. Plasma Membrane–Cortical Cytoskeleton Attachment
4. Cell Cortex Remodeling
4.1. Microarray Studies in the Drosophila Cell Wound Repair Model
4.2. RNAseq Studies in the Wounded MCF7 Cancer Cell Model
5. Relationship to Diseases and Infections
6. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nakamura, M.; Dominguez, A.; Decker, J.R.; Hull, A.J.; Verboon, J.M.; Parkhurst, S.M. Into the breach: How cells cope with wounds. Open Biol. 2018, 8, 180135. [Google Scholar] [CrossRef] [PubMed]
- Sonnemann, K.J.; Bement, W.M. Wound Repair: Toward Understanding and Integration of Single-Cell and Multicellular Wound Responses. Annu. Rev. Cell Dev. Biol. 2011, 27, 237–263. [Google Scholar] [CrossRef] [PubMed]
- Abreu-Blanco, M.T.; Verboon, J.M.; Parkhurst, S.M. Single cell wound repair: Dealing with life’s little traumas. Bioarchitecture 2011, 1, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Andrews, N.W.; Almeida, P.E.; Corrotte, M. Damage control: Cellular mechanisms of plasma membrane repair. Trends Cell Biol. 2014, 24, 734–742. [Google Scholar] [CrossRef]
- Andrews, N.W.; Corrotte, M. Plasma membrane repair. Curr. Biol. 2018, 28, R392–R397. [Google Scholar] [CrossRef]
- McNeil, P.L.; Terasaki, M. Coping with the inevitable: How cells repair a torn surface membrane. Nat. Cell Biol. 2001, 3, E124–E129. [Google Scholar] [CrossRef]
- McNeil, P.L.; Kirchhausen, T. An emergency response team for membrane repair. Nat. Rev. Mol. Cell Biol. 2005, 6, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Cooper, S.T.; McNeil, P.L. Membrane Repair: Mechanisms and Pathophysiology. Physiol. Rev. 2015, 95, 1205–1240. [Google Scholar] [CrossRef] [PubMed]
- Abreu-Blanco, M.T.; Verboon, J.M.; Parkhurst, S.M. Cell wound repair in Drosophila occurs through three distinct phases of membrane and cytoskeletal remodeling. J. Cell Biol. 2011, 193, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Bement, W.M.; Mandato, C.A.; Kirsch, M.N. Wound-induced assembly and closure of an actomyosin purse string in Xenopus oocytes. Curr. Biol. 1999, 9, 579–587. [Google Scholar] [CrossRef]
- Merriam, R.W.; Christensen, K. A contractile ring-like mechanism in wound healing and soluble factors affecting structural stability in the cortex of Xenopus eggs and oocytes. Development 1983, 75, 11–20. [Google Scholar] [CrossRef]
- Yumura, S.; Hashima, S.; Muranaka, S. Myosin II does not contribute to wound repair in Dictyostelium cells. Biol. Open 2014, 3, 966–973. [Google Scholar] [CrossRef] [PubMed]
- Terasaki, M.; Miyake, K.; McNeil, P.L. Large Plasma Membrane Disruptions Are Rapidly Resealed by Ca2+-dependent Vesicle–Vesicle Fusion Events. J. Cell Biol. 1997, 139, 63–74. [Google Scholar] [CrossRef]
- Xu, S.; Chisholm, A.D. A Gαq-Ca2+ Signaling Pathway Promotes Actin-Mediated Epidermal Wound Closure in C. elegans. Curr. Biol. 2011, 21, 1960–1967. [Google Scholar] [CrossRef] [PubMed]
- Steinhardt, R.A.; Bi, G.; Alderton, J.M. Cell Membrane Resealing by a Vesicular Mechanism Similar to Neurotransmitter Release. Science 1994, 263, 390–393. [Google Scholar] [CrossRef]
- Heitmann, A.S.B.; Zanjani, A.A.H.; Klenow, M.B.; Mularski, A.; Sønder, S.L.; Lund, F.W.; Boye, T.L.; Dias, C.; Bendix, P.M.; Simonsen, A.C.; et al. Phenothiazines alter plasma membrane properties and sensitize cancer cells to injury by inhibiting annexin-mediated repair. J. Biol. Chem. 2021, 297, 101012. [Google Scholar] [CrossRef] [PubMed]
- Pervin, M.S.; Itoh, G.; Talukder, S.U.; Fujimoto, K.; Morimoto, Y.V.; Tanaka, M.; Ueda, M.; Yumura, S. A study of wound repair in Dictyostelium cells by using novel laserporation. Sci. Rep. 2018, 8, 7969. [Google Scholar] [CrossRef]
- McNeil, P.; Vogel, S.; Miyake, K.; Terasaki, M. Patching plasma membrane disruptions with cytoplasmic membrane. J. Cell Sci. 2000, 113, 1891–1902. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Verboon, J.M.; Allen, T.E.; Abreu-Blanco, M.T.; Liu, R.; Dominguez, A.N.M.; Delrow, J.J.; Parkhurst, S.M. Autocrine insulin pathway signaling regulates actin dynamics in cell wound repair. PLoS Genet. 2020, 16, e1009186. [Google Scholar] [CrossRef] [PubMed]
- Davenport, N.R.; Sonnemann, K.J.; Eliceiri, K.W.; Bement, W.M. Membrane dynamics during cellular wound repair. Mol. Biol. Cell 2016, 27, 2272–2285. [Google Scholar] [CrossRef] [PubMed]
- Golding, A.E.; Visco, I.; Bieling, P.; Bement, W.M. Extraction of active RhoGTPases by RhoGDI regulates spatiotemporal patterning of RhoGTPases. eLife 2019, 8, e50471. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.G.; Miller, A.L.; Vaughan, E.; Yu, H.-Y.E.; Penkert, R.; Bement, W.M. Integration of Single and Multicellular Wound Responses. Curr. Biol. 2009, 19, 1389–1395. [Google Scholar] [CrossRef] [PubMed]
- Marg, A.; Schoewel, V.; Timmel, T.; Schulze, A.; Shah, C.; Daumke, O.; Spuler, S. Sarcolemmal Repair Is a Slow Process and Includes EHD2. Traffic 2012, 13, 1286–1294. [Google Scholar] [CrossRef]
- Demonbreun, A.R.; Quattrocelli, M.; Barefield, D.; Allen, M.V.; Swanson, K.E.; McNally, E.M. An actin-dependent annexin complex mediates plasma membrane repair in muscle. J. Cell Biol. 2016, 213, 705–718. [Google Scholar] [CrossRef] [PubMed]
- Burkel, B.M.; Benink, H.A.; Vaughan, E.M.; von Dassow, G.; Bement, W.M. A Rho GTPase Signal Treadmill Backs a Contractile Array. Dev. Cell 2012, 23, 384–396. [Google Scholar] [CrossRef] [PubMed]
- Abreu-Blanco, M.T.; Verboon, J.M.; Parkhurst, S.M. Coordination of Rho Family GTPase Activities to Orchestrate Cytoskeleton Responses during Cell Wound Repair. Curr. Biol. 2014, 24, 144–155. [Google Scholar] [CrossRef]
- Nakamura, M.; Verboon, J.; Parkhurst, S.M. Prepatterning by RhoGEFs governs Rho GTPase spatiotemporal dynamics during wound repair. J. Cell Biol. 2017, 216, 3959–3969. [Google Scholar] [CrossRef] [PubMed]
- Holland, A.; Dowling, P.; Zweyer, M.; Swandulla, D.; Henry, M.; Clynes, M.; Ohlendieck, K. Proteomic profiling of cardiomyopathic tissue from the aged mdx model of Duchenne muscular dystrophy reveals a drastic decrease in laminin, nidogen and annexin. Proteomics 2013, 13, 2312–2323. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Aoki, M.; Illa, I.; Wu, C.; Fardeau, M.; Angelini, C.; Serrano, C.; Urtizberea, J.A.; Hentati, F.; Ben Hamida, M.; et al. Dysferlin, a novel skeletal muscle gene, is mutated in Miyoshi myopathy and limb girdle muscular dystrophy. Nat. Genet. 1998, 20, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Howard, A.C.; McNeil, A.K.; Xiong, F.; Xiong, W.-C.; McNeil, P.L. A Novel Cellular Defect in Diabetes: Membrane repair failure. Diabetes 2011, 60, 3034–3043. [Google Scholar] [CrossRef] [PubMed]
- Kamai, T.; Tsujii, T.; Arai, K.; Takagi, K.; Asami, H.; Ito, Y.; Oshima, H. Significant association of Rho/ROCK pathway with invasion and metastasis of bladder cancer. Clin. Cancer Res. 2003, 9, 2632–2641. [Google Scholar] [PubMed]
- Gerke, V.; Creutz, C.E.; Moss, S.E. Annexins: Linking Ca2+ signalling to membrane dynamics. Nat. Rev. Mol. Cell Biol. 2005, 6, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Benaud, C.; Le Dez, G.; Mironov, S.; Galli, F.; Reboutier, D.; Prigent, C. Annexin A2 is required for the early steps of cytokinesis. EMBO Rep. 2015, 16, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Gabel, M.; Delavoie, F.; Demais, V.; Royer, C.; Bailly, Y.; Vitale, N.; Bader, M.-F.; Chasserot-Golaz, S. Annexin A2–dependent actin bundling promotes secretory granule docking to the plasma membrane and exocytosis. J. Cell Biol. 2015, 210, 785–800. [Google Scholar] [CrossRef] [PubMed]
- Bouter, A.; Carmeille, R.; Gounou, C.; Bouvet, F.; Degrelle, S.; Evain-Brion, D.; Brisson, A. Review: Annexin-A5 and cell membrane repair. Placenta 2015, 36 (Suppl. S1), S43–S49. [Google Scholar] [CrossRef] [PubMed]
- Bouvet, F.; Ros, M.; Bonedeau, E.; Croissant, C.; Frelin, L.; Saltel, F.; Moreau, V.; Bouter, A. Defective membrane repair machinery impairs survival of invasive cancer cells. Sci. Rep. 2020, 10, 21821. [Google Scholar] [CrossRef]
- Bendix, P.M.; Simonsen, A.C.; Florentsen, C.D.; Häger, S.C.; Mularski, A.; Zanjani, A.A.H.; Moreno-Pescador, G.; Klenow, M.B.; Sønder, S.L.; Danielsen, H.M.; et al. Interdisciplinary Synergy to Reveal Mechanisms of Annexin-Mediated Plasma Membrane Shaping and Repair. Cells 2020, 9, 1029. [Google Scholar] [CrossRef]
- Lauritzen, S.P.; Boye, T.L.; Nylandsted, J. Annexins are instrumental for efficient plasma membrane repair in cancer cells. Semin. Cell Dev. Biol. 2015, 45, 32–38. [Google Scholar] [CrossRef]
- Koerdt, S.N.; Ashraf, A.P.; Gerke, V. Annexins and plasma membrane repair. Curr. Top. Membr. 2019, 84, 43–65. [Google Scholar] [CrossRef]
- Bittel, D.C.; Chandra, G.; Tirunagri, L.M.S.; Deora, A.B.; Medikayala, S.; Scheffer, L.; Defour, A.; Jaiswal, J.K. Annexin A2 Mediates Dysferlin Accumulation and Muscle Cell Membrane Repair. Cells 2020, 9, 1919. [Google Scholar] [CrossRef]
- Lennon, N.J.; Kho, A.; Bacskai, B.J.; Perlmutter, S.L.; Hyman, B.T.; Brown, R.H. Dysferlin Interacts with Annexins A1 and A2 and Mediates Sarcolemmal Wound-healing. J. Biol. Chem. 2003, 278, 50466–50473. [Google Scholar] [CrossRef] [PubMed]
- Reymann, A.-C.; Staniscia, F.; Erzberger, A.; Salbreux, G.; Grill, S.W. Cortical flow aligns actin filaments to form a furrow. eLife 2016, 5, e17807. [Google Scholar] [CrossRef] [PubMed]
- Heath, J.P.; Holifield, B.F. On the mechanisms of cortical actin flow and its role in cytoskeletal organisation of fibroblasts. Symp. Soc. Exp. Biol. 1993, 47, 35–56. [Google Scholar]
- Bray, D.; White, J.G. Cortical Flow in Animal Cells. Science 1988, 239, 883–888. [Google Scholar] [CrossRef] [PubMed]
- Moe, A.; Holmes, W.; Golding, A.E.; Zola, J.; Swider, Z.T.; Edelstein-Keshet, L.; Bement, W. Cross talk-dependent cortical patterning of Rho GTPases during cell repair. Mol. Biol. Cell 2021, 32, 1417–1432. [Google Scholar] [CrossRef] [PubMed]
- Chhabra, E.S.; Higgs, H.N. The Many Faces of Actin: Matching Assembly Factors with Cellular Structures. Nat. Cell Biol. 2007, 9, 1110–1121. [Google Scholar] [CrossRef]
- Faix, J.; Grosse, R. Staying in Shape with Formins. Dev. Cell 2006, 10, 693–706. [Google Scholar] [CrossRef]
- Goode, B.L.; Eck, M.J. Mechanism and Function of Formins in the Control of Actin Assembly. Annu. Rev. Biochem. 2007, 76, 593–627. [Google Scholar] [CrossRef]
- Achard, V.; Martiel, J.-L.; Michelot, A.; Guérin, C.; Reymann, A.-C.; Blanchoin, L.; Boujemaa-Paterski, R. A “Primer”-Based Mechanism Underlies Branched Actin Filament Network Formation and Motility. Curr. Biol. 2010, 20, 423–428. [Google Scholar] [CrossRef]
- Akin, O.; Mullins, R.D. Capping Protein Increases the Rate of Actin-Based Motility by Promoting Filament Nucleation by the Arp2/3 Complex. Cell 2008, 133, 841–851. [Google Scholar] [CrossRef]
- Li, Y.; Christensen, J.; Homa, K.E.; Hocky, G.; Fok, A.; Sees, J.A.; Voth, G.A.; Kovar, D.R. The F-actin bundler α-actinin Ain1 is tailored for ring assembly and constriction during cytokinesis in fission yeast. Mol. Biol. Cell 2016, 27, 1821–1833. [Google Scholar] [CrossRef] [PubMed]
- Takenawa, T.; Suetsugu, S. The WASP-WAVE protein network: Connecting the membrane to the cytoskeleton. Nat. Rev. Mol. Cell Biol. 2007, 8, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Millard, T.H.; Sharp, S.J.; Machesky, L.M. Signalling to actin assembly via the WASP (Wiskott-Aldrich syndrome protein)-family proteins and the Arp2/3 complex. Biochem. J. 2004, 380, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Campellone, K.G.; Welch, M.D. A nucleator arms race: Cellular control of actin assembly. Nat. Rev. Mol. Cell Biol. 2010, 11, 237–251. [Google Scholar] [CrossRef] [PubMed]
- Campellone, K.G. Cytoskeleton-modulating effectors of enteropathogenic and enterohaemorrhagic Escherichia coli: Tir, EspFU and actin pedestal assembly. FEBS J. 2010, 277, 2390–2402. [Google Scholar] [CrossRef] [PubMed]
- Burianek, L.E.; Soderling, S.H. Under lock and key: Spatiotemporal regulation of WASP family proteins coordinates separate dynamic cellular processes. Semin. Cell Dev. Biol. 2013, 24, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Massaad, M.J.; Ramesh, N.; Geha, R.S. Wiskott-Aldrich syndrome: A comprehensive review. Ann. N. Y. Acad. Sci. 2013, 1285, 26–43. [Google Scholar] [CrossRef]
- Oda, A.; Eto, K. WASPs and WAVEs: From molecular function to physiology in hematopoietic cells. Semin. Cell Dev. Biol. 2013, 24, 308–313. [Google Scholar] [CrossRef]
- Alekhina, O.; Burstein, E.; Billadeau, D.D. Cellular functions of WASP family proteins at a glance. J. Cell Sci. 2017, 130, 2235–2241. [Google Scholar] [CrossRef]
- Ennomani, H.; Letort, G.; Guérin, C.; Martiel, J.-L.; Cao, W.; Nédélec, F.; De La Cruz, E.M.; Théry, M.; Blanchoin, L. Architecture and Connectivity Govern Actin Network Contractility. Curr. Biol. 2016, 26, 616–626. [Google Scholar] [CrossRef]
- Hui, J.; Nakamura, M.; Dubrulle, J.; Parkhurst, S.M. Cell wound repair requires the coordinated action of linear and branched actin nucleation factors. bioRxiv 2022. [Google Scholar] [CrossRef]
- Mishima, M.; Kaitna, S.; Glotzer, M. Central Spindle Assembly and Cytokinesis Require a Kinesin-like Protein/RhoGAP Complex with Microtubule Bundling Activity. Dev. Cell 2002, 2, 41–54. [Google Scholar] [CrossRef]
- Pollard, T.D.; O’Shaughnessy, B. Molecular Mechanism of Cytokinesis. Annu. Rev. Biochem. 2019, 88, 661–689. [Google Scholar] [CrossRef]
- White, E.A.; Glotzer, M. Centralspindlin: At the heart of cytokinesis. Cytoskeleton 2012, 69, 882–892. [Google Scholar] [CrossRef] [PubMed]
- Somers, W.; Saint, R. A RhoGEF and Rho Family GTPase-Activating Protein Complex Links the Contractile Ring to Cortical Microtubules at the Onset of Cytokinesis. Dev. Cell 2003, 4, 29–39. [Google Scholar] [CrossRef]
- Adams, R.R.; Tavares, A.A.; Salzberg, A.; Bellen, H.J.; Glover, D.M. pavarotti encodes a kinesin-like protein required to organize the central spindle and contractile ring for cytokinesis. Genes Dev. 1998, 12, 1483–1494. [Google Scholar] [CrossRef] [PubMed]
- Sommi, P.; Ananthakrishnan, R.; Cheerambathur, D.K.; Kwon, M.; Morales-Mulia, S.; Brust-Mascher, I.; Mogilner, A. A mitotic kinesin-6, Pav-KLP, mediates interdependent cortical reorganization and spindle dynamics in Drosophila embryos. J. Cell Sci. 2010, 123, 1862–1872. [Google Scholar] [CrossRef]
- Nakamura, M.; Verboon, J.M.; Prentiss, C.L.; Parkhurst, S.M. The kinesin-like protein Pavarotti functions noncanonically to regulate actin dynamics. J. Cell Biol. 2020, 219, e201912117. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, A.; Murakami, H.; Asai, N.; Morone, N.; Watanabe, T.; Kawai, K.; Murakumo, Y.; Usukura, J.; Kaibuchi, K.; Takahashi, M. Akt/PKB Regulates Actin Organization and Cell Motility via Girdin/APE. Dev. Cell 2005, 9, 389–402. [Google Scholar] [CrossRef]
- Houssin, E.; Tepass, U.; Laprise, P. Girdin-mediated interactions between cadherin and the actin cytoskeleton are required for epithelial morphogenesis in Drosophila. Development 2015, 142, 1777–1784. [Google Scholar] [CrossRef]
- Ghiglione, C.; Jouandin, P.; Cérézo, D.; Noselli, S. The Drosophila insulin pathway controls Profilin expression and dynamic actin-rich protrusions during collective cell migration. Development 2018, 145, dev.161117. [Google Scholar] [CrossRef] [PubMed]
- Xue, B.; Robinson, R.C. Guardians of the actin monomer. Eur. J. Cell Biol. 2013, 92, 316–332. [Google Scholar] [CrossRef]
- Blanchoin, L.; Boujemaa-Paterski, R.; Sykes, C.; Plastino, J. Actin Dynamics, Architecture, and Mechanics in Cell Motility. Physiol. Rev. 2014, 94, 235–263. [Google Scholar] [CrossRef] [PubMed]
- Michelot, A.; Drubin, D.G. Building Distinct Actin Filament Networks in a Common Cytoplasm. Curr. Biol. 2011, 21, R560–R569. [Google Scholar] [CrossRef]
- Tojkander, S.; Gateva, G.; Lappalainen, P. Actin stress fibers–assembly, dynamics and biological roles. J. Cell Sci. 2012, 125, 1855–1864. [Google Scholar] [CrossRef] [PubMed]
- Murrell, M.P.; Oakes, P.; Lenz, M.; Gardel, M.L. Forcing cells into shape: The mechanics of actomyosin contractility. Nat. Rev. Mol. Cell Biol. 2015, 16, 486–498. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, D.A.; Mullins, R.D. Cell mechanics and the cytoskeleton. Nature 2010, 463, 485–492. [Google Scholar] [CrossRef]
- Henson, J.H.; Nazarian, R.; Schulberg, K.L.; Trabosh, V.A.; Kolnik, S.E.; Burns, A.R.; McPartland, K.J. Wound Closure in the Lamellipodia of Single Cells: Mediation by Actin Polymerization in the Absence of an Actomyosin Purse String. Mol. Biol. Cell 2002, 13, 1001–1014. [Google Scholar] [CrossRef] [PubMed]
- Kučera, O.; Siahaan, V.; Janda, D.; Dijkstra, S.H.; Pilátová, E.; Zatecka, E.; Diez, S.; Braun, M.; Lansky, Z. Anillin propels myosin-independent constriction of actin rings. Nat. Commun. 2021, 12, 4595. [Google Scholar] [CrossRef] [PubMed]
- Lenz, M.; Thoresen, T.; Gardel, M.L.; Dinner, A.R. Contractile Units in Disordered Actomyosin Bundles Arise from F-Actin Buckling. Phys. Rev. Lett. 2012, 108, 238107. [Google Scholar] [CrossRef] [PubMed]
- Murrell, M.P.; Gardel, M.L. F-actin buckling coordinates contractility and severing in a biomimetic actomyosin cortex. Proc. Natl. Acad. Sci. USA 2012, 109, 20820–20825. [Google Scholar] [CrossRef] [PubMed]
- Senger, F.; Pitaval, A.; Ennomani, H.; Kurzawa, L.; Blanchoin, L.; Théry, M. Spatial integration of mechanical forces by α-actinin establishes actin network symmetry. J. Cell Sci. 2019, 132, jcs.236604. [Google Scholar] [CrossRef] [PubMed]
- Zumdieck, A.; Kruse, K.; Bringmann, H.; Hyman, A.A.; Jülicher, F. Stress Generation and Filament Turnover during Actin Ring Constriction. PLoS ONE 2007, 2, e696. [Google Scholar] [CrossRef] [PubMed]
- Mendes Pinto, I.; Rubinstein, B.; Kucharavy, A.; Unruh, J.R.; Li, R. Actin Depolymerization Drives Actomyosin Ring Contraction during Budding Yeast Cytokinesis. Dev. Cell 2012, 22, 1247–1260. [Google Scholar] [CrossRef] [PubMed]
- Mandato, C.A.; Bement, W.M. Contraction and polymerization cooperate to assemble and close actomyosin rings around Xenopus oocyte wounds. J. Cell Biol. 2001, 154, 785–798. [Google Scholar] [CrossRef]
- Benink, H.A.; Bement, W.M. Concentric zones of active RhoA and Cdc42 around single cell wounds. J. Cell Biol. 2005, 168, 429–439. [Google Scholar] [CrossRef]
- Tanvir, I.O.; Itoh, G.; Adachi, H.; Yumura, S. Dynamics of Myosin II Filaments during Wound Repair in Dividing Cells. Cells 2021, 10, 1229. [Google Scholar] [CrossRef]
- Vogel, S.K.; Petrasek, Z.; Heinemann, F.; Schwille, P. Myosin motors fragment and compact membrane-bound actin filaments. eLife 2013, 2, e00116. [Google Scholar] [CrossRef]
- Chew, T.G.; Huang, J.; Palani, S.; Sommese, R.; Kamnev, A.; Hatano, T.; Gu, Y.; Oliferenko, S.; Sivaramakrishnan, S.; Balasubramanian, M.K. Actin turnover maintains actin filament homeostasis during cytokinetic ring contraction. J. Cell Biol. 2017, 216, 2657–2667. [Google Scholar] [CrossRef]
- Thiyagarajan, S.; Wang, S.; O’Shaughnessy, B. A node organization in the actomyosin contractile ring generates tension and aids stability. Mol. Biol. Cell 2017, 28, 3286–3297. [Google Scholar] [CrossRef]
- Wang, S.; O’Shaughnessy, B. Anchoring of actin to the plasma membrane enables tension production in the fission yeast cytokinetic ring. Mol. Biol. Cell 2019, 30, 2053–2064. [Google Scholar] [CrossRef] [PubMed]
- Blackwood, R.; Ernst, J.D. Characterization of Ca2+-dependent phospholipid binding, vesicle aggregation and membrane fusion by annexins. Biochem. J. 1990, 266, 195–200. [Google Scholar] [CrossRef]
- Ratheesh, A.; Yap, A. A bigger picture: Classical cadherins and the dynamic actin cytoskeleton. Nat. Rev. Mol. Cell Biol. 2012, 13, 673–679. [Google Scholar] [CrossRef]
- Häger, S.C.; Dias, C.; Sønder, S.L.; Olsen, A.V.; da Piedade, I.; Heitmann, A.S.B.; Papaleo, E.; Nylandsted, J. Short-term transcriptomic response to plasma membrane injury. Sci. Rep. 2021, 11, 19141. [Google Scholar] [CrossRef]
- Hunter, C.; Sung, P.; Schejter, E.D.; Wieschaus, E. Conserved Domains of the Nullo Protein Required for Cell-Surface Localization and Formation of Adherens Junctions. Mol. Biol. Cell 2002, 13, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Hunter, C.; Wieschaus, E. Regulated Expression of nullo Is Required for the Formation of Distinct Apical and Basal Adherens Junctions in the Drosophila Blastoderm. J. Cell Biol. 2000, 150, 391–402. [Google Scholar] [CrossRef] [PubMed]
- Simpson, L.; Wieschaus, E. Zygotic activity of the nullo locus is required to stabilize the actin-myosin network during cellularization in Drosophila. Development 1990, 110, 851–863. [Google Scholar] [CrossRef] [PubMed]
- Berleth, T.; Burri, M.; Thoma, G.; Bopp, D.; Richstein, S.; Frigerio, G.; Noll, M.; Nüsslein-Volhard, C. The role of localization of bicoid RNA in organizing the anterior pattern of the Drosophila embryo. EMBO J. 1988, 7, 1749–1756. [Google Scholar] [CrossRef] [PubMed]
- Nüsslein-Volhard, C.; Frohnhöfer, H.G.; Lehmann, R. Determination of Anteroposterior Polarity in Drosophila. Science 1987, 238, 1675–1681. [Google Scholar] [CrossRef] [PubMed]
- Marcey, D.; Watkins, W.; Hazelrigg, T. The temporal and spatial distribution pattern of maternal exuperantia protein: Evidence for a role in establishment but not maintenance of bicoid mRNA localization. EMBO J. 1991, 10, 4259–4266. [Google Scholar] [CrossRef] [PubMed]
- Irion, U.; St Johnston, D. bicoid RNA localization requires specific binding of an endosomal sorting complex. Nature 2007, 445, 554–558. [Google Scholar] [CrossRef] [PubMed]
- Pokrywka, N.; Stephenson, E. Microtubules mediate the localization of bicoid RNA during Drosophila oogenesis. Development 1991, 113, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.Y.; Hengst, U.; Cox, L.J.; Macosko, E.Z.; Jeromin, A.; Urquhart, E.R.; Jaffrey, S. Local translation of RhoA regulates growth cone collapse. Nat. 2005, 436, 1020–1024. [Google Scholar] [CrossRef] [PubMed]
- Chatzifrangkeskou, M.; Yadin, D.; Marais, T.; Chardonnet, S.; Cohen-Tannoudji, M.; Mougenot, N.; Schmitt, A.; Crasto, S.; Di Pasquale, E.; Macquart, C.; et al. Cofilin-1 phosphorylation catalyzed by ERK1/2 alters cardiac actin dynamics in dilated cardiomyopathy caused by lamin A/C gene mutation. Hum. Mol. Genet. 2018, 27, 3060–3078. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Varshney, B.; Chatterjee, S.; Ray, K. Somatic ERK activation during transit amplification is essential for maintaining the synchrony of germline divisions in Drosophila testis. Open Biol. 2018, 8, 180033. [Google Scholar] [CrossRef]
- Liao, P.-H.; Hsu, H.-H.; Chen, T.-S.; Chen, M.-C.; Day, C.-H.; Tu, C.-C.; Lin, Y.-M.; Tsai, F.-J.; Kuo, W.-W.; Huang, C.-Y. Phosphorylation of cofilin-1 by ERK confers HDAC inhibitor resistance in hepatocellular carcinoma cells via decreased ROS-mediated mitochondria injury. Oncogene 2017, 36, 1978–1990. [Google Scholar] [CrossRef]
- Sønder, S.L.; Häger, S.C.; Heitmann, A.S.B.; Frankel, L.B.; Dias, C.; Simonsen, A.C.; Nylandsted, J. Restructuring of the plasma membrane upon damage by LC3-associated macropinocytosis. Sci. Adv. 2021, 7, eabg1969. [Google Scholar] [CrossRef]
- Roubinet, C.; Tran, P.T.; Piel, M. Common mechanisms regulating cell cortex properties during cell division and cell migration. Cytoskeleton 2012, 69, 957–972. [Google Scholar] [CrossRef]
- Clarke, M.S.F.; Caldwell, R.W.; Chiao, H.; Miyake, K.; McNeil, P.L. Contraction-Induced Cell Wounding and Release of Fibroblast Growth Factor in Heart. Circ. Res. 1995, 76, 927–934. [Google Scholar] [CrossRef]
- Coulombe, P.A.; Hutton, M.; Letal, A.; Hebert, A.; Paller, A.S.; Fuchs, E. Point mutations in human keratin 14 genes of epidermolysis bullosa simplex patients: Genetic and functional analyses. Cell 1991, 66, 1301–1311. [Google Scholar] [CrossRef]
- Petrof, B.J.; Shrager, J.B.; Stedman, H.H.; Kelly, A.M.; Sweeney, H.L. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc. Natl. Acad. Sci. USA 1993, 90, 3710–3714. [Google Scholar] [CrossRef] [PubMed]
- Trump, B.F.; Berezesky, I.K. Calcium-mediated cell injury and cell death. FASEB J. 1995, 9, 219–228. [Google Scholar] [CrossRef] [PubMed]
- McNeil, P.L.; Steinhardt, R.A. Plasma Membrane Disruption: Repair, Prevention, Adaptation. Annu. Rev. Cell Dev. Biol. 2003, 19, 697–731. [Google Scholar] [CrossRef]
- Bansal, D.; Miyake, K.; Vogel, S.S.; Groh, S.; Chen, C.-C.; Williamson, R.; McNeil, P.L.; Campbell, K.P. Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature 2003, 423, 168–172. [Google Scholar] [CrossRef]
- Han, R.; Bansal, D.; Miyake, K.; Muniz, V.P.; Weiss, R.M.; McNeil, P.L.; Campbell, K.P. Dysferlin-mediated membrane repair protects the heart from stress-induced left ventricular injury. J. Clin. Investig. 2007, 117, 1805–1813. [Google Scholar] [CrossRef]
- Minetti, C.; Sotgia, F.; Bruno, C.; Scartezzini, P.; Broda, P.; Bado, M.; Masetti, E.; Mazzocco, M.; Egeo, A.; Donati, M.A.; et al. Mutations in the caveolin-3 gene cause autosomal dominant limb-girdle muscular dystrophy. Nat. Genet. 1998, 18, 365–368. [Google Scholar] [CrossRef] [PubMed]
- Nigro, V.; Piluso, G. Spectrum of muscular dystrophies associated with sarcolemmal-protein genetic defects. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2015, 1852, 585–593. [Google Scholar] [CrossRef] [PubMed]
- Nigro, V.; Savarese, M. Genetic basis of limb-girdle muscular dystrophies: The 2014 update. Acta Myol. 2014, 33, 1. [Google Scholar] [PubMed]
- D’Souza, D.M.; Al-Sajee, D.; Hawke, T.J. Diabetic myopathy: Impact of diabetes mellitus on skeletal muscle progenitor cells. Front. Physiol. 2013, 4, 379. [Google Scholar] [CrossRef]
- Huynh, C.; Roth, D.; Ward, D.M.; Kaplan, J.; Andrews, N.W. Defective lysosomal exocytosis and plasma membrane repair in Chediak–Higashi/beige cells. Proc. Natl. Acad. Sci. USA 2004, 101, 16795–16800. [Google Scholar] [CrossRef]
- Brem, H.; Tomic-Canic, M. Cellular and molecular basis of wound healing in diabetes. J. Clin. Investig. 2007, 117, 1219–1222. [Google Scholar] [CrossRef]
- Wild, S.; Roglic, G.; Green, A.; Sicree, R.; King, H. Global Prevalence of Diabetes: Estimates for the year 2000 and projections for 2030. Diabetes Care 2004, 27, 1047–1053. [Google Scholar] [CrossRef] [PubMed]
- Burgess, J.L.; Wyant, W.A.; Abujamra, B.A.; Kirsner, R.S.; Jozic, I. Diabetic Wound-Healing Science. Medicina 2021, 57, 1072. [Google Scholar] [CrossRef]
- Jaiswal, J.K.; Lauritzen, S.P.; Scheffer, L.; Sakaguchi, M.; Bunkenborg, J.; Simon, S.M.; Kallunki, T.; Jäättelä, M.; Nylandsted, J. S100A11 is required for efficient plasma membrane repair and survival of invasive cancer cells. Nat. Commun. 2014, 5, 3795. [Google Scholar] [CrossRef] [PubMed]
- Boye, T.L.; Maeda, K.; Pezeshkian, W.; Sønder, S.L.; Haeger, S.C.; Gerke, V.; Simonsen, A.C.; Nylandsted, J. Annexin A4 and A6 induce membrane curvature and constriction during cell membrane repair. Nat. Commun. 2017, 8, 1623. [Google Scholar] [CrossRef] [PubMed]
- Carmeille, R.; Degrelle, S.A.; Plawinski, L.; Bouvet, F.; Gounou, C.; Evain-Brion, D.; Brisson, A.R.; Bouter, A. Annexin-A5 promotes membrane resealing in human trophoblasts. Biochim. Biophys. Acta 2015, 1853, 2033–2044. [Google Scholar] [CrossRef]
- Husmann, M.; Beckmann, E.; Boller, K.; Kloft, N.; Tenzer, S.; Bobkiewicz, W.; Neukirch, C.; Bayley, H.; Bhakdi, S. Elimination of a bacterial pore-forming toxin by sequential endocytosis and exocytosis. FEBS Lett. 2009, 583, 337–344. [Google Scholar] [CrossRef]
- Mesquita, F.S.; Brito, C.; Cabanes, D.; Sousa, S. Control of cytoskeletal dynamics during cellular responses to pore forming toxins. Commun. Integr. Biol. 2017, 10, e1349582. [Google Scholar] [CrossRef]
- Brito, C.; Cabanes, D.; Mesquita, F.S.; Sousa, S. Mechanisms protecting host cells against bacterial pore-forming toxins. Cell. Mol. Life Sci. 2019, 76, 1319–1339. [Google Scholar] [CrossRef]
- Keyel, P.A.; Loultcheva, L.; Roth, R.; Salter, R.D.; Watkins, S.; Yokoyama, W.M.; Heuser, J.E. Streptolysin O clearance through sequestration into blebs that bud passively from the plasma membrane. J. Cell Sci. 2011, 124, 2414–2423. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, A.J.; Maiuri, P.; Lafaurie-Janvore, J.; Divoux, S.; Piel, M.; Perez, F. ESCRT Machinery Is Required for Plasma Membrane Repair. Science 2014, 343, 1247136. [Google Scholar] [CrossRef] [PubMed]
- Lata, K.; Singh, M.; Chatterjee, S.; Chattopadhyay, K. Membrane Dynamics and Remodelling in Response to the Action of the Membrane-Damaging Pore-Forming Toxins. J. Membr. Biol. 2022, 255, 161–173. [Google Scholar] [CrossRef] [PubMed]
- Georgescu, M.; Gheorghe, I.; Curutiu, C.; Lazar, V.; Bleotu, C.; Chifiriuc, M.-C. Virulence and resistance features of Pseudomonas aeruginosa strains isolated from chronic leg ulcers. BMC Infect. Dis. 2016, 16 (Suppl. S1), 92. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, Y.; Mengist, H.; Shi, C.; Zhang, C.; Wang, B.; Li, T.; Huang, Y.; Xu, Y.; Jin, T. Structural Basis of the Pore-Forming Toxin/Membrane Interaction. Toxins 2021, 13, 128. [Google Scholar] [CrossRef]
- Bischofberger, M.; Iacovache, I.; van der Goot, F.G. Pathogenic Pore-Forming Proteins: Function and Host Response. Cell Host Microbe 2012, 12, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Peraro, M.D.; van der Goot, G. Pore-forming toxins: Ancient, but never really out of fashion. Nat. Rev. Genet. 2016, 14, 77–92. [Google Scholar] [CrossRef] [PubMed]
- Houthaeve, G.; De Smedt, S.C.; Braeckmans, K.; De Vos, W.H. The cellular response to plasma membrane disruption for nanomaterial delivery. Nano Converg. 2022, 9, 6. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Singh, D.; Helfield, B. An Overview of Cell Membrane Perforation and Resealing Mechanisms for Localized Drug Delivery. Pharmaceutics 2022, 14, 886. [Google Scholar] [CrossRef] [PubMed]
- Jia, C.; Shi, J.; Han, T.; Yu, A.C.; Qin, P. Spatiotemporal Dynamics and Mechanisms of Actin Cytoskeletal Re-modeling in Cells Perforated by Ultrasound-Driven Microbubbles. Ultrasound Med. Biol. 2022, 48, 760–777. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hui, J.; Stjepić, V.; Nakamura, M.; Parkhurst, S.M. Wrangling Actin Assemblies: Actin Ring Dynamics during Cell Wound Repair. Cells 2022, 11, 2777. https://doi.org/10.3390/cells11182777
Hui J, Stjepić V, Nakamura M, Parkhurst SM. Wrangling Actin Assemblies: Actin Ring Dynamics during Cell Wound Repair. Cells. 2022; 11(18):2777. https://doi.org/10.3390/cells11182777
Chicago/Turabian StyleHui, Justin, Viktor Stjepić, Mitsutoshi Nakamura, and Susan M. Parkhurst. 2022. "Wrangling Actin Assemblies: Actin Ring Dynamics during Cell Wound Repair" Cells 11, no. 18: 2777. https://doi.org/10.3390/cells11182777
APA StyleHui, J., Stjepić, V., Nakamura, M., & Parkhurst, S. M. (2022). Wrangling Actin Assemblies: Actin Ring Dynamics during Cell Wound Repair. Cells, 11(18), 2777. https://doi.org/10.3390/cells11182777