
A Novel m6A-Related LncRNA Signature for Predicting Prognosis, Chemotherapy and Immunotherapy Response in Patients with Lung Adenocarcinoma


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Acquisition
2.2. WGCNA
2.3. Unsupervised Clustering for 9 LncRNAs and Principal Component Analysis (PCA)
2.4. Gene Set Variation Analysis (GSVA)
2.5. Identification of Differentially Expressed Genes (DEGs) between LncRNA Subtypes
2.6. Establishment of LncRNA Score
2.7. Mutation Profiles
2.8. Prediction of Chemotherapeutic Drugs
2.9. Statistical Analysis
3. Results
3.1. m6A-Related LncRNAs Associated with the Prognosis of LUAD
3.2. Three LncRNA Clusters Were Highly Consistent with the Three Immune Phenotypes
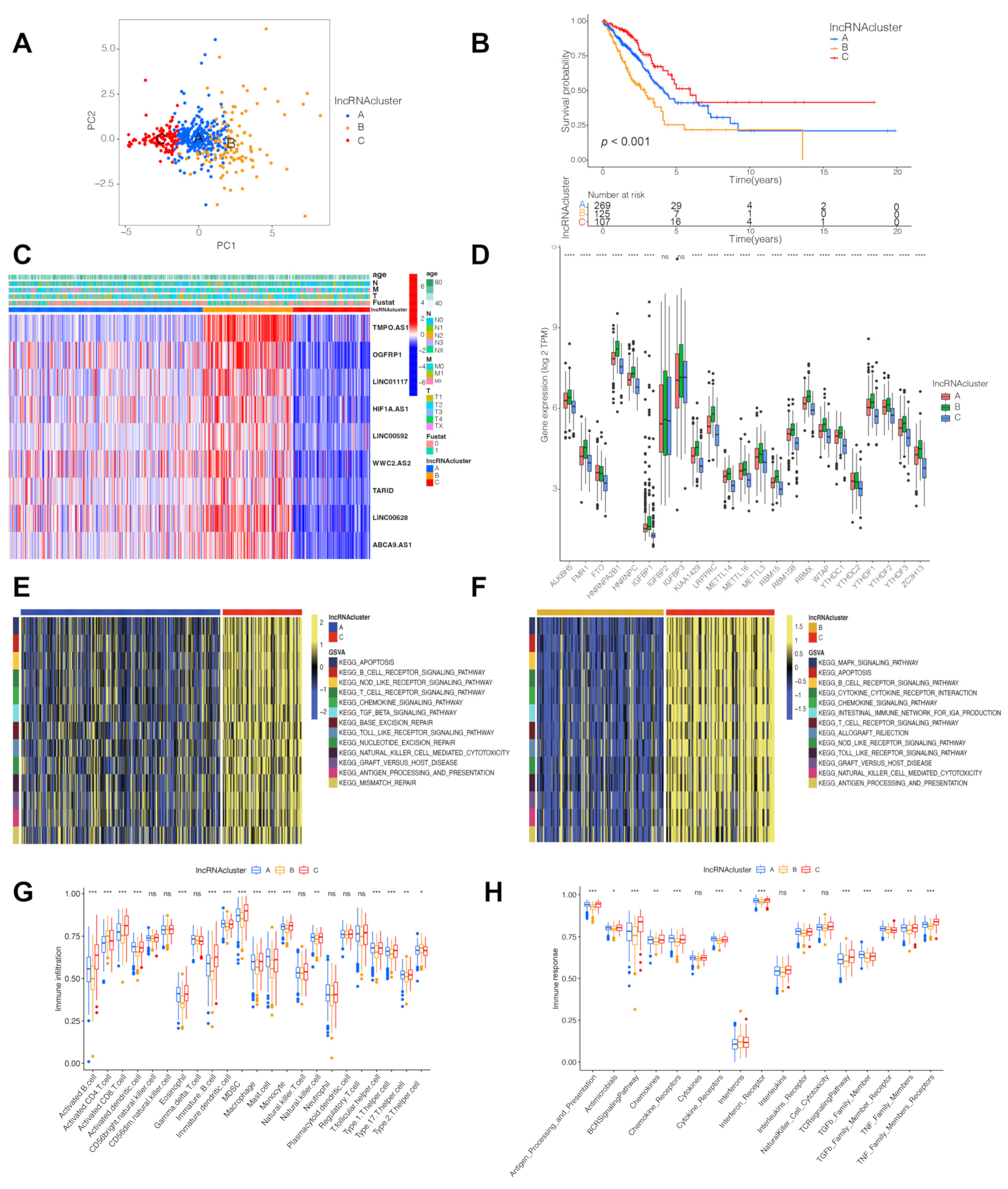
3.3. Identification of LncRNA-Related Gene Subtypes and Construction of LncRNA Score
3.4. LncRNA Score Associated with Immune Checkpoints

3.5. LncRNA Score Predicted Immunotherapeutic Benefits
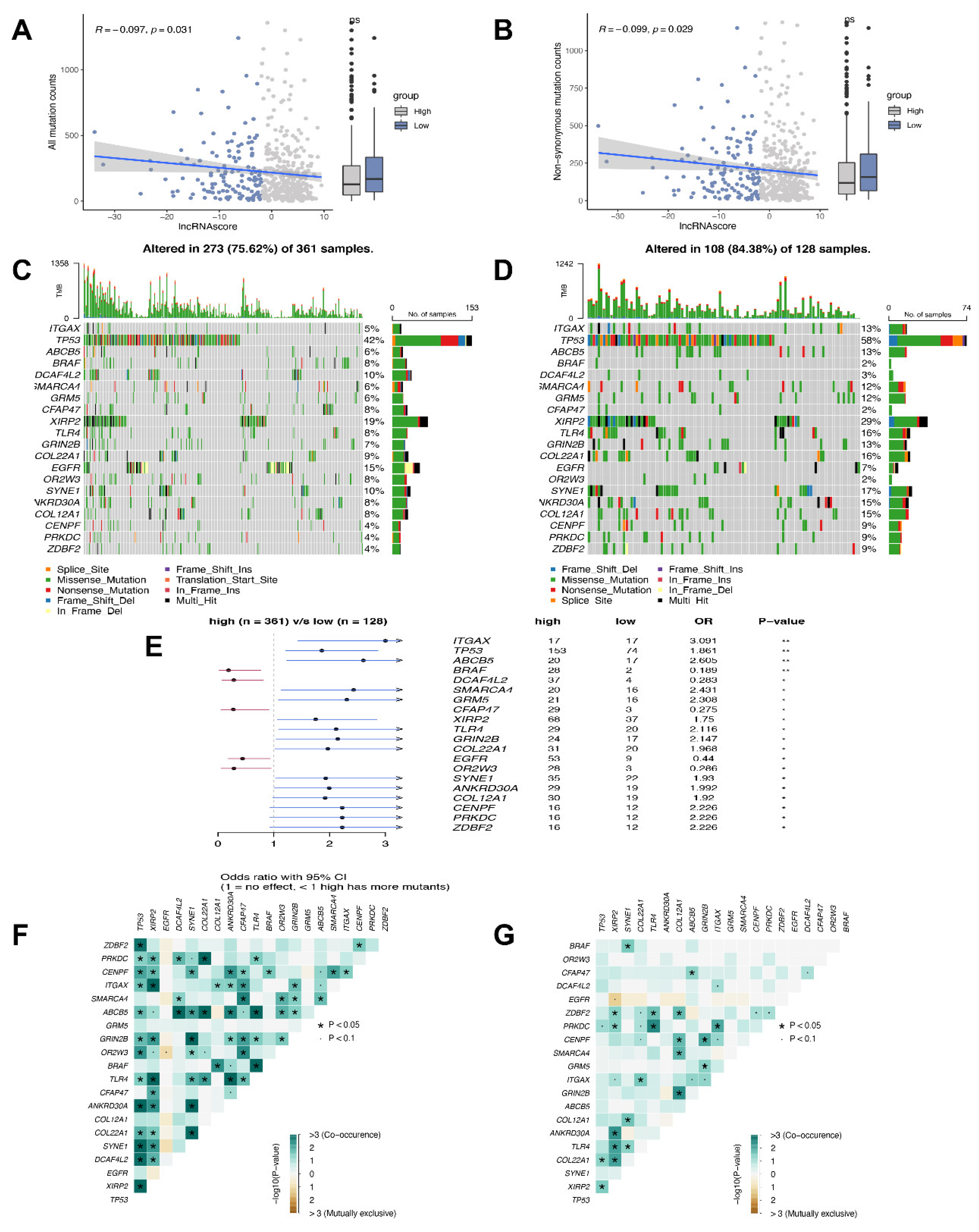
3.6. Mutation Status in the High and Low LncRNA Score Groups
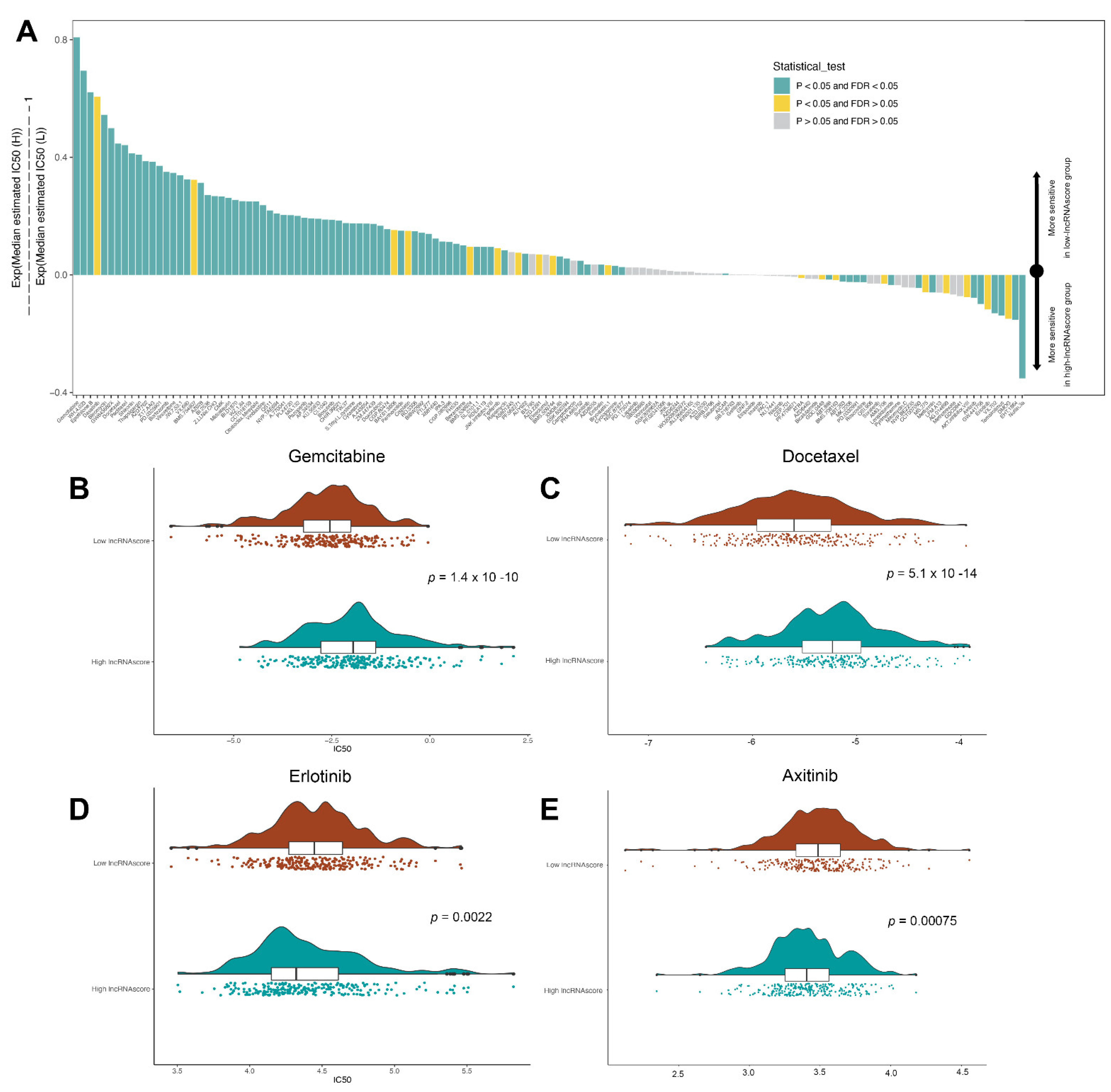
3.7. LncRNA Score Predicted the Sensitivity to Chemotherapeutic Drugs
3.8. LncRNA Score Was Correlated with MiRNA and Post-Transcriptional Regulation

4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Zappa, C.; Mousa, S.A. Non-small cell lung cancer: Current treatment and future advances. Transl. Lung Cancer Res. 2016, 5, 288–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walder, D.; O’Brien, M. Looking back and to the future: Are we improving ‘cure’ in non-small cell lung cancer? Eur. J. Cancer 2017, 75, 192–194. [Google Scholar] [CrossRef] [PubMed]
- Boo, S.H.; Kim, Y.K. The emerging role of RNA modifications in the regulation of mRNA stability. Exp. Mol. Med. 2020, 52, 400–408. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.; Chai, P.; Jia, R.; Fan, X. Novel insight into the regulatory roles of diverse RNA modifications: Re-defining the bridge between transcription and translation. Mol. Cancer 2020, 19, 78. [Google Scholar] [CrossRef]
- Wu, Y.; Yang, X.; Chen, Z.; Tian, L.; Jiang, G.; Chen, F.; Li, J.; An, P.; Lu, L.; Luo, N.; et al. m(6)A-induced lncRNA RP11 triggers the dissemination of colorectal cancer cells via upregulation of Zeb1. Mol. Cancer 2019, 18, 87. [Google Scholar] [CrossRef]
- Fang, S.; Shen, Y.; Chen, B.; Wu, Y.; Jia, L.; Li, Y.; Zhu, Y.; Yan, Y.; Li, M.; Chen, R.; et al. H3K27me3 induces multidrug resistance in small cell lung cancer by affecting HOXA1 DNA methylation via regulation of the lncRNA HOTAIR. Ann. Transl. Med. 2018, 6, 440. [Google Scholar] [CrossRef]
- Wang, P.; Xu, J.; Wang, Y.; Cao, X. An interferon-independent lncRNA promotes viral replication by modulating cellular metabolism. Science 2017, 358, 1051–1055. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Liu, X.; Cui, X.; Tan, Y.; Wang, Q.; Wang, Y.; Xu, C.; Fang, C.; Kang, C. LncRNA PRADX-mediated recruitment of PRC2/DDX5 complex suppresses UBXN1 expression and activates NF-kappaB activity, promoting tumorigenesis. Theranostics 2021, 11, 4516–4530. [Google Scholar] [CrossRef]
- Qian, X.; Yang, J.; Qiu, Q.; Li, X.; Jiang, C.; Li, J.; Dong, L.; Ying, K.; Lu, B.; Chen, E.; et al. LCAT3, a novel m6A-regulated long non-coding RNA, plays an oncogenic role in lung cancer via binding with FUBP1 to activate c-MYC. J. Hematol. Oncol. 2021, 14, 112. [Google Scholar] [CrossRef]
- Kabbout, M.; Garcia, M.M.; Fujimoto, J.; Liu, D.D.; Woods, D.; Chow, C.W.; Mendoza, G.; Momin, A.A.; James, B.P.; Solis, L.; et al. ETS2 mediated tumor suppressive function and MET oncogene inhibition in human non-small cell lung cancer. Clin. Cancer Res. 2013, 19, 3383–3395. [Google Scholar] [CrossRef] [Green Version]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E., III.; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef] [Green Version]
- Feng, X.; Li, L.; Wagner, E.J.; Li, W. TC3A: The Cancer 3′ UTR Atlas. Nucleic Acids Res. 2018, 46, D1027–D1030. [Google Scholar] [CrossRef]
- Xiang, Y.; Ye, Y.; Lou, Y.; Yang, Y.; Cai, C.; Zhang, Z.; Mills, T.; Chen, N.-Y.; Kim, Y.; Ozguc, F.M.; et al. Comprehensive Characterization of Alternative Polyadenylation in Human Cancer. J. Natl. Cancer Inst. 2018, 110, 379–389. [Google Scholar] [CrossRef]
- Wilkerson, M.D.; Hayes, D.N. ConsensusClusterPlus: A class discovery tool with confidence assessments and item tracking. Bioinformatics 2010, 26, 1572–1573. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Wu, Q.; Li, B.; Wang, D.; Wang, L.; Zhou, Y.L. m(6)A regulator-mediated methylation modification patterns and tumor microenvironment infiltration characterization in gastric cancer. Mol. Cancer 2020, 19, 53. [Google Scholar] [CrossRef]
- Geeleher, P.; Cox, N.J.; Huang, R.S. Clinical drug response can be predicted using baseline gene expression levels and in vitro drug sensitivity in cell lines. Genome Biol. 2014, 15, R47. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Yu, Y.; Li, H.; Hu, Q.; Chen, X.; He, Y.; Xue, C.; Ren, F.; Ren, Z.; Li, J.; et al. Long non-coding RNA PVT1 promotes tumor progression by regulating the miR-143/HK2 axis in gallbladder cancer. Mol. Cancer 2019, 18, 33. [Google Scholar] [CrossRef] [Green Version]
- Shen, D.; Ding, L.; Lu, Z.; Wang, R.; Yu, C.; Wang, H.; Zheng, Q.; Wang, X.; Xu, W.; Yu, X.; et al. METTL14-mediated Lnc-LSG1 m6A modification inhibits clear cell renal cell carcinoma metastasis via regulating ESRP2 ubiquitination. Mol. Ther. Nucleic Acids 2022, 27, 547–561. [Google Scholar] [CrossRef]
- Desbois, M.; Udyavar, A.R.; Ryner, L.; Kozlowski, C.; Guan, Y.; Durrbaum, M.; Lu, S.; Fortin, J.-P.; Koeppen, H.; Ziai, J.; et al. Integrated digital pathology and transcriptome analysis identifies molecular mediators of T-cell exclusion in ovarian cancer. Nat. Commun. 2020, 11, 5583. [Google Scholar] [CrossRef]
- Hegde, P.S.; Karanikas, V.; Evers, S. The Where, the When, and the How of Immune Monitoring for Cancer Immunotherapies in the Era of Checkpoint Inhibition. Clin. Cancer Res. 2016, 22, 1865–1874. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Ren, W.; Wang, W.; Xia, J.; Gou, L.; Liu, M.; Qan, Q.; Zhou, L.; Weng, Y.; He, T.; et al. Inhibitor of beta-catenin and TCF (ICAT) promotes cervical cancer growth and metastasis by disrupting E-cadherin/beta-catenin complex. Oncol. Rep. 2017, 38, 2597–2606. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Jiao, Z.; Ji, Y.; Zhang, S. Elevated TUBA1A Might Indicate the Clinical Outcomes of Patients with Gastric Cancer, Being Associated with the Infiltration of Macrophages in the Tumor Immune Microenvironment. J. Gastrointest. Liver Dis. 2020, 29, 509–522. [Google Scholar] [CrossRef]
- Shin, J.; Shin, Y.; Oh, S.M.; Yang, H.; Yu, W.J.; Lee, J.P.; Huh, S.-O.; Lee, S.-H.; Suh, Y.-H.; Chung, S.; et al. MiR-29b controls fetal mouse neurogenesis by regulating ICAT-mediated Wnt/beta-catenin signaling. Cell Death Dis. 2014, 5, e1473. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Song, X.; Li, Y.; Chen, B.; Zhao, W.; Wang, L.; Zhang, H.; Liu, Y.; Han, D.; Zhang, N.; et al. LncRNA BCRT1 promotes breast cancer progression by targeting miR-1303/PTBP3 axis. Mol. Cancer 2020, 19, 85. [Google Scholar] [CrossRef]
- Zhu, P.; He, F.; Hou, Y.; Tu, G.; Li, Q.; Jin, T.; Zeng, H.; Qin, Y.; Wan, X.; Qiao, Y.; et al. A novel hypoxic long noncoding RNA KB-1980E6.3 maintains breast cancer stem cell stemness via interacting with IGF2BP1 to facilitate c-Myc mRNA stability. Oncogene 2021, 40, 1609–1627. [Google Scholar] [CrossRef] [PubMed]
- Ban, Y.; Tan, P.; Cai, J.; Li, J.; Hu, M.; Zhou, Y.; Mei, Y.; Tan, Y.; Li, X.; Zeng, Z.; et al. LNCAROD is stabilized by m6A methylation and promotes cancer progression via forming a ternary complex with HSPA1A and YBX1 in head and neck squamous cell carcinoma. Mol. Oncol. 2020, 14, 1282–1296. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Niu, N.; Li, P.; Zhai, L.; Xiao, K.; Chen, W.; Zhuang, X. LncRNA OGFRP1 acts as an oncogene in NSCLC via miR-4640-5p/eIF5A axis. Cancer Cell Int. 2021, 21, 425. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.A.; Yarchoan, M.; Jaffee, E.; Swanton, C.; Quezada, S.A.; Stenzinger, A.; Peters, S. Development of tumor mutation burden as an immunotherapy biomarker: Utility for the oncology clinic. Ann. Oncol. 2019, 30, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Gong, Y.; Fan, Z.; Luo, G.; Huang, Q.; Deng, S.; Cheng, H.; Jin, K.; Ni, Q.; Yu, X.; et al. Molecular alterations and targeted therapy in pancreatic ductal adenocarcinoma. J. Hematol. Oncol. 2020, 13, 130. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Liu, Z.; Li, M.; Chen, C.; Wang, X. Immunogenomics Analysis Reveals that TP53 Mutations Inhibit Tumor Immunity in Gastric Cancer. Transl. Oncol. 2018, 11, 1171–1187. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; Du, N.; Huang, T.; Guo, J.; Mo, X.; Yuan, T.; Chen, Y.; Ye, T.; Xu, C.; Wang, W.; et al. TP53 Mutation as Potential Negative Predictor for Response of Anti-CTLA-4 Therapy in Metastatic Melanoma. eBioMedicine 2018, 32, 119–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyu, H.; Li, M.; Jiang, Z.; Liu, Z.; Wang, X. Correlate the TP53 Mutation and the HRAS Mutation with Immune Signatures in Head and Neck Squamous Cell Cancer. Comput. Struct. Biotechnol. J. 2019, 17, 1020–1030. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.Y.; Zhong, W.Z.; Zhang, X.C.; Su, J.; Xie, Z.; Liu, S.Y.; Tu, H.-Y.; Chen, H.-J.; Sun, Y.-L.; Zhou, Q.; et al. Potential Predictive Value of TP53 and KRAS Mutation Status for Response to PD-1 Blockade Immunotherapy in Lung Adenocarcinoma. Clin. Cancer Res. 2017, 23, 3012–3024. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen, Y.; Wang, S.; Wu, Y. A Novel m6A-Related LncRNA Signature for Predicting Prognosis, Chemotherapy and Immunotherapy Response in Patients with Lung Adenocarcinoma. Cells 2022, 11, 2399. https://doi.org/10.3390/cells11152399
Shen Y, Wang S, Wu Y. A Novel m6A-Related LncRNA Signature for Predicting Prognosis, Chemotherapy and Immunotherapy Response in Patients with Lung Adenocarcinoma. Cells. 2022; 11(15):2399. https://doi.org/10.3390/cells11152399
Chicago/Turabian StyleShen, Yefeng, Shaochun Wang, and Yuanzhou Wu. 2022. "A Novel m6A-Related LncRNA Signature for Predicting Prognosis, Chemotherapy and Immunotherapy Response in Patients with Lung Adenocarcinoma" Cells 11, no. 15: 2399. https://doi.org/10.3390/cells11152399
APA StyleShen, Y., Wang, S., & Wu, Y. (2022). A Novel m6A-Related LncRNA Signature for Predicting Prognosis, Chemotherapy and Immunotherapy Response in Patients with Lung Adenocarcinoma. Cells, 11(15), 2399. https://doi.org/10.3390/cells11152399