
ENO2 Promotes Colorectal Cancer Metastasis by Interacting with the LncRNA CYTOR and Activating YAP1-Induced EMT

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Reagents
2.2. TCGA and Gene Expression Omnibus (GEO) Analysis
2.3. Quantitative Real-Time PCR (qRT–PCR)
2.4. Western Blotting and Immunoprecipitation
2.5. Construction of the ENO2 and CYTOR Knockout (KO) Cell Lines
2.6. Construction of ENO2 Overexpression and Mutant Plasmids
2.7. Cell Viability and Transwell Assays
2.8. Metabolic Flux Analysis
2.9. Glucose Consumption, Lactate Production and Enolase Activity Assays
2.10. Cell Immunofluorescence (IF) and Immunohistochemistry (IHC)
2.11. RNA Immunoprecipitation (RIP)
2.12. RNA-Seq and Gene Set Enrichment Analysis (GSEA)
2.13. Mouse Metastasis Model
2.14. Statistical Analysis
3. Results
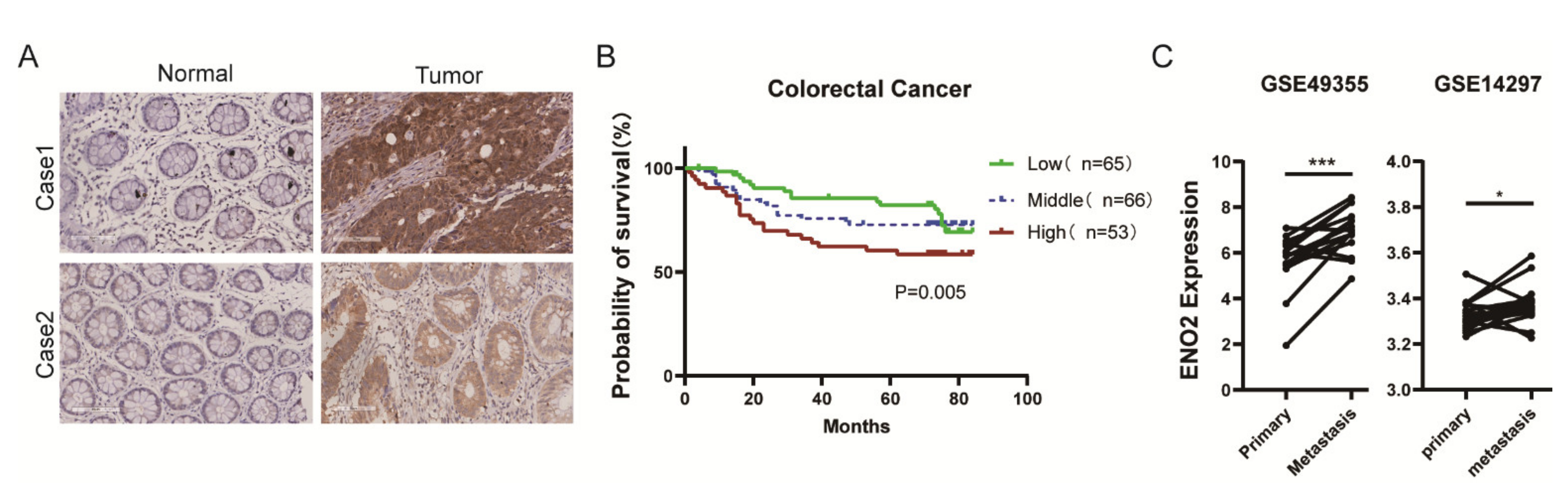
3.1. ENO2 Was Generally Highly Expressed in CRC Tissues and Negatively Correlated with Prognosis
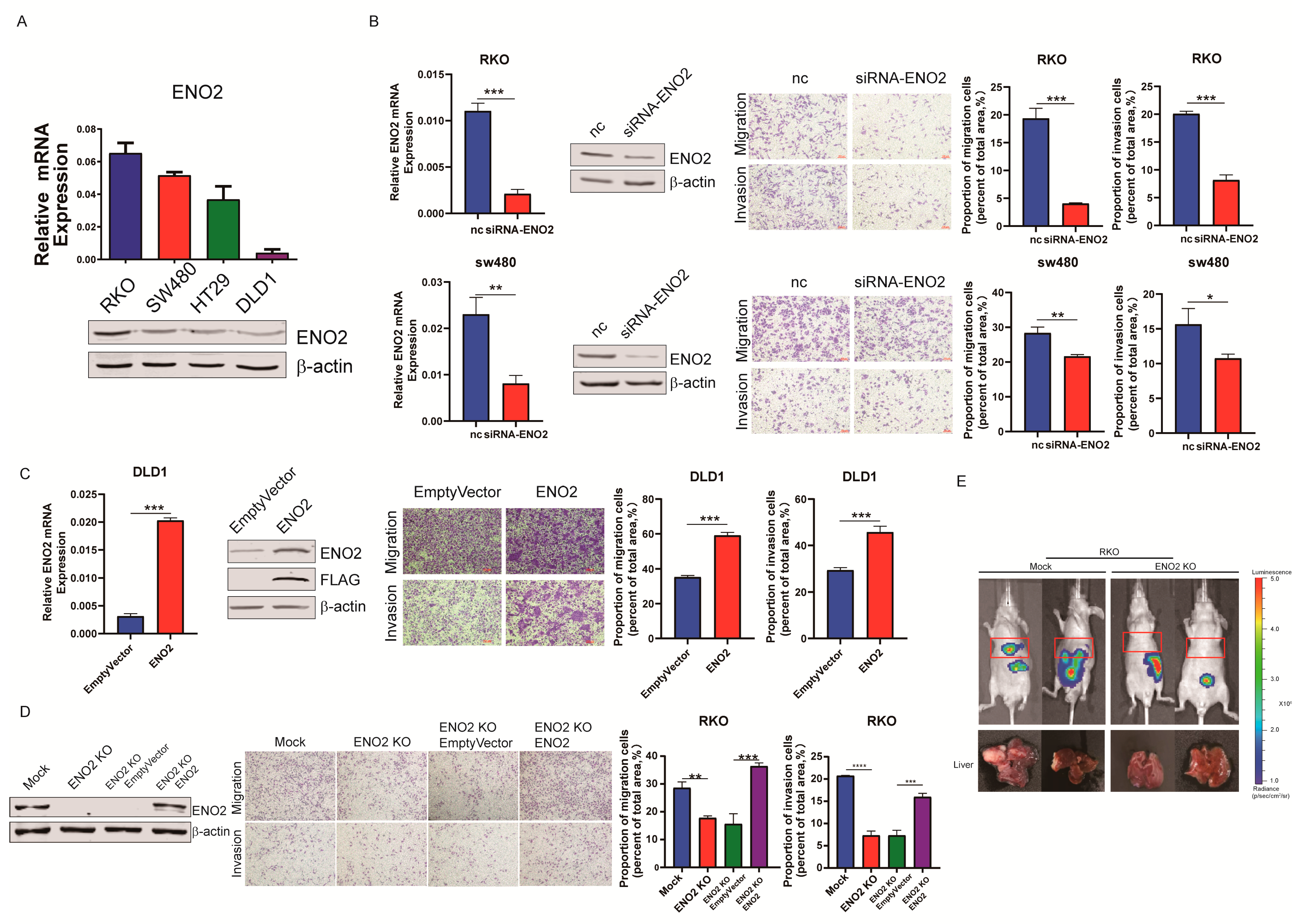
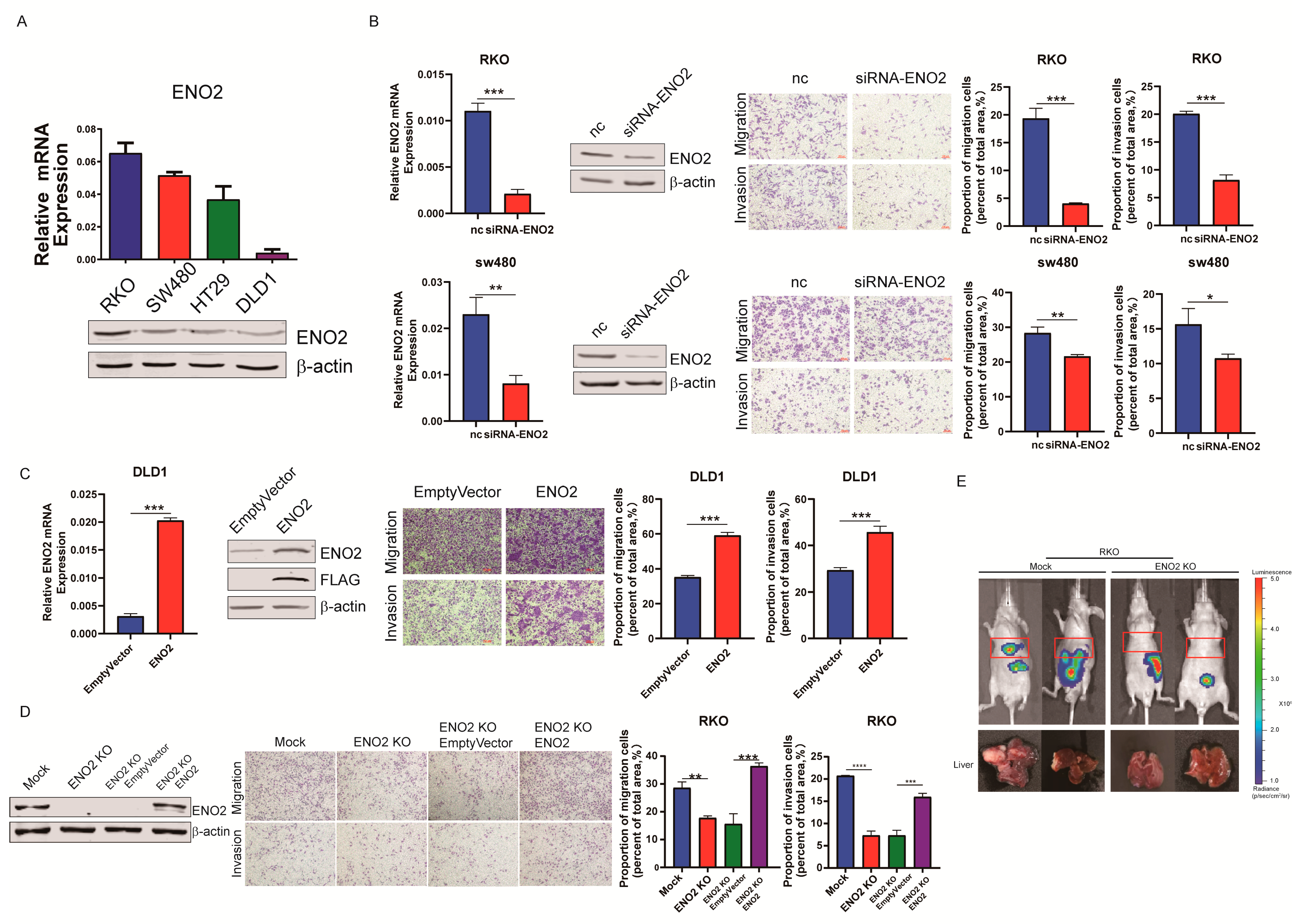
3.2. ENO2 Promoted CRC Cells’ Migration and Invasion
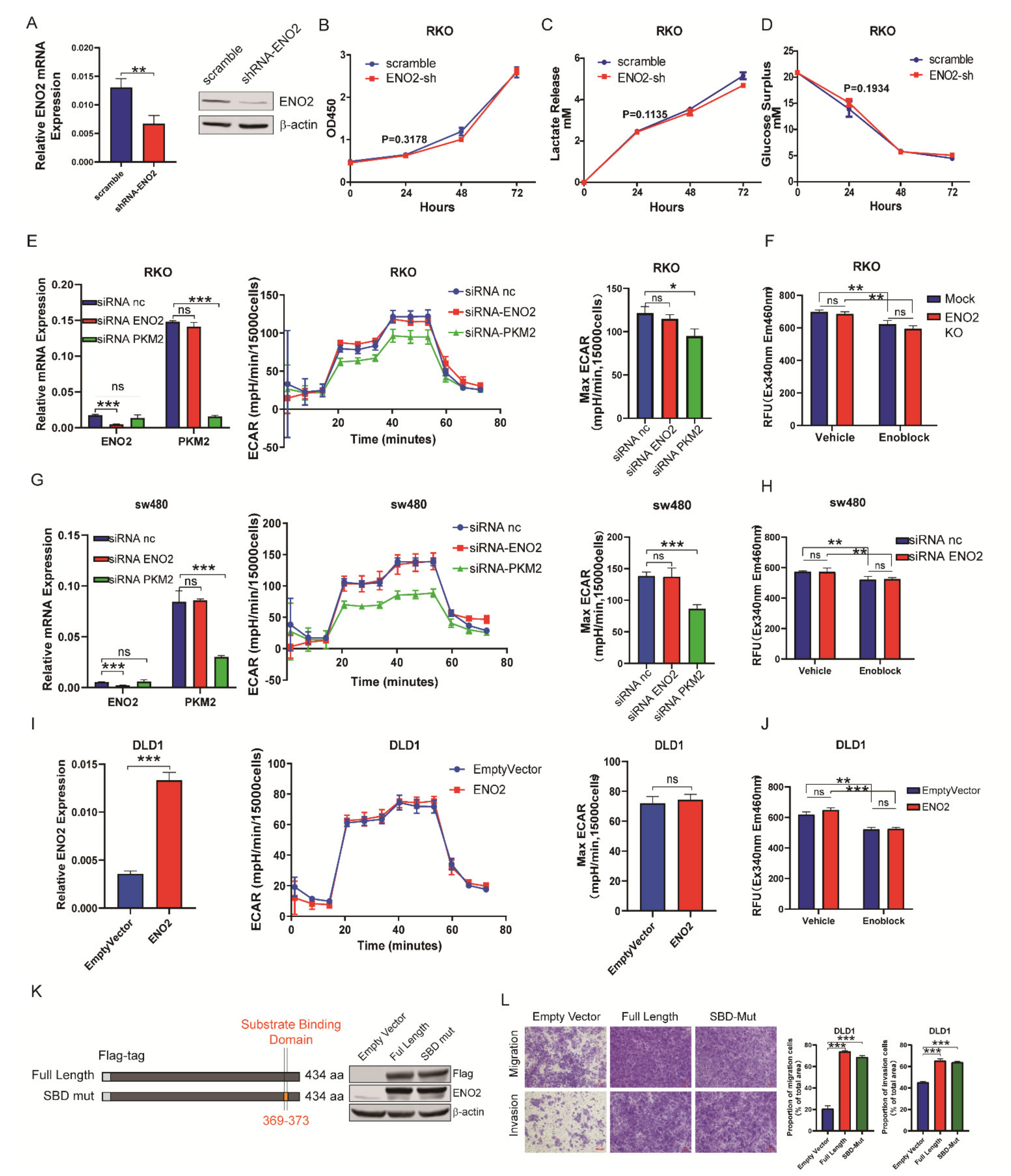
3.3. ENO2 Promoted Metastasis Independently of Altering the Glycolytic Rate in CRC
3.4. ENO2 Induced EMT by Interacting with the lncRNA CYTOR
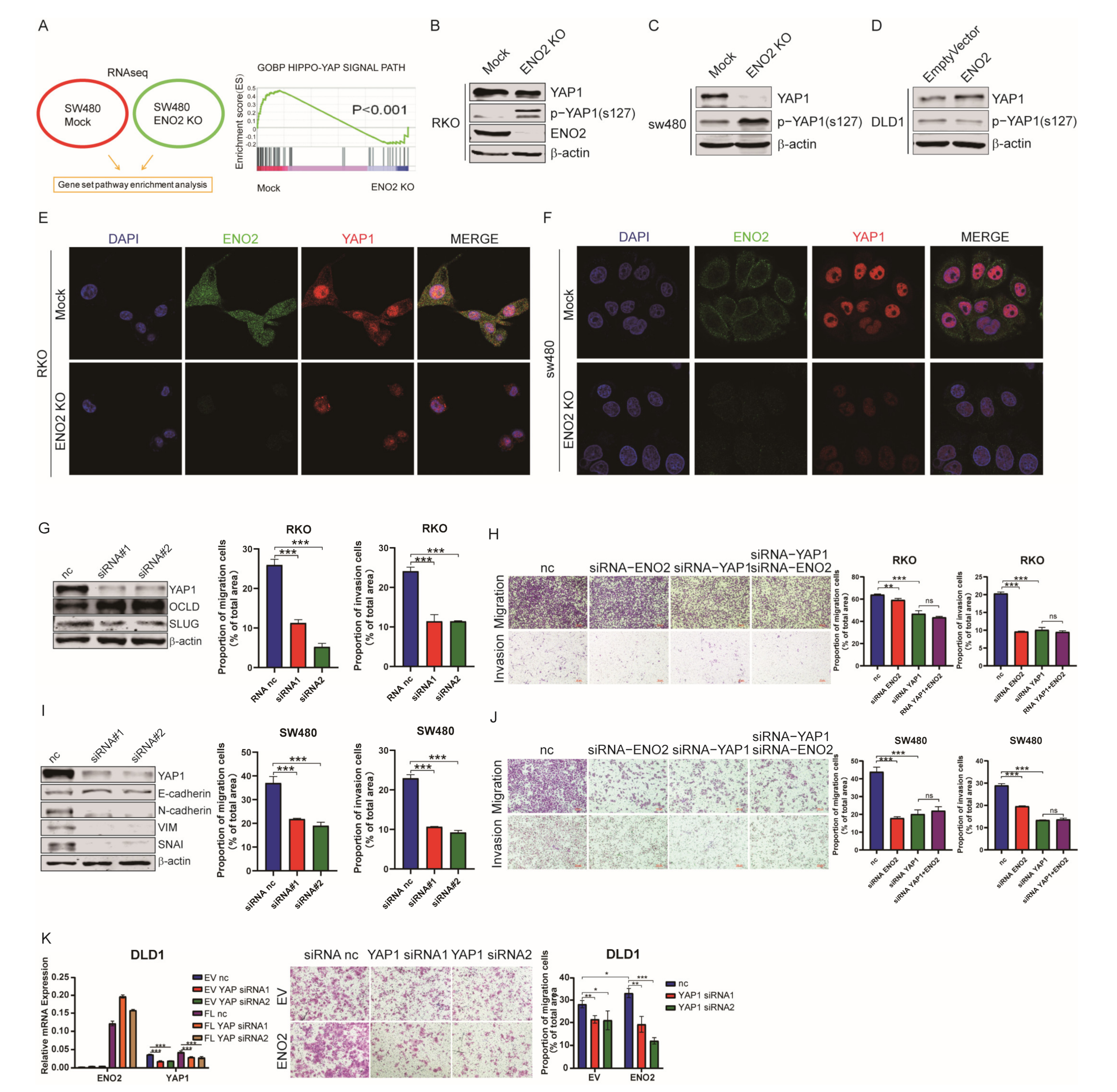
3.5. ENO2 Activated YAP1 by Suppressing YAP1 Phosphorylation
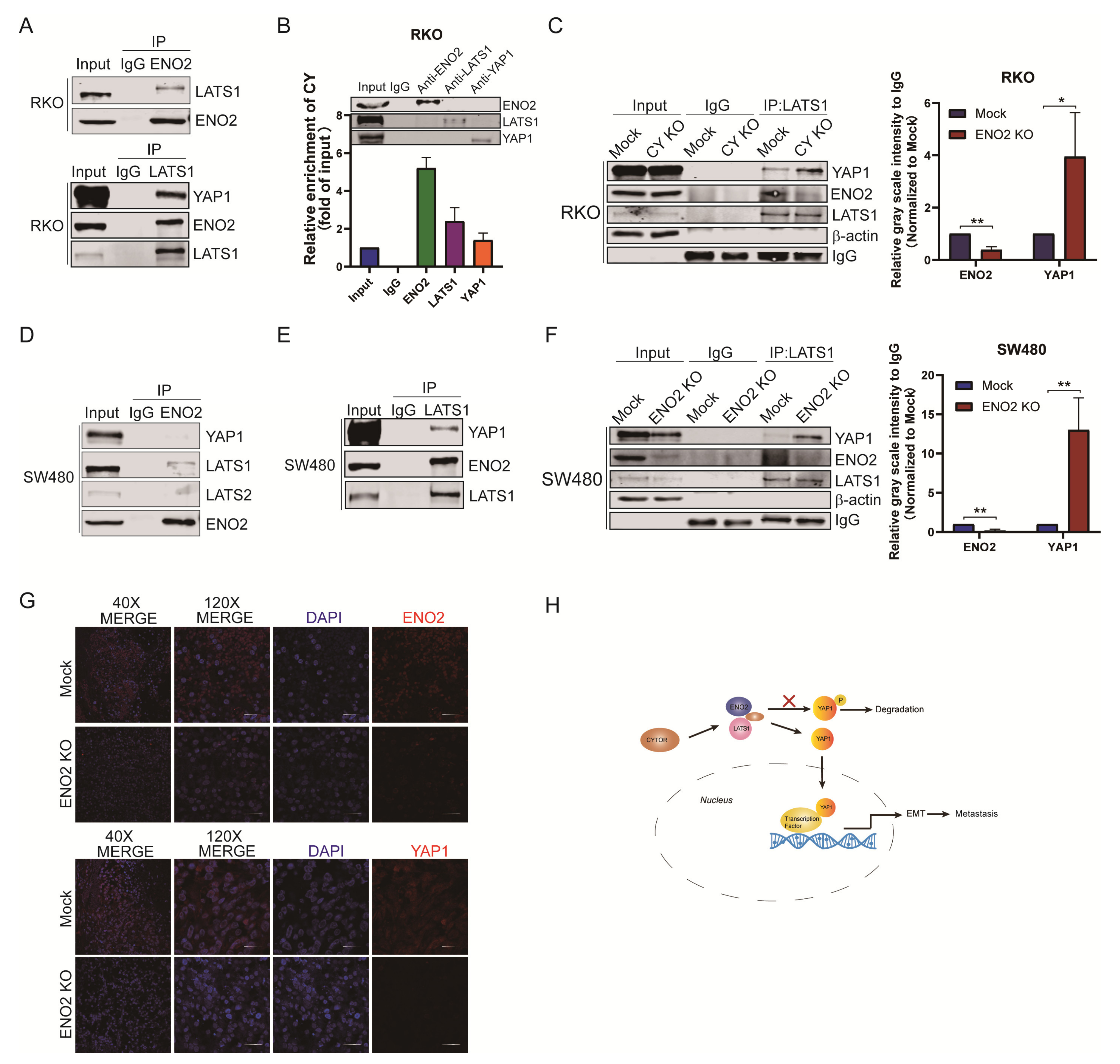
3.6. CYTOR Mediated the Interaction between ENO2 and LATS1 to Inhibit the Degradation of YAP1
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohlsson, B.; Palsson, B. Follow-up after colorectal cancer surgery. Acta Oncol. 2003, 42, 816–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, T.; Liu, H.; Liang, Z.; Wang, F.; Zhou, C.; Zheng, X.; Zhang, Y.; Song, Y.; Hu, J.; He, X.; et al. Tumor-intrinsic CD47 signal regulates glycolysis and promotes colorectal cancer cell growth and metastasis. Theranostics 2020, 10, 4056–4072. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Hay, E.D. The mesenchymal cell, its role in the embryo, and the remarkable signaling mechanisms that create it. Dev. Dyn. 2005, 233, 706–720. [Google Scholar] [CrossRef]
- Wang, X.; Yu, H.; Sun, W.; Kong, J.; Zhang, L.; Tang, J.; Wang, J.; Xu, E.; Lai, M.; Zhang, H. The long non-coding RNA CYTOR drives colorectal cancer progression by interacting with NCL and Sam68. Mol. Cancer 2018, 17, 110. [Google Scholar] [CrossRef] [Green Version]
- Prinz, R.A.; Marangos, P.J. Use of neuron-specific enolase as a serum marker for neuroendocrine neoplasms. Surgery 1982, 92, 887–889. [Google Scholar]
- Liu, C.C.; Wang, H.; Wang, W.D.; Wang, L.; Liu, W.J.; Wang, J.H.; Geng, Q.R.; Lu, Y. ENO2 Promotes Cell Proliferation, Glycolysis, and Glucocorticoid-Resistance in Acute Lymphoblastic Leukemia. Cell. Physiol. Biochem. 2018, 46, 1525–1535. [Google Scholar] [CrossRef]
- Carney, D.N.; Marangos, P.J.; Ihde, D.C.; Bunn, P.A., Jr.; Cohen, M.H.; Minna, J.D.; Gazdar, A.F. Serum neuron-specific enolase: A marker for disease extent and response to therapy of small-cell lung cancer. Lancet 1982, 1, 583–585. [Google Scholar] [CrossRef]
- Reed, G.H.; Poyner, R.R.; Larsen, T.M.; Wedekind, J.E.; Rayment, I. Structural and mechanistic studies of enolase. Curr. Opin. Struct. Biol. 1996, 6, 736–743. [Google Scholar] [CrossRef]
- Zheng, Y.; Wu, C.; Yang, J.; Zhao, Y.; Jia, H.; Xue, M.; Xu, D.; Yang, F.; Fu, D.; Wang, C.; et al. Insulin-like growth factor 1-induced enolase 2 deacetylation by HDAC3 promotes metastasis of pancreatic cancer. Signal Transduct. Target. Ther. 2020, 5, 53. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Wang, M.; Dai, Y.; Wei, X. Kruppel-like factor 12 suppresses bladder cancer growth through transcriptionally inhibition of enolase 2. Gene 2021, 769, 145338. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Chen, C.; Ding, Q.; Zhao, Y.; Wang, Z.; Chen, J.; Jiang, Z.; Zhang, Y.; Xu, G.; Zhang, J.; et al. METTL3-mediated m(6)A modification of HDGF mRNA promotes gastric cancer progression and has prognostic significance. Gut 2020, 69, 1193–1205. [Google Scholar] [CrossRef]
- Yukimoto, R.; Nishida, N.; Hata, T.; Fujino, S.; Ogino, T.; Miyoshi, N.; Takahashi, H.; Uemura, M.; Satoh, T.; Hirofumi, Y.; et al. Specific activation of glycolytic enzyme enolase 2 in BRAF V600E-mutated colorectal cancer. Cancer Sci. 2021, 112, 2884–2894. [Google Scholar] [CrossRef]
- Muller, F.L.; Colla, S.; Aquilanti, E.; Manzo, V.E.; Genovese, G.; Lee, J.; Eisenson, D.; Narurkar, R.; Deng, P.; Nezi, L.; et al. Passenger deletions generate therapeutic vulnerabilities in cancer. Nature 2012, 488, 337–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, D.W.; Kim, W.H.; Park, S.H.; Lee, J.; Kim, J.; Su, D.; Ha, H.H.; Chang, Y.T.; Williams, D.R. A unique small molecule inhibitor of enolase clarifies its role in fundamental biological processes. ACS Chem. Biol. 2013, 8, 1271–1282. [Google Scholar] [CrossRef]
- Paludo, G.P.; Lorenzatto, K.R.; Bonatto, D.; Ferreira, H.B. Systems biology approach reveals possible evolutionarily conserved moonlighting functions for enolase. Comput. Biol. Chem. 2015, 58, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Liu, F.; Zheng, H.; Wu, Q.; Liu, S. IncRNA ZFAS1 contributes to the radioresistance of nasopharyngeal carcinoma cells by sponging hsa-miR-7-5p to upregulate ENO2. Cell Cycle 2021, 20, 126–141. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Dang, C.V. Multifaceted roles of glycolytic enzymes. Trends Biochem. Sci. 2005, 30, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Jung, D.W.; Kim, W.H.; Williams, D.R. Chemical genetics and its application to moonlighting in glycolytic enzymes. Biochem. Soc. Trans. 2014, 42, 1756–1761. [Google Scholar] [CrossRef]
- Piccolo, S.; Dupont, S.; Cordenonsi, M. The biology of YAP/TAZ: Hippo signaling and beyond. Physiol. Rev. 2014, 94, 1287–1312. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Feldmann, G.; Huang, J.; Wu, S.; Zhang, N.; Comerford, S.A.; Gayyed, M.F.; Anders, R.A.; Maitra, A.; Pan, D. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell 2007, 130, 1120–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Wu, S.; Barrera, J.; Matthews, K.; Pan, D. The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila Homolog of YAP. Cell 2005, 122, 421–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, B.; Wei, X.; Li, W.; Udan, R.S.; Yang, Q.; Kim, J.; Xie, J.; Ikenoue, T.; Yu, J.; Li, L.; et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007, 21, 2747–2761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Shi, S.; Guo, Z.; Zhang, X.; Han, S.; Yang, A.; Wen, W.; Zhu, Q. Overexpression of YAP and TAZ is an independent predictor of prognosis in colorectal cancer and related to the proliferation and metastasis of colon cancer cells. PLoS ONE 2013, 8, e65539. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Li, L.; Tumaneng, K.; Wang, C.Y.; Guan, K.L. A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF(beta-TRCP). Genes Dev. 2010, 24, 72–85. [Google Scholar] [CrossRef] [Green Version]
- Lv, C.; Su, Q.; Liang, Y.; Hu, J.; Yuan, S. Oncolytic vaccine virus harbouring the IL-24 gene suppresses the growth of lung cancer by inducing apoptosis. Biochem. Biophys. Res. Commun. 2016, 476, 21–28. [Google Scholar] [CrossRef]
- Han, F.; Zhang, L.; Chen, C.; Wang, Y.; Zhang, Y.; Qian, L.; Sun, W.; Zhou, D.; Yang, B.; Zhang, H.; et al. GLTSCR1 Negatively Regulates BRD4-Dependent Transcription Elongation and Inhibits CRC Metastasis. Adv. Sci. 2019, 6, 1901114. [Google Scholar] [CrossRef] [Green Version]
- Joseph, J.; Cruz-Sanchez, F.F.; Carreras, J. Enolase activity and isoenzyme distribution in human brain regions and tumors. J. Neurochem. 1996, 66, 2484–2490. [Google Scholar] [CrossRef]
- Pan, X.; Wu, H.; Chen, G.; Li, W. Prognostic Value of Enolase Gene Family in Colon Cancer. Med. Sci. Monit. 2020, 26, e922980. [Google Scholar] [CrossRef]
- Sun, C.; Liu, M.; Zhang, W.; Wang, S.; Qian, G.; Wang, M.; Zhang, G. Overexpression of enolase 2 is associated with worsened prognosis and increased glycikolysis in papillary renal cell carcinoma. J. Cell. Physiol. 2021, 236, 3821–3831. [Google Scholar] [CrossRef] [PubMed]
- Filipp, F.V. Cancer metabolism meets systems biology: Pyruvate kinase isoform PKM2 is a metabolic master regulator. J. Carcinog. 2013, 12, 14. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.D.; Fan, F.; Camp, E.R.; van Buren, G.; Liu, W.; Somcio, R.; Gray, M.J.; Cheng, H.; Hoff, P.M.; Ellis, L.M. Chronic oxaliplatin resistance induces epithelial-to-mesenchymal transition in colorectal cancer cell lines. Clin. Cancer Res. 2006, 12, 4147–4153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Lai, Q.; He, C.; Fang, Y.; Yan, Q.; Zhang, Y.; Wang, X.; Gu, C.; Wang, Y.; Ye, L.; et al. RUNX1 promotes tumour metastasis by activating the Wnt/beta-catenin signalling pathway and EMT in colorectal cancer. J. Exp. Clin. Cancer Res. 2019, 38, 334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Bao, J.; Yao, J.; Li, J. lncRNA USP2-AS1 promotes colon cancer progression by modulating Hippo/YAP1 signaling. Am. J. Transl. Res. 2020, 12, 5670–5682. [Google Scholar] [PubMed]
- Yu, Y.; Yang, J.; Li, Q.; Xu, B.; Lian, Y.; Miao, L. LINC00152: A pivotal oncogenic long non-coding RNA in human cancers. Cell Prolif. 2017, 50, e12349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leonard, P.G.; Satani, N.; Maxwell, D.; Lin, Y.H.; Hammoudi, N.; Peng, Z.; Pisaneschi, F.; Link, T.M.; Lee, G.R.; Sun, D.; et al. SF2312 is a natural phosphonate inhibitor of enolase. Nat. Chem. Biol. 2016, 12, 1053–1058. [Google Scholar] [CrossRef]
- Pisaneschi, F.; Lin, Y.H.; Leonard, P.G.; Satani, N.; Yan, V.C.; Hammoudi, N.; Raghavan, S.; Link, T.M.; Georgiou, D.K.; Czako, B.; et al. The 3S Enantiomer Drives Enolase Inhibitory Activity in SF2312 and Its Analogues. Molecules 2019, 24, 2510. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Zhang, Y.; Yang, J.; Zhan, H.; Zhou, Z.; Jiang, Y.; Shi, X.; Fan, X.; Zhang, J.; Luo, W.; et al. Zinc-Dependent Regulation of ZEB1 and YAP1 Coactivation Promotes Epithelial-Mesenchymal Transition Plasticity and Metastasis in Pancreatic Cancer. Gastroenterology 2021, 160, 1771–1783. [Google Scholar] [CrossRef]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP/TAZ at the Roots of Cancer. Cancer Cell 2016, 29, 783–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lv, C.; Yu, H.; Wang, K.; Chen, C.; Tang, J.; Han, F.; Mai, M.; Ye, K.; Lai, M.; Zhang, H. ENO2 Promotes Colorectal Cancer Metastasis by Interacting with the LncRNA CYTOR and Activating YAP1-Induced EMT. Cells 2022, 11, 2363. https://doi.org/10.3390/cells11152363
Lv C, Yu H, Wang K, Chen C, Tang J, Han F, Mai M, Ye K, Lai M, Zhang H. ENO2 Promotes Colorectal Cancer Metastasis by Interacting with the LncRNA CYTOR and Activating YAP1-Induced EMT. Cells. 2022; 11(15):2363. https://doi.org/10.3390/cells11152363
Chicago/Turabian StyleLv, Chunwei, Hongfei Yu, Keyi Wang, Chaoyi Chen, Jinlong Tang, Fengyan Han, Minglang Mai, Kehong Ye, Maode Lai, and Honghe Zhang. 2022. "ENO2 Promotes Colorectal Cancer Metastasis by Interacting with the LncRNA CYTOR and Activating YAP1-Induced EMT" Cells 11, no. 15: 2363. https://doi.org/10.3390/cells11152363
APA StyleLv, C., Yu, H., Wang, K., Chen, C., Tang, J., Han, F., Mai, M., Ye, K., Lai, M., & Zhang, H. (2022). ENO2 Promotes Colorectal Cancer Metastasis by Interacting with the LncRNA CYTOR and Activating YAP1-Induced EMT. Cells, 11(15), 2363. https://doi.org/10.3390/cells11152363