
Structure of an Intranucleosomal DNA Loop That Senses DNA Damage during Transcription

 , and
, and
Abstract
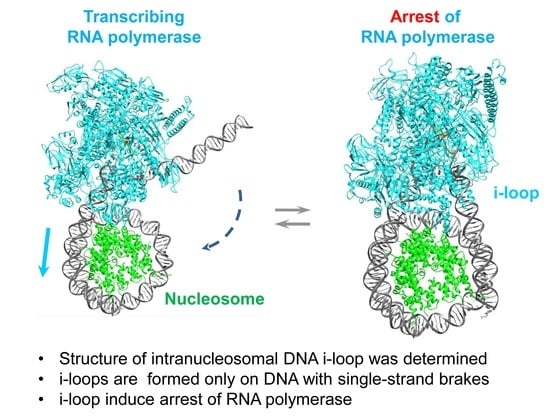
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. DNA Templates
2.2. Nucleosome Assembly
2.3. Protein Purifications
2.4. Transcription
2.5. DNaseI and Hydroxyl Radical Footprinting
2.6. Electron Microscopy and Image Processing
2.7. Modeling the RNAP–Nucleosome Complex EC+20
3. Results
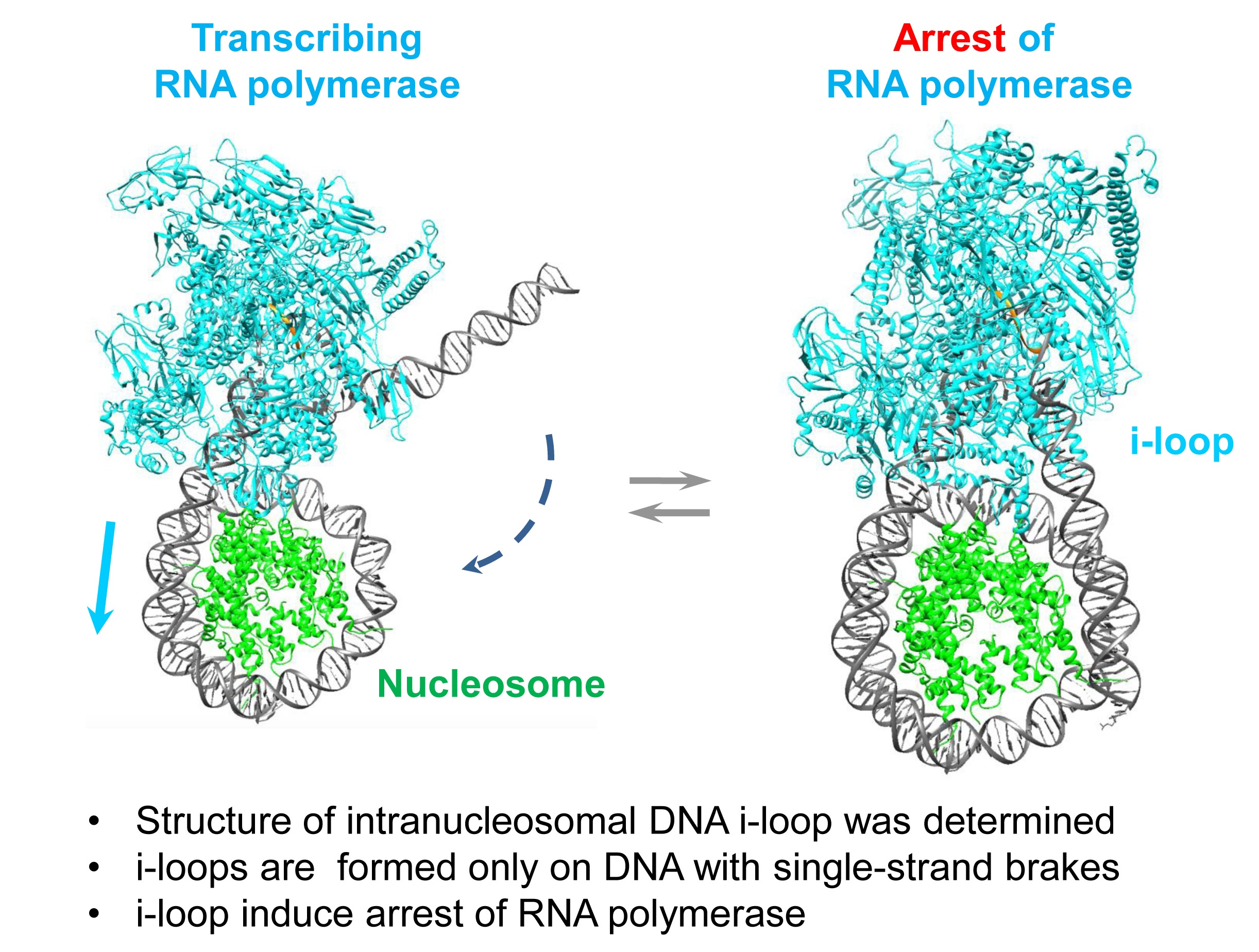
3.1. Experimental Strategy
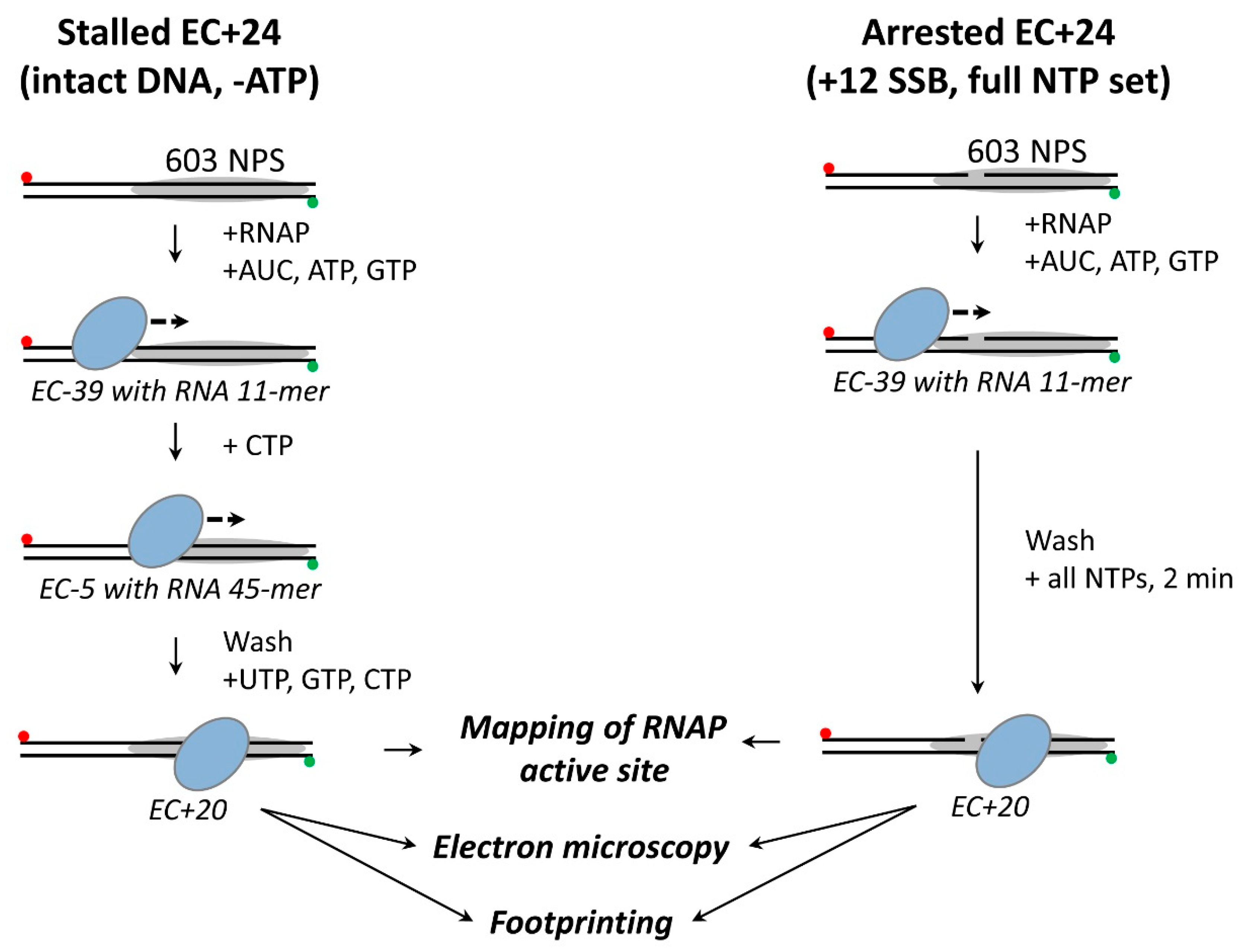
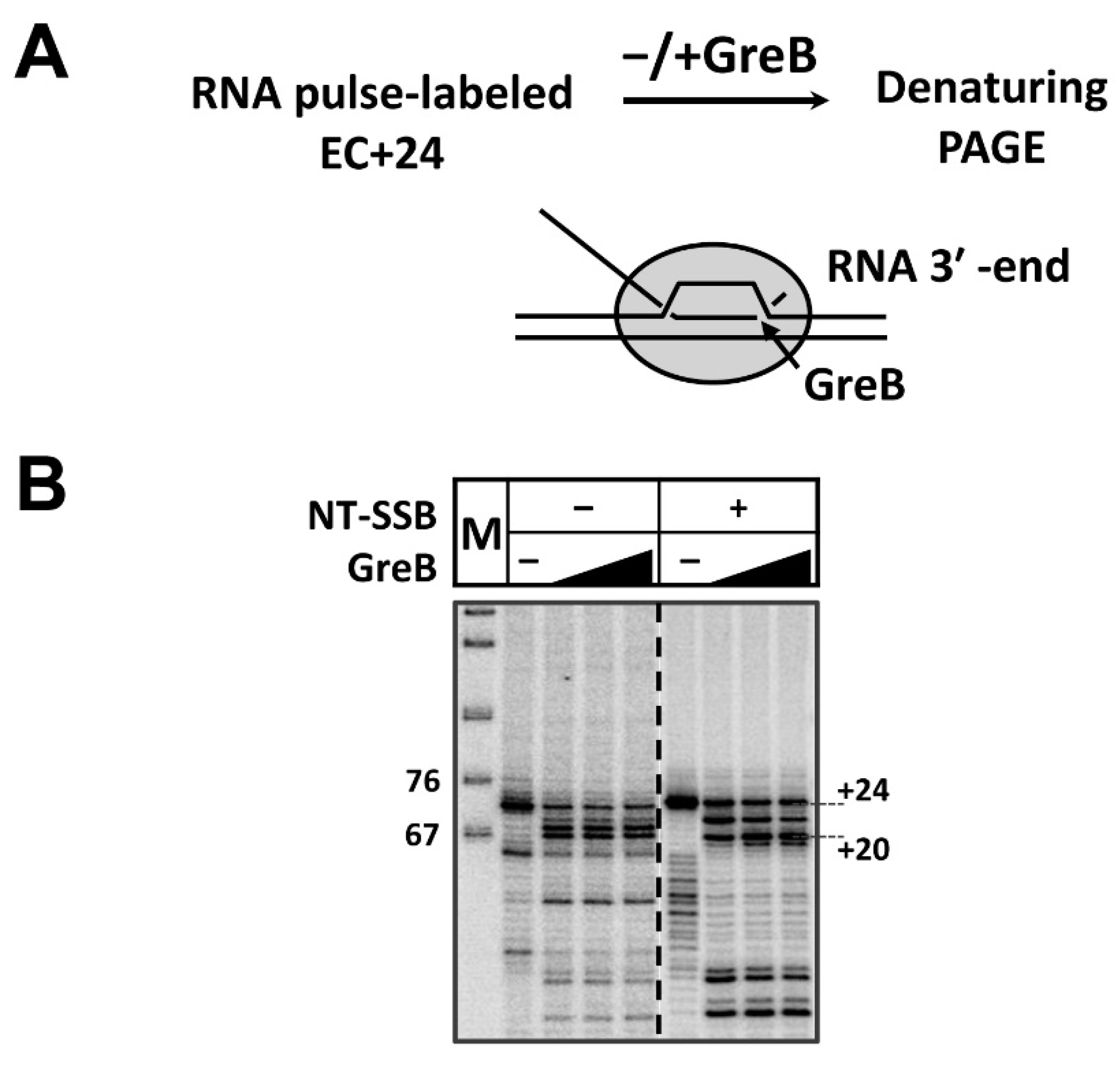
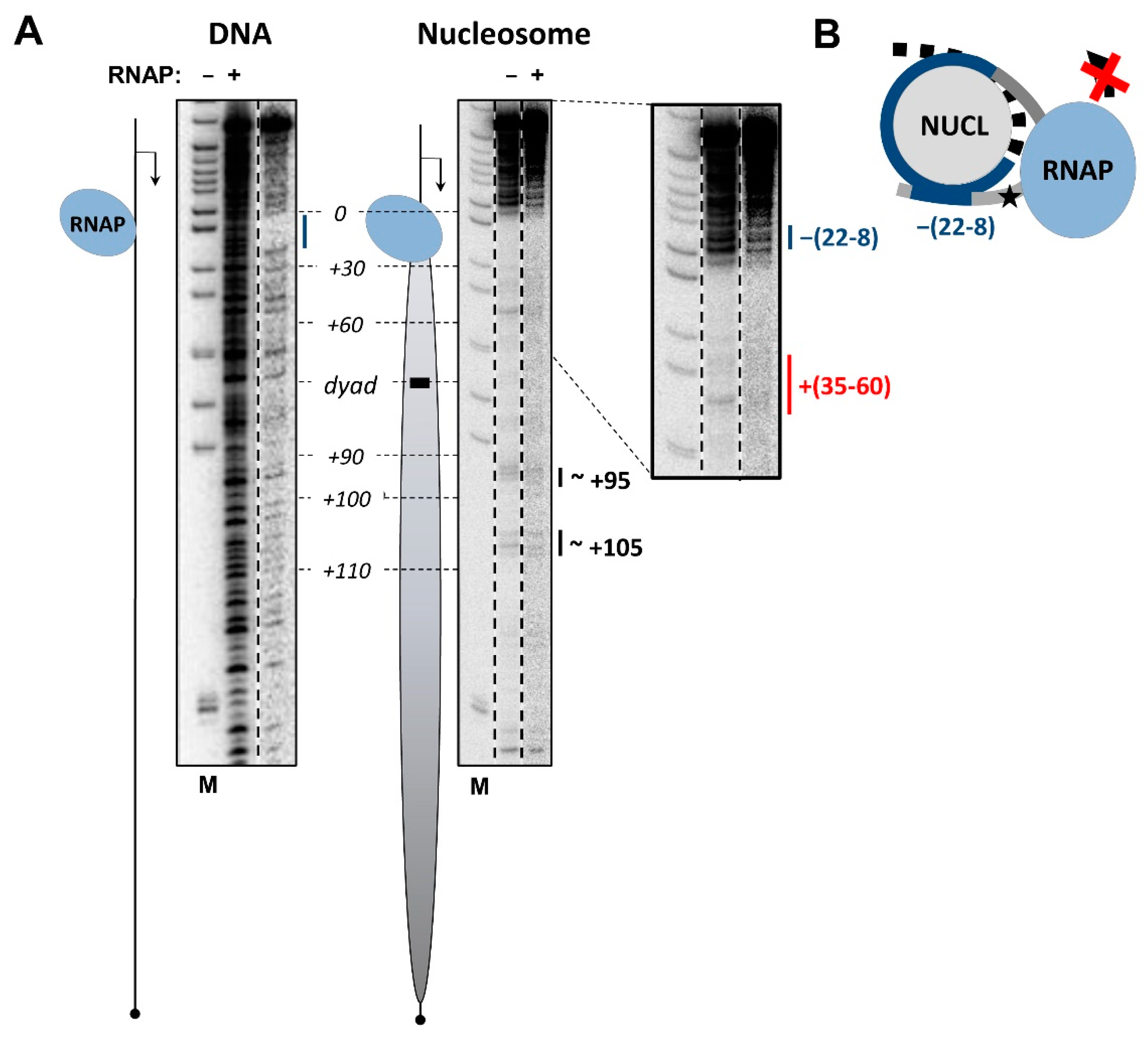
3.2. RNAP Backtracks to Positions +(20–22) after Stalling/Arrest at the Position +24
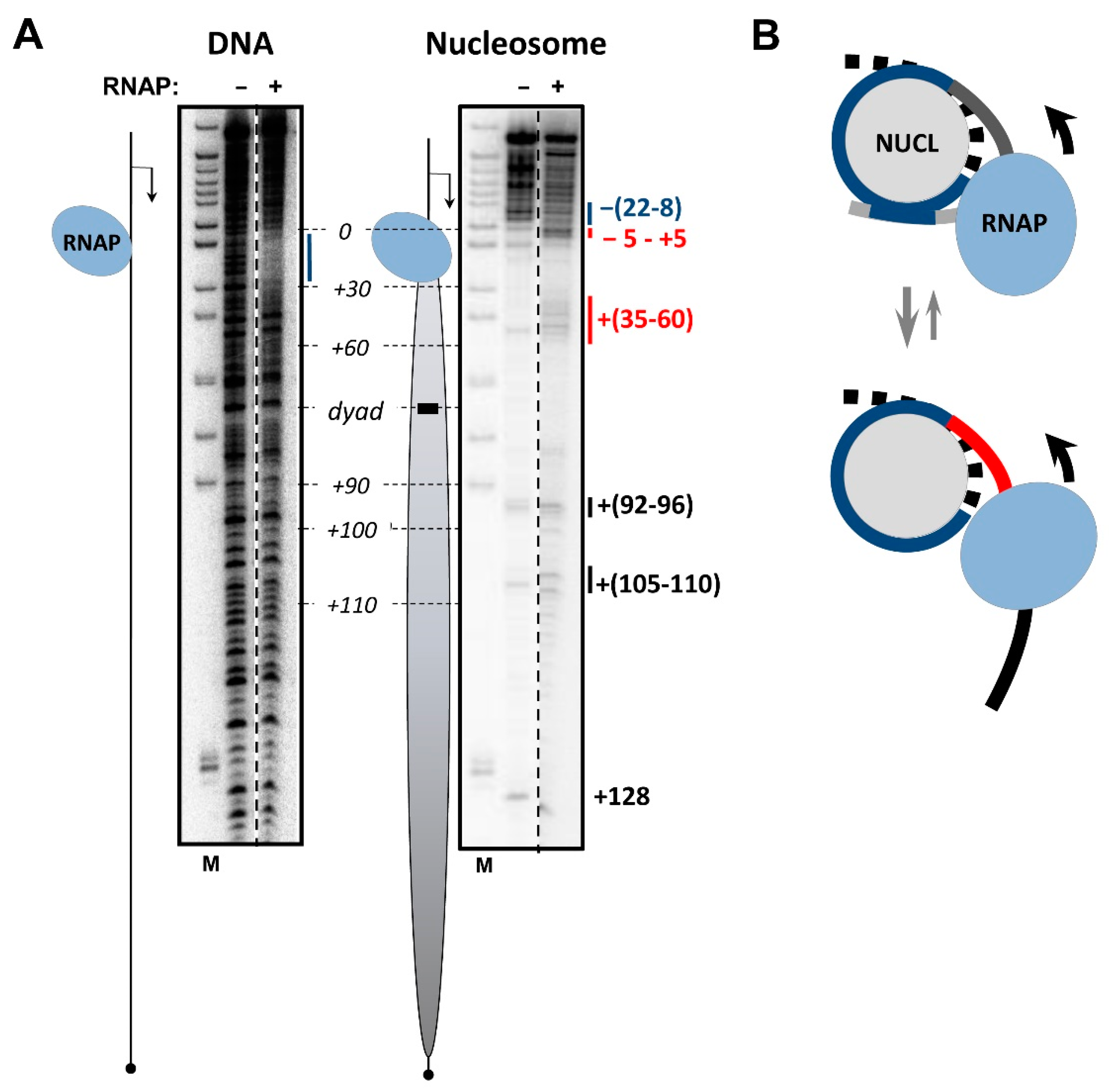
3.3. The i-Loop Is Efficiently Formed during Transcription through the Nucleosome Containing a Nick: Analysis by Footprinting
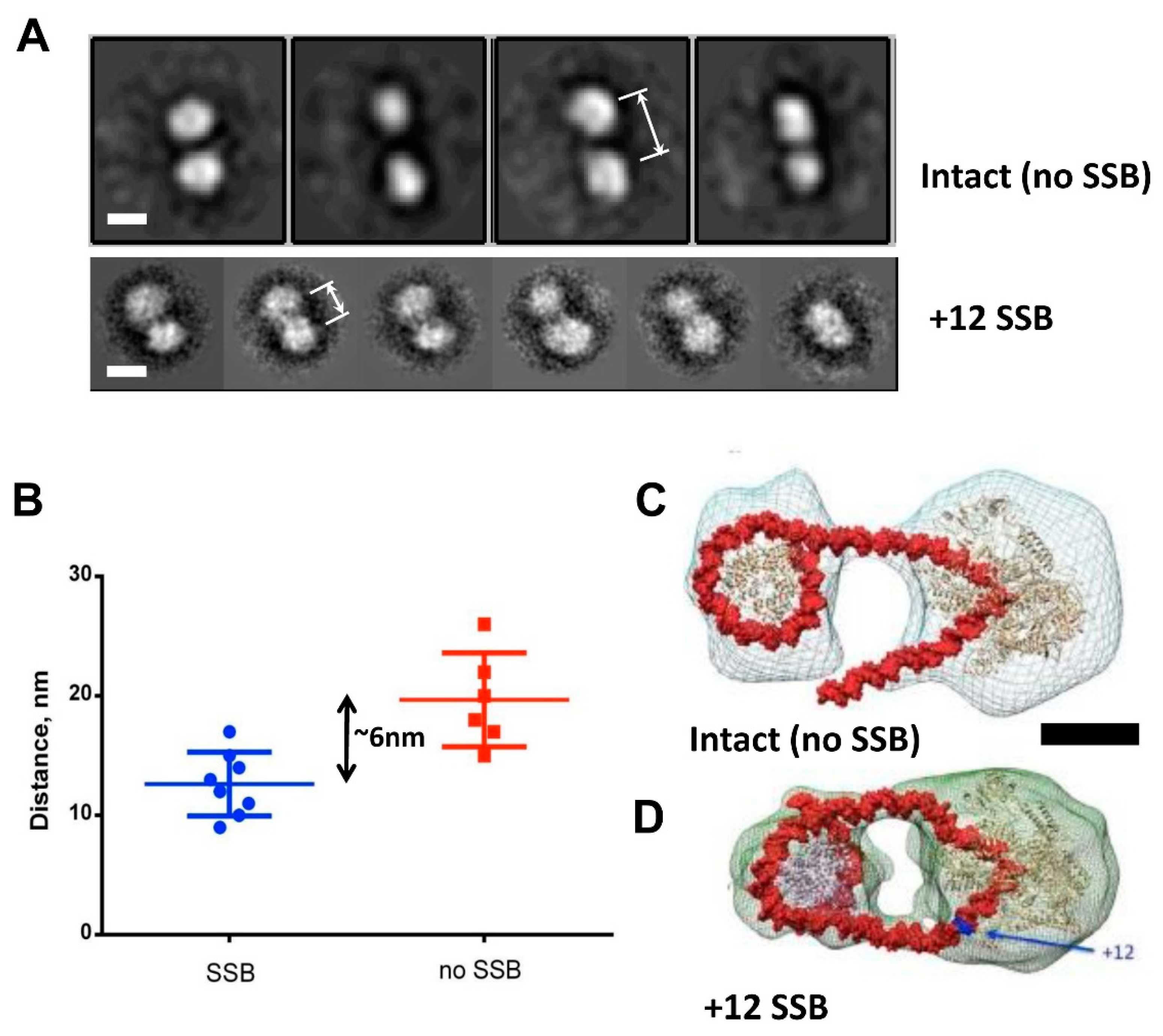
3.4. DNA Is Partially Uncoiled from the Nucleosome in Intact EC+20: Analysis by Electron Microscopy
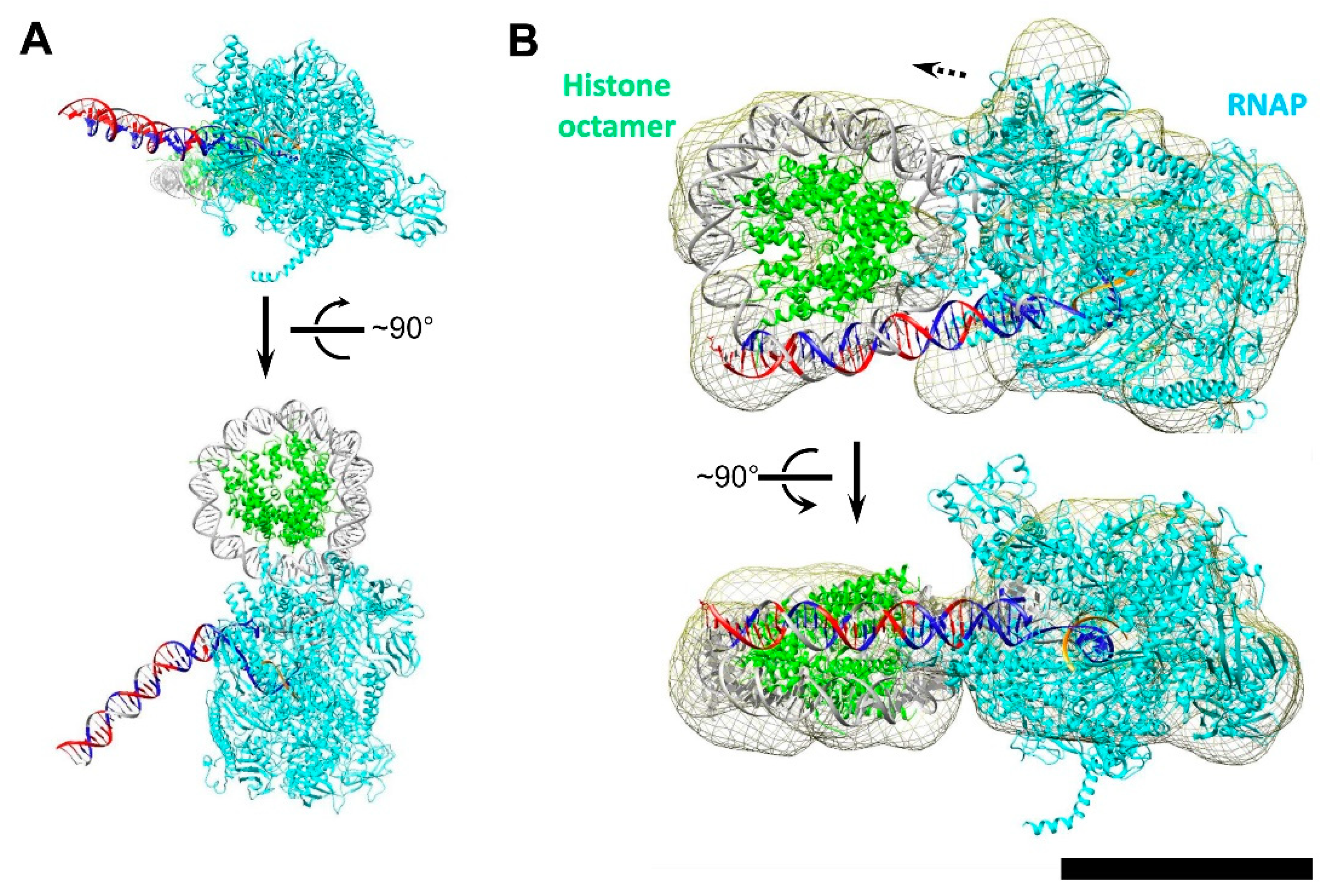
3.5. Modeling the i-Loop
3.6. Mechanism of i-Loop Formation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tubbs, A.; Nussenzweig, A. Endogenous DNA damage as a source of genomic instability in cancer. Cell 2017, 168, 644–656. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, T. Instability and decay of the primary structure of DNA. Nature 1993, 362, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Pogozelski, W.K.; Tullius, T.D. Oxidative strand scission of nucleic acids: Routes initiated by hydrogen abstraction from the sugar moiety. Chem. Rev. 1998, 98, 1089–1108. [Google Scholar] [CrossRef]
- Caldecott, K.W. DNA single-strand break repair and human genetic disease. Trends Cell Biol. 2022, 32, 733–745. [Google Scholar] [CrossRef]
- Caldecott, K.W.; Ward, M.E.; Nussenzweig, A. The threat of programmed DNA damage to neuronal genome integrity and plasticity. Nat. Genet. 2022, 54, 115–120. [Google Scholar] [CrossRef] [PubMed]
- McKinnon, P.J.; Caldecott, K.W. DNA strand break repair and human genetic disease. Annu. Rev. Genom. Hum. Genet. 2007, 8, 37–55. [Google Scholar] [CrossRef] [PubMed]
- McKinnon, P.J. DNA repair deficiency and neurological disease. Nat. Rev. Neurosci. 2009, 10, 100–112. [Google Scholar] [CrossRef]
- Nassour, J.; Martien, S.; Martin, N.; Deruy, E.; Tomellini, E.; Malaquin, N.; Bouali, F.; Sabatier, L.; Wernert, N.; Pinte, S.; et al. Defective DNA single-strand break repair is responsible for senescence and neoplastic escape of epithelial cells. Nat. Commun. 2016, 7, 10399. [Google Scholar] [CrossRef]
- Collin, G.; Huna, A.; Warnier, M.; Flaman, J.M.; Bernard, D. Transcriptional repression of DNA repair genes is a hallmark and a cause of cellular senescence. Cell Death Dis. 2018, 9, 259. [Google Scholar] [CrossRef]
- Wu, W.; Hill, S.E.; Nathan, W.J.; Paiano, J.; Callen, E.; Wang, D.; Shinoda, K.; van Wietmarschen, N.; Colon-Mercado, J.M.; Zong, D.; et al. Neuronal enhancers are hotspots for DNA single-strand break repair. Nature 2021, 593, 440–444. [Google Scholar] [CrossRef]
- Dileep, V.; Tsai, L.H. Neuronal enhancers get a break. Neuron 2021, 109, 1766–1768. [Google Scholar] [CrossRef] [PubMed]
- Reid, D.A.; Reed, P.J.; Schlachetzki, J.C.M.; Nitulescu, II; Chou, G.; Tsui, E.C.; Jones, J.R.; Chandran, S.; Lu, A.T.; McClain, C.A.; et al. Incorporation of a nucleoside analog maps genome repair sites in postmitotic human neurons. Science 2021, 372, 91–94. [Google Scholar] [CrossRef]
- Reynolds, J.J.; Stewart, G.S. A nervous predisposition to unrepaired DNA double strand breaks. DNA Repair 2013, 12, 588–599. [Google Scholar] [CrossRef]
- Welch, G.; Tsai, L.H. Mechanisms of DNA damage-mediated neurotoxicity in neurodegenerative disease. EMBO Rep. 2022, 23, e54217. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Raj, J.; Li, J.; Ha, A.; Hossain, M.A.; Richardson, C.; Mukherjee, P.; Yan, S. APE1 senses DNA single-strand breaks for repair and signaling. Nucleic Acids Res. 2020, 48, 1925–1940. [Google Scholar] [CrossRef] [PubMed]
- Caldecott, K.W. Single-strand break repair and genetic disease. Nat. Rev. Genet. 2008, 9, 619–631. [Google Scholar] [CrossRef] [PubMed]
- Weinfeld, M.; Chaudhry, M.A.; D’Amours, D.; Pelletier, J.D.; Poirier, G.G.; Povirk, L.F.; Lees-Miller, S.P. Interaction of DNA-dependent protein kinase and poly(ADP-ribose) polymerase with radiation-induced DNA strand breaks. Radiat. Res. 1997, 148, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Le Cam, E.; Fack, F.; Ménissier-de Murcia, J.; Cognet, J.A.; Barbin, A.; Sarantoglou, V.; Révet, B.; Delain, E.; de Murcia, G. Conformational analysis of a 139 base-pair DNA fragment containing a single-stranded break and its interaction with human poly(ADP-ribose) polymerase. J. Mol. Biol. 1994, 235, 1062–1071. [Google Scholar] [CrossRef]
- Caldecott, K.W. DNA single-strand break repair. Exp. Cell Res. 2014, 329, 2–8. [Google Scholar] [CrossRef]
- Caldecott, K.W. Protein ADP-ribosylation and the cellular response to DNA strand breaks. DNA Repair 2014, 19, 108–113. [Google Scholar] [CrossRef]
- Iyama, T.; Wilson, D.M. DNA repair mechanisms in dividing and non-dividing cells. DNA Repair 2013, 12, 620–636. [Google Scholar] [CrossRef] [PubMed]
- Davis, L.; Maizels, N. Homology-directed repair of DNA nicks via pathways distinct from canonical double-strand break repair. Proc. Natl. Acad. Sci. USA 2014, 111, E924-932. [Google Scholar] [CrossRef] [PubMed]
- Biechele-Speziale, D.J.; Sutton, T.B.; Delaney, S. Obstacles and opportunities for base excision repair in chromatin. DNA Repair 2022, 116, 103345. [Google Scholar] [CrossRef]
- Bennett, L.; Madders, E.; Parsons, J.L. HECTD1 promotes base excision repair in nucleosomes through chromatin remodelling. Nucleic Acids Res. 2020, 48, 1301–1313. [Google Scholar] [CrossRef]
- Ukraintsev, A.A.; Belousova, E.A.; Kutuzov, M.M.; Lavrik, O.I. Study of Interaction of the PARP Family DNA-Dependent Proteins with Nucleosomes Containing DNA Intermediates of the Initial Stages of BER Process. Biochemistry 2022, 87, 331–345. [Google Scholar] [CrossRef] [PubMed]
- Kutuzov, M.M.; Belousova, E.A.; Kurgina, T.A.; Ukraintsev, A.A.; Vasil’eva, I.A.; Khodyreva, S.N.; Lavrik, O.I. The contribution of PARP1, PARP2 and poly(ADP-ribosyl)ation to base excision repair in the nucleosomal context. Sci. Rep. 2021, 11, 4849. [Google Scholar] [CrossRef]
- Kathe, S.D.; Shen, G.-P.; Wallace, S.S. Single-stranded breaks in DNA but not oxidative DNA base damages block transcriptional elongation by RNA polymerase II in HeLa cell nuclear extracts. J. Biol. Chem. 2004, 279, 18511–18520. [Google Scholar] [CrossRef]
- Neil, A.J.; Belotserkovskii, B.P.; Hanawalt, P.C. Transcription blockage by bulky end termini at single-strand breaks in the DNA template: Differential effects of 5′ and 3′ adducts. Biochemistry 2012, 51, 8964–8970. [Google Scholar] [CrossRef]
- Li, S.; Smerdon, M.J. Dissecting transcription-coupled and global genomic repair in the chromatin of yeast GAL1-10 genes. J. Biol. Chem. 2004, 279, 14418–14426. [Google Scholar] [CrossRef]
- Belotserkovskii, B.P.; Neil, A.J.; Saleh, S.S.; Shin, J.H.S.; Mirkin, S.M.; Hanawalt, P.C. Transcription blockage by homopurine DNA sequences: Role of sequence composition and single-strand breaks. Nucleic Acids Res. 2013, 41, 1817–1828. [Google Scholar] [CrossRef]
- Bielas, J.H. Non-transcribed strand repair revealed in quiescent cells. Mutagenesis 2006, 21, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Smolle, M.; Workman, J.L.; Venkatesh, S. reSETting chromatin during transcription elongation. Epigenetics 2013, 8, 10–15. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Das, C.; Tyler, J.K. Histone exchange and histone modifications during transcription and aging. Biochim. Biophys. Acta 2013, 1819, 332–342. [Google Scholar] [CrossRef] [PubMed]
- Zentner, G.E.; Henikoff, S. Regulation of nucleosome dynamics by histone modifications. Nat. Struct. Mol. Biol 2013, 20, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Hartzog, G.A.; Speer, J.L.; Lindstrom, D.L. Transcript elongation on a nucleoprotein template. Biochim. Biophys. Acta 2002, 1577, 276–286. [Google Scholar] [CrossRef]
- Kristjuhan, A.; Svejstrup, J.Q. Evidence for distinct mechanisms facilitating transcript elongation through chromatin in vivo. EMBO J. 2004, 23, 4243–4252. [Google Scholar] [CrossRef] [PubMed]
- Schwabish, M.A.; Struhl, K. Evidence for eviction and rapid deposition of histones upon transcriptional elongation by RNA polymerase II. Mol. Cell. Biol. 2004, 24, 10111–10117. [Google Scholar] [CrossRef]
- Lee, C.-K.; Shibata, Y.; Rao, B.; Strahl, B.D.; Lieb, J.D. Evidence for nucleosome depletion at active regulatory regions genome-wide. Nat. Genet. 2004, 36, 900–905. [Google Scholar] [CrossRef]
- Izban, M.G.; Luse, D.S. Factor-stimulated RNA polymerase II transcribes at physiological elongation rates on naked DNA but very poorly on chromatin templates. J. Biol. Chem. 1992, 267, 13647–13655. [Google Scholar] [CrossRef]
- Kireeva, M.L.; Walter, W.; Tchernajenko, V.; Bondarenko, V.; Kashlev, M.; Studitsky, V.M. Nucleosome remodeling induced by RNA polymerase II: Loss of the H2A/H2B dimer during transcription. Mol. Cell 2002, 9, 541–552. [Google Scholar] [CrossRef]
- Churchman, L.S.; Weissman, J.S. Nascent transcript sequencing visualizes transcription at nucleotide resolution. Nature 2011, 469, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Weber, C.M.; Ramachandran, S.; Henikoff, S. Nucleosomes are context-specific, H2A.Z-modulated barriers to RNA polymerase. Mol. Cell 2014, 53, 819–830. [Google Scholar] [CrossRef] [PubMed]
- Cole, H.A.; Ocampo, J.; Iben, J.R.; Chereji, R.V.; Clark, D.J. Heavy transcription of yeast genes correlates with differential loss of histone H2B relative to H4 and queued RNA polymerases. Nucleic Acids Res. 2014, 42, 12512–12522. [Google Scholar] [CrossRef] [PubMed]
- Kulaeva, O.I.; Gaykalova, D.A.; Pestov, N.A.; Golovastov, V.V.; Vassylyev, D.G.; Artsimovitch, I.; Studitsky, V.M. Mechanism of chromatin remodeling and recovery during passage of RNA polymerase II. Nat. Struct. Mol. Biol. 2009, 16, 1272–1278. [Google Scholar] [CrossRef]
- Pestov, N.A.; Gerasimova, N.S.; Kulaeva, O.I.; Studitsky, V.M. Structure of transcribed chromatin is a sensor of DNA damage. Sci. Adv. 2015, 1, e1500021. [Google Scholar] [CrossRef]
- Gerasimova, N.S.; Pestov, N.A.; Kulaeva, O.I.; Clark, D.J.; Studitsky, V.M. Transcription-induced DNA supercoiling: New roles of intranucleosomal DNA loops in DNA repair and transcription. Transcription 2016, 7, 91–95. [Google Scholar] [CrossRef][Green Version]
- Lowary, P.T.; Widom, J. New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J. Mol. Biol. 1998, 276, 19–42. [Google Scholar] [CrossRef]
- Walter, W.; Kireeva, M.L.; Studitsky, V.M.; Kashlev, M. Bacterial polymerase and yeast polymerase II use similar mechanisms for transcription through nucleosomes. J. Biol. Chem. 2003, 278, 36148–36156. [Google Scholar] [CrossRef]
- Kireeva, M.L.; Komissarova, N.; Waugh, D.S.; Kashlev, M. The 8-nucleotide-long RNA:DNA hybrid is a primary stability determinant of the RNA polymerase II elongation complex. J. Biol. Chem. 2000, 275, 6530–6536. [Google Scholar] [CrossRef]
- Artsimovitch, I.; Svetlov, V.; Murakami, K.S.; Landick, R. Co-overexpression of Escherichia coli RNA polymerase subunits allows isolation and analysis of mutant enzymes lacking lineage-specific sequence insertions. J. Biol. Chem. 2003, 278, 12344–12355. [Google Scholar] [CrossRef]
- Vassylyeva, M.N.; Svetlov, V.; Dearborn, A.D.; Klyuyev, S.; Artsimovitch, I.; Vassylyev, D.G. The carboxy-terminal coiled-coil of the RNA polymerase beta’-subunit is the main binding site for Gre factors. EMBO Rep. 2007, 8, 1038–1043. [Google Scholar] [CrossRef]
- Dixon, W.J.; Hayes, J.J.; Levin, J.R.; Weidner, M.F.; Dombroski, B.A.; Tullius, T.D. Hydroxyl radical footprinting. Methods Enzymol. 1991, 208, 380–413. [Google Scholar] [CrossRef] [PubMed]
- Gerasimova, N.S.; Studitsky, V.M. Hydroxyl radical footprinting of fluorescently labeled DNA. Mosc. Univ. Biol. Sci. Bull. 2016, 71, 93–96. [Google Scholar] [CrossRef]
- Walter, W.; Kireeva, M.L.; Tchernajenko, V.; Kashlev, M.; Studitsky, V.M. Assay of the fate of the nucleosome during transcription by RNA polymerase II. Meth. Enzymol. 2003, 371, 564–577. [Google Scholar] [CrossRef]
- Mastronarde, D.N. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 2005, 152, 36–51. [Google Scholar] [CrossRef] [PubMed]
- Kremer, J.R.; Mastronarde, D.N.; McIntosh, J.R. Computer visualization of three-dimensional image data using IMOD. J. Struct. Biol. 1996, 116, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Ludtke, S.J.; Baldwin, P.R.; Chiu, W. EMAN: Semiautomated software for high-resolution single-particle reconstructions. J. Struct. Biol. 1999, 128, 82–97. [Google Scholar] [CrossRef]
- Scheres, S.H.; Valle, M.; Nunez, R.; Sorzano, C.O.; Marabini, R.; Herman, G.T.; Carazo, J.M. Maximum-likelihood multi-reference refinement for electron microscopy images. J. Mol. Biol. 2005, 348, 139–149. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Barnes, C.O.; Calero, M.; Malik, I.; Graham, B.W.; Spahr, H.; Lin, G.; Cohen, A.E.; Brown, I.S.; Zhang, Q.; Pullara, F.; et al. Crystal structure of a transcribing RNA polymerase II complex reveals a complete transcription bubble. Mol. Cell 2015, 59, 258–269. [Google Scholar] [CrossRef]
- Vasudevan, D.; Chua, E.Y.D.; Davey, C.A. Crystal structures of nucleosome core particles containing the ‘601’ strong positioning sequence. J. Mol. Biol 2010, 403, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.; Wang, Y.; Steitz, T.A. The mechanism of E. coli RNA polymerase regulation by ppGpp is suggested by the structure of their complex. Mol. Cell 2013, 50, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.J.; Olson, W.K. 3DNA: A versatile, integrated software system for the analysis, rebuilding and visualization of three-dimensional nucleic-acid structures. Nat. Protoc. 2008, 3, 1213–1227. [Google Scholar] [CrossRef] [PubMed]
- Shaytan, A.K.; Xiao, H.; Armeev, G.A.; Wu, C.; Landsman, D.; Panchenko, A.R. Hydroxyl-radical footprinting combined with molecular modeling identifies unique features of DNA conformation and nucleosome positioning. Nucleic Acids Res. 2017, 45, 9229–9243. [Google Scholar] [CrossRef] [PubMed]
- Thåström, A.; Lowary, P.T.; Widlund, H.R.; Cao, H.; Kubista, M.; Widom, J. Sequence motifs and free energies of selected natural and non-natural nucleosome positioning DNA sequences. J. Mol. Biol. 1999, 288, 213–229. [Google Scholar] [CrossRef]
- Bondarenko, V.A.; Steele, L.M.; Ujvári, A.; Gaykalova, D.A.; Kulaeva, O.I.; Polikanov, Y.S.; Luse, D.S.; Studitsky, V.M. Nucleosomes can form a polar barrier to transcript elongation by RNA polymerase II. Mol. Cell 2006, 24, 469–479. [Google Scholar] [CrossRef]
- Chang, H.-W.; Kulaeva, O.I.; Shaytan, A.K.; Kibanov, M.; Kuznedelov, K.; Severinov, K.V.; Kirpichnikov, M.P.; Clark, D.J.; Studitsky, V.M. Analysis of the mechanism of nucleosome survival during transcription. Nucleic Acids Res. 2014, 42, 1619–1627. [Google Scholar] [CrossRef]
- Gaykalova, D.A.; Kulaeva, O.I.; Volokh, O.; Shaytan, A.K.; Hsieh, F.-K.; Kirpichnikov, M.P.; Sokolova, O.S.; Studitsky, V.M. Structural analysis of nucleosomal barrier to transcription. Proc. Natl. Acad. Sci. USA 2015, 112, E5787–E5795. [Google Scholar] [CrossRef]
- Bintu, L.; Kopaczynska, M.; Hodges, C.; Lubkowska, L.; Kashlev, M.; Bustamante, C. The elongation rate of RNA polymerase determines the fate of transcribed nucleosomes. Nat. Struct. Mol. Biol. 2011, 18, 1394–1399. [Google Scholar] [CrossRef]
- Chen, Z.; Gabizon, R.; Brown, A.I.; Lee, A.; Song, A.; Diaz-Celis, C.; Kaplan, C.D.; Koslover, E.F.; Yao, T.; Bustamante, C. High-resolution and high-accuracy topographic and transcriptional maps of the nucleosome barrier. eLife 2019, 8, e48281. [Google Scholar] [CrossRef]
- Rudnizky, S.; Khamis, H.; Ginosar, Y.; Goren, E.; Melamed, P.; Kaplan, A. Extended and dynamic linker histone-DNA Interactions control chromatosome compaction. Mol. Cell 2021, 81, 3410–3421.e4. [Google Scholar] [CrossRef] [PubMed]
- Khobta, A.; Lingg, T.; Schulz, I.; Warken, D.; Kitsera, N.; Epe, B. Mouse CSB protein is important for gene expression in the presence of a single-strand break in the non-transcribed DNA strand. DNA Repair 2010, 9, 985–993. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gerasimova, N.S.; Volokh, O.I.; Pestov, N.A.; Armeev, G.A.; Kirpichnikov, M.P.; Shaytan, A.K.; Sokolova, O.S.; Studitsky, V.M. Structure of an Intranucleosomal DNA Loop That Senses DNA Damage during Transcription. Cells 2022, 11, 2678. https://doi.org/10.3390/cells11172678
Gerasimova NS, Volokh OI, Pestov NA, Armeev GA, Kirpichnikov MP, Shaytan AK, Sokolova OS, Studitsky VM. Structure of an Intranucleosomal DNA Loop That Senses DNA Damage during Transcription. Cells. 2022; 11(17):2678. https://doi.org/10.3390/cells11172678
Chicago/Turabian StyleGerasimova, Nadezhda S., Olesya I. Volokh, Nikolay A. Pestov, Grigory A. Armeev, Mikhail P. Kirpichnikov, Alexey K. Shaytan, Olga S. Sokolova, and Vasily M. Studitsky. 2022. "Structure of an Intranucleosomal DNA Loop That Senses DNA Damage during Transcription" Cells 11, no. 17: 2678. https://doi.org/10.3390/cells11172678
APA StyleGerasimova, N. S., Volokh, O. I., Pestov, N. A., Armeev, G. A., Kirpichnikov, M. P., Shaytan, A. K., Sokolova, O. S., & Studitsky, V. M. (2022). Structure of an Intranucleosomal DNA Loop That Senses DNA Damage during Transcription. Cells, 11(17), 2678. https://doi.org/10.3390/cells11172678