
The Proteome Signatures of Fibroblasts from Patients with Severe, Intermediate and Mild Spinal Muscular Atrophy Show Limited Overlap

 ,
,  ,
,  ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
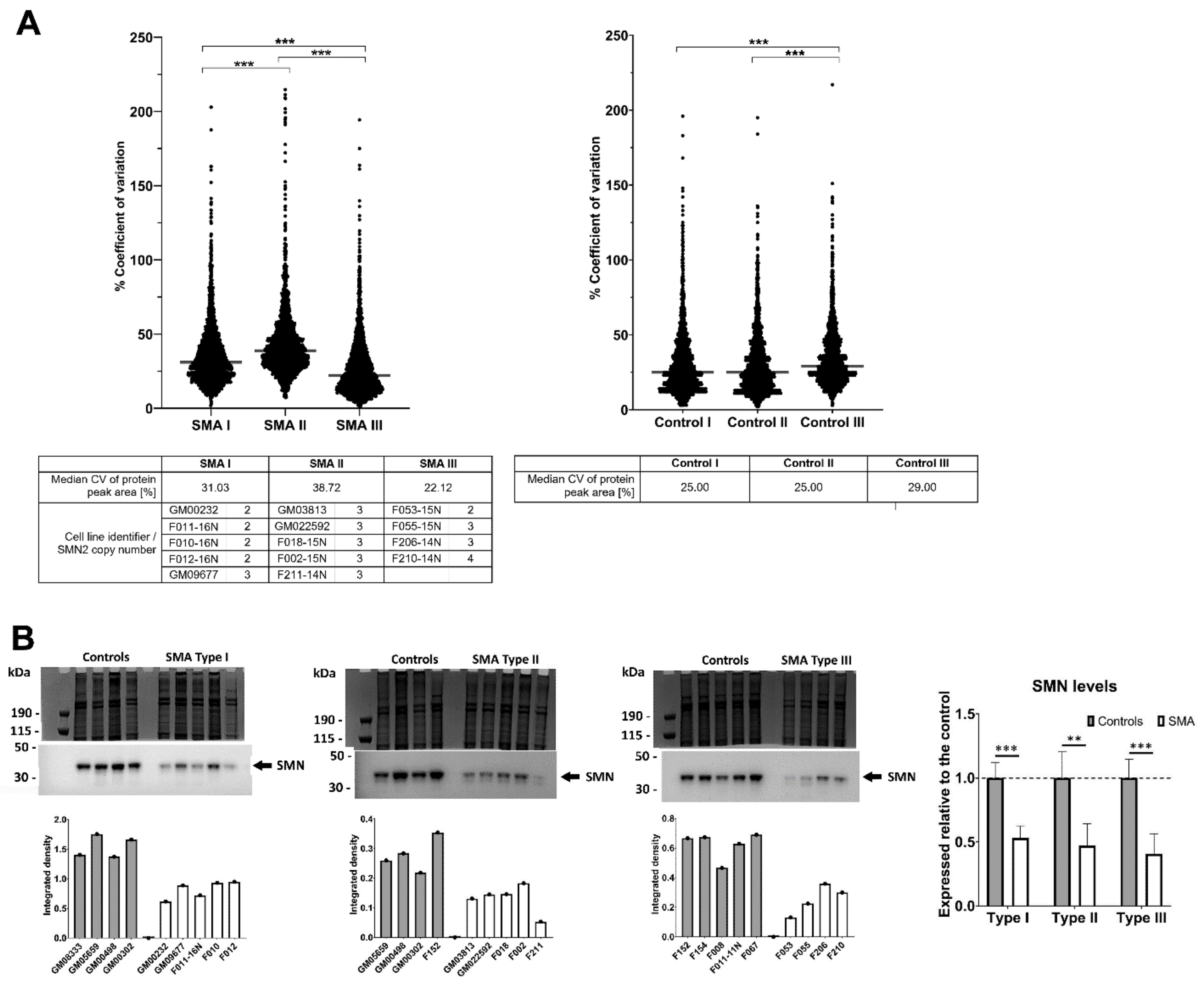
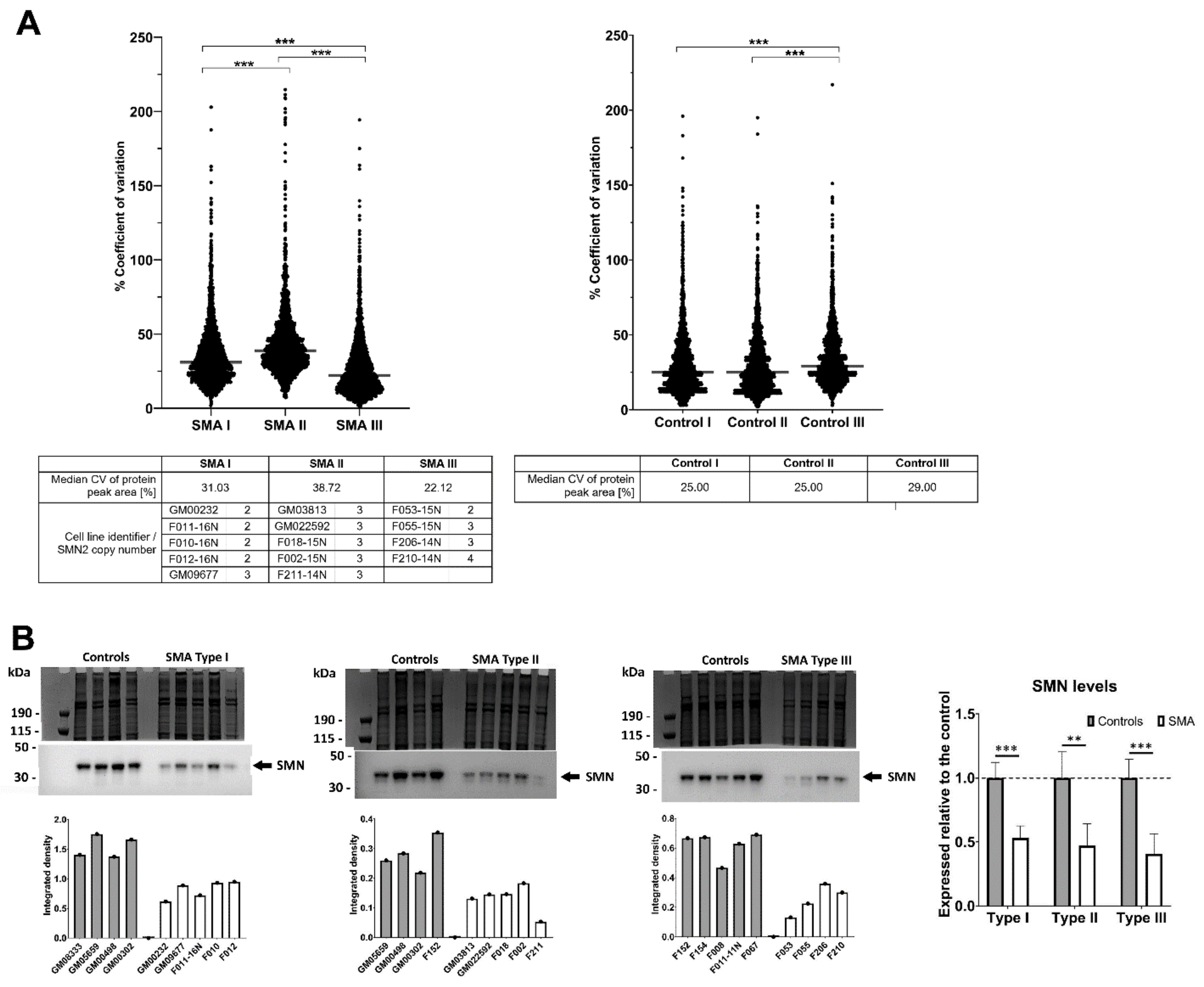
2.1. Fibroblasts from SMA II Patients Have Greater Variability in Their Proteomic Profiles Compared to Fibroblasts from Patients with SMA I and III
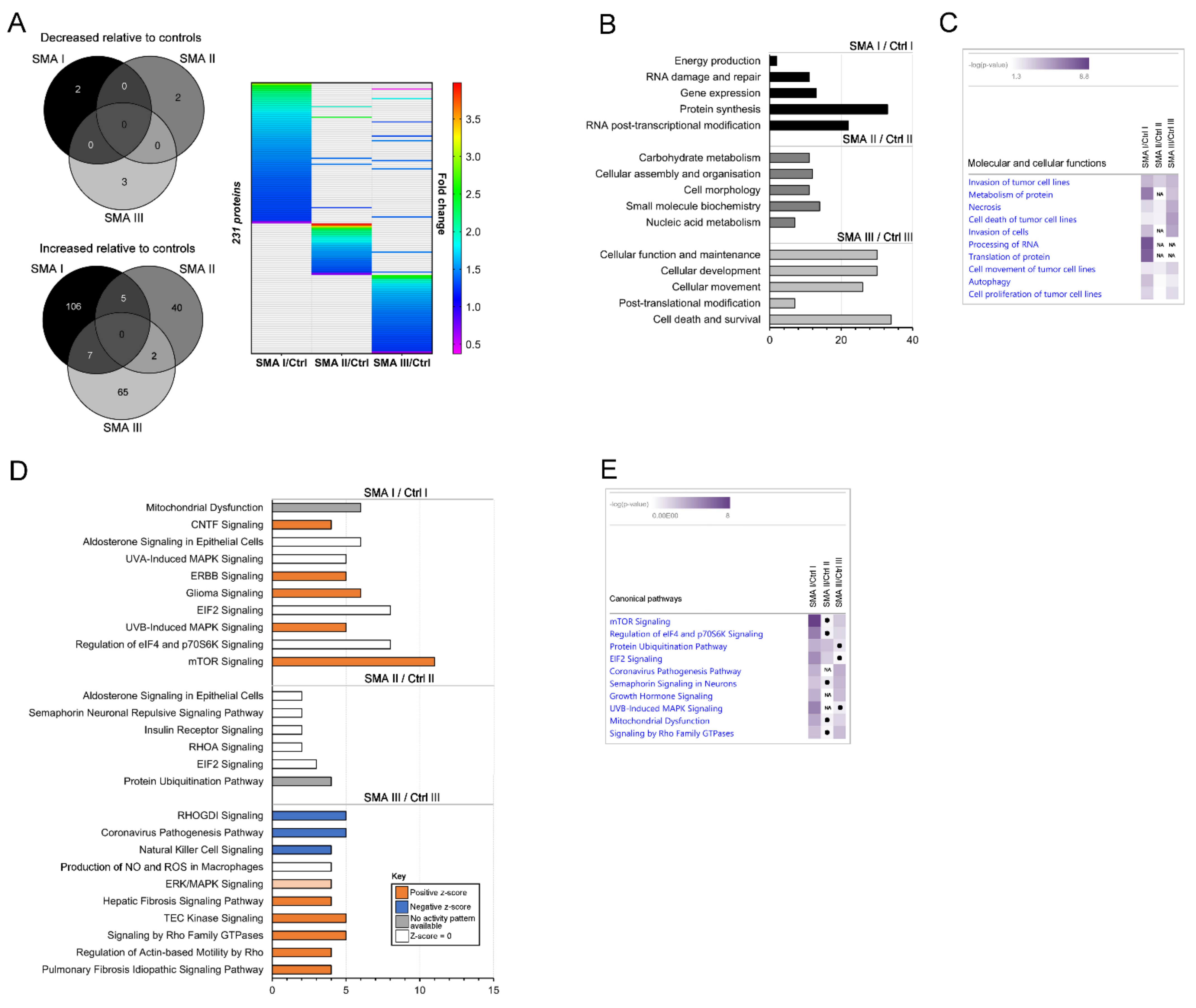
2.2. Differentially Expressed Proteomic Profiles of SMA I, II and III Patient Fibroblasts Show Little Overlap
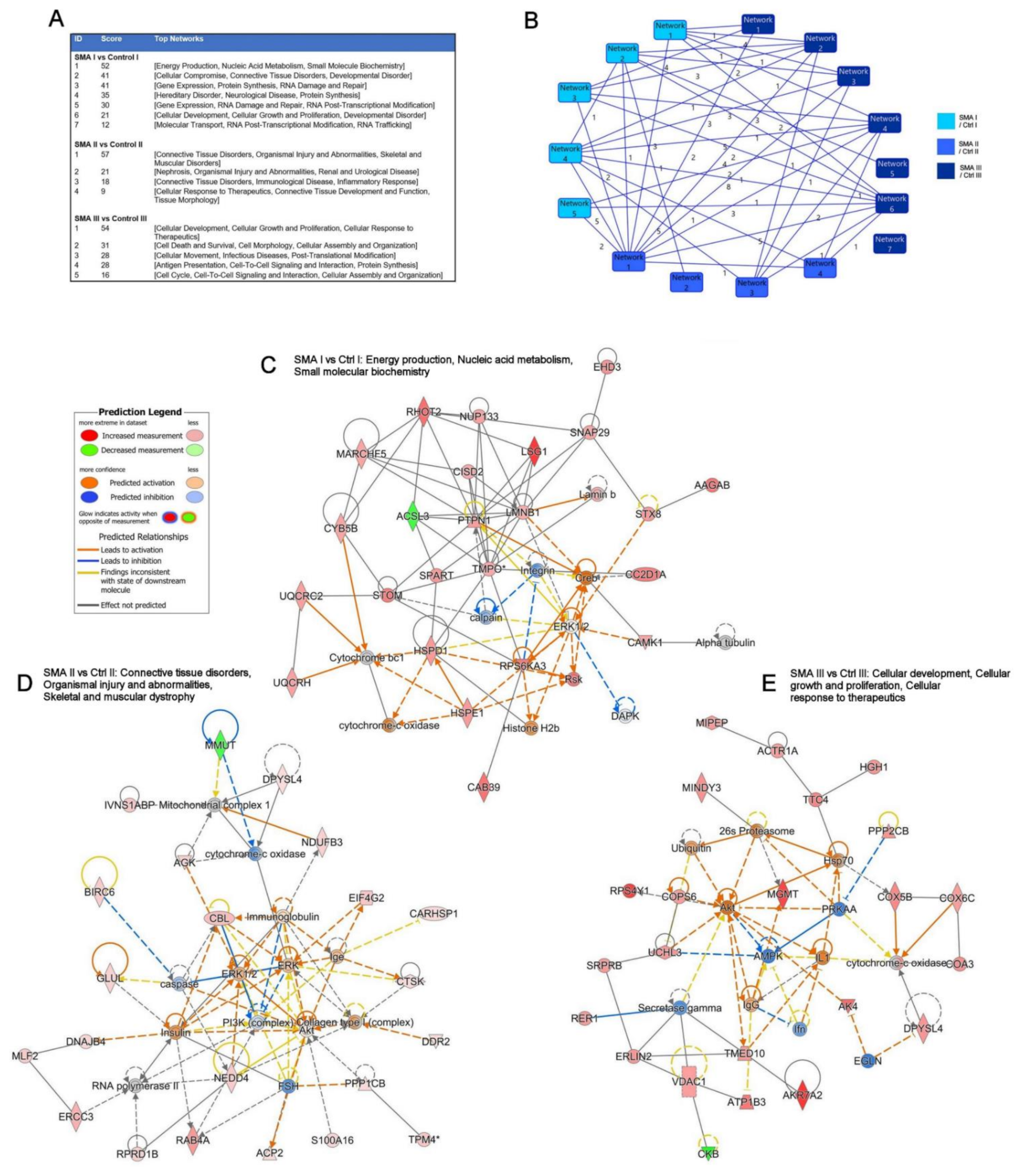
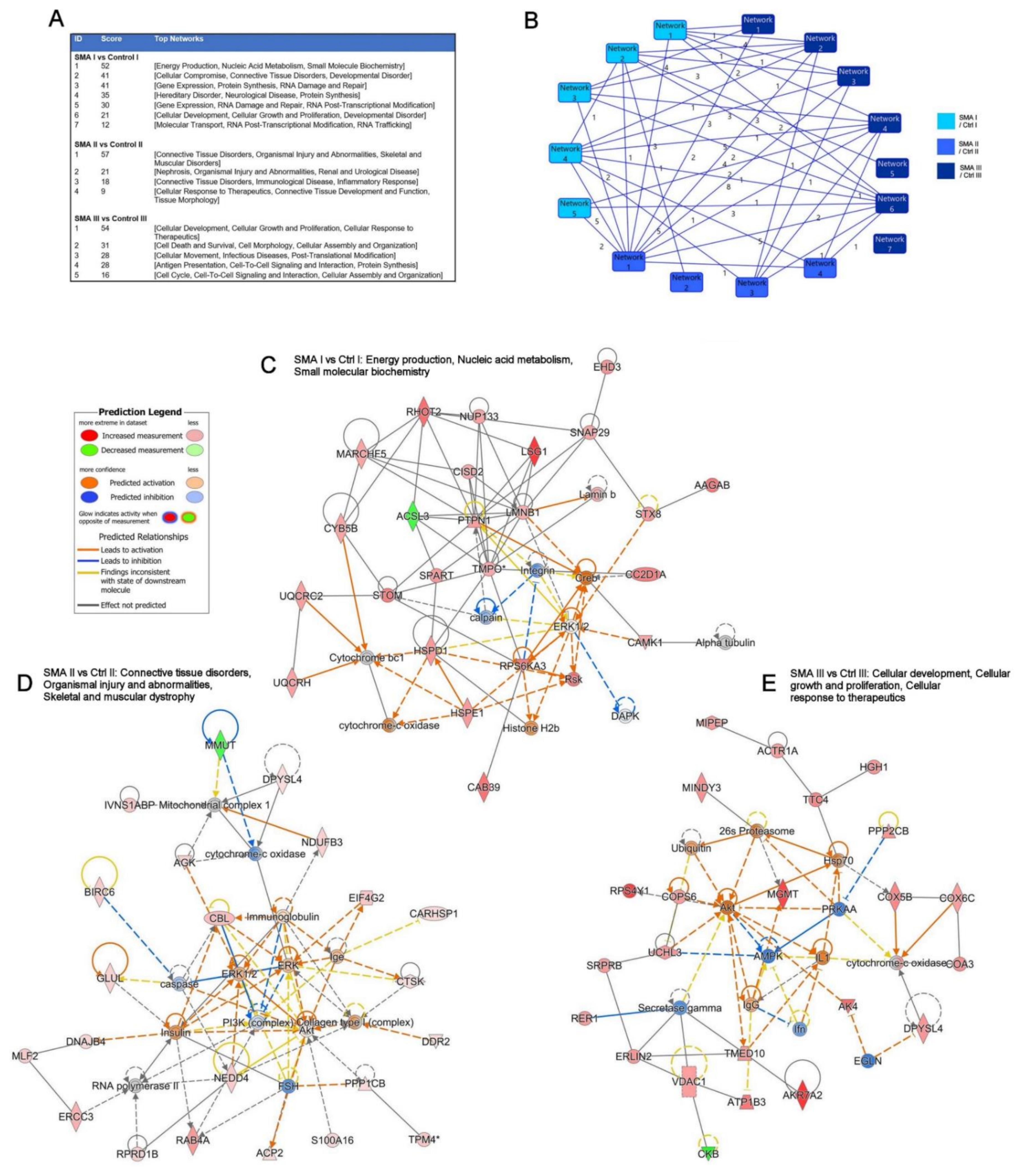
2.3. Relevance of SMA I-III Fibroblast Proteomic Data to Other Biological Datasets
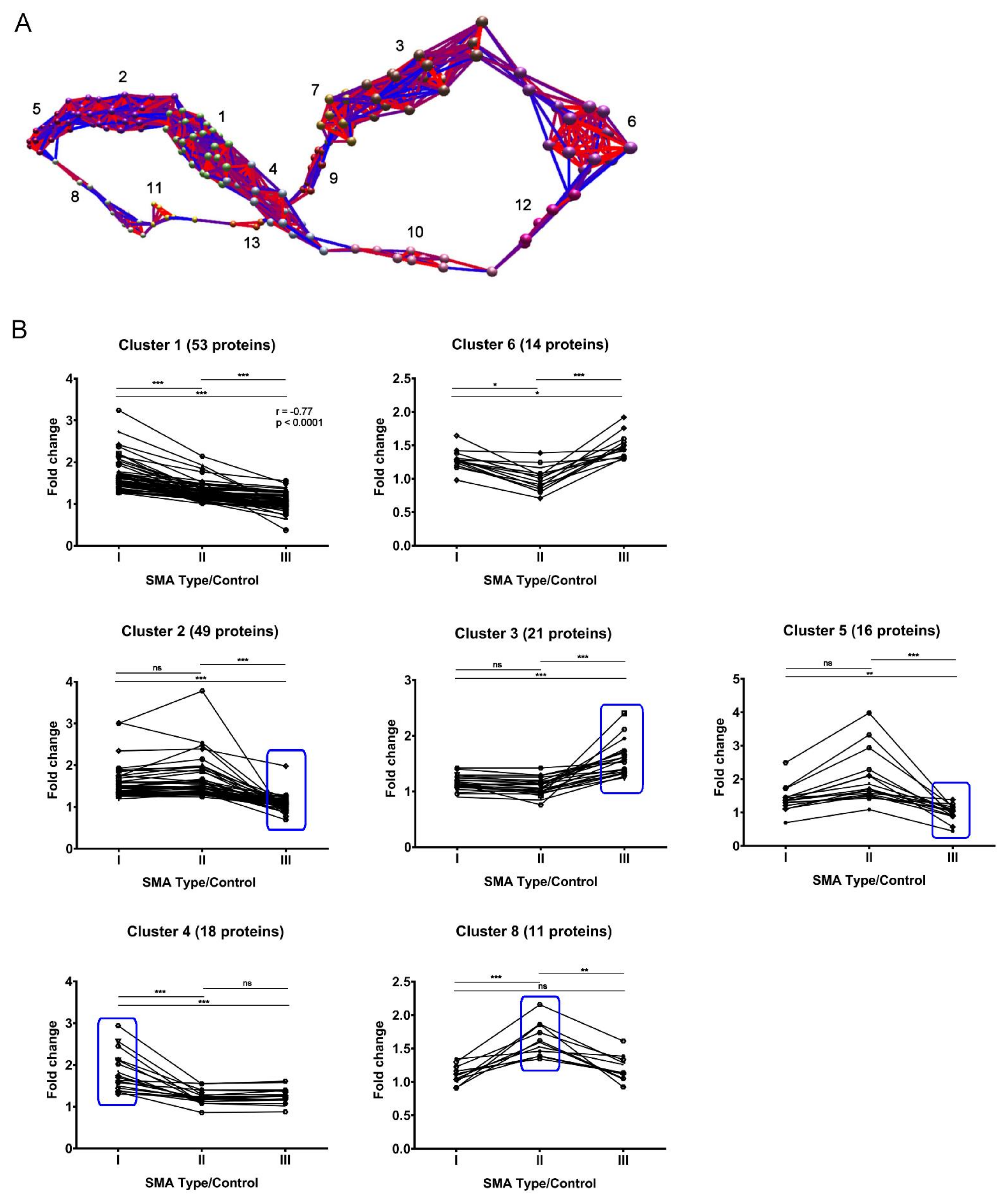
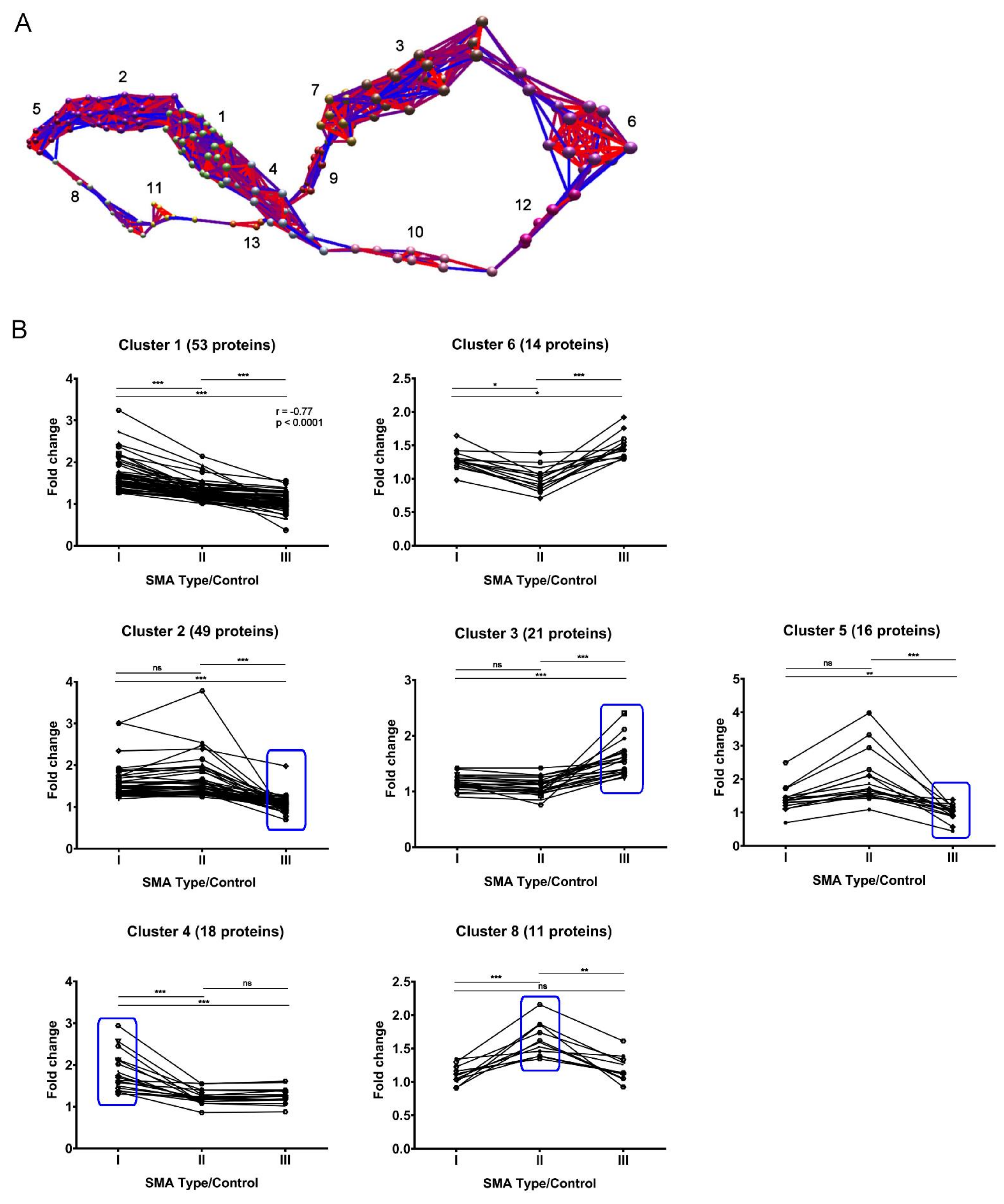
2.4. Expression Clustering Analysis Identifies Molecular Profiles That Discriminate and/or Correlate between Different SMA Severities
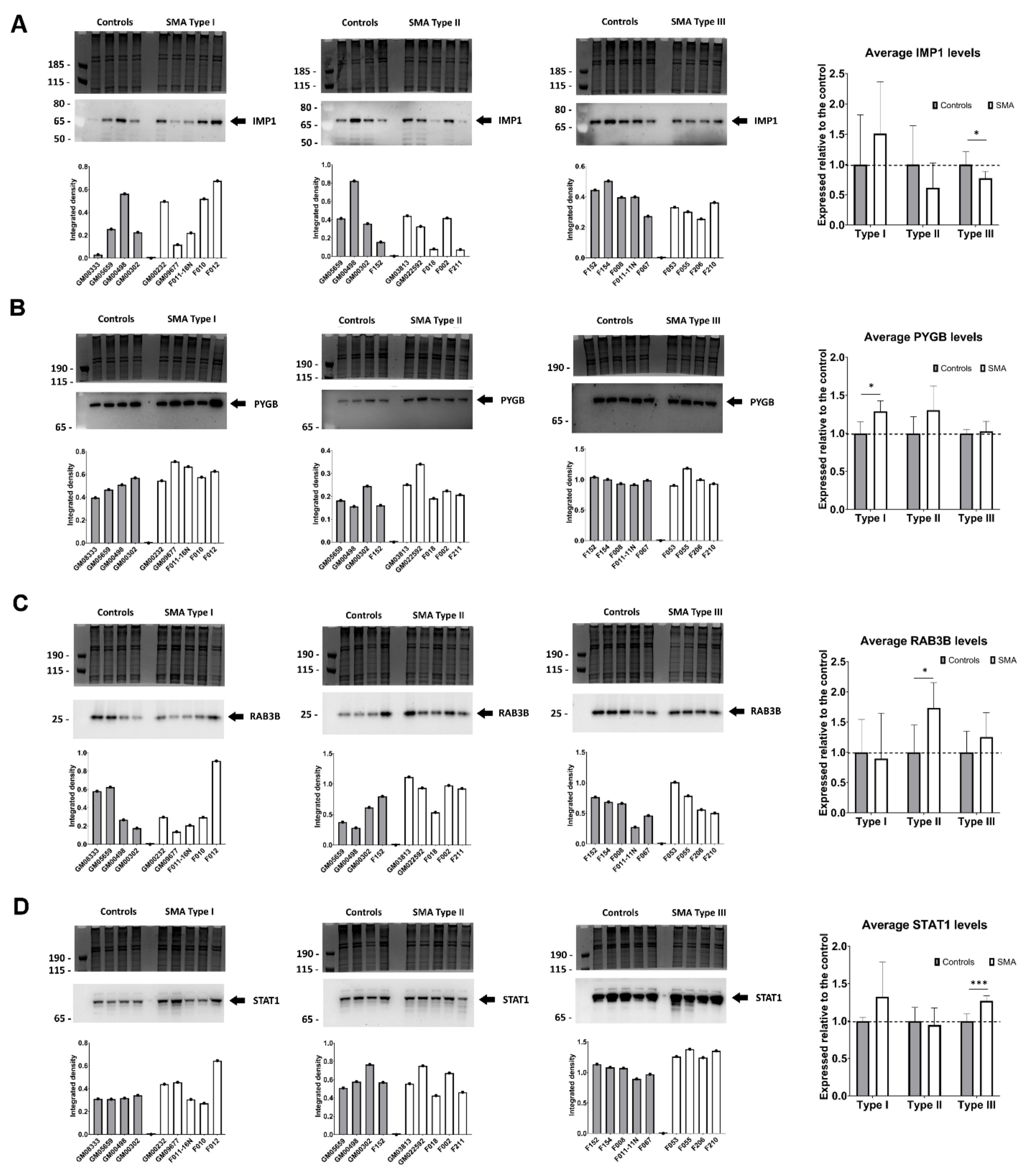
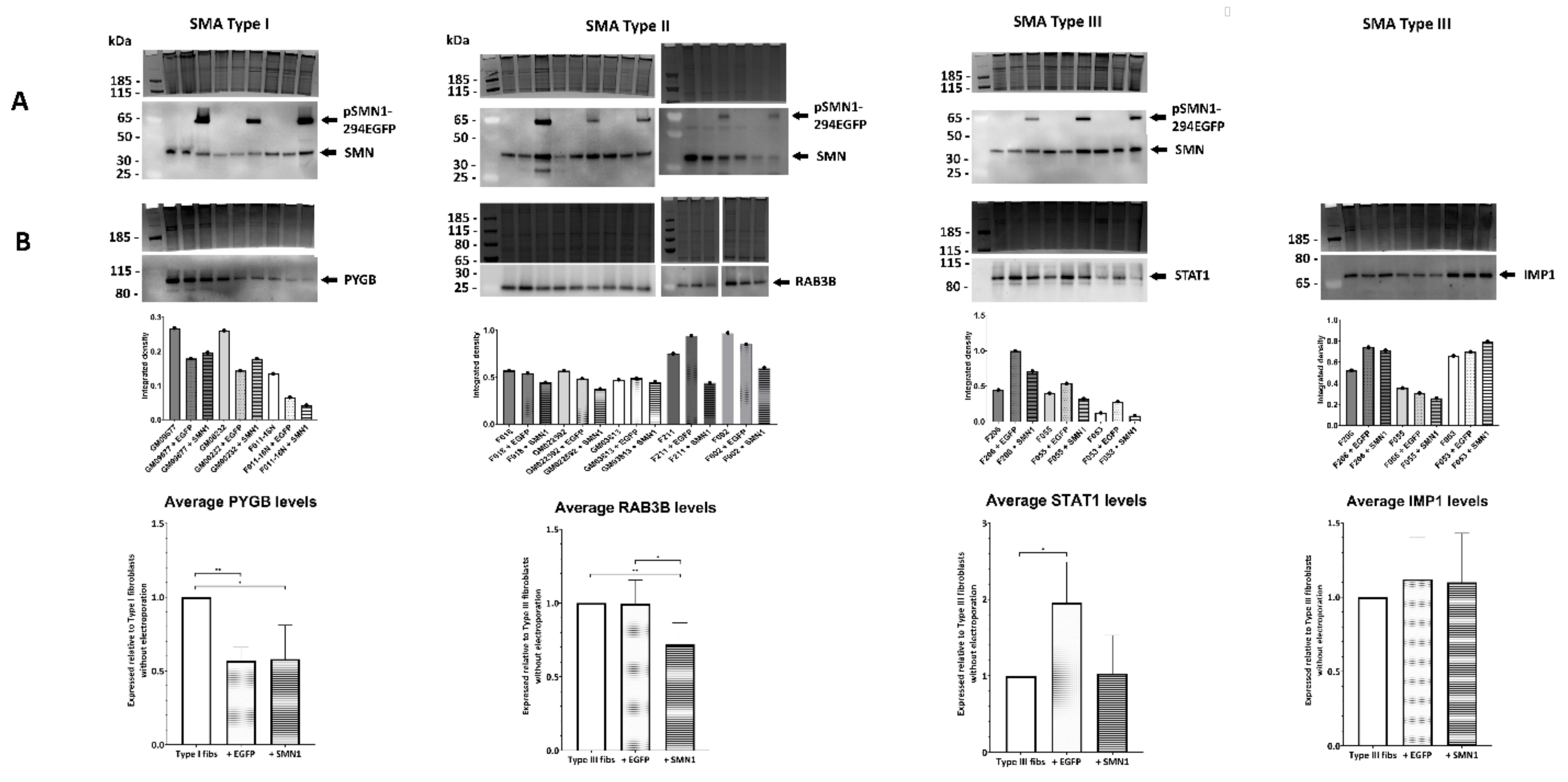
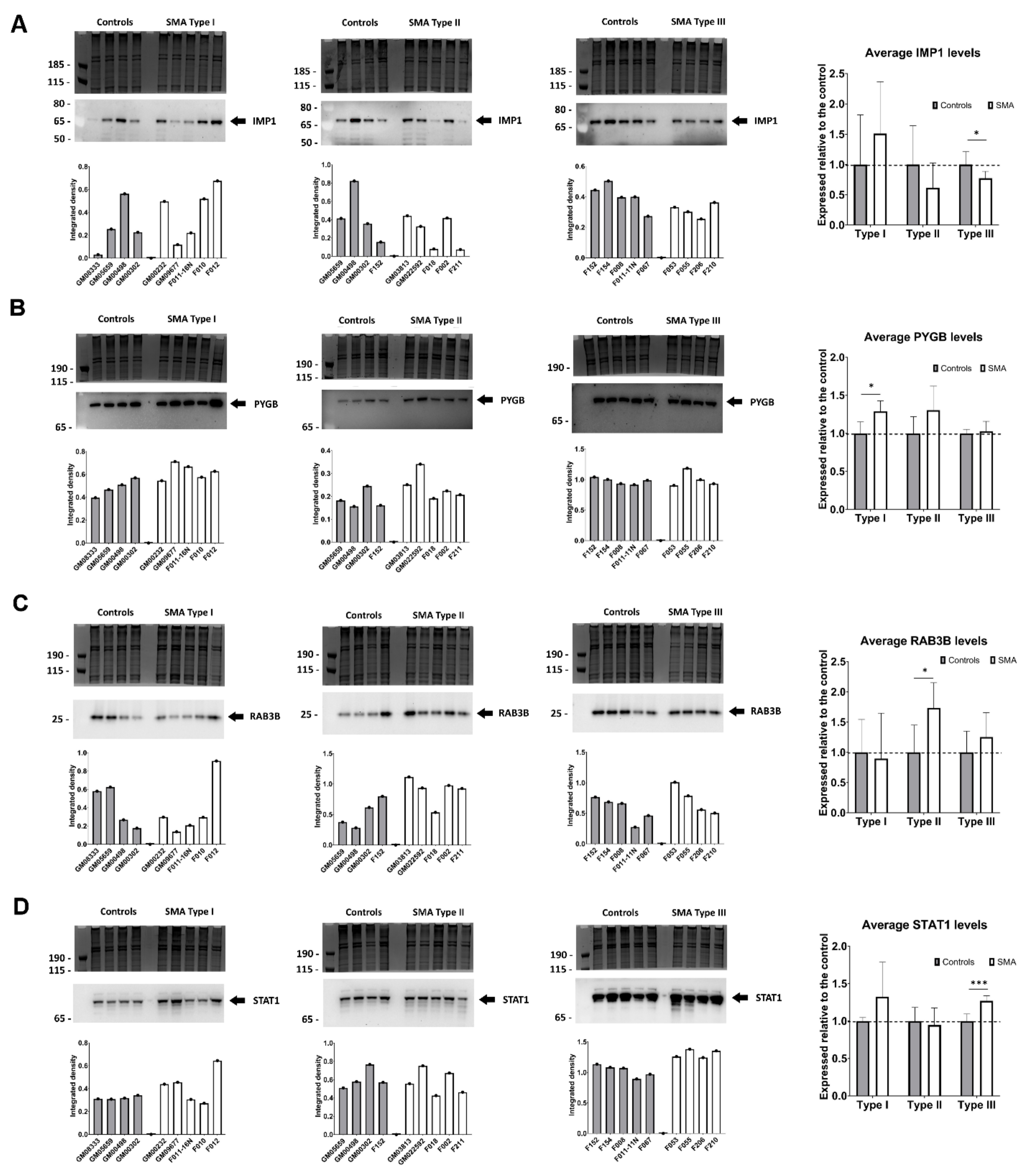
2.5. Quantitative Western Blotting Verifies Potential Utility of Fibroblast Biomarkers for Distinguishing SMA Severities
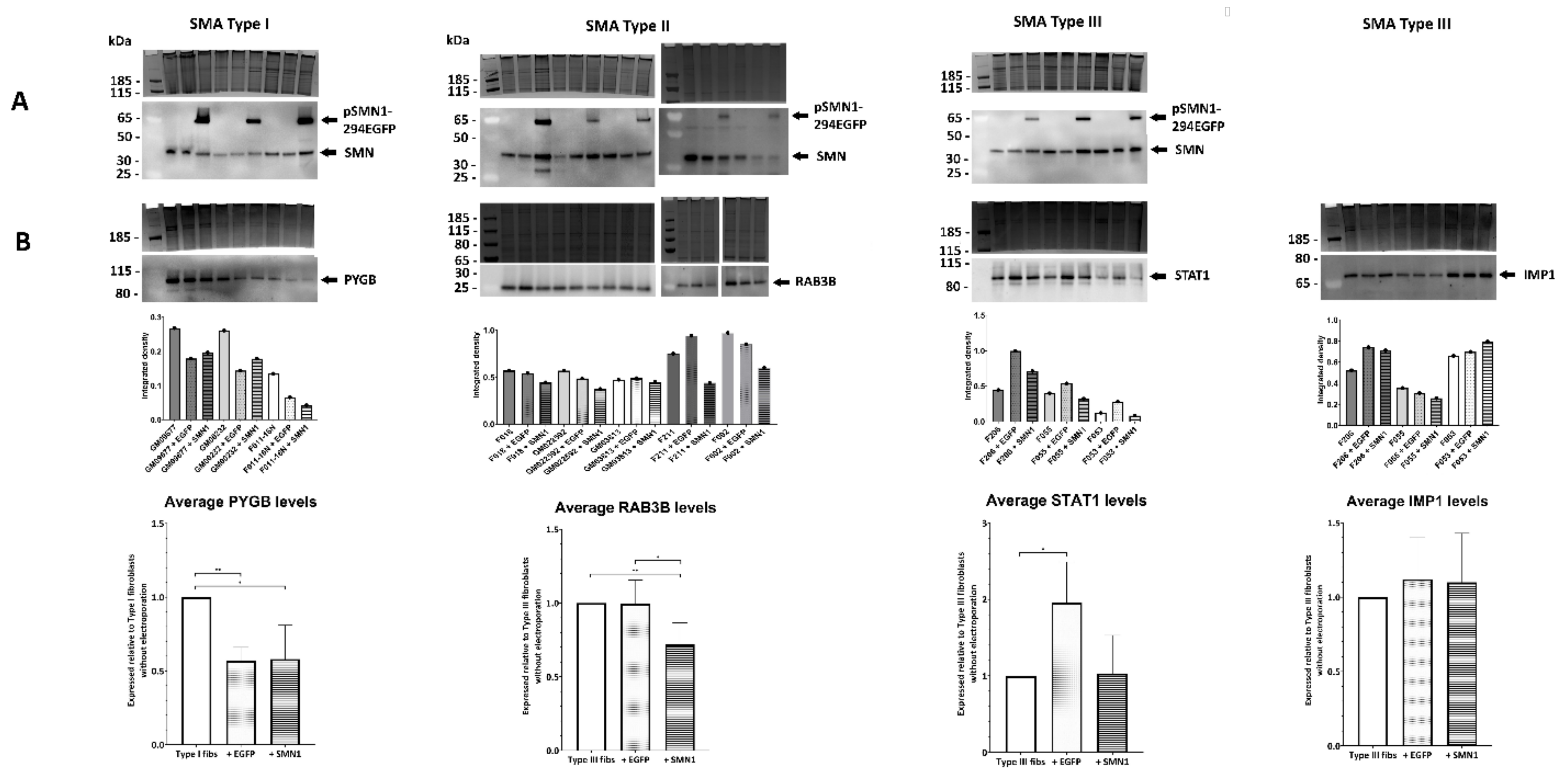
2.6. Transient Transfection of SMA Fibroblasts with EGFP-SMN1 Reduces Levels of RAB3B in Type II Fibroblasts
3. Discussion
4. Materials and Methods
4.1. Patient Cell Lines
4.2. Cell Culture
4.3. Transient Transfection of Fibroblasts Cells
4.4. DNA Extraction
4.5. SMN2 Copy Number Determination Using Droplet Digital™ PCR (ddPCR™)
4.6. Protein Extraction for Mass Spectrometry and Western Blot Analysis
4.7. Preparation of Samples for IDA and SWATH Mass Spectrometry
4.8. Mass Spectrometry Data Acquisition
4.9. Bioinformatics Analysis
4.10. BioLayout Express3D
4.11. Western Blot Analysis
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| eIF2 and eIF4 | Eukaryotic initiation factor 2 and 4 |
| IMP1 | Insulin-like growth factor 2 mRNA-binding protein 1 |
| IPA | Ingenuity Pathway Analysis |
| mTOR | Mammalian target of rapamycin |
| PYGB | Glycogen phosphorylase, brain form |
| RAB3B | Ras-related protein Rab-3B |
| RHO | Ras homologous protein family |
| SMA | Spinal muscular atrophy |
| SMN | Survival of motor neuron |
| STAT1 | Signal transducer and activator of transcription 1-alpha/beta |
| SWATH-MS | Sequential Window Acquisition of all Theoretical Mass Spectra |
References
- Mercuri, E.; Finkel, R.S.; Muntoni, F.; Wirth, B.; Montes, J.; Main, M.; Mazzone, E.; Vitale, M.; Snyder, B.; Quijano-Roy, S.; et al. Diagnosis and Management of Spinal Muscular Atrophy: Part 1: Recommendations for Diagnosis, Rehabilitation, Orthopedic and Nutritional Care. Neuromuscul. Disord. 2018, 28, 103–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finkel, R.S.; Mercuri, E.; Meyer, O.H.; Simonds, A.K.; Schroth, M.K.; Graham, R.J.; Kirschner, J.; Iannaccone, S.T.; Crawford, T.O.; Woods, S.; et al. Diagnosis and Management of Spinal Muscular Atrophy: Part 2: Pulmonary and Acute Care; Medications, Supplements and Immunizations; Other Organ Systems; and Ethics. Neuromuscul. Disord. 2018, 28, 197–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeo, C.J.J.; Darras, B.T. Overturning the Paradigm of Spinal Muscular Atrophy as Just a Motor Neuron Disease. Pediatr. Neurol. 2020, 109, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Jha, N.N.; Feng, Z.; Faleiro, M.R.; Chiriboga, C.A.; Wei-Lapierre, L.; Dirksen, R.T.; Ko, C.P.; Monani, U.R. Muscle-Specific SMN Reduction Reveals Motor Neuron–Independent Disease in Spinal Muscular Atrophy Models. J. Clin. Investig. 2020, 130, 1271–1287. [Google Scholar] [CrossRef] [Green Version]
- Wijngaarde, C.A.; Blank, A.C.; Stam, M.; Wadman, R.I.; Van Den Berg, L.H.; Van Der Pol, W.L. Cardiac Pathology in Spinal Muscular Atrophy: A Systematic Review. Orphanet J. Rare Dis. 2017, 12, 67. [Google Scholar] [CrossRef]
- Šoltić, D.; Shorrock, H.K.; Allardyce, H.; Wilson, E.L.; Holt, I.; Synowsky, S.A.; Shirran, S.L.; Parson, S.H.; Gillingwater, T.H.; Fuller, H.R. Lamin A/C Dysregulation Contributes to Cardiac Pathology in a Mouse Model of Severe Spinal Muscular Atrophy. Hum. Mol. Genet. 2019, 28, 3515–3527. [Google Scholar] [CrossRef]
- Somers, E.; Lees, R.D.; Hoban, K.; Sleigh, J.N.; Zhou, H.; Muntoni, F.; Talbot, K.; Gillingwater, T.H.; Parson, S.H. Vascular Defects and Spinal Cord Hypoxia in Spinal Muscular Atrophy. Ann. Neurol. 2016, 79, 217–230. [Google Scholar] [CrossRef] [Green Version]
- Deguise, M.; Baranello, G.; Mastella, C.; Beauvais, A.; Michaud, J.; Leone, A.; De Amicis, R.; Battezzati, A.; Dunham, C.; Selby, K.; et al. Abnormal Fatty Acid Metabolism Is a Core Component of Spinal Muscular Atrophy. Ann. Clin. Transl. Neurol. 2019, 6, 1519–1532. [Google Scholar] [CrossRef] [Green Version]
- Allardyce, H.; Kuhn, D.; Hernandez-Gerez, E.; Hensel, N.; Huang, Y.-T.; Faller, K.; Gillingwater, T.H.; Quondamatteo, F.; Claus, P.; Parson, S.H. Renal Pathology in a Mouse Model of Severe Spinal Muscular Atrophy Is Associated with Downregulation of Glial Cell-Line Derived Neurotrophic Factor (GDNF). Hum. Mol. Genet. 2020, 29, 2365–2378. [Google Scholar] [CrossRef]
- Nery, F.C.; Siranosian, J.J.; Rosales, I.; Deguise, M.O.; Sharma, A.; Muhtaseb, A.W.; Nwe, P.; Johnstone, A.J.; Zhang, R.; Fatouraei, M.; et al. Impaired Kidney Structure and Function in Spinal Muscular Atrophy. Neurol. Genet. 2019, 5, e353. [Google Scholar] [CrossRef] [Green Version]
- Lefebvre, S.; Bürglen, L.; Reboullet, S.; Clermont, O.; Burlet, P.; Viollet, L.; Benichou, B.; Cruaud, C.; Millasseau, P.; Zeviani, M.; et al. Identification and Characterization of a Spinal Muscular Atrophy-Determining Gene. Cell 1995, 80, 155–165. [Google Scholar] [CrossRef] [Green Version]
- Lorson, C.L.; Strasswimmer, J.; Yao, J.-M.; Baleja, J.D.; Hahnen, E.; Wirth, B.; Le, T.; Burghes, A.H.M.; Androphy, E.J. SMN Oligomerization Defect Correlates with Spinal Muscular Atrophy Severity. Nat. Genet. 1998, 19, 63–66. [Google Scholar] [CrossRef]
- Pellizzoni, L.; Charroux, B.; Dreyfuss, G. SMN Mutants of Spinal Muscular Atrophy Patients Are Defective in Binding to SnRNP Proteins. Proc. Natl. Acad. Sci. USA 1999, 96, 11167–11172. [Google Scholar] [CrossRef] [Green Version]
- Wirth, B.; Karakaya, M.; Kye, M.J.; Mendoza-Ferreira, N. Twenty-Five Years of Spinal Muscular Atrophy Research: From Phenotype to Genotype to Therapy, and What Comes Next. Annu. Rev. Genom. Hum. Genet. 2020, 21, 231–261. [Google Scholar] [CrossRef] [Green Version]
- Calucho, M.; Bernal, S.; Alías, L.; March, F.; Venceslá, A.; Rodríguez-Álvarez, F.J.; Aller, E.; Fernández, R.M.; Borrego, S.; Millán, J.M.; et al. Correlation between SMA Type and SMN2 Copy Number Revisited: An Analysis of 625 Unrelated Spanish Patients and a Compilation of 2834 Reported Cases. Neuromuscul. Disord. 2018, 28, 208–215. [Google Scholar] [CrossRef]
- Ramdas, S.; Servais, L. New Treatments in Spinal Muscular Atrophy: An Overview of Currently Available Data. Expert Opin. Pharmacother. 2020, 21, 307–315. [Google Scholar] [CrossRef]
- Schorling, D.C.; Pechmann, A.; Kirschner, J. Advances in Treatment of Spinal Muscular Atrophy—New Phenotypes, New Challenges, New Implications for Care. J. Neuromuscul. Dis. 2020, 7, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Le, T.T.; McGovern, V.L.; Alwine, I.E.; Wang, X.; Massoni-Laporte, A.; Rich, M.M.; Burghes, A.H.M. Temporal Requirement for High SMN Expression in SMA Mice. Hum. Mol. Genet. 2011, 20, 3578–3591. [Google Scholar] [CrossRef] [Green Version]
- Kariya, S.; Obis, T.; Garone, C.; Akay, T.; Sera, F.; Iwata, S.; Homma, S.; Monani, U.R. Requirement of Enhanced Survival Motoneuron Protein Imposed during Neuromuscular Junction Maturation. J. Clin. Investig. 2014, 124, 785–800. [Google Scholar] [CrossRef]
- Lutz, C.M.; Kariya, S.; Patruni, S.; Osborne, M.A.; Liu, D.; Henderson, C.E.; Li, D.K.; Pellizzoni, L.; Rojas, J.; Valenzuela, D.M.; et al. Postsymptomatic Restoration of SMN Rescues the Disease Phenotype in a Mouse Model of Severe Spinal Muscular Atrophy. J. Clin. Investig. 2011, 121, 3029–3041. [Google Scholar] [CrossRef]
- Foust, K.D.; Wang, X.; McGovern, V.L.; Braun, L.; Bevan, A.K.; Haidet, A.M.; Le, T.T.; Morales, P.R.; Rich, M.M.; Burghes, A.H.M.; et al. Rescue of the Spinal Muscular Atrophy Phenotype in a Mouse Model by Early Postnatal Delivery of SMN. Nat. Biotechnol. 2010, 28, 271–274. [Google Scholar] [CrossRef] [Green Version]
- Bowerman, M.; Murray, L.M.; Beauvais, A.; Pinheiro, B.; Kothary, R. A Critical Smn Threshold in Mice Dictates Onset of an Intermediate Spinal Muscular Atrophy Phenotype Associated with a Distinct Neuromuscular Junction Pathology. Neuromuscul. Disord. 2012, 22, 263–276. [Google Scholar] [CrossRef]
- Hensel, N.; Brickwedde, H.; Tsaknakis, K.; Grages, A.; Braunschweig, L.; Lüders, K.A.; Lorenz, H.M.; Lippross, S.; Walter, L.M.; Tavassol, F.; et al. Altered Bone Development with Impaired Cartilage Formation Precedes Neuromuscular Symptoms in Spinal Muscular Atrophy. Hum. Mol. Genet. 2020, 29, 2662–2673. [Google Scholar] [CrossRef]
- Sheng, L.; Wan, B.; Feng, P.; Sun, J.; Rigo, F.; Frank Bennett, C.; Akerman, M.; Krainer, A.R.; Hua, Y. Downregulation of Survivin Contributes to Cell-Cycle Arrest during Postnatal Cardiac Development in a Severe Spinal Muscular Atrophy Mouse Model. Hum. Mol. Genet. 2018, 27, 486–498. [Google Scholar] [CrossRef]
- Schreml, J.; Riessland, M.; Paterno, M.; Garbes, L.; Robach, K.; Ackermann, B.; Krämer, J.; Somers, E.; Parson, S.H.; Heller, R.; et al. Severe SMA Mice Show Organ Impairment That Cannot Be Rescued by Therapy with the HDACi JNJ-26481585. Eur. J. Hum. Genet. 2013, 21, 643–652. [Google Scholar] [CrossRef] [Green Version]
- Crawford, T.O.; Paushkin, S.V.; Kobayashi, D.T.; Forrest, S.J.; Joyce, C.L.; Finkel, R.S.; Kaufmann, P.; Swoboda, K.J.; Tiziano, D.; Lomastro, R.; et al. Evaluation of SMN Protein, Transcript, and Copy Number in the Biomarkers for Spinal Muscular Atrophy (BforSMA) Clinical Study. PLoS ONE 2012, 7, e33572. [Google Scholar] [CrossRef] [Green Version]
- Sivaramakrishnan, M.; McCarthy, K.D.; Campagne, S.; Huber, S.; Meier, S.; Augustin, A.; Heckel, T.; Meistermann, H.; Hug, M.N.; Birrer, P.; et al. Binding to SMN2 Pre-MRNA-Protein Complex Elicits Specificity for Small Molecule Splicing Modifiers. Nat. Commun. 2017, 8, 1476. [Google Scholar] [CrossRef]
- Vatine, G.D.; Al-Ahmad, A.; Barriga, B.K.; Svendsen, S.; Salim, A.; Garcia, L.; Garcia, V.J.; Ho, R.; Yucer, N.; Qian, T.; et al. Modeling Psychomotor Retardation Using IPSCs from MCT8-Deficient Patients Indicates a Prominent Role for the Blood-Brain Barrier. Cell Stem Cell 2017, 20, 831–843.e5. [Google Scholar] [CrossRef] [Green Version]
- Theocharidis, A.; van Dongen, S.; Enright, A.J.; Freeman, T.C. Network Visualization and Analysis of Gene Expression Data Using BioLayout Express(3D). Nat. Protoc. 2009, 4, 1535–1550. [Google Scholar] [CrossRef]
- Boraldi, F.; Bini, L.; Liberatori, S.; Armini, A.; Pallini, V.; Tiozzo, R.; Pasquali-Ronchetti, I.; Quaglino, D. Proteome Analysis of Dermal Fibroblasts Cultured in Vitro from Human Healthy Subjects of Different Ages. Proteomics 2003, 3, 917–929. [Google Scholar] [CrossRef]
- Ignjatovic, V.; Lai, C.; Summerhayes, R.; Mathesius, U.; Tawfilis, S.; Perugini, M.A.; Monagle, P. Age-Related Differences in Plasma Proteins: How Plasma Proteins Change from Neonates to Adults. PLoS ONE 2011, 6, e17213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjelosevic, S.; Pascovici, D.; Ping, H.; Karlaftis, V.; Zaw, T.; Song, X.; Molloy, M.P.; Monagle, P.; Ignjatovic, V. Quantitative Age-Specific Variability of Plasma Proteins in Healthy Neonates, Children and Adults. Mol. Cell. Proteom. 2017, 16, 924–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wirth, B.; Mendoza-Ferreira, N.; Torres-Benito, L. Spinal Muscular Atrophy Disease Modifiers. In Spinal Muscular Atrophy: Disease Mechanisms and Therapy; Sumner, C.J., Paushkin, S., Ko, C.P., Eds.; Academic Press: Cambridge, MA, USA, 2017; pp. 191–210. ISBN 9780128036853. [Google Scholar]
- Oprea, G.E.; Krober, S.; McWhorter, M.L.; Rossoll, W.; Muller, S.; Krawczak, M.; Bassell, G.J.; Beattie, C.E.; Wirth, B. Plastin 3 Is a Protective Modifier of Autosomal Recessive Spinal Muscular Atrophy. Science 2008, 320, 524–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riessland, M.; Kaczmarek, A.; Schneider, S.; Swoboda, K.J.; Löhr, H.; Bradler, C.; Grysko, V.; Dimitriadi, M.; Hosseinibarkooie, S.; Torres-Benito, L.; et al. Neurocalcin Delta Suppression Protects against Spinal Muscular Atrophy in Humans and across Species by Restoring Impaired Endocytosis. Am. J. Hum. Genet. 2017, 100, 297–315. [Google Scholar] [CrossRef] [Green Version]
- Wadman, R.I.; Jansen, M.D.; Curial, C.A.D.; Groen, E.J.N.; Stam, M.; Wijngaarde, C.A.; Medic, J.; Sodaar, P.; Van Eijk, K.R.; Huibers, M.M.H.; et al. Analysis of FUS, PFN2, TDP-43, and PLS3 as Potential Disease Severity Modifiers in Spinal Muscular Atrophy. Neurol. Genet. 2020, 6, e386. [Google Scholar] [CrossRef] [Green Version]
- Also-Rallo, E.; Alías, L.; Martínez-Hernández, R.; Caselles, L.; Barceló, M.J.; Baiget, M.; Bernal, S.; Tizzano, E.F. Treatment of Spinal Muscular Atrophy Cells with Drugs That Upregulate SMN Expression Reveals Inter- and Intra-Patient Variability. Eur. J. Hum. Genet. 2011, 19, 1059–1065. [Google Scholar] [CrossRef]
- Wadman, R.I.; Stam, M.; Jansen, M.; Van Der Weegen, Y.; Wijngaarde, C.A.; Harschnitz, O.; Sodaar, P.; Braun, K.P.J.; Dooijes, D.; Lemmink, H.H.; et al. A Comparative Study of SMN Protein and MRNA in Blood and Fibroblasts in Patients with Spinal Muscular Atrophy and Healthy Controls. PLoS ONE 2016, 11, e0167087. [Google Scholar] [CrossRef]
- Millino, C.; Fanin, M.; Vettori, A.; Laveder, P.; Mostacciuolo, M.; Angelini, C.; Lanfranchi, G. Different Atrophy-Hypertrophy Transcription Pathways in Muscles Affected by Severe and Mild Spinal Muscular Atrophy. BMC Med. 2009, 7, 14. [Google Scholar] [CrossRef]
- Ghazalpour, A.; Bennett, B.; Petyuk, V.A.; Orozco, L.; Hagopian, R.; Mungrue, I.N.; Farber, C.R.; Sinsheimer, J.; Kang, H.M.; Furlotte, N.; et al. Comparative Analysis of Proteome and Transcriptome Variation in Mouse. PLoS Genet. 2011, 7, e1001393. [Google Scholar] [CrossRef] [Green Version]
- Simic, G.; Mladinov, M.; Simic, D.S.; Milosevic, N.J.; Islam, A.; Pajtak, A.; Barisic, N.; Sertic, J.; Lucassen, P.J.; Hof, P.R.; et al. Abnormal Motoneuron Migration, Differentiation, and Axon Outgrowth in Spinal Muscular Atrophy. Acta Neuropathol. 2008, 115, 313–326. [Google Scholar] [CrossRef] [Green Version]
- Araujo, A.; Araujo, M.; Swoboda, K.J. Vascular Perfusion Abnormalities in Infants with Spinal Muscular Atrophy. J. Pediatr. 2009, 155, 292–294. [Google Scholar] [CrossRef] [Green Version]
- Deguise, M.-O.; Beauvais, A.; Schneider, B.L.; Kothary, R. Blood Flow to the Spleen Is Altered in a Mouse Model of Spinal Muscular Atrophy. J. Neuromuscul. Dis. 2020, 7, 315–322. [Google Scholar] [CrossRef]
- Chaytow, H.; Huang, Y.T.; Gillingwater, T.H.; Faller, K.M.E. The Role of Survival Motor Neuron Protein (SMN) in Protein Homeostasis. Cell. Mol. Life Sci. 2018, 75, 3877–3894. [Google Scholar] [CrossRef] [Green Version]
- Acsadi, G.; Lee, I.; Li, X.; Khaidakov, M.; Pecinova, A.; Parker, G.C.; Hüttemann, M. Mitochondrial Dysfunction in a Neural Cell Model of Spinal Muscular Atrophy. J. Neurosci. Res. 2009, 87, 2748–2756. [Google Scholar] [CrossRef]
- Thelen, M.P.; Wirth, B.; Kye, M.J. Mitochondrial Defects in the Respiratory Complex I Contribute to Impaired Translational Initiation via ROS and Energy Homeostasis in SMA Motor Neurons. Acta Neuropathol. Commun. 2020, 8, 223. [Google Scholar] [CrossRef]
- Miller, N.; Shi, H.; Zelikovich, A.S.; Ma, Y.C. Motor Neuron Mitochondrial Dysfunction in Spinal Muscular Atrophy. Hum. Mol. Genet. 2016, 25, 3395–3406. [Google Scholar] [CrossRef] [Green Version]
- Ripolone, M.; Ronchi, D.; Violano, R.; Vallejo, D.; Fagiolari, G.; Barca, E.; Lucchini, V.; Colombo, I.; Villa, L.; Berardinelli, A.; et al. Impaired Muscle Mitochondrial Biogenesis and Myogenesis in Spinal Muscular Atrophy. JAMA Neurol. 2015, 72, 666–675. [Google Scholar] [CrossRef]
- Wishart, T.M.; Mutsaers, C.A.; Riessland, M.; Reimer, M.M.; Hunter, G.; Hannam, M.L.; Eaton, S.L.; Fuller, H.R.; Roche, S.L.; Somers, E.; et al. Dysregulation of Ubiquitin Homeostasis and Beta-Catenin Signalling Promote Spinal Muscular Atrophy. J. Clin. Investig. 2014, 124, 1821–1834. [Google Scholar] [CrossRef] [Green Version]
- Yan, L.L.; Zaher, H.S. How Do Cells Cope with RNA Damage and Its Consequences? J. Biol. Chem. 2019, 294, 15158–15171. [Google Scholar] [CrossRef] [Green Version]
- Kye, M.J.; Niederst, E.D.; Wertz, M.H.; Gonçalves, I.D.C.G.; Akten, B.; Dover, K.Z.; Peters, M.; Riessland, M.; Neveu, P.; Wirth, B.; et al. SMN Regulates Axonal Local Translation via MiR-183/MTOR Pathway. Hum. Mol. Genet. 2014, 23, 6318–6331. [Google Scholar] [CrossRef]
- Li, Y.J.; Chen, T.H.; Wu, Y.Z.; Tseng, Y.H. Metabolic and Nutritional Issues Associated with Spinal Muscular Atrophy. Nutrients 2020, 12, 3842. [Google Scholar] [CrossRef]
- Deguise, M.O.; Chehade, L.; Kothary, R. Metabolic Dysfunction in Spinal Muscular Atrophy. Int. J. Mol. Sci. 2021, 22, 5913. [Google Scholar] [CrossRef]
- Liu, Q.; Fischer, U.; Wang, F.; Dreyfuss, G. The Spinal Muscular Atrophy Disease Gene Product, SMN, and Its Associated Protein SIP1 Are in a Complex with Spliceosomal SnRNP Proteins. Cell 1997, 90, 1013–1021. [Google Scholar] [CrossRef] [Green Version]
- Ning, K.; Drepper, C.; Valori, C.F.; Ahsan, M.; Wyles, M.; Higginbottom, A.; Herrmann, T.; Shaw, P.; Azzouz, M.; Sendtner, M. PTEN Depletion Rescues Axonal Growth Defect and Improves Survival in SMN-Deficient Motor Neurons. Hum. Mol. Genet. 2010, 19, 3159–3168. [Google Scholar] [CrossRef] [Green Version]
- Gabanella, F.; Barbato, C.; Fiore, M.; Petrella, C.; De Vincentiis, M.; Greco, A.; Minni, A.; Corbi, N.; Passananti, C.; Grazia Di Certo, M.; et al. Fine-Tuning of MTOR MRNA and Nucleolin Complexes by SMN. Cells 2021, 10, 3015. [Google Scholar] [CrossRef]
- Zou, T.; Yang, X.; Pan, D.; Huang, J.; Sahin, M.; Zhou, J. SMN Deficiency Reduces Cellular Ability to Form Stress Granules, Sensitizing Cells to Stress. Cell. Mol. Neurobiol. 2011, 31, 541–550. [Google Scholar] [CrossRef]
- Bowerman, M.; Beauvais, A.; Anderson, C.L.; Kothary, R. Rho-Kinase Inactivation Prolongs Survival of an Intermediate SMA Mouse Model. Hum. Mol. Genet. 2010, 19, 1468–1478. [Google Scholar] [CrossRef]
- Caraballo-Miralles, V.; Cardona-Rossinyol, A.; Garcera, A.; Villalonga, P.; Soler, R.M.; Olmos, G.; Lladó, J. SMN Deficiency Attenuates Migration of U87MG Astroglioma Cells through the Activation of RhoA. Mol. Cell. Neurosci. 2012, 49, 282–289. [Google Scholar] [CrossRef]
- Rademacher, S.; Verheijen, B.M.; Hensel, N.; Peters, M.; Bora, G.; Brandes, G.; de Sá, R.V.; Heidrich, N.; Fischer, S.; Brinkmann, H.; et al. Metalloprotease-Mediated Cleavage of PlexinD1 and Its Sequestration to Actin Rods in the Motoneuron Disease Spinal Muscular Atrophy (SMA). Hum. Mol. Genet. 2017, 26, 3946–3959. [Google Scholar] [CrossRef]
- Fallini, C.; Rouanet, J.P.; Donlin-Asp, P.G.; Guo, P.; Zhang, H.; Singer, R.H.; Rossoll, W.; Bassell, G.J. Dynamics of Survival of Motor Neuron (SMN) Protein Interaction with the MRNA-Binding Protein IMP1 Facilitates Its Trafficking into Motor Neuron Axons. Dev. Neurobiol. 2014, 74, 319–332. [Google Scholar] [CrossRef]
- Chung, C.Y.; Koprich, J.B.; Hallett, P.J.; Isacson, O. Functional Enhancement and Protection of Dopaminergic Terminals by RAB3B Overexpression. Proc. Natl. Acad. Sci. USA 2009, 106, 22474–22479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, C.P.; Cao, X. Structure, Function, and Regulation of STAT Proteins. Mol. Biosyst. 2006, 2, 536–550. [Google Scholar] [CrossRef] [PubMed]
- Ebert, A.D.; Yu, J.; Rose Jr, F.F.; Mattis, V.B.; Lorson, C.L.; Thomson, J.A.; Svendsen, C.N. Induced Pluripotent Stem Cells from a Spinal Muscular Atrophy Patient. Nature 2009, 457, 277–280. [Google Scholar] [CrossRef] [PubMed]
- Hentschel, A.; Czech, A.; Münchberg, U.; Freier, E.; Schara-Schmidt, U.; Sickmann, A.; Reimann, J.; Roos, A. Protein Signature of Human Skin Fibroblasts Allows the Study of the Molecular Etiology of Rare Neurological Diseases. Orphanet J. Rare Dis. 2021, 16, 73. [Google Scholar] [CrossRef] [PubMed]
- Bergeijk, J.; Rydel-Könecke, K.; Grothe, C.; Claus, P. The Spinal Muscular Atrophy Gene Product Regulates Neurite Outgrowth: Importance of the C Terminus. FASEB J. 2007, 21, 1492–1502. [Google Scholar] [CrossRef]
- Holt, I.; Clements, L.; Manilal, S.; Brown, S.C.; Morris, G.E. The R482Q Lamin A/C Mutation That Causes Lipodystrophy Does Not Prevent Nuclear Targeting of Lamin A in Adipocytes or Its Interaction with Emerin. Eur. J. Hum. Genet. 2001, 9, 204–208. [Google Scholar] [CrossRef]
- Reiter, L.; Rinner, O.; Picotti, P.; Hüttenhain, R.; Beck, M.; Brusniak, M.-Y.; Hengartner, M.O.; Aebersold, R. MProphet: A General and Flexible Data Model and Algorithm for Automated SRM Data Processing and Statistical Error Estimation. MCP Submitt. 2011, 8, 430–435. [Google Scholar]
- Ludwig, C.; Gillet, L.; Rosenberger, G.; Amon, S.; Collins, B.C.; Aebersold, R. Data-independent Acquisition-based SWATH-MS for Quantitative Proteomics: A Tutorial. Mol. Syst. Biol. 2018, 14, e8126. [Google Scholar] [CrossRef]
- Heberle, H.; Meirelles, V.G.; da Silva, F.R.; Telles, G.P.; Minghim, R. InteractiVenn: A Web-Based Tool for the Analysis of Sets through Venn Diagrams. BMC Bioinform. 2015, 16, 169. [Google Scholar] [CrossRef]
- Krämer, A.; Green, J.; Pollard, J.; Tugendreich, S. Causal Analysis Approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef]
- Tugendreich, S.M. “Understanding Biological Mechanisms in Transcriptomics or Proteomics Datasets with Ingenuity Pathway Analysis (IPA) and Analysis Match.” Biological Mechanisms in Omics Datasets with QIAGEN IPA. QIAGEN, January 2019. Available online: https://www.qiagen.pathfactory.com/pathway-analysis/ipa-analysis-match-w?lx=lVyQNh&xs=201104 (accessed on 28 July 2022).
- Šoltić, D.; Bowerman, M.; Stock, J.; Shorrock, H.; Gillingwater, T.; Fuller, H. Multi-Study Proteomic and Bioinformatic Identification of Molecular Overlap between Amyotrophic Lateral Sclerosis (ALS) and Spinal Muscular Atrophy (SMA). Brain Sci. 2018, 8, 212. [Google Scholar] [CrossRef] [Green Version]
- Abràmoff, M.D.; Magalhães, P.J.; Ram, S.J. Image Processing with ImageJ. Biophotonics Int. 2004, 11, 36–41. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Perez-Riverol, Y.; Bai1, J.; Bandla, C.; García-Seisdedos, D.; Hewapathirana, S.; Kamatchinathan, S.; Kundu, D.J.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The PRIDE database resources in 2022: A hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022, 50, D543–D552. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brown, S.J.; Kline, R.A.; Synowsky, S.A.; Shirran, S.L.; Holt, I.; Sillence, K.A.; Claus, P.; Wirth, B.; Wishart, T.M.; Fuller, H.R. The Proteome Signatures of Fibroblasts from Patients with Severe, Intermediate and Mild Spinal Muscular Atrophy Show Limited Overlap. Cells 2022, 11, 2624. https://doi.org/10.3390/cells11172624
Brown SJ, Kline RA, Synowsky SA, Shirran SL, Holt I, Sillence KA, Claus P, Wirth B, Wishart TM, Fuller HR. The Proteome Signatures of Fibroblasts from Patients with Severe, Intermediate and Mild Spinal Muscular Atrophy Show Limited Overlap. Cells. 2022; 11(17):2624. https://doi.org/10.3390/cells11172624
Chicago/Turabian StyleBrown, Sharon J., Rachel A. Kline, Silvia A. Synowsky, Sally L. Shirran, Ian Holt, Kelly A. Sillence, Peter Claus, Brunhilde Wirth, Thomas M. Wishart, and Heidi R. Fuller. 2022. "The Proteome Signatures of Fibroblasts from Patients with Severe, Intermediate and Mild Spinal Muscular Atrophy Show Limited Overlap" Cells 11, no. 17: 2624. https://doi.org/10.3390/cells11172624
APA StyleBrown, S. J., Kline, R. A., Synowsky, S. A., Shirran, S. L., Holt, I., Sillence, K. A., Claus, P., Wirth, B., Wishart, T. M., & Fuller, H. R. (2022). The Proteome Signatures of Fibroblasts from Patients with Severe, Intermediate and Mild Spinal Muscular Atrophy Show Limited Overlap. Cells, 11(17), 2624. https://doi.org/10.3390/cells11172624