
Canonical Hedgehog Pathway and Noncanonical GLI Transcription Factor Activation in Cancer

Abstract
:1. Introduction
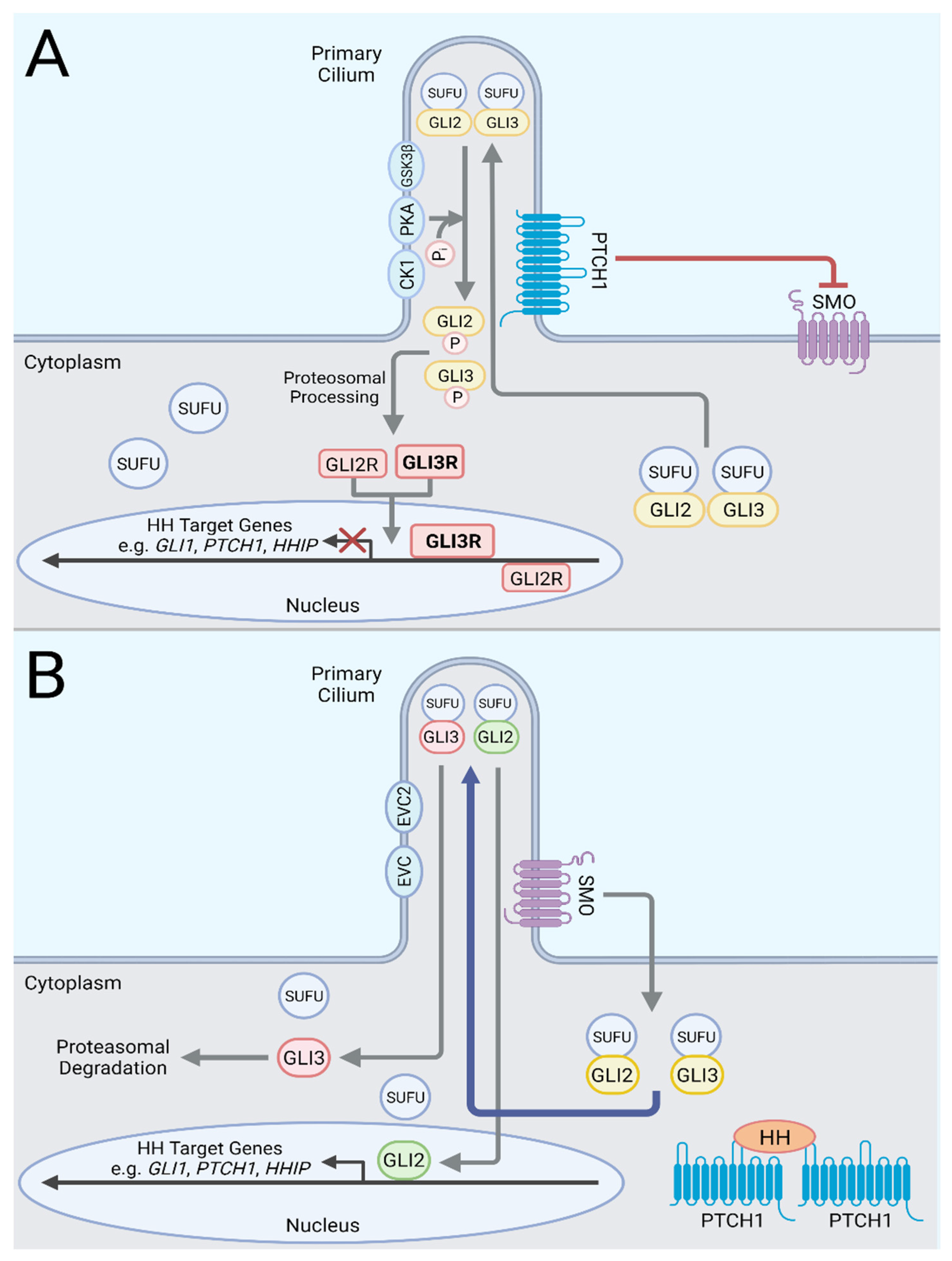
2. Hedgehog Signaling Pathway
3. Modes of Canonical Hedgehog Pathway Activation in Cancers
4. Cancers with Ligand-Independent Canonical Pathway Activation
4.1. Basal Cell Carcinoma
4.2. Medulloblastoma
4.3. Rhabdomyosarcoma/Rhabdomyoma
4.4. Ameloblastoma
4.5. Meningioma
5. Cancers with Ligand-Dependent Canonical Hedgehog Pathway Activation in Cancer
5.1. Tumor-Suppressive Action of Ligand-Dependent Paracrine Canonical Hh Signaling Pathway
5.1.1. Bladder Cancer
5.1.2. Pancreas Cancer
5.1.3. Colon Cancer
5.1.4. Prostate Cancer
5.1.5. Lung Adenocarcinoma
5.2. Tumor–Promoting Action of Ligand-Dependent Canonical Hh Signaling Pathway
5.2.1. Acute Myeloid Leukemia
5.2.2. Small Cell Lung Cancer
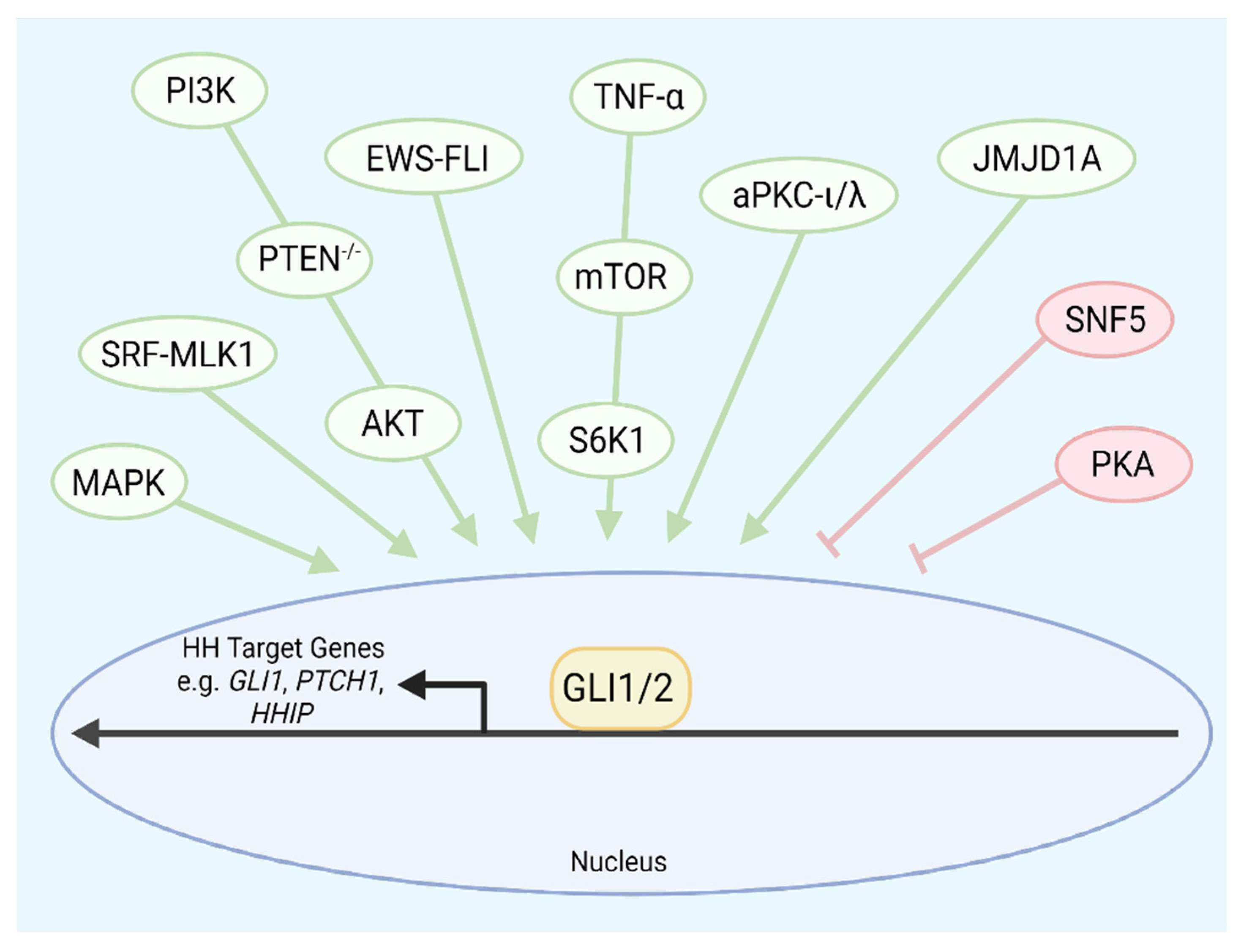
6. Cancers with Noncanonical Activation of GLI Transcription Factors
6.1. Esophageal Cancer
6.2. Glioblastoma
6.3. Lung Squamous Cell Carcinoma
6.4. Malignant Rhabdoid Tumor
6.5. Ewing Sarcoma
6.6. Vismodegib-Resistant Basal Cell Carcinoma
6.7. Rhabdomyosarcoma and Lung Adenocarcinoma
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nüsslein-Volhard, C.; Wieschaus, E. Mutations affecting segment number and polarity in Drosophila. Nature 1980, 287, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Echelard, Y.; Epstein, D.J.; St Jacques, B.; Shen, L.; Mohler, J.; McMahon, J.A.; McMahon, A.P. Sonic hedgehog, a mem-ber of a family of putative signaling molecules, is implicated in the regulation of CNS polarity. Cell 1993, 75, 1417–1430. [Google Scholar] [CrossRef]
- Krauss, S.; Concordet, J.-P.; Ingham, P. A functionally conserved homolog of the Drosophila segment polarity gene hh is expressed in tissues with polarizing activity in zebrafish embryos. Cell 1993, 75, 1431–1444. [Google Scholar] [CrossRef]
- Riddle, R.D.; Johnson, R.L.; Laufer, E.; Tabin, C. Sonic hedgehog mediates the polarizing activity of the ZPA. Cell 1993, 75, 1401–1416. [Google Scholar] [CrossRef]
- Pietrobono, S.; Gagliardi, S.; Stecca, B. Non-canonical Hedgehog Signaling Pathway in Cancer: Activation of GLI Tran-scription Factors Beyond Smoothened. Front. Genet. 2019, 10, 556. [Google Scholar] [CrossRef] [PubMed]
- Sasai, N.; Toriyama, M.; Kondo, T. Hedgehog Signal and Genetic Disorders. Front. Genet. 2019, 10, 1103. [Google Scholar] [CrossRef] [PubMed]
- Kalff-Suske, M.; Wild, A.; Topp, J.; Wessling, M.; Jacobsen, E.-M.; Bornholdt, D.; Engel, H.; Heuer, H.; Aalfs, C.M.; Ausems, M.G.E.M.; et al. Point Mutations Throughout the GLI3 Gene Cause Greig Cephalopolysyndactyly Syndrome. Hum. Mol. Genet. 1999, 8, 1769–1777. [Google Scholar] [CrossRef] [PubMed]
- Roessler, E.; Belloni, E.; Gaudenz, K.; Jay, P.; Berta, P.; Scherer, S.W.; Tsui, L.-C.; Muenke, M. Mutations in the human Sonic Hedgehog gene cause holoprosencephaly. Nat. Genet. 1996, 14, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Belloni, E.; Muenke, M.; Roessler, E.; Traverse, G.; Siegel-Bartelt, J.; Frumkin, A.; Mitchell, H.; Donis-Keller, H.; Helms, C.; Hing, A.; et al. Identification of Sonic hedgehog as a candidate gene responsible for holoprosencephaly. Nat. Genet. 1996, 14, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Graham, J.M.; Olney, A.H., Jr.; Biesecker, L.G. GLI3 frameshift mutations cause autosomal dominant Pallis-ter-Hall syndrome. Nat. Genet. 1997, 15, 266–268. [Google Scholar] [CrossRef]
- Briscoe, J.; Thérond, P.P. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat. Rev. Mol. Cell Biol. 2013, 14, 416–429. [Google Scholar] [CrossRef] [PubMed]
- Gailani, M.R.; Bale, S.J.; Leffell, D.J.; DiGiovanna, J.J.; Peck, G.L.; Poliak, S.; Drum, M.A.; Pastakia, B.; McBride, O.W.; Kase, R.; et al. Developmental defects in gorlin syndrome related to a putative tumor suppressor gene on chromosome 9. Cell 1992, 69, 111–117. [Google Scholar] [CrossRef]
- Gailani, M.R.; Ståhle-Bäckdahl, M.; Leffell, D.J.; Glyn, M.; Zaphiropoulos, P.; Undén, A.B.; Dean, M.; Brash, D.E.; Bale, A.E.; Toftgård, R. The role of the human homologue of Drosophila patched in sporadic basal cell carcinomas. Nat. Genet. 1996, 14, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Hahn, H.; Wicking, C.; Zaphiropoulos, P.G.; Gailani, M.R.; Shanley, S.; Chidambaram, A.; Vorechovsky, I.; Holmberg, E.; Unden, A.B.; Gillies, S.; et al. Mutations of the Human Homolog of Drosophila patched in the Nevoid Basal Cell Carcinoma Syndrome. Cell 1996, 85, 841–851. [Google Scholar] [CrossRef]
- Teglund, S.; Toftgård, R. Hedgehog beyond medulloblastoma and basal cell carcinoma. Biochim. Biophys. Acta 2010, 1805, 181–208. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Zhang, Y.; Sun, B.; McMahon, A.P.; Wang, Y. Hedgehog Signaling: From Basic Biology to Cancer Therapy. Cell Chem. Biol. 2017, 24, 252–280. [Google Scholar] [CrossRef] [PubMed]
- Taipale, J.; Cooper, M.K.; Maiti, T.; Beachy, P.A. Patched acts catalytically to suppress the activity of Smoothened. Nature 2002, 418, 892–896. [Google Scholar] [CrossRef]
- Qi, X.; Liu, H.; Thompson, B.; McDonald, J.; Zhang, C.; Li, X. Cryo-EM structure of oxysterol-bound human Smoothened coupled to a heterotrimeric Gi. Nature 2019, 571, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, I.; Liang, J.; Hedeen, D.; Roberts, K.J.; Zhang, Y.; Ha, B.; Latorraca, N.R.; Faust, B.; Dror, R.O.; Beachy, P.A.; et al. Smoothened stimulation by membrane sterols drives Hedgehog pathway activity. Nature 2019, 571, 284–288. [Google Scholar] [CrossRef] [PubMed]
- Myers, B.R.; Neahring, L.; Zhang, Y.; Roberts, K.J.; Beachy, P.A. Rapid, direct activity assays for Smoothened reveal Hedgehog pathway regulation by membrane cholesterol and extracellular sodium. Proc. Natl. Acad. Sci. USA 2017, 114, E11141–E11150. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Bulkley, D.; Xin, Y.; Roberts, K.J.; Asarnow, D.E.; Sharma, A.; Myers, B.R.; Cho, W.; Cheng, Y.; Beachy, P.A. Structural Basis for Cholesterol Transport-like Activity of the Hedgehog Receptor Patched. Cell 2018, 175, 1352–1364.e14. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Li, X. Mechanistic Insights into the Generation and Transduction of Hedgehog Signaling. Trends Biochem. Sci. 2020, 45, 397–410. [Google Scholar] [CrossRef]
- Huang, P.; Nedelcu, D.; Watanabe, M.; Jao, C.; Kim, Y.; Liu, J.; Salic, A. Cellular Cholesterol Directly Activates Smooth-ened in Hedgehog Signaling. Cell 2016, 166, 1176–1187.e14. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Schmiege, P.; Coutavas, E.; Li, X. Two Patched molecules engage distinct sites on Hedgehog yielding a signal-ing-competent complex. Science 2018, 362, 6410. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Schmiege, P.; Coutavas, E.; Wang, J.; Li, X. Structures of human Patched and its complex with native pal-mitoylated sonic hedgehog. Nature 2018, 560, 128–132. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Sasai, N.; Ma, G.; Yue, T.; Jia, J.; Briscoe, J.; Jiang, J. Sonic Hedgehog Dependent Phosphorylation by CK1α and GRK2 Is Required for Ciliary Accumulation and Activation of Smoothened. PLoS Biol. 2011, 9, e1001083. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Lee, R.T.H.; Pusapati, G.V.; Iyu, A.; Rohatgi, R.; Ingham, P.W. An essential role for Grk2 in Hedgehog signalling downstream of Smoothened. EMBO Rep. 2016, 17, 739–752. [Google Scholar] [CrossRef] [PubMed]
- Rohatgi, R.; Milenkovic, L.; Scott, M.P. Patched1 Regulates Hedgehog Signaling at the Primary Cilium. Science 2007, 317, 372–376. [Google Scholar] [CrossRef]
- Kovacs, J.J.; Whalen, E.J.; Liu, R.; Xiao, K.; Kim, J.; Chen, M.; Wang, J.; Chen, W.; Lefkowitz, R.J. Beta-arrestin-mediated localization of smoothened to the primary cilium. Science 2008, 320, 1777–1781. [Google Scholar] [CrossRef] [PubMed]
- Kogerman, P.; Grimm, T.; Kogerman, L.; Krause, D.R.; Undén, A.B.; Sandstedt, B.; Toftgård, R.; Zaphiropoulos, P. Mammalian Suppressor-of-Fused modulates nuclear–cytoplasmic shuttling of GLI-1. Nat. Cell Biol. 1999, 1, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Li, Y. Evidence for the direct involvement of {beta}TrCP in Gli3 protein processing. Proc. Natl. Acad. Sci. USA 2006, 103, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Wang, B. A Novel Protein-processing Domain in Gli2 and Gli3 Differentially Blocks Complete Protein Degradation by the Proteasome. J. Biol. Chem. 2007, 282, 10846–10852. [Google Scholar] [CrossRef] [PubMed]
- Niewiadomski, P.; Kong, J.H.; Ahrends, R.; Ma, Y.; Humke, E.W.; Khan, S.; Teruel, M.N.; Novitch, B.G.; Rohatgi, R. Gli Protein Activity Is Controlled by Multisite Phosphorylation in Vertebrate Hedgehog Signaling. Cell Rep. 2013, 6, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Bai, C.B.; Joyner, A.L.; Wang, B. Sonic hedgehog Signaling Regulates Gli2 Transcriptional Activity by Suppressing Its Processing and Degradation. Mol. Cell. Biol. 2006, 26, 3365–3377. [Google Scholar] [CrossRef]
- Humke, E.W.; Dorn, K.V.; Milenkovic, L.; Scott, M.P.; Rohatgi, R. The output of Hedgehog signaling is controlled by the dynamic association between Suppressor of Fused and the Gli proteins. Genes Dev. 2010, 24, 670–682. [Google Scholar] [CrossRef] [PubMed]
- Tukachinsky, H.; Lopez, L.V.; Salic, A. A mechanism for vertebrate Hedgehog signaling: Recruitment to cilia and dissoci-ation of SuFu-Gli protein complexes. J. Cell Biol. 2010, 191, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Shen, L.; Law, K.; Zhang, Z.; Liu, X.; Hua, H.; Li, S.; Huang, H.; Yue, S.; Hui, C.-C.; et al. Suppressor of Fused Chaperones Gli Proteins to Generate Transcriptional Responses to Sonic Hedgehog Signaling. Mol. Cell. Biol. 2017, 37, e00421-16. [Google Scholar] [CrossRef] [PubMed]
- Dorn, K.V.; Hughes, C.E.; Rohatgi, R. A Smoothened-Evc2 Complex Transduces the Hedgehog Signal at Primary Cilia. Dev. Cell 2012, 23, 823–835. [Google Scholar] [CrossRef]
- Yang, C.; Chen, W.; Chen, Y.; Jiang, J. Smoothened transduces Hedgehog signal by forming a complex with Evc/Evc2. Cell Res. 2012, 22, 1593–1604. [Google Scholar] [CrossRef] [PubMed]
- Caparros-Martin, J.A.; Valencia, M.; Reytor, E.; Pacheco, M.; Fernandez, M.; Perez-Aytes, A.; Gean, E.; Lapunzina, P.; Peters, H.; Goodship, J.A.; et al. The ciliary Evc/Evc2 complex interacts with Smo and controls Hedgehog pathway activity in chon-drocytes by regulating Sufu/Gli3 dissociation and Gli3 trafficking in primary cilia. Hum. Mol. Genet. 2013, 22, 124–139. [Google Scholar] [CrossRef] [PubMed]
- Arveseth, C.D.; Happ, J.T.; Hedeen, D.S.; Zhu, J.-F.; Capener, J.L.; Shaw, D.K.; Deshpande, I.; Liang, J.; Xu, J.; Stubben, S.L.; et al. Smoothened transduces Hedgehog signals via activity-dependent sequestration of PKA catalytic subunits. PLoS Biol. 2021, 19, e3001191. [Google Scholar] [CrossRef]
- Kinzler, K.W.; Ruppert, J.M.; Bigner, S.H.; Vogelstein, B. The GLI gene is a member of the Kruppel family of zinc finger proteins. Nature 1988, 332, 371–374. [Google Scholar] [CrossRef]
- Hui, C.-C.; Angers, S. Gli Proteins in Development and Disease. Annu. Rev. Cell Dev. Biol. 2011, 27, 513–537. [Google Scholar] [CrossRef] [PubMed]
- Kinzler, K.W.; Vogelstein, B. The GLI gene encodes a nuclear protein which binds specific sequences in the human genome. Mol. Cell Biol. 1990, 10, 634–642. [Google Scholar] [PubMed]
- Dierks, C.; Grbic, J.; Zirlik, K.; Beigi, R.; Englund, N.P.; Guo, G.-R.; Veelken, H.; Engelhardt, M.; Mertelsmann, R.; Kelleher, J.F.; et al. Essential role of stromally induced hedgehog signaling in B-cell malignancies. Nat. Med. 2007, 13, 944–951. [Google Scholar] [CrossRef]
- Reifenberger, J.; Wolter, M.; Knobbe, C.B.; Kohler, B.; Schonicke, A.; Scharwachter, C.; Kumar, K.; Blaschke, B.; Ruzicka, T.; Reifenberger, G. Somatic mutations in the PTCH, SMOH, SUFUH and TP53 genes in sporadic basal cell carcinomas. Br. J. Dermatol. 2005, 152, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Lindström, E.; Shimokawa, T.; Toftgård, R.; Zaphiropoulos, P. PTCH mutations: Distribution and analyses. Hum. Mutat. 2006, 27, 215–219. [Google Scholar] [CrossRef]
- Johnson, R.L.; Rothman, A.L.; Xie, J.; Goodrich, L.V.; Bare, J.W.; Bonifas, J.M.; Quinn, A.G.; Myers, R.M.; Cox, D.R.; Epstein, E.H.; et al. Human Homolog of patched, a Candidate Gene for the Basal Cell Nevus Syndrome. Science 1996, 272, 1668–1671. [Google Scholar] [CrossRef]
- Xie, J.; Murone, M.; Luoh, S.M.; Ryan, A.; Gu, Q.; Zhang, C.; Bonifas, J.M.; Lam, C.W.; Hynes, M.; Goddard, A.; et al. Acti-vating Smoothened mutations in sporadic basal-cell carcinoma. Nature 1998, 391, 90–92. [Google Scholar] [CrossRef] [PubMed]
- Aszterbaum, M.; Rothman, A.; Fisher, M.; Xie, J.; Bonifas, J.M.; Zhang, X.; Epstein, E.H.; Johnson, R.L.; Scott, M.P. Identification of Mutations in the Human PATCHED Gene in Sporadic Basal Cell Carcinomas and in Patients with the Basal Cell Nevus Syndrome. J. Investig. Dermatol. 1998, 110, 885–888. [Google Scholar] [CrossRef] [PubMed]
- Goodrich, L.V.; Milenković, L.; Higgins, K.M.; Scott, M.P. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science 1997, 277, 1109–1113. [Google Scholar] [CrossRef] [PubMed]
- Wetmore, C.; Eberhart, D.E.; Curran, T. Loss of p53 but not ARF accelerates medulloblastoma in mice heterozygous for patched. Cancer Res. 2001, 61, 513–516. [Google Scholar] [PubMed]
- Berman, D.M.; Karhadkar, S.S.; Hallahan, A.R.; Pritchard, J.I.; Eberhart, C.G.; Watkins, D.N.; Chen, J.K.; Cooper, M.K.; Taipale, J.; Olson, J.M.; et al. Medulloblastoma Growth Inhibition by Hedgehog Pathway Blockade. Science 2002, 297, 1559–1561. [Google Scholar] [CrossRef]
- Lee, Y.; Kawagoe, R.; Sasai, K.; Li, Y.; Russell, H.R.; Curran, T.; McKinnon, P.J. Loss of suppressor-of-fused function promotes tumorigenesis. Oncogene 2007, 26, 6442–6447. [Google Scholar] [CrossRef]
- Mao, J.; Ligon, K.L.; Rakhlin, E.Y.; Thayer, S.P.; Bronson, R.T.; Rowitch, D.; McMahon, A.P. A Novel Somatic Mouse Model to Survey Tumorigenic Potential Applied to the Hedgehog Pathway. Cancer Res. 2006, 66, 10171–10178. [Google Scholar] [CrossRef]
- Jones, D.T.W.; Jaeger, N.; Kool, M.; Zichner, T.; Hutter, B.; Sultan, M.; Cho, Y.-J.; Pugh, T.; Hovestadt, V.; Stütz, A.M.; et al. Dissecting the genomic complexity underlying medulloblastoma. Nature 2012, 488, 100–105. [Google Scholar] [CrossRef]
- Pugh, T.J.; Weeraratne, S.D.; Archer, T.C.; Krummel, D.A.P.; Auclair, D.; Bochicchio, J.; Carneiro, M.O.; Carter, S.L.; Cibulskis, K.; Erlich, R.L.; et al. Medulloblastoma exome sequencing uncovers subtype-specific somatic mutations. Nature 2012, 488, 106–110. [Google Scholar] [CrossRef]
- Robinson, G.; Parker, M.; Kranenburg, T.A.; Lu, C.; Chen, X.; Ding, L.; Phoenix, T.N.; Hedlund, E.; Wei, L.; Zhu, X.; et al. Novel mutations target distinct subgroups of medulloblastoma. Nature 2012, 488, 43–48. [Google Scholar] [CrossRef]
- Tostar, U.; Malm, C.J.; Meis-Kindblom, J.M.; Kindblom, L.G.; Toftgard, R.; Unden, A.B. Deregulation of the hedgehog signalling pathway: A possible role for the PTCH and SUFU genes in human rhabdomyoma and rhabdomyosarcoma development. J. Pathol. 2006, 208, 17–25. [Google Scholar] [CrossRef]
- Bridge, J.A.; Liu, J.; Weibolt, V.; Baker, K.S.; Perry, D.; Kruger, R.; Qualman, S.; Barr, F.; Sorensen, P.; Triche, T.; et al. Novel genomic imbalances in embryonal rhabdomyosarcoma revealed by comparative genomic hybridization and fluorescence in situ hybridization: An Intergroup Rhabdomyosarcoma Study. Genes Chromosom. Cancer 2000, 27, 337–344. [Google Scholar] [CrossRef]
- Hettmer, S.; Teot, A.L.; Van Hummelen, P.; MacConaill, L.; Bronson, R.T.; Dall’Osso, C.; Mao, J.; McMahon, A.P.; Gruber, P.J.; Grier, E.H.; et al. Mutations in Hedgehog pathway genes in fetal rhabdomyomas. J. Pathol. 2013, 231, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, R.T.; McClary, A.C.; Myers, B.R.; Biscocho, J.; Neahring, L.; Kwei, A.K.; Qu, K.; Gong, X.; Ng, T.; Jones, C.D.; et al. Identification of recurrent SMO and BRAF mutations in ameloblastomas. Nat. Genet. 2014, 46, 722–725. [Google Scholar] [CrossRef] [PubMed]
- Youngblood, M.W.; Duran, D.; Montejo, J.D.; Li, C.; Omay, S.B.; Ozduman, K.; Sheth, A.H.; Zhao, A.Y.; Tyrtova, E.; Miyagishima, D.F.; et al. Correlations between genomic subgroup and clinical features in a cohort of more than 3000 meningiomas. J. Neurosurg. 2019, 133, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Clark, V.E.; Erson-Omay, E.Z.; Serin, A.; Yin, J.; Cotney, J.; Ozduman, K.; Avsar, T.; Li, J.; Murray, P.B.; Henegariu, O.; et al. Genomic analysis of non-NF2 meningiomas reveals mutations in TRAF7, KLF4, AKT1, and SMO. Science 2013, 339, 1077–1080. [Google Scholar] [CrossRef]
- Brastianos, P.K.; Horowitz, P.M.; Santagata, S.; Jones, R.T.; McKenna, A.; Getz, G.; Ligon, K.L.; Palescandolo, E.; Van Hummelen, P.; Ducar, M.D.; et al. Genomic sequencing of meningiomas identifies oncogenic SMO and AKT1 mutations. Nat. Genet. 2013, 45, 285–289. [Google Scholar] [CrossRef]
- Clark, V.E.; Harmanci, A.S.; Bai, H.; Youngblood, M.W.; Lee, T.I.; Baranoski, J.F.; Ercan-Sencicek, A.G.; Abraham, B.J.; Weintraub, A.S.; Hnisz, D.; et al. Recurrent somatic mutations in POLR2A define a distinct subset of meningiomas. Nat. Genet. 2016, 48, 1253–1259. [Google Scholar] [CrossRef]
- Watkins, D.N.; Berman, D.M.; Burkholder, S.G.; Wang, B.; Beachy, P.A.; Baylin, S.B. Hedgehog signalling within airway epithelial progenitors and in small-cell lung cancer. Nature 2003, 422, 313–317. [Google Scholar] [CrossRef]
- Park, K.-S.; Martelotto, L.G.; Peifer, M.; Sos, M.L.; Karnezis, A.N.; Mahjoub, M.R.; Bernard, K.; Conklin, J.F.; Szczepny, A.; Yuan, J.; et al. A crucial requirement for Hedgehog signaling in small cell lung cancer. Nat. Med. 2011, 17, 1504–1508. [Google Scholar] [CrossRef]
- Szczepny, A.; Rogers, S.; Jayasekara, W.S.N.; Park, K.; McCloy, R.A.; Cochrane, C.; Ganju, V.; Cooper, W.A.; Sage, J.; Peacock, C.D.; et al. The role of canonical and non-canonical Hedgehog signaling in tumor progression in a mouse model of small cell lung cancer. Oncogene 2017, 36, 5544–5550. [Google Scholar] [CrossRef]
- Zahreddine, H.A.; Culjkovic-Kraljacic, B.; Assouline, S.; Gendron, P.; Romeo, A.A.; Morris, S.J.; Cormack, G.; Jaquith, J.B.; Cerchietti, L.; Cocolakis, E.; et al. The sonic hedgehog factor GLI1 imparts drug resistance through inducible glucuronidation. Nature 2014, 511, 90–93. [Google Scholar] [CrossRef]
- Li, X.; Chen, F.; Zhu, Q.; Ding, B.; Zhong, Q.; Huang, K.; Jiang, X.; Wang, Z.; Yin, C.; Zhu, Y.; et al. Gli-1/PI3K/AKT/NF-kB pathway mediates resistance to radiation and is a target for reversion of responses in refractory acute myeloid leukemia cells. Oncotarget 2016, 7, 33004–33015. [Google Scholar] [CrossRef] [PubMed]
- Norsworthy, K.J.; By, K.; Subramaniam, S.; Zhuang, L.; Del Valle, P.L.; Przepiorka, D.; Shen, Y.-L.; Sheth, C.M.; Liu, C.; Leong, R.; et al. FDA Approval Summary: Glasdegib for Newly Diagnosed Acute Myeloid Leukemia. Clin. Cancer Res. 2019, 25, 6021–6025. [Google Scholar] [CrossRef] [PubMed]
- Shin, K.; Lim, A.; Odegaard, J.I.; Honeycutt, J.D.; Kawano, S.; Hsieh, M.H.; Beachy, P.A. Cellular origin of bladder neo-plasia and tissue dynamics of its progression to invasive carcinoma. Nat. Cell Biol. 2014, 16, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Shin, K.; Lim, A.; Zhao, C.; Sahoo, D.; Pan, Y.; Spiekerkoetter, E.; Liao, J.C.; Beachy, P.A. Hedgehog Signaling Restrains Bladder Cancer Progression by Eliciting Stromal Production of Urothelial Differentiation Factors. Cancer Cell 2014, 26, 521–533. [Google Scholar] [CrossRef]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.; Perera, R.M.; Wang, H.; Wu, D.C.; Liu, X.S.; Han, S.; Fitamant, J.; Jones, P.D.; Ghanta, K.S.; Kawano, S.; et al. Stro-mal response to Hedgehog signaling restrains pancreatic cancer progression. Proc. Natl. Acad. Sci. USA 2014, 111, E3091–E3100. [Google Scholar] [CrossRef]
- Gerling, M.; Büller, N.V.J.A.; Kirn, L.M.; Joost, S.; Frings, O.; Englert, B.; Bergström, Å.; Kuiper, R.V.; Blaas, L.; Wielenga, M.C.B.; et al. Stromal Hedgehog signalling is downregulated in colon cancer and its restoration restrains tumour growth. Nat. Commun. 2016, 7, 12321. [Google Scholar] [CrossRef]
- Lee, J.J.; Rothenberg, M.E.; Seeley, E.S.; Zimdahl, B.; Kawano, S.; Lu, W.-J.; Shin, K.; Sakata-Kato, T.; Chen, J.K.; Diehn, M.; et al. Control of inflammation by stromal Hedgehog pathway activation restrains colitis. Proc. Natl. Acad. Sci. USA 2016, 113, E7545–E7553. [Google Scholar] [CrossRef]
- Yang, Z.; Peng, Y.-C.; Gopalan, A.; Gao, D.; Chen, Y.; Joyner, A.L. Stromal Hedgehog signaling maintains smooth muscle and hampers micro-invasive prostate cancer. Dis. Models Mech. 2016, 10, 39–52. [Google Scholar] [CrossRef]
- Kasiri, S.; Chen, B.; Wilson, A.N.; Reczek, A.; Mazambani, S.; Gadhvi, J.; Noel, E.; Marriam, U.; Mino, B.; Lu, W.; et al. Stromal Hedgehog pathway activation by IHH suppresses lung adenocarcinoma growth and metastasis by limiting reactive oxygen species. Oncogene 2020, 39, 3258–3275. [Google Scholar] [CrossRef]
- Bakshi, A.; Chaudhary, S.C.; Rana, M.; Elmets, C.A.; Athar, M. Basal cell carcinoma pathogenesis and therapy involving hedgehog signaling and beyond. Mol. Carcinog. 2017, 56, 2543–2557. [Google Scholar] [CrossRef] [PubMed]
- Grachtchouk, M.; Mo, R.; Yu, S.; Zhang, X.; Sasaki, H.; Hui, C.C.; Dlugosz, A.A. Basal cell carcinomas in mice overex-pressing Gli2 in skin. Nat. Genet. 2000, 24, 216–217. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, M.; Unden, A.B.; Krause, D.; Malmqwist, U.; Raza, K.; Zaphiropoulos, P.G.; Toftgard, R. Induction of basal cell carcinomas and trichoepitheliomas in mice overexpressing GLI-1. Proc. Natl. Acad. Sci. USA 2000, 97, 3438–3443. [Google Scholar] [CrossRef]
- Nitzki, F.; Becker, M.; Frommhold, A.; Schulz-Schaeffer, W.; Hahn, H. Patched knockout mouse models of Basal cell car-cinoma. J. Skin Cancer 2012, 2012, 907543. [Google Scholar] [CrossRef] [PubMed]
- Migden, M.; Farberg, A.S.; Dummer, R.; Squittieri, N.; Hanke, C.W. A Review of Hedgehog Inhibitors Sonidegib and Vismodegib for Treatment of Advanced Basal Cell Carcinoma. J. Drugs Dermatol. 2021, 20, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Robarge, K.D.; Brunton, S.A.; Castanedo, G.M.; Cui, Y.; Dina, M.S.; Goldsmith, R.; Gould, S.E.; Guichert, O.; Gunzner, J.L.; Halladay, J.; et al. GDC-0449—A potent inhibitor of the hedgehog pathway. Bioorg. Med. Chem. Lett. 2009, 19, 5576–5581. [Google Scholar] [CrossRef] [PubMed]
- Sekulic, A.; Migden, M.R.; Oro, A.E.; Dirix, L.; Lewis, K.D.; Hainsworth, J.D.; Solomon, J.A.; Yoo, S.; Arron, S.T.; Friedlander, P.A.; et al. Efficacy and Safety of Vismodegib in Advanced Basal-Cell Carcinoma. N. Engl. J. Med. 2012, 366, 2171–2179. [Google Scholar] [CrossRef]
- Sekulic, A.; Migden, M.R.; Lewis, K.; Hainsworth, J.D.; Solomon, J.A.; Yoo, S.; Arron, S.T.; Friedlander, P.A.; Marmur, E.; Rudin, C.M.; et al. Pivotal ERIVANCE basal cell carcinoma (BCC) study: 12-month update of efficacy and safety of vismo-degib in advanced BCC. J. Am. Acad. Dermatol. 2015, 72, 1021–1026.e8. [Google Scholar] [CrossRef]
- Basset-Seguin, N.; Hauschild, A.; Grob, J.-J.; Kunstfeld, R.; Dréno, B.; Mortier, L.; Ascierto, A.P.; Licitra, L.; Dutriaux, C.; Thomas, L.; et al. Vismodegib in patients with advanced basal cell carcinoma (STEVIE): A pre-planned interim analysis of an international, open-label trial. Lancet Oncol. 2015, 16, 729–736. [Google Scholar] [CrossRef]
- Tang, J.Y.; Mackay-Wiggan, J.M.; Aszterbaum, M.; Yauch, R.L.; Lindgren, J.; Chang, K.; Coppola, C.; Chanana, A.M.; Marji, J.; Bickers, D.R.; et al. Inhibiting the Hedgehog Pathway in Patients with the Basal-Cell Nevus Syndrome. N. Engl. J. Med. 2012, 366, 2180–2188. [Google Scholar] [CrossRef]
- Pan, S.; Wu, X.; Jiang, J.; Gao, W.; Wan, Y.; Cheng, D.; Han, D.; Liu, J.; Englund, N.P.; Wang, Y.; et al. Discovery of NVP-LDE225, a Potent and Selective Smoothened Antagonist. ACS Med. Chem. Lett. 2010, 1, 130–134. [Google Scholar] [CrossRef] [PubMed]
- Migden, M.R.; Guminski, A.; Gutzmer, R.; Dirix, L.; Lewis, K.D.; Combemale, P.; Herd, R.M.; Kudchadkar, R.; Trefzer, U.; Gogov, S.; et al. Treatment with two different doses of sonidegib in patients with locally advanced or metastatic basal cell carcinoma (BOLT): A multicentre, randomised, double-blind phase 2 trial. Lancet Oncol. 2015, 16, 716–728. [Google Scholar] [CrossRef]
- Axelson, M.; Liu, K.; Jiang, X.; He, K.; Wang, J.; Zhao, H.; Kufrin, D.; Palmby, T.; Dong, Z.; Russell, A.M.; et al. U.S. Food and Drug Administration Approval: Vismodegib for Recurrent, Locally Advanced, or Metastatic Basal Cell Carcinoma. Clin. Cancer Res. 2013, 19, 2289–2293. [Google Scholar] [CrossRef] [PubMed]
- Casey, D.; Demko, S.; Shord, S.; Zhao, H.; Chen, H.; He, K.; Putman, A.; Helms, W.; Keegan, P.; Pazdur, R. FDA Ap-proval Summary: Sonidegib for Locally Advanced Basal Cell Carcinoma. Clin. Cancer Res. 2017, 23, 2377–2381. [Google Scholar] [CrossRef] [PubMed]
- Atwood, S.X.; Sarin, K.Y.; Whitson, R.J.; Li, J.R.; Kim, G.; Rezaee, M.; Ally, M.S.; Kim, J.; Yao, C.; Chang, A.L.S.; et al. Smoothened Variants Explain the Majority of Drug Resistance in Basal Cell Carcinoma. Cancer Cell 2015, 27, 342–353. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, H.J.; Pau, G.; Dijkgraaf, G.J.; Basset-Seguin, N.; Modrusan, Z.; Januario, T.; Tsui, V.; Durham, A.B.; Dlugosz, A.A.; Haverty, P.M.; et al. Genomic Analysis of Smoothened Inhibitor Resistance in Basal Cell Carcinoma. Cancer Cell 2015, 27, 327–341. [Google Scholar] [CrossRef] [PubMed]
- Yauch, R.L.; Dijkgraaf, G.J.P.; Alicke, B.; Januario, T.; Ahn, C.P.; Holcomb, T.; Pujara, K.; Stinson, J.; Callahan, C.A.; Tang, T.; et al. Smoothened Mutation Confers Resistance to a Hedgehog Pathway Inhibitor in Medulloblastoma. Science 2009, 326, 572–574. [Google Scholar] [CrossRef]
- Reifenberger, J.; Wolter, M.; Weber, R.G.; Megahed, M.; Ruzicka, T.; Lichter, P.; Reifenberger, G. Missense mutations in SMOH in sporadic basal cell carcinomas of the skin and primitive neuroectodermal tumors of the central nervous system. Cancer Res. 1998, 58, 1798–1803. [Google Scholar]
- Taipale, J.; Chen, J.; Cooper, M.K.; Wang, B.; Mann, R.K.; Milenkovic, L.; Scott, M.P.; Beachy, P.A. Effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature 2000, 406, 1005–1009. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2014–2018. Neuro Oncol. 2021, 23, iii1–iii105. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.C.; Fuller, C.; Hogg, T.L.; Dalton, J.; Finkelstein, D.; Lau, C.C.; Chintagumpala, M.; Adesina, A.; Ashley, D.M.; Kellie, S.J.; et al. Genomics Identifies Medulloblastoma Subgroups That Are Enriched for Specific Genetic Alterations. J. Clin. Oncol. 2006, 24, 1924–1931. [Google Scholar] [CrossRef]
- Kool, M.; Koster, J.; Bunt, J.; Hasselt, N.E.; Lakeman, A.; Van Sluis, P.; Troost, D.; Meeteren, N.S.-V.; Caron, H.N.; Cloos, J.; et al. Integrated Genomics Identifies Five Medulloblastoma Subtypes with Distinct Genetic Profiles, Pathway Signatures and Clinicopathological Features. PLoS ONE 2008, 3, e3088. [Google Scholar] [CrossRef] [PubMed]
- Northcott, P.A.; Korshunov, A.; Witt, H.; Hielscher, T.; Eberhart, C.G.; Mack, S.C.; Bouffet, E.; Clifford, S.C.; Hawkins, C.E.; French, P.; et al. Medulloblastoma Comprises Four Distinct Molecular Variants. J. Clin. Oncol. 2011, 29, 1408–1414. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.D.; Northcott, P.A.; Korshunov, A.; Remke, M.; Cho, Y.-J.; Clifford, S.C.; Eberhart, C.G.; Parsons, D.W.; Rutkowski, S.; Gajjar, A.; et al. Molecular subgroups of medulloblastoma: The current consensus. Acta Neuropathol. 2011, 123, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Kool, M.; Korshunov, A.; Remke, M.; Jones, D.T.; Schlanstein, M.; Northcott, P.A.; Cho, Y.J.; Koster, J.; Schouten-van Meeteren, A.; van Vuurden, D.; et al. Molecular subgroups of medulloblastoma: An international meta-analysis of transcrip-tome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol. Berl. 2012, 123, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Northcott, P.A.; Hielscher, T.; Dubuc, A.; Mack, S.C.; Shih, D.J.H.; Remke, M.; Al-Halabi, H.; Albrecht, S.; Jabado, N.; Eberhart, C.G.; et al. Pediatric and adult sonic hedgehog medulloblastomas are clinically and molecularly distinct. Acta Neuropathol. 2011, 122, 231–240. [Google Scholar] [CrossRef]
- Dahmane, N.; Altaba, I.A.R. Sonic hedgehog regulates the growth and patterning of the cerebellum. Development 1999, 126, 3089–3100. [Google Scholar] [CrossRef] [PubMed]
- Wallace, V.A. Purkinje-cell-derived Sonic hedgehog regulates granule neuron precursor cell proliferation in the developing mouse cerebellum. Curr. Biol. 1999, 9, 445–448. [Google Scholar] [CrossRef]
- Wechsler-Reya, R.J.; Scott, M.P. Control of Neuronal Precursor Proliferation in the Cerebellum by Sonic Hedgehog. Neuron 1999, 22, 103–114. [Google Scholar] [CrossRef]
- Li, Y.; Song, Q.; Day, B.W. Phase I and phase II sonidegib and vismodegib clinical trials for the treatment of paediatric and adult MB patients: A systemic review and meta-analysis. Acta Neuropathol. Commun. 2019, 7, 123–128. [Google Scholar] [CrossRef] [PubMed]
- LoRusso, P.M.; Rudin, C.M.; Reddy, J.C.; Tibes, R.; Weiss, G.J.; Borad, M.J.; Hann, C.L.; Brahmer, J.R.; Chang, I.; Darbonne, W.C.; et al. Phase I Trial of Hedgehog Pathway Inhibitor Vismodegib (GDC-0449) in Patients with Refractory, Locally Advanced or Metastatic Solid Tumors. Clin. Cancer Res. 2011, 17, 2502–2511. [Google Scholar] [CrossRef] [PubMed]
- Gajjar, A.; Stewart, C.F.; Ellison, D.W.; Kaste, S.; Kun, L.E.; Packer, R.J.; Goldman, S.; Chintagumpala, M.; Wallace, D.; Takebe, N.; et al. Phase I study of vismodegib in children with recurrent or refractory medulloblastoma: A pediatric brain tumor consortium study. Clin. Cancer Res. 2013, 19, 6305–6312. [Google Scholar] [CrossRef] [PubMed]
- Rodon, J.; Tawbi, H.A.; Thomas, A.L.; Stoller, R.G.; Turtschi, C.P.; Baselga, J.; Sarantopoulos, J.; Mahalingam, D.; Shou, Y.; Moles, M.A.; et al. A Phase I, Multicenter, Open-Label, First-in-Human, Dose-Escalation Study of the Oral Smoothened Inhibitor Sonidegib (LDE225) in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2014, 20, 1900–1909. [Google Scholar] [CrossRef] [PubMed]
- Robinson, G.W.; Orr, B.A.; Wu, G.; Gururangan, S.; Lin, T.; Qaddoumi, I.; Packer, R.J.; Goldman, S.; Prados, M.D.; Desjardins, A.; et al. Vismodegib Exerts Targeted Efficacy Against Recurrent Sonic Hedgehog–Subgroup Medulloblastoma: Results From Phase II Pediatric Brain Tumor Consortium Studies PBTC-025B and PBTC-032. J. Clin. Oncol. 2015, 33, 2646–2654. [Google Scholar] [CrossRef]
- Kieran, M.W.; Chisholm, J.; Casanova, M.; Brandes, A.A.; Aerts, I.; Bouffet, E.; Bailey, S.; Leary, S.; MacDonald, T.J.; Mechinaud, F.; et al. Phase I study of oral sonidegib (LDE225) in pediatric brain and solid tumors and a phase II study in children and adults with relapsed medulloblastoma. Neuro Oncol. 2017, 19, 1542–1552. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.A.; Guicherit, O.M.; Zaharian, B.I.; Xu, Y.; Chai, L.; Wichterle, H.; Kon, C.; Gatchalian, C.; Porter, J.A.; Rubin, L.L.; et al. Identification of a small molecule inhibitor of the hedgehog signaling pathway: Effects on basal cell carcinoma-like lesions. Proc. Natl. Acad. Sci. USA 2003, 100, 4616–4621. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H.; Ng, J.M.; Curran, T. Transient Inhibition of the Hedgehog Pathway in Young Mice Causes Permanent Defects in Bone Structure. Cancer Cell 2008, 13, 249–260. [Google Scholar] [CrossRef] [PubMed]
- Robinson, G.W.; Kaste, S.C.; Chemaitilly, W.; Bowers, D.C.; Laughton, S.; Smith, A.; Gottardo, N.G.; Partap, S.; Bendel, A.; Wright, K.D.; et al. Irreversible growth plate fusions in children with medulloblastoma treated with a targeted hedgehog pathway inhibitor. Oncotarget 2017, 8, 69295–69302. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, R.; Fuchs, J.; Rodeberg, D. Rhabdomyosarcoma. Semin. Pediatr. Surg. 2016, 25, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Gorlin, R.J. Nevoid basal-cell carcinoma syndrome. Med. Baltim. 1987, 66, 98–113. [Google Scholar] [CrossRef] [PubMed]
- Pressey, J.G.; Anderson, J.R.; Crossman, D.; Lynch, J.C.; Barr, F.G. Hedgehog pathway activity in pediatric embryonal rhabdomyosarcoma and undifferentiated sarcoma: A report from the Children’s Oncology Group. Pediatr. Blood Cancer 2011, 57, 930–938. [Google Scholar] [CrossRef]
- Paulson, V.; Chandler, G.; Rakheja, D.; Galindo, R.L.; Wilson, K.; Amatruda, J.F.; Cameron, S. High-resolution array CGH identifies common mechanisms that drive embryonal rhabdomyosarcoma pathogenesis. Genes Chromosom. Cancer 2011, 50, 397–408. [Google Scholar] [CrossRef]
- Ragazzini, P.; Gamberi, G.; Pazzaglia, L.; Serra, M.; Magagnoli, G.; Ponticelli, F.; Ferrari, C.; Ghinelli, C.; Alberghini, M.; Ber-toni, F.; et al. Amplification of CDK4, MDM2, SAS and GLI genes in leiomyosarcoma, alveolar and embryonal rhabdomyo-sarcoma. Histol. Histopathol. 2004, 19, 401–411. [Google Scholar] [PubMed]
- Oue, T.; Yoneda, A.; Uehara, S.; Yamanaka, H.; Fukuzawa, M. Increased expression of the hedgehog signaling pathway in pediatric solid malignancies. J. Pediatr. Surg. 2010, 45, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Shern, J.F.; Chen, L.; Chmielecki, J.; Wei, J.S.; Patidar, R.; Rosenberg, M.; Ambrogio, L.; Auclair, D.; Wang, J.; Song, Y.K.; et al. Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic ax-is in fusion-positive and fusion-negative tumors. Cancer Discov. 2014, 4, 216–231. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Stewart, E.; Shelat, A.A.; Qu, C.; Bahrami, A.; Hatley, M.; Wu, G.; Bradley, C.; McEvoy, J.; Pappo, A.; et al. Tar-geting oxidative stress in embryonal rhabdomyosarcoma. Cancer Cell. 2013, 24, 710–724. [Google Scholar] [CrossRef] [PubMed]
- Effiom, O.A.; Ogundana, O.M.; Akinshipo, A.O.; Akintoye, S.O. Ameloblastoma: Current etiopathological concepts and management. Oral Dis. 2017, 24, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Dassule, H.; Lewis, P.; Bei, M.; Maas, R.; McMahon, A. Sonic hedgehog regulates growth and morphogenesis of the tooth. Development 2000, 127, 4775–4785. [Google Scholar] [CrossRef]
- Dassule, H.R.; McMahon, A.P. Analysis of Epithelial–Mesenchymal Interactions in the Initial Morphogenesis of the Mammalian Tooth. Dev. Biol. 1998, 202, 215–227. [Google Scholar] [CrossRef]
- Barreto, D.; Gomez, R.; Bale, A.; Boson, W.; De Marco, L. PTCH Gene Mutations in Odontogenic Keratocysts. J. Dent. Res. 2000, 79, 1418–1422. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Chen, X.-M.; Sun, Z.-J.; Bian, Z.; Fan, M.-W.; Chen, Z. Epithelial expression of SHH signaling pathway in odontogenic tumors. Oral Oncol. 2006, 42, 398–408. [Google Scholar] [CrossRef] [PubMed]
- Barreto, D.C.; Bale, A.E.; De Marco, L.; Gomez, R.S. Immunolocalization of PTCH protein in odontogenic cysts and tumors. J. Dent. Res. 2002, 81, 757–760. [Google Scholar] [CrossRef] [PubMed]
- Heikinheimo, K.; Jee, K.J.; Niini, T.; Aalto, Y.; Happonen, R.P.; Leivo, I.; Knuutila, S. Gene expression profiling of amelo-blastoma and human tooth germ by means of a cDNA microarray. J. Dent. Res. 2002, 81, 525–530. [Google Scholar] [CrossRef]
- Gurgel, C.A.S.; Buim, M.E.C.; Carvalho, K.C.; Sales, C.B.S.; Reis, M.G.; De Souza, R.O.; Valverde, L.D.F.; De Azevedo, R.A.; Dos Santos, J.N.; Soares, F.A.; et al. Transcriptional profiles of SHH pathway genes in keratocystic odontogenic tumor and ameloblastoma. J. Oral Pathol. Med. 2014, 43, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Cioffi, G.; Gittleman, H.; Patil, N.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2012–2016. Neuro Oncol. 2019, 21, v1–v100. [Google Scholar] [CrossRef]
- Roberts, K.J.; Kershner, A.M.; Beachy, P.A. The Stromal Niche for Epithelial Stem Cells: A Template for Regeneration and a Brake on Malignancy. Cancer Cell 2017, 32, 404–410. [Google Scholar] [CrossRef]
- Lenis, A.T.; Lec, P.M.; Chamie, K.; Mshs, M.D. Bladder Cancer: A Review. JAMA 2020, 324, 1980–1991. [Google Scholar] [CrossRef]
- Linnenbach, A.J.; Pressler, L.B.; Seng, B.A.; Kimmel, B.S.; Tomaszewski, J.E.; Malkowicz, S.B. Characterization of chro-mosome 9 deletions in transitional cell carcinoma by microsatellite assay. Hum. Mol. Genet. 1993, 2, 1407–1411. [Google Scholar] [CrossRef]
- Spruck, C.H., 3rd; Ohneseit, P.F.; Gonzalez-Zulueta, M.; Esrig, D.; Miyao, N.; Tsai, Y.C.; Lerner, S.P.; Schmutte, C.; Yang, A.S.; Cote, R.; et al. Two molecular pathways to transitional cell carcinoma of the bladder. Cancer Res. 1994, 54, 784–788. [Google Scholar]
- McGarvey, T.W.; Maruta, Y.; Tomaszewski, E.J.; Linnenbach, A.J.; Malkowicz, S.B. PTCH gene mutations in invasive transitional cell carcinoma of the bladder. Oncogene 1998, 17, 1167–1172. [Google Scholar] [CrossRef]
- Habuchi, T.; Devlin, J.; Elder, P.A.; Knowles, M.A. Detailed deletion mapping of chromosome 9q in bladder cancer: Evi-dence for two tumour suppressor loci. Oncogene 1995, 11, 1671–1674. [Google Scholar]
- Robertson, A.G.; Kim, J.; Al-Ahmadie, H.; Bellmunt, J.; Guo, G.; Cherniack, A.D.; Hinoue, T.; Laird, P.W.; Hoadley, K.A.; Akbani, R.; et al. Comprehensive Molecular Characterization of Muscle-Invasive Bladder Cancer. Cell 2017, 171, 540–556.e25. [Google Scholar] [CrossRef] [PubMed]
- Shin, K.; Lee, J.; Guo, N.; Kim, J.; Lim, A.; Qu, L.; Mysorekar, I.U.; Beachy, P.A. Hedgehog/Wnt feedback supports re-generative proliferation of epithelial stem cells in bladder. Nature 2011, 472, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Pignot, G.; Vieillefond, A.; Vacher, S.; Zerbib, M.; Debre, B.; Lidereau, R.; Amsellem-Ouazana, D.; Bieche, I. Hedgehog pathway activation in human transitional cell carcinoma of the bladder. Br. J. Cancer 2012, 106, 1177–1186. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.-M.; Gingras, M.-C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.C.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef]
- Cao, L.; Huang, C.; Cui Zhou, D.; Hu, Y.; Lih, T.M.; Savage, S.R.; Krug, K.; Clark, D.J.; Schnaubelt, M.; Chen, L.; et al. Pro-teogenomic characterization of pancreatic ductal adenocarcinoma. Cell 2021, 184, 5031–5052.e26. [Google Scholar] [CrossRef]
- Thayer, S.P.; di Magliano, M.P.; Heiser, P.W.; Nielsen, C.M.; Roberts, D.J.; Lauwers, G.Y.; Qi, Y.P.; Gysin, S.; Castillo, C.F.-D.; Yajnik, V.; et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature 2003, 425, 851–856. [Google Scholar] [CrossRef]
- Berman, D.M.; Karhadkar, S.S.; Maitra, A.; de Oca, R.M.; Gerstenblith, M.R.; Briggs, K.; Parker, A.R.; Shimada, Y.; Eshleman, J.R.; Watkins, D.N.; et al. Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours. Nature 2003, 425, 846–851. [Google Scholar] [CrossRef]
- Yauch, R.L.; Gould, S.E.; Scales, S.J.; Tang, T.; Tian, H.; Ahn, C.P.; Marshall, D.; Fu, L.; Januario, T.; Kallop, D.; et al. A para-crine requirement for hedgehog signalling in cancer. Nature 2008, 455, 406–410. [Google Scholar] [CrossRef]
- Tian, H.; Callahan, C.A.; DuPree, K.J.; Darbonne, W.C.; Ahn, C.P.; Scales, S.J.; de Sauvage, F.J. Hedgehog signaling is restricted to the stromal compartment during pancreatic carcinogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 4254–4259. [Google Scholar] [CrossRef] [PubMed]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Cald-well, M.E.; Allard, D.; et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef] [PubMed]
- Catenacci, D.V.T.; Junttila, M.R.; Karrison, T.; Bahary, N.; Horiba, M.N.; Nattam, S.R.; Marsh, R.; Wallace, J.; Kozloff, M.; Rajdev, L.; et al. Randomized Phase Ib/II Study of Gemcitabine Plus Placebo or Vismodegib, a Hedgehog Pathway Inhibitor, in Patients with Metastatic Pancreatic Cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin Oncol. 2015, 33, 4284–4292. [Google Scholar] [CrossRef] [PubMed]
- De Jesus-Acosta, A.; Sugar, E.A.; O’Dwyer, P.J.; Ramanathan, R.K.; Von Hoff, D.D.; Rasheed, Z.; Zheng, L.; Begum, A.; Anders, R.; Maitra, A.; et al. Phase 2 study of vismodegib, a hedgehog inhibitor, combined with gemcitabine and nab-paclitaxel in patients with untreated metastatic pancreatic adenocarcinoma. Br. J. Cancer 2019, 122, 498–505. [Google Scholar] [CrossRef]
- Kim, E.J.; Sahai, V.; Abel, E.V.; Griffith, K.A.; Greenson, J.K.; Takebe, N.; Khan, G.N.; Blau, J.L.; Craig, R.; Balis, U.G.; et al. Pilot Clinical Trial of Hedgehog Pathway Inhibitor GDC-0449 (Vismodegib) in Combination with Gemcitabine in Patients with Metastatic Pancreatic Adenocarcinoma. Clin. Cancer Res. 2014, 20, 5937–5945. [Google Scholar] [CrossRef]
- Tremblay, M.R.; Lescarbeau, A.; Grogan, M.J.; Tan, E.; Lin, G.; Austad, B.C.; Yu, L.-C.; Behnke, M.L.; Nair, S.J.; Hagel, M.; et al. Discovery of a Potent and Orally Active Hedgehog Pathway Antagonist (IPI-926). J. Med. Chem. 2009, 52, 4400–4418. [Google Scholar] [CrossRef]
- Brunton, S.A.; Stibbard, J.H.; Rubin, L.L.; Guicherit, O.M.; Kruse, L.I.; Price, S.; di Lucrezia, R.; MacKinnon, C.H.; Avery, A.; Park, Y.; et al. Potent agonists of the Hedgehog signaling pathway. Bioorg. Med. Chem. Lett. 2009, 19, 4308–4311. [Google Scholar] [CrossRef]
- Ozdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef]
- Walton, K.D.; Gumucio, D.L. Hedgehog Signaling in Intestinal Development and Homeostasis. Annu. Rev. Physiol. 2021, 83, 359–380. [Google Scholar] [CrossRef]
- Berlin, J.; Bendell, J.C.; Hart, L.L.; Firdaus, I.; Gore, I.; Hermann, R.C.; Mulcahy, M.F.; Zalupski, M.M.; Mackey, H.M.; Yauch, R.L.; et al. A Randomized Phase II Trial of Vismodegib versus Placebo with FOLFOX or FOLFIRI and Bevacizumab in Patients with Previously Untreated Metastatic Colorectal Cancer. Clin. Cancer Res. 2013, 19, 258–267. [Google Scholar] [CrossRef]
- Freestone, S.H.; Marker, P.; Grace, O.C.; Tomlinson, D.C.; Cunha, G.R.; Harnden, P.; Thomson, A.A. Sonic hedgehog regulates prostatic growth and epithelial differentiation. Dev. Biol. 2003, 264, 352–362. [Google Scholar] [CrossRef] [PubMed]
- Lim, A.; Shin, K.; Zhao, C.; Kawano, S.; Beachy, P.A. Spatially restricted Hedgehog signalling regulates HGF-induced branching of the adult prostate. Nat. Cell Biol. 2014, 16, 1135–1145. [Google Scholar] [CrossRef] [PubMed]
- Le, V.; He, Y.; Aldahl, J.; Hooker, E.; Yu, E.-J.; Olson, A.; Kim, W.K.; Lee, D.-H.; Wong, M.; Sheng, R.; et al. Loss of androgen signaling in mesenchymal sonic hedgehog responsive cells diminishes prostate development, growth, and regeneration. PLoS Genet. 2020, 16, e1008588. [Google Scholar] [CrossRef] [PubMed]
- Karhadkar, S.S.; Bova, G.S.; Abdallah, N.; Dhara, S.; Gardner, D.R.; Maitra, A.; Isaacs, J.T.; Berman, D.M.; Beachy, P.A. Hedgehog signalling in prostate regeneration, neoplasia and metastasis. Nature 2004, 431, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Pepicelli, C.V.; Dibble, C.C.; Catbagan, W.; Zarycki, J.L.; Laciak, R.; Gipp, J.; Shaw, A.; Lamm, M.L.G.; Munoz, A.; et al. Hedgehog Signaling Promotes Prostate Xenograft Tumor Growth. Endocrinology 2004, 145, 3961–3970. [Google Scholar] [CrossRef]
- Shaw, A.; Attia, S.; Bushman, W. Prostate stromal and urogenital sinus mesenchymal cell lines for investigations of stromal–epithelial interactions. Differentiation 2008, 76, 599–605. [Google Scholar] [CrossRef] [PubMed]
- Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins. A Study of Vismodegib in Men with Metastatic CRPC with Ac-cessible Metastatic Lesions for Tumor Biopsy. Available online: https://ClinicalTrials.gov/show/NCT02115828 (accessed on 12 June 2022).
- Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins. A Pre-Surgical Study of LDE225 in Men with High-Risk Localized Prostate Cancer. Available online: https://ClinicalTrials.gov/show/NCT02111187 (accessed on 12 June 2022).
- Pepicelli, C.V.; Lewis, P.M.; McMahon, A.P. Sonic hedgehog regulates branching morphogenesis in the mammalian lung. Curr. Biol. 1998, 8, 1083–1086. [Google Scholar] [CrossRef]
- Litingtung, Y.; Lei, L.; Westphal, H.; Chiang, C. Sonic hedgehog is essential to foregut development. Nat. Genet. 1998, 20, 58–61. [Google Scholar] [CrossRef] [PubMed]
- Bitgood, M.J.; McMahon, A.P. Hedgehog and Bmp genes are coexpressed at many diverse sites of cell-cell interaction in the mouse embryo. Dev. Biol. 1995, 172, 126–138. [Google Scholar] [CrossRef] [PubMed]
- Bellusci, S.; Furuta, Y.; Rush, M.; Henderson, R.; Winnier, G.; Hogan, B. Involvement of Sonic hedgehog (Shh) in mouse embryonic lung growth and morphogenesis. Development 1997, 124, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Kugler, M.C.; Loomis, C.A.; Zhao, Z.; Cushman, J.C.; Liu, L.; Munger, J.S. Sonic Hedgehog Signaling Regulates Myofi-broblast Function during Alveolar Septum Formation in Murine Postnatal Lung. Am. J. Respir. Cell Mol. Biol. 2017, 57, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Rankin, S.A.; Han, L.; McCracken, K.W.; Kenny, A.P.; Anglin, C.T.; Grigg, E.A.; Crawford, C.M.; Wells, J.M.; Shannon, J.M.; Zorn, A.M. A Retinoic Acid-Hedgehog Cascade Coordinates Mesoderm-Inducing Signals and Endoderm Competence during Lung Specification. Cell Rep. 2016, 16, 66–78. [Google Scholar] [CrossRef] [PubMed]
- Ericson, J.; Morton, S.; Kawakami, A.; Roelink, H.; Jessell, T.M. Two critical periods of Sonic Hedgehog signaling re-quired for the specification of motor neuron identity. Cell 1996, 87, 661–673. [Google Scholar] [CrossRef]
- Wellbrock, J.; Latuske, E.; Köhler, J.; Wagner, K.; Stamm, H.; Vettorazzi, E.; Vohwinkel, G.; Klokow, M.; Uibeleisen, R.; Ehm, P.; et al. Expression of Hedgehog Pathway Mediator GLI Represents a Negative Prognostic Marker in Human Acute Myeloid Leukemia and Its Inhibition Exerts Antileukemic Effects. Clin. Cancer Res. 2015, 21, 2388–2398. [Google Scholar] [CrossRef]
- Dohner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute Myeloid Leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef] [PubMed]
- Terao, T.; Minami, Y. Targeting Hedgehog (Hh) Pathway for the Acute Myeloid Leukemia Treatment. Cells 2019, 8, 312. [Google Scholar] [CrossRef] [PubMed]
- Lau, B.W.; Huh, K.; Madero-Marroquin, R.; De Marchi, F.; Lim, Y.; Wang, Q.; Lobo, F.; Marchionni, L.; Smith, D.B.; DeZern, A.; et al. Hedgehog/GLI1 activation leads to leukemic transformation of myelodysplastic syndrome in vivo and GLI1 inhibi-tion results in antitumor activity. Oncogene 2019, 38, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.; Gondek, L.; Li, L.; Wang, Q.; Ma, H.; Chang, E.; Huso, D.L.; Foerster, S.; Marchionni, L.; McGovern, K.; et al. Inte-gration of Hedgehog and mutant FLT3 signaling in myeloid leukemia. Sci. Transl. Med. 2015, 7, 291ra296. [Google Scholar] [CrossRef] [PubMed]
- Munchhof, M.J.; Li, Q.; Shavnya, A.; Borzillo, G.V.; Boyden, T.L.; Jones, C.S.; Lagreca, S.D.; Martinez-Alsina, L.; Patel, N.; Pelletier, K.; et al. Discovery of PF-04449913, a Potent and Orally Bioavailable Inhibitor of Smoothened. ACS Med. Chem. Lett. 2011, 3, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Minami, Y.; Minami, H.; Miyamoto, T.; Yoshimoto, G.; Kobayashi, Y.; Munakata, W.; Onishi, Y.; Kobayashi, M.; Ikuta, M.; Chan, G.; et al. Phase I study of glasdegib (PF-04449913), an oral smoothened inhibitor, in Japanese patients with select hematologic malignancies. Cancer Sci. 2017, 108, 1628–1633. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, G.; Oehler, V.G.; Papayannidis, C.; Courtney, R.; Shaik, M.N.; Zhang, X.; O’Connell, A.; McLachlan, K.R.; Zheng, X.; Radich, J.; et al. Treatment with PF-04449913, an oral smoothened antagonist, in patients with myeloid malignancies: A phase 1 safety and pharmacokinetics study. Lancet Haematol. 2015, 2, e339–e346. [Google Scholar] [CrossRef]
- Cortes, J.E.; Heidel, F.H.; Hellmann, A.; Fiedler, W.; Smith, B.D.; Robak, T.; Montesinos, P.; Pollyea, D.A.; DesJardins, P.; Ottmann, O.; et al. Randomized comparison of low dose cytarabine with or without glasdegib in patients with newly diag-nosed acute myeloid leukemia or high-risk myelodysplastic syndrome. Leukemia 2019, 33, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Shallis, R.M.; Bewersdorf, J.P.; Boddu, P.C.; Zeidan, A.M. Hedgehog pathway inhibition as a therapeutic target in acute myeloid leukemia. Expert Rev. Anticancer Ther. 2019, 19, 717–729. [Google Scholar] [CrossRef] [PubMed]
- Kalemkerian, G.P.; Schneider, B.J. Advances in Small Cell Lung Cancer. Hematol. Oncol. Clin. N. Am. 2017, 31, 143–156. [Google Scholar] [CrossRef]
- Cooper, M.K.; Porter, J.A.; Young, K.E.; Beachy, P.A. Teratogen-mediated inhibition of target tissue response to Shh signaling. Science 1998, 280, 1603–1607. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.K.; Taipale, J.; Cooper, M.K.; Beachy, P.A. Inhibition of Hedgehog signaling by direct binding of cyclopamine to Smoothened. Genes Dev. 2002, 16, 2743–2748. [Google Scholar] [CrossRef] [PubMed]
- Vestergaard, J.; Pedersen, M.W.; Pedersen, N.; Ensinger, C.; Tümer, Z.; Tommerup, N.; Poulsen, H.S.; Larsen, L.A. Hedgehog signaling in small-cell lung cancer: Frequent in vivo but a rare event in vitro. Lung Cancer 2006, 52, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Meuwissen, R.; Linn, S.C.; Linnoila, R.; Zevenhoven, J.; Mooi, W.J.; Berns, A. Induction of small cell lung cancer by somatic inactivation of both Trp53 and Rb1 in a conditional mouse model. Cancer Cell 2003, 4, 181–189. [Google Scholar] [CrossRef]
- Pietanza, M.C.; Litvak, A.M.; Varghese, A.M.; Krug, L.M.; Fleisher, M.; Teitcher, J.B.; Holodny, A.; Sima, C.S.; Woo, K.M.; Ng, K.K.; et al. A phase I trial of the Hedgehog inhibitor, sonidegib (LDE225), in combination with etoposide and cisplatin for the initial treatment of extensive stage small cell lung cancer. Lung Cancer 2016, 99, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Belani, C.P.; Dahlberg, S.; Rudin, C.; Fleisher, M.; Chen, H.X.; Takebe, N.; Velasco, M.R.; Tester, W.J.; Sturtz, K.; Hann, C.L.; et al. Vismodegib or cixutumumab in combination with standard chemotherapy for patients with extensive-stage small cell lung cancer: A trial of the ECOG-ACRIN Cancer Research Group (E1508). Cancer 2016, 122, 2371–2378. [Google Scholar] [CrossRef] [PubMed]
- Shevde, L.A.; Samant, R.S. Nonclassical hedgehog-gli signaling and its clinical implications. Int. J. Cancer 2013, 135, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ding, Q.; Yen, C.-J.; Xia, W.; Izzo, J.G.; Lang, J.-Y.; Li, C.-W.; Hsu, J.L.; Miller, S.A.; Wang, X.; et al. The Crosstalk of mTOR/S6K1 and Hedgehog Pathways. Cancer Cell 2012, 21, 374–387. [Google Scholar] [CrossRef]
- Gruber Filbin, M.; Dabral, S.K.; Pazyra-Murphy, M.F.; Ramkissoon, S.; Kung, A.L.; Pak, E.; Chung, J.; Theisen, M.A.; Sun, Y.; Franchetti, Y.; et al. Coordinate activation of Shh and PI3K signaling in PTEN-deficient glioblastoma: New therapeutic opportunities. Nat. Med. 2013, 19, 1518–1523. [Google Scholar] [CrossRef] [PubMed]
- Kasiri, S.; Shao, C.; Chen, B.; Wilson, A.N.; Yenerall, P.; Timmons, B.C.; Girard, L.; Tian, H.; Behrens, C.; Wistuba, I.I.; et al. GLI1 Blockade Potentiates the Antitumor Activity of PI3K Antagonists in Lung Squamous Cell Carcinoma. Cancer Res. 2017, 77, 4448–4459. [Google Scholar] [CrossRef] [PubMed]
- Jagani, Z.; Mora-Blanco, E.L.; Sansam, C.G.; McKenna, E.S.; Wilson, B.; Chen, D.; Klekota, J.; Tamayo, P.; Nguyen, P.T.L.; Tolstorukov, M.; et al. Loss of the tumor suppressor Snf5 leads to aberrant activation of the Hedgehog-Gli pathway. Nat. Med. 2010, 16, 1429–1433. [Google Scholar] [CrossRef] [PubMed]
- Zwerner, J.P.; Joo, J.; Warner, K.L.; Christensen, L.; Hu-Lieskovan, S.; Triche, T.J.; A May, W. The EWS/FLI1 oncogenic transcription factor deregulates GLI1. Oncogene 2007, 27, 3282–3291. [Google Scholar] [CrossRef] [PubMed]
- Joo, J.; Christensen, L.; Warner, K.; States, L.; Kang, H.-G.; Vo, K.; Lawlor, E.R.; May, W.A. GLI1 Is a Central Mediator of EWS/FLI1 Signaling in Ewing Tumors. PLoS ONE 2009, 4, e7608. [Google Scholar] [CrossRef] [PubMed]
- Beauchamp, E.; Bulut, G.; Abaan, O.; Chen, K.; Merchant, A.; Matsui, W.; Endo, Y.; Rubin, J.S.; Toretsky, J.; Üren, A. GLI1 Is a Direct Transcriptional Target of EWS-FLI1 Oncoprotein. J. Biol. Chem. 2009, 284, 9074–9082. [Google Scholar] [CrossRef]
- Beauchamp, E.M.; Ringer, L.; Bulut, G.; Sajwan, K.P.; Hall, M.D.; Lee, Y.-C.; Peaceman, D.; Özdemirli, M.; Rodriguez, O.; Macdonald, T.J.; et al. Arsenic trioxide inhibits human cancer cell growth and tumor development in mice by blocking Hedgehog/GLI pathway. J. Clin. Investig. 2011, 121, 148–160. [Google Scholar] [CrossRef] [PubMed]
- Atwood, S.X.; Li, M.; Lee, A.; Tang, J.Y.; Oro, A.E. GLI activation by atypical protein kinase C iota/lambda regulates the growth of basal cell carcinomas. Nature 2013, 494, 484–488. [Google Scholar] [CrossRef]
- Whitson, R.J.; Lee, A.; Urman, N.M.; Mirza, A.; Yao, C.Y.; Brown, A.S.; Li, J.R.; Shankar, G.; Fry, M.A.; Atwood, S.X.; et al. Noncanonical hedgehog pathway activation through SRF–MKL1 promotes drug resistance in basal cell carcinomas. Nat. Med. 2018, 24, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Schneider, P.; Bayo-Fina, J.M.; Singh, R.; Kumar Dhanyamraju, P.; Holz, P.; Baier, A.; Fendrich, V.; Ramaswamy, A.; Baumeister, S.; Martinez, E.D.; et al. Identification of a novel actin-dependent signal transducing module allows for the targeted degradation of GLI1. Nat. Commun. 2015, 6, 8023. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Wang, L.S.; Chen, X.L.; Gatalica, Z.; Qiu, S.; Liu, Z.; Stoner, G.; Zhang, H.; Weiss, H.; Xie, J. Hedgehog signal-ing activation in the development of squamous cell carcinoma and adenocarcinoma of esophagus. Int. J. Biochem. Mol. Biol. 2012, 3, 46–57. [Google Scholar]
- Wirsching, H.G.; Galanis, E.; Weller, M. Glioblastoma. Handb. Clin. Neurol. 2016, 134, 381–397. [Google Scholar]
- Kinzler, K.W.; Bigner, S.H.; Bigner, D.D.; Trent, J.M.; Law, M.L.; O’Brien, S.J.; Wong, A.J.; Vogelstein, B. Identification of an Amplified, Highly Expressed Gene in a Human Glioma. Science 1987, 236, 70–73. [Google Scholar] [CrossRef]
- Ehtesham, M.; Sarangi, A.; Valadez, J.G.; Chanthaphaychith, S.; Becher, M.W.; Abel, T.W.; Thompson, R.C.; Cooper, M.K. Ligand-dependent activation of the hedgehog pathway in glioma progenitor cells. Oncogene 2007, 26, 5752–5761. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Dai, B.; Kang, S.-H.; Ban, K.; Huang, F.-J.; Lang, F.F.; Aldape, K.D.; Xie, T.-X.; Pelloski, C.E.; Xie, K.; et al. FoxM1B Is Overexpressed in Human Glioblastomas and Critically Regulates the Tumorigenicity of Glioma Cells. Cancer Res. 2006, 66, 3593–3602. [Google Scholar] [CrossRef]
- Hsieh, A.; Ellsworth, R.; Hsieh, D. Hedgehog/GLI1 regulates IGF dependent malignant behaviors in glioma stem cells. J. Cell. Physiol. 2010, 226, 1118–1127. [Google Scholar] [CrossRef]
- Maira, S.-M.; Pecchi, S.; Huang, A.; Burger, M.; Knapp, M.; Sterker, D.; Schnell, C.; Guthy, D.; Nagel, T.; Wiesmann, M.; et al. Identification and Characterization of NVP-BKM120, an Orally Available Pan-Class I PI3-Kinase Inhibitor. Mol. Cancer Ther. 2012, 11, 317–328. [Google Scholar] [CrossRef]
- Burger, M.T.; Pecchi, S.; Wagman, A.; Ni, Z.-J.; Knapp, M.; Hendrickson, T.; Atallah, G.; Pfister, K.; Zhang, Y.; Bartulis, S.; et al. Identification of NVP-BKM120 as a Potent, Selective, Orally Bioavailable Class I PI3 Kinase Inhibitor for Treating Cancer. ACS Med. Chem. Lett. 2011, 2, 774–779. [Google Scholar] [CrossRef]
- Herbst, R.S.; Morgensztern, D.; Boshoff, C. The biology and management of non-small cell lung cancer. Nature 2018, 553, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Fujita, E.; Khoroku, Y.; Urase, K.; Tsukahara, T.; Momoi, M.Y.; Kumagai, H.; Takemura, T.; Kuroki, T.; Momoi, T. In-volvement of Sonic hedgehog in the cell growth of LK-2 cells, human lung squamous carcinoma cells. Biochem. Biophys. Res. Commun. 1997, 238, 658–664. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lee, J.J.; Kim, J.; Gardner, D.; Beachy, P.A. Arsenic antagonizes the Hedgehog pathway by preventing ciliary accumulation and reducing stability of the Gli2 transcriptional effector. Proc. Natl. Acad. Sci. USA 2010, 107, 13432–13437. [Google Scholar] [CrossRef] [PubMed]
- Foulkes, W.D.; Kamihara, J.; Evans, D.G.R.; Brugières, L.; Bourdeaut, F.; Molenaar, J.J.; Walsh, M.F.; Brodeur, G.M.; Diller, L. Cancer Surveillance in Gorlin Syndrome and Rhabdoid Tumor Predisposition Syndrome. Clin. Cancer Res. 2017, 23, e62–e67. [Google Scholar] [CrossRef]
- Versteege, I.; Sevenet, N.; Lange, J.; Rousseau-Merck, M.F.; Ambros, P.; Handgretinger, R.; Aurias, A.; Delattre, O. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature 1998, 394, 203–206. [Google Scholar] [CrossRef]
- Hargreaves, D.C.; Crabtree, G.R. ATP-dependent chromatin remodeling: Genetics, genomics and mechanisms. Cell Res. 2011, 21, 396–420. [Google Scholar] [CrossRef]
- Hyman, J.M.; Firestone, A.J.; Heine, V.M.; Zhao, Y.; Ocasio, C.A.; Han, K.; Sun, M.; Rack, P.G.; Sinha, S.; Wu, J.J.; et al. Small-molecule inhibitors reveal multiple strategies for Hedgehog pathway blockade. Proc. Natl. Acad. Sci. USA 2009, 106, 14132–14137. [Google Scholar] [CrossRef]
- Choi, E.Y.; Gardner, J.M.; Lucas, D.R.; McHugh, J.B.; Patel, R.M. Ewing sarcoma. Semin. Diagn. Pathol. 2014, 31, 39–47. [Google Scholar] [CrossRef]
- Sbaraglia, M.; Righi, A.; Gambarotti, M.; Dei Tos, A.P. Ewing sarcoma and Ewing-like tumors. Virchows. Arch. 2020, 476, 109–119. [Google Scholar] [CrossRef]
- Lauth, M.; Bergström, Å.; Shimokawa, T.; Toftgård, R. Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proc. Natl. Acad. Sci. USA 2007, 104, 8455–8460. [Google Scholar] [CrossRef]
- Sinx, K.A.E.; Roemen, G.; van Zutven, V.; Janssen, R.; Speel, E.M.; Steijlen, P.M.; van Geel, M.; Mosterd, K. Vismodeg-ib-resistant basal cell carcinomas in basal cell nevus syndrome: Clinical approach and genetic analysis. JAAD Case Rep. 2018, 4, 408–411. [Google Scholar] [CrossRef] [PubMed]
- Callahan, C.A.; Ofstad, T.; Horng, L.; Wang, J.K.; Zhen, H.H.; Coulombe, P.A.; Oro, A.E. MIM/BEG4, a Sonic hedge-hog-responsive gene that potentiates Gli-dependent transcription. Genes Dev. 2004, 18, 2724–2729. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chang, J.; Varghese, D.; Dellinger, M.; Kumar, S.; Best, A.M.; Ruiz, J.; Bruick, R.; Peña-Llopis, S.; Xu, J.; et al. A small molecule modulates Jumonji histone demethylase activity and selectively inhibits cancer growth. Nat. Commun. 2013, 4, 1–13. [Google Scholar] [CrossRef]
- Dusek, O.C.; Hadden, M.K. Targeting the GLI family of transcription factors for the development of anti-cancer drugs. Expert Opin. Drug Discov. 2020, 16, 289–302. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Grob, J.J.; Stroyakovskiy, D.; Karaszewska, B.; Hauschild, A.; Levchenko, E.; Chiarion Sileni, V.; Schachter, J.; Garbe, C.; Bondarenko, I.; et al. Five-Year Outcomes with Dabrafenib plus Trametinib in Metastatic Melanoma. N. Engl. J. Med. 2019, 381, 626–636. [Google Scholar] [CrossRef] [PubMed]
- Planchard, D.; Smit, E.F.; Groen, H.J.M.; Mazieres, J.; Besse, B.; Helland, A.; Giannone, V.; D’Amelio, A.M., Jr.; Zhang, P.; Mookerjee, B.; et al. Dabrafenib plus trametinib in patients with previously untreated BRAF(V600E)-mutant metastatic non-small-cell lung cancer: An open-label, phase 2 trial. Lancet Oncol. 2017, 18, 1307–1316. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Cancer | Hedgehog Signaling Type | Cause of Pathway Activation | Reference |
|---|---|---|---|
| Basal Cell Carcinoma | Ligand-Independent Canonical Oncogenic | Primarily inactivating PTCH1 mutations, secondarily SMO activating mutations (~10%) | [13,14,46,47,48,49,50] |
| Medulloblastoma | Ligand-Independent Canonical Oncogenic | Loss of function mutations in PTCH1 and SUFU, gain of function mutations in SMO | [51,52,53,54,55,56,57,58] |
| Rhabdomyosarcoma/Rhabdomyoma | Ligand-Independent Canonical Oncogenic | Loss of function mutations in PTCH1 and activating mutations in SMO | [55,59,60,61] |
| Ameloblastoma | Ligand-Independent Canonical Oncogenic | SMO activating mutations | [62] |
| Meningioma | Ligand-Independent Canonical Oncogenic | SMO and SUFU mutations | [63,64,65,66] |
| Small Cell Lung Cancer | Ligand-Dependent Canonical Oncogenic | Overexpression and loss of SHH and loss of SMO in autochthonous tumor cells modulate tumor growth | [67,68,69] |
| Acute Myeloid Leukemia | Ligand-Dependent Canonical Oncogenic | GLI1 upregulation in chemotherapy- and radiation-resistant AML. SMO antagonists re-sensitize cells to therapy. SMO antagonism with low dose cytarabine improves overall survival in older AML patients. Source of Hh ligand is unknown currently. | [70,71,72] |
| Bladder | Ligand-Dependent Canonical Tumor Suppressive | Stromal Hh pathway activation by SHH from tumor epithelia inhibits tumor growth by secretion of BMP4 and BMP5 differentiation factors to tumor epithelia | [73,74] |
| Pancreas | Ligand-Dependent Canonical Tumor Suppressive | Stromal Hh pathway activation by SHH from tumor epithelia inhibits tumor growth, metastases, and increases formation of well differentiated pancreas cancers | [75,76] |
| Colon | Ligand-Dependent Canonical Tumor Suppressive | Deletion of IHH from intestinal epithelia increased tumor burden. Genetic loss or pharmacologic inhibition of stromal SMO increased tumor burden | [77,78] |
| Prostate | Ligand-Dependent Canonical Tumor Suppressive | IHH secretion from prostate tumor epithelia activates stromal Hh pathway to inhibit tumor growth | [79] |
| Lung Adenocarcinoma | Ligand-Dependent Canonical Tumor Suppressive | IHH secretion from lung tumor epithelia activates stromal Hh pathway to inhibit tumor growth and metastasis | [80] |
| Cancer | Hedgehog Signaling Type | Cause of Pathway Activation | Reference |
|---|---|---|---|
| Esophageal Adenocarcinoma | Noncanonical GLI Activation Oncogenic | TNF-α-mediated activation of mTOR promotes GLI1 activity | [194] |
| Glioblastoma | Noncanonical GLI Activation Oncogenic | PI3K-mTOR activity increases GLI1 and GLI2 expression | [195] |
| Lung Squamous Cell Carcinoma | Noncanonical GLI Activation Oncogenic | Increased PI3K activity from PIK3CA amplification drives GLI1 expression and activity | [196] |
| Malignant Rhabdoid Tumor | Noncanonical GLI Activation Oncogenic | Loss of SNF5 leads to elevated GLI1 expression and activity | [197] |
| Ewing-Sarcoma | Noncanonical GLI Activation Oncogenic | EWS-FLI1 complex binds to the GLI1 gene promoter region to induce GLI1 transcription | [198,199,200,201] |
| Vismodegib-resistant Basal Cell Carcinoma | Noncanonical GLI Activation Oncogenic | aPKC-ι/λ and MIM promote GLI activation downstream of SMO. SRF and co-regulator MKL1 potentiate GLI1 activation | [202,203] |
| Rhabdomyosarcoma, Lung Adenocarcinoma | Noncanonical GLI Activation Oncogenic | JMJD1A and co-regulator MKL1 stabilize GLI1 | [204] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suchors, C.; Kim, J. Canonical Hedgehog Pathway and Noncanonical GLI Transcription Factor Activation in Cancer. Cells 2022, 11, 2523. https://doi.org/10.3390/cells11162523
Suchors C, Kim J. Canonical Hedgehog Pathway and Noncanonical GLI Transcription Factor Activation in Cancer. Cells. 2022; 11(16):2523. https://doi.org/10.3390/cells11162523
Chicago/Turabian StyleSuchors, Chamey, and James Kim. 2022. "Canonical Hedgehog Pathway and Noncanonical GLI Transcription Factor Activation in Cancer" Cells 11, no. 16: 2523. https://doi.org/10.3390/cells11162523
APA StyleSuchors, C., & Kim, J. (2022). Canonical Hedgehog Pathway and Noncanonical GLI Transcription Factor Activation in Cancer. Cells, 11(16), 2523. https://doi.org/10.3390/cells11162523