
New Insights into Hippo/YAP Signaling in Fibrotic Diseases


Abstract
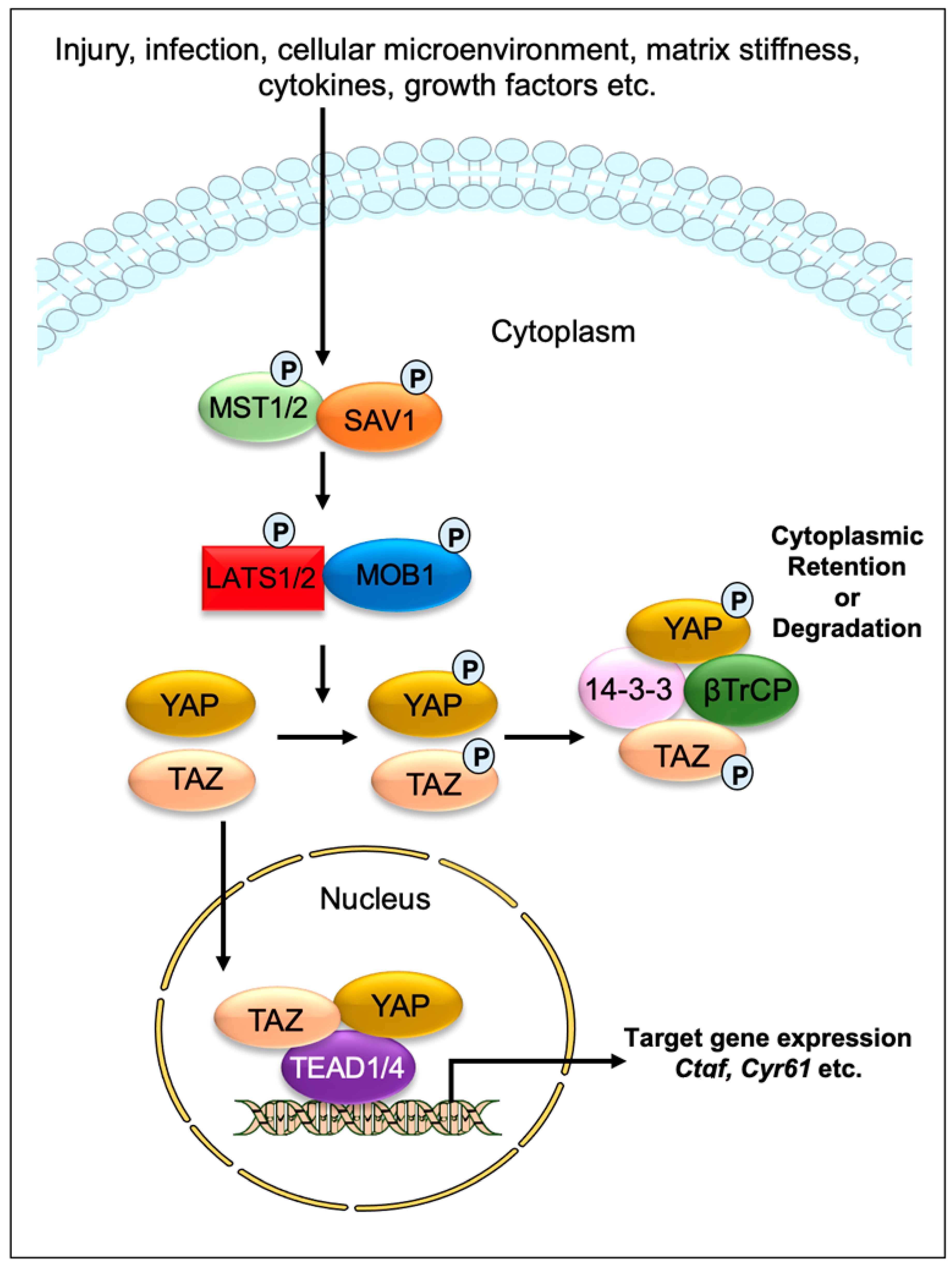
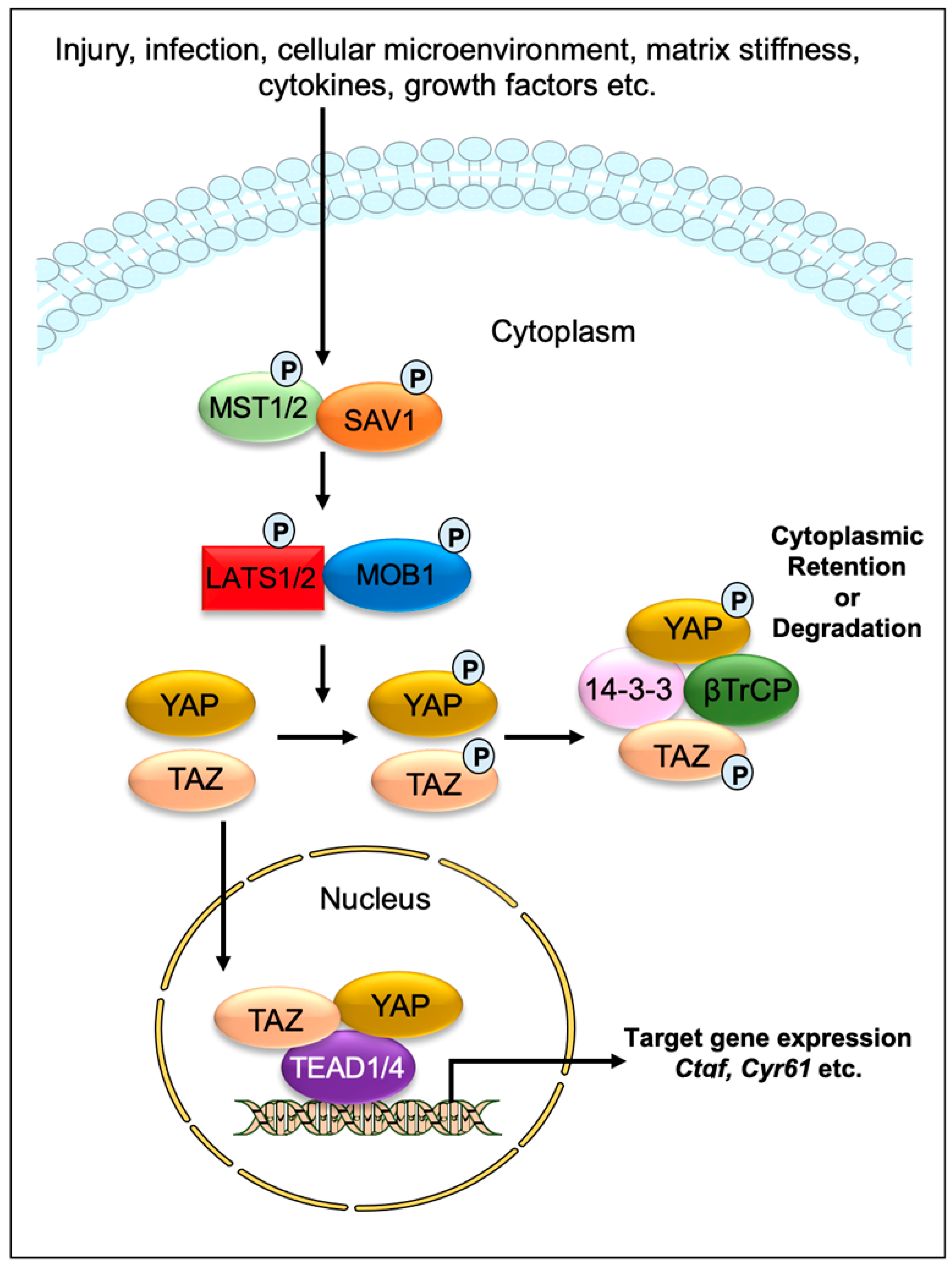
:1. Introduction
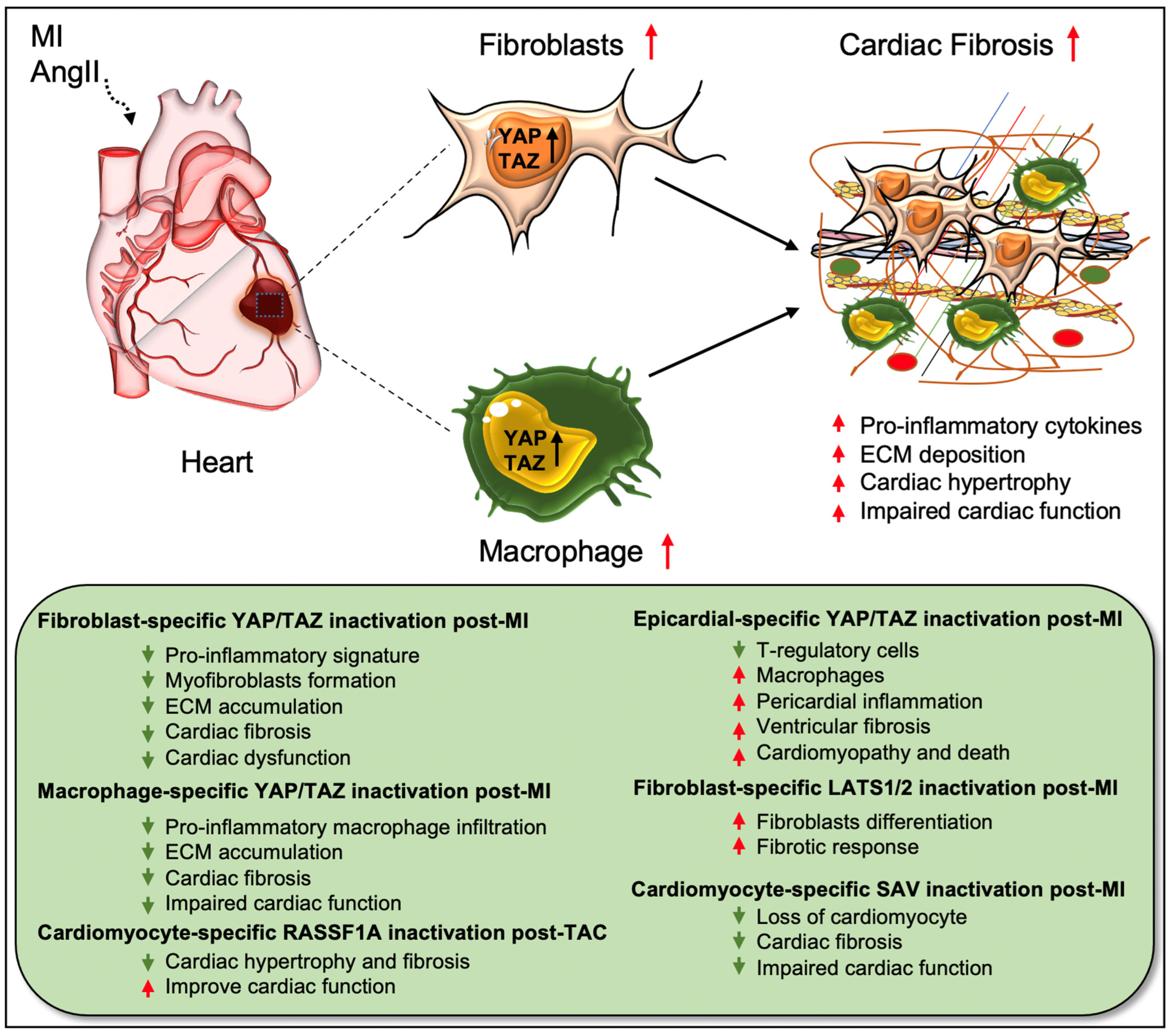
2. Hippo Signaling Pathway in Cardiac Fibrosis
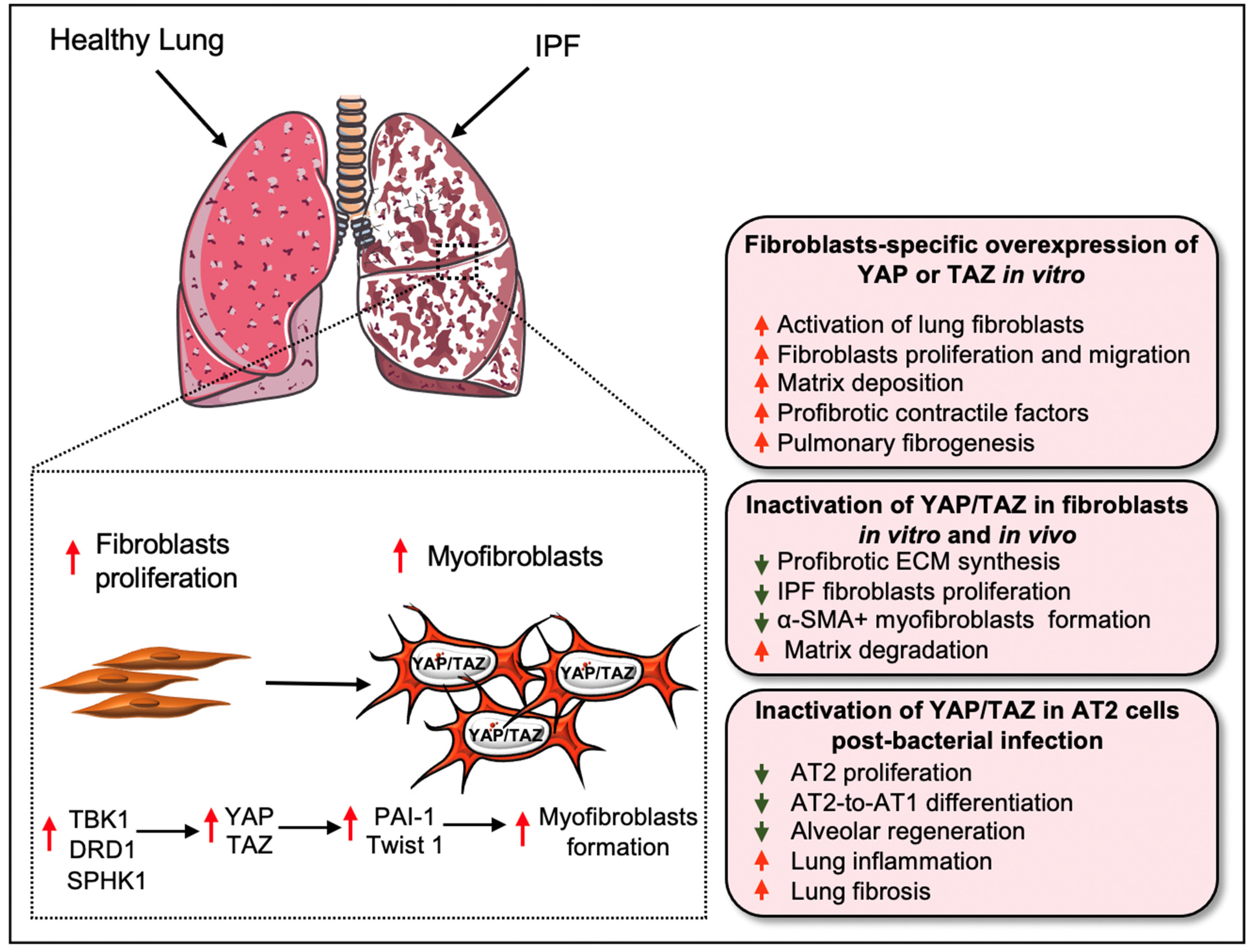
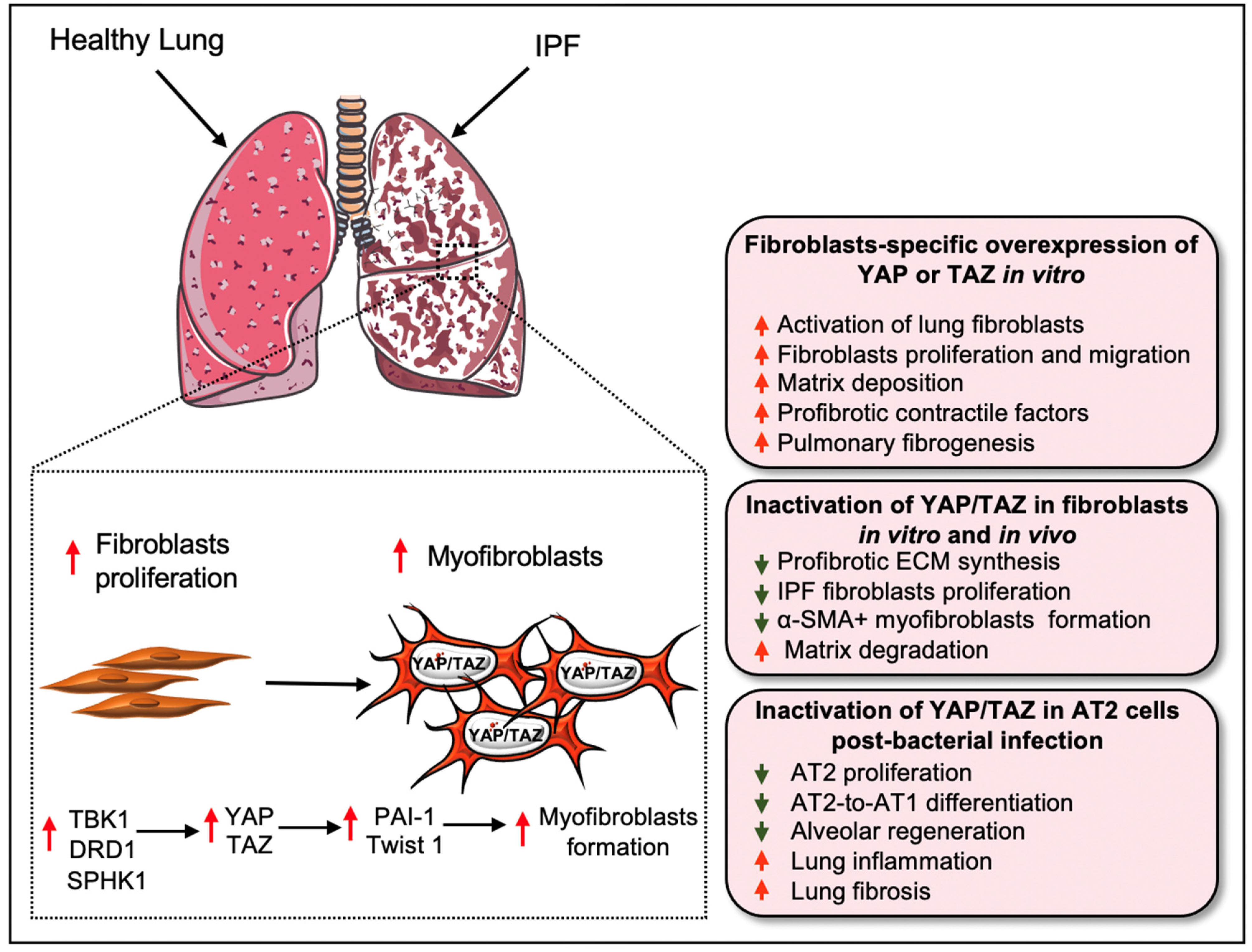
3. Hippo Signaling Pathway in Pulmonary Fibrosis
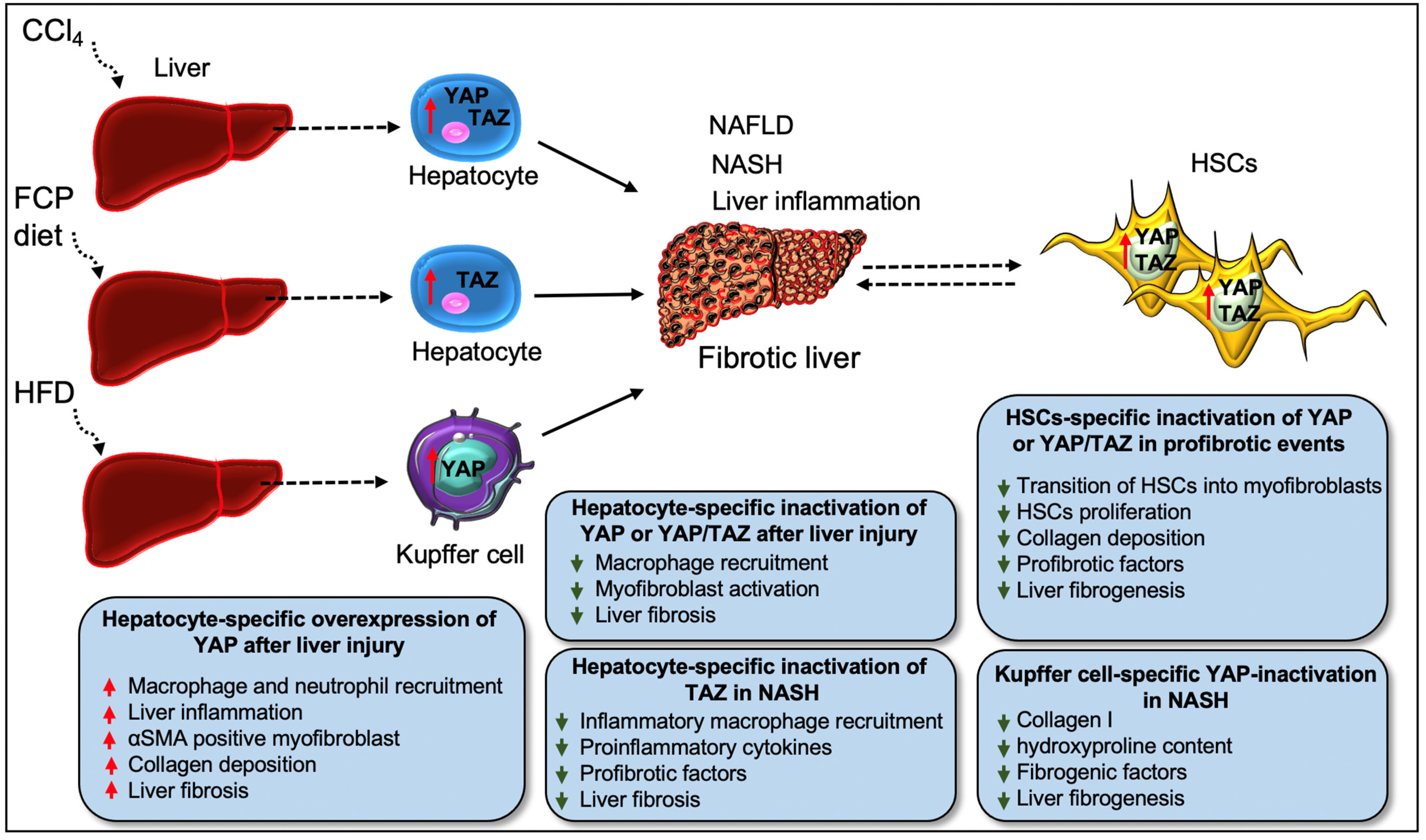
4. Hippo Signaling Pathway in Liver Fibrosis
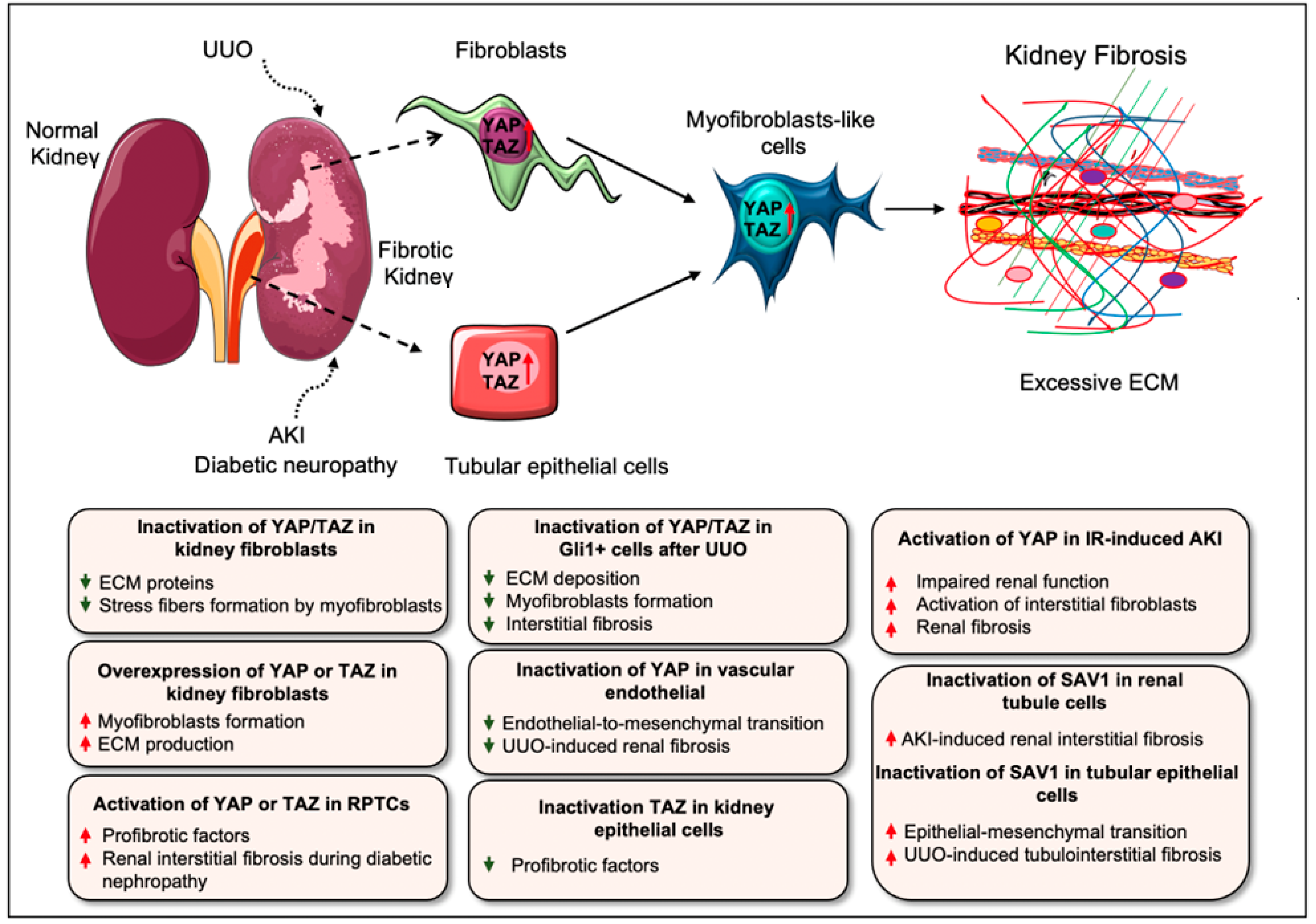
5. Hippo Signaling Pathway in Renal Fibrosis
6. Hippo Signaling Pathway in Skin Fibrosis
7. Hippo Signaling Pathway in Fibrosis of Other Organs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name of the Compounds | Mode of Actin | Experimental Study |
|---|---|---|
| Verteporfin [13,15,17,18,94,95,97] | Interference of YAP/TAZ-TEAD complex | |
| XMU-MP-1 [39] | MST1/2 blocker | TAC-induced cardiac fibrosis in mice. |
| Qishen granules [43] | Disrupt YAP/TAZ expression | Reduce the fibrogenic protein expression in HF-hearts of the rat. |
| SKI [27] | Inducing proteasomal degradation of TAZ through the interaction with LIMD1 | Inhibits the transition of primary rat cardiac fibroblast to myofibroblasts. |
| Lovastatin [44] | Suppression of YAP/TAZ signaling | Alleviates the AngII-induced cardiac fibrosis in mice. |
| Sulfur dioxide (SO2) [45] | Inhibits MST1/2 | Prevent the myocardial fibrosis in diabetic rats |
| Melatonin [69] | interrupting the translocation of YAP into the nucleus |
|
| Icariin [70] | Inhibition of YAP function | Prevents the bleomycin-induced PF in rats. |
| Simvastatin [67] | Modulates YAP localization | Reduces established fibrosis in bleomycin-challenged mouse IPF. |
| PP242 [16] | Inhibits mTORC2 and suppresses YAP/TAZ | Inhibits the fibroblasts activation in UUO nephropathy induced mouse kidney fibrosis |
| FSLLRY-NH2 [104] | Selectively inhibits YAP activity | Blocks the endothelial-to-mesenchymal transition and reduces the UUO-induced kidney fibrosis. |
| Kindlin-2 siRNA [108] | Induces the degradation of MOB1 and promotes the nuclear translocation of YAP | Attenuates UUO-induced renal fibrosis in mice. |
| Exosomes [105] | Promotes the βTrCP-mediated ubiquitination and degradation of YAP | Attenuates UUO-induced renal fibrosis. |
| Fluphenazine dihydrochloride, (a DRD2 antagonist) [100] | Suppress YAP activity and induce the type I interferon signaling | Reduces CCl4-induced liver fibrosis. |
| LPA [97] | YAP activator | Prevent IR-induced liver fibrogenesis in mice. |
| Dihydrexidine (a DRD1 agonist) [7] | Blocks YAP/TAZ | Reverse HSCs activation and bile duct ligation-induced hepatic fibrosis in mice |
| Tricyclic antidepressants [14] | Inhibit acid ceramidase and YAP/TAZ activity | Reduces human HSCs-activation |
| ω-3 PUFAs [98] | Promoting YAP/TAZ degradation in a proteasome-dependent manner |
|
| RhoA siRNA [9] | Reduces YAP/TAZ expression and augments the LATS1/2 activity | Halt the liver fibrosis |
| Morin [99] | Reduces YAP/TAZ expression while activating the MST1 and LATS1 expression |
|
| Dimethyl fumarate (DMF) [110] | Inhibition of YAP/TAZ |
|
| Y27632 [115] | Inhibition of ROCK1 | Inhibits YAP/TAZ-dependent activation of fibrotic events in intestinal fibroblasts and DSS-induced chronic colitis mice. |
8. Conclusions and Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Rockey, D.C.; Bell, P.D.; Hill, J.A. Fibrosis—A common pathway to organ injury and failure. N. Engl. J. Med. 2015, 372, 1138–1149. [Google Scholar] [CrossRef] [PubMed]
- Gabbiani, G. The myofibroblast: A key cell for wound healing and fibrocontractive diseases. Prog. Clin. Biol. Res. 1981, 54, 183–194. [Google Scholar] [PubMed]
- Gabbiani, G. The myofibroblast in wound healing and fibrocontractive diseases. J. Pathol. 2003, 200, 500–503. [Google Scholar] [CrossRef] [PubMed]
- Piersma, B.; Bank, R.A.; Boersema, M. Signaling in Fibrosis: TGF-beta, WNT, and YAP/TAZ Converge. Front Med. 2015, 2, 59. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Lagares, D.; Choi, K.M.; Stopfer, L.; Marinkovic, A.; Vrbanac, V.; Probst, C.K.; Hiemer, S.E.; Sisson, T.H.; Horowitz, J.C.; et al. Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L344–L357. [Google Scholar] [CrossRef] [Green Version]
- Link, P.A.; Choi, K.M.; Diaz Espinosa, A.M.; Jones, D.L.; Gao, A.Y.; Haak, A.J.; Tschumperlin, D.J. Combined control of the fibroblast contractile program by YAP and TAZ. Am. J. Physiol. Lung Cell. Mol. Physiol. 2022, 322, L23–L32. [Google Scholar] [CrossRef]
- Haak, A.J.; Kostallari, E.; Sicard, D.; Ligresti, G.; Choi, K.M.; Caporarello, N.; Jones, D.L.; Tan, Q.; Meridew, J.; Diaz Espinosa, A.M.; et al. Selective YAP/TAZ inhibition in fibroblasts via dopamine receptor D1 agonism reverses fibrosis. Sci. Transl. Med. 2019, 11, eaau6296. [Google Scholar] [CrossRef]
- Mooring, M.; Fowl, B.H.; Lum, S.Z.C.; Liu, Y.; Yao, K.; Softic, S.; Kirchner, R.; Bernstein, A.; Singhi, A.D.; Jay, D.G.; et al. Hepatocyte Stress Increases Expression of Yes-Associated Protein and Transcriptional Coactivator With PDZ-Binding Motif in Hepatocytes to Promote Parenchymal Inflammation and Fibrosis. Hepatology 2020, 71, 1813–1830. [Google Scholar] [CrossRef]
- Wang, X.; Cai, B.; Yang, X.; Sonubi, O.O.; Zheng, Z.; Ramakrishnan, R.; Shi, H.; Valenti, L.; Pajvani, U.B.; Sandhu, J.; et al. Cholesterol Stabilizes TAZ in Hepatocytes to Promote Experimental Non-alcoholic Steatohepatitis. Cell Metab. 2020, 31, 969–986.e967. [Google Scholar] [CrossRef]
- Wang, X.; Zheng, Z.; Caviglia, J.M.; Corey, K.E.; Herfel, T.M.; Cai, B.; Masia, R.; Chung, R.T.; Lefkowitch, J.H.; Schwabe, R.F.; et al. Hepatocyte TAZ/WWTR1 Promotes Inflammation and Fibrosis in Nonalcoholic Steatohepatitis. Cell Metab. 2016, 24, 848–862. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Zhang, R.; Zhan, Y.; Zheng, J. Resveratrol Inhibits Hepatic Stellate Cell Activation via the Hippo Pathway. Mediat. Inflamm. 2021, 2021, 3399357. [Google Scholar] [CrossRef] [PubMed]
- Du, K.; Hyun, J.; Premont, R.T.; Choi, S.S.; Michelotti, G.A.; Swiderska-Syn, M.; Dalton, G.D.; Thelen, E.; Rizi, B.S.; Jung, Y.; et al. Hedgehog-YAP Signaling Pathway Regulates Glutaminolysis to Control Activation of Hepatic Stellate Cells. Gastroenterology 2018, 154, 1465–1479.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.X.; Yao, Y.; Bu, F.T.; Chen, Y.; Wu, Y.T.; Yang, Y.; Chen, X.; Zhu, Y.; Wang, Q.; Pan, X.Y.; et al. Blockade of YAP alleviates hepatic fibrosis through accelerating apoptosis and reversion of activated hepatic stellate cells. Mol. Immunol. 2019, 107, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Alsamman, S.; Christenson, S.A.; Yu, A.; Ayad, N.M.E.; Mooring, M.S.; Segal, J.M.; Hu, J.K.; Schaub, J.R.; Ho, S.S.; Rao, V.; et al. Targeting acid ceramidase inhibits YAP/TAZ signaling to reduce fibrosis in mice. Sci. Transl. Med. 2020, 12, eaay8798. [Google Scholar] [CrossRef] [PubMed]
- Liang, M.; Yu, M.; Xia, R.; Song, K.; Wang, J.; Luo, J.; Chen, G.; Cheng, J. Yap/Taz Deletion in Gli(+) Cell-Derived Myofibroblasts Attenuates Fibrosis. J. Am. Soc. Nephrol. 2017, 28, 3278–3290. [Google Scholar] [CrossRef] [Green Version]
- Gui, Y.; Li, J.; Lu, Q.; Feng, Y.; Wang, M.; He, W.; Yang, J.; Dai, C. Yap/Taz mediates mTORC2-stimulated fibroblast activation and kidney fibrosis. J. Biol. Chem. 2018, 293, 16364–16375. [Google Scholar] [CrossRef] [Green Version]
- Szeto, S.G.; Narimatsu, M.; Lu, M.; He, X.; Sidiqi, A.M.; Tolosa, M.F.; Chan, L.; De Freitas, K.; Bialik, J.F.; Majumder, S.; et al. YAP/TAZ Are Mechanoregulators of TGF-beta-Smad Signaling and Renal Fibrogenesis. J. Am. Soc. Nephrol. 2016, 27, 3117–3128. [Google Scholar] [CrossRef] [Green Version]
- Mia, M.M.; Cibi, D.M.; Binte Abdul Ghani, S.A.; Singh, A.; Tee, N.; Sivakumar, V.; Bogireddi, H.; Cook, S.A.; Mao, J.; Singh, M.K. Loss of Yap/taz in cardiac fibroblasts attenuates adverse remodeling and improves cardiac function. Cardiovasc. Res. 2022, 118, 1785–1804. [Google Scholar] [CrossRef]
- Francisco, J.; Zhang, Y.; Jeong, J.I.; Mizushima, W.; Ikeda, S.; Ivessa, A.; Oka, S.; Zhai, P.; Tallquist, M.D.; Del Re, D.P. Blockade of Fibroblast YAP Attenuates Cardiac Fibrosis and Dysfunction Through MRTF-A Inhibition. JACC Basic Transl. Sci. 2020, 5, 931–945. [Google Scholar] [CrossRef]
- Leach, J.P.; Heallen, T.; Zhang, M.; Rahmani, M.; Morikawa, Y.; Hill, M.C.; Segura, A.; Willerson, J.T.; Martin, J.F. Hippo pathway deficiency reverses systolic heart failure after infarction. Nature 2017, 550, 260–264. [Google Scholar] [CrossRef] [Green Version]
- Halder, G.; Johnson, R.L. Hippo signaling: Growth control and beyond. Development 2011, 138, 9–22. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.; Tumaneng, K.; Guan, K.L. The Hippo pathway in organ size control, tissue regeneration and stem cell self-renewal. Nat. Cell Biol. 2011, 13, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Mia, M.M.; Singh, M.K. The Hippo Signaling Pathway in Cardiac Development and Diseases. Front. Cell Dev. Biol. 2019, 7, 211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, F.X.; Zhao, B.; Guan, K.L. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell 2015, 163, 811–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, R.; Fallon, T.R.; Saladi, S.V.; Pardo-Saganta, A.; Villoria, J.; Mou, H.; Vinarsky, V.; Gonzalez-Celeiro, M.; Nunna, N.; Hariri, L.P.; et al. Yap tunes airway epithelial size and architecture by regulating the identity, maintenance, and self-renewal of stem cells. Dev. Cell. 2014, 30, 151–165. [Google Scholar] [CrossRef] [Green Version]
- Szymaniak, A.D.; Mahoney, J.E.; Cardoso, W.V.; Varelas, X. Crumbs3-Mediated Polarity Directs Airway Epithelial Cell Fate through the Hippo Pathway Effector Yap. Dev. Cell. 2015, 34, 283–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landry, N.M.; Rattan, S.G.; Filomeno, K.L.; Meier, T.W.; Meier, S.C.; Foran, S.J.; Meier, C.F.; Koleini, N.; Fandrich, R.R.; Kardami, E.; et al. SKI activates the Hippo pathway via LIMD1 to inhibit cardiac fibroblast activation. Basic Res. Cardiol. 2021, 116, 25. [Google Scholar] [CrossRef]
- Francisco, J.; Zhang, Y.; Nakada, Y.; Jeong, J.I.; Huang, C.Y.; Ivessa, A.; Oka, S.; Babu, G.J.; Del Re, D.P. AAV-mediated YAP expression in cardiac fibroblasts promotes inflammation and increases fibrosis. Sci. Rep. 2021, 11, 10553. [Google Scholar] [CrossRef]
- Mia, M.M.; Cibi, D.M.; Abdul Ghani, S.A.B.; Song, W.; Tee, N.; Ghosh, S.; Mao, J.; Olson, E.N.; Singh, M.K. YAP/TAZ deficiency reprograms macrophage phenotype and improves infarct healing and cardiac function after myocardial infarction. PLoS Biol. 2020, 18, e3000941. [Google Scholar] [CrossRef]
- Song, K.; Kwon, H.; Han, C.; Chen, W.; Zhang, J.; Ma, W.; Dash, S.; Gandhi, C.R.; Wu, T. Yes-Associated Protein in Kupffer Cells Enhances the Production of Proinflammatory Cytokines and Promotes the Development of Nonalcoholic Steatohepatitis. Hepatology 2020, 72, 72–87. [Google Scholar] [CrossRef]
- Anorga, S.; Overstreet, J.M.; Falke, L.L.; Tang, J.; Goldschmeding, R.G.; Higgins, P.J.; Samarakoon, R. Deregulation of Hippo-TAZ pathway during renal injury confers a fibrotic maladaptive phenotype. FASEB J. 2018, 32, 2644–2657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; You, H.; Li, Y.; Xu, Y.; He, Q.; Harris, R.C. EGF Receptor-Dependent YAP Activation Is Important for Renal Recovery from AKI. J. Am. Soc. Nephrol. 2018, 29, 2372–2385. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, X.; He, Q.; Bulus, N.; Fogo, A.B.; Zhang, M.Z.; Harris, R.C. YAP Activation in Renal Proximal Tubule Cells Drives Diabetic Renal Interstitial Fibrogenesis. Diabetes 2020, 69, 2446–2457. [Google Scholar] [CrossRef] [PubMed]
- Mia, M.M.; Singh, M.K. Emerging roles of the Hippo signaling pathway in modulating immune response and inflammation-driven tissue repair and remodeling. FEBS J. 2022; in press. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Hill, M.C.; Li, L.; Deshmukh, V.; Martin, T.J.; Wang, J.; Martin, J.F. Hippo pathway deletion in adult resting cardiac fibroblasts initiates a cell state transition with spontaneous and self-sustaining fibrosis. Genes Dev. 2019, 33, 1491–1505. [Google Scholar] [CrossRef] [Green Version]
- Leung, J.Y.; Wilson, H.L.; Voltzke, K.J.; Williams, L.A.; Lee, H.J.; Wobker, S.E.; Kim, W.Y. Sav1 Loss Induces Senescence and Stat3 Activation Coinciding with Tubulointerstitial Fibrosis. Mol. Cell. Biol. 2017, 37, e00565-16. [Google Scholar] [CrossRef] [Green Version]
- Shao, D.; Zhai, P.; Hu, C.; Mukai, R.; Sciarretta, S.; Del Re, D.; Sadoshima, J. Lats2 promotes heart failure by stimulating p53-mediated apoptosis during pressure overload. Sci. Rep. 2021, 11, 23469. [Google Scholar] [CrossRef]
- Gokey, J.J.; Sridharan, A.; Xu, Y.; Green, J.; Carraro, G.; Stripp, B.R.; Perl, A.T.; Whitsett, J.A. Active epithelial Hippo signaling in idiopathic pulmonary fibrosis. JCI Insight 2018, 3, e98738. [Google Scholar] [CrossRef] [Green Version]
- Triastuti, E.; Nugroho, A.B.; Zi, M.; Prehar, S.; Kohar, Y.S.; Bui, T.A.; Stafford, N.; Cartwright, E.J.; Abraham, S.; Oceandy, D. Pharmacological inhibition of Hippo pathway, with the novel kinase inhibitor XMU-MP-1, protects the heart against adverse effects during pressure overload. Br. J. Pharmacol. 2019, 176, 3956–3971. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Tolosa, M.F.; Zhang, T.; Goru, S.K.; Ulloa Severino, L.; Misra, P.S.; McEvoy, C.M.; Caldwell, L.; Szeto, S.G.; Gao, F.; et al. Myofibroblast YAP/TAZ activation is a key step in organ fibrogenesis. JCI Insight 2022, 7, e146243. [Google Scholar] [CrossRef]
- Caliari, S.R.; Perepelyuk, M.; Cosgrove, B.D.; Tsai, S.J.; Lee, G.Y.; Mauck, R.L.; Wells, R.G.; Burdick, J.A. Stiffening hydrogels for investigating the dynamics of hepatic stellate cell mechanotransduction during myofibroblast activation. Sci. Rep. 2016, 6, 21387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharifi-Sanjani, M.; Berman, M.; Goncharov, D.; Alhamaydeh, M.; Avolio, T.G.; Baust, J.; Chang, B.; Kobir, A.; Ross, M.; St Croix, C.; et al. Yes-Associated Protein (Yap) Is Up-Regulated in Heart Failure and Promotes Cardiac Fibroblast Proliferation. Int. J. Mol. Sci. 2021, 22, 6164. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, J.; Gao, S.; Lin, W.; Gao, P.; Gao, K.; Zhang, Y.; Du, K.; Yang, X.; Wang, W.; et al. Antiapoptosis and Antifibrosis Effects of Qishen Granules on Heart Failure Rats via Hippo Pathway. Biomed. Res. Int. 2019, 2019, 1642575. [Google Scholar] [CrossRef] [Green Version]
- Wu, P.; Liu, Z.; Zhao, T.; Xia, F.; Gong, L.; Zheng, Z.; Chen, Z.; Yang, T.; Duan, Q. Lovastatin attenuates angiotensin II induced cardiovascular fibrosis through the suppression of YAP/TAZ signaling. Biochem. Biophys. Res. Commun. 2019, 512, 736–741. [Google Scholar] [CrossRef]
- Liu, M.; Liu, S.; Tan, W.; Tang, F.; Long, J.; Li, Z.; Liang, B.; Chu, C.; Yang, J. Gaseous signalling molecule SO2 via HippoMST pathway to improve myocardial fibrosis of diabetic rats. Mol. Med. Rep. 2017, 16, 8953–8963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramjee, V.; Li, D.; Manderfield, L.J.; Liu, F.; Engleka, K.A.; Aghajanian, H.; Rodell, C.B.; Lu, W.; Ho, V.; Wang, T.; et al. Epicardial YAP/TAZ orchestrate an immunosuppressive response following myocardial infarction. J. Clin. Investig. 2017, 127, 899–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Re, D.P.; Matsuda, T.; Zhai, P.; Gao, S.; Clark, G.J.; Van Der Weyden, L.; Sadoshima, J. Proapoptotic Rassf1A/Mst1 signaling in cardiac fibroblasts is protective against pressure overload in mice. J. Clin. Investig. 2010, 120, 3555–3567. [Google Scholar] [CrossRef] [Green Version]
- Mia, M.M.; Chelakkot-Govindalayathil, A.L.; Singh, M.K. Targeting NF2-Hippo/Yap signaling pathway for cardioprotection after ischemia/reperfusion injury. Ann. Transl. Med. 2016, 4, 545. [Google Scholar] [CrossRef] [Green Version]
- Lucas, A.; Yasa, J.; Lucas, M. Regeneration and repair in the healing lung. Clin. Transl. Immunol. 2020, 9, e1152. [Google Scholar] [CrossRef]
- Barkauskas, C.E.; Cronce, M.J.; Rackley, C.R.; Bowie, E.J.; Keene, D.R.; Stripp, B.R.; Randell, S.H.; Noble, P.W.; Hogan, B.L. Type 2 alveolar cells are stem cells in adult lung. J. Clin. Investig. 2013, 123, 3025–3036. [Google Scholar] [CrossRef]
- Vaughan, A.E.; Brumwell, A.N.; Xi, Y.; Gotts, J.E.; Brownfield, D.G.; Treutlein, B.; Tan, K.; Tan, V.; Liu, F.C.; Looney, M.R.; et al. Lineage-negative progenitors mobilize to regenerate lung epithelium after major injury. Nature 2015, 517, 621–625. [Google Scholar] [CrossRef] [PubMed]
- Lederer, D.J.; Martinez, F.J. Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2018, 378, 1811–1823. [Google Scholar] [CrossRef] [PubMed]
- Barratt, S.L.; Creamer, A.; Hayton, C.; Chaudhuri, N. Idiopathic Pulmonary Fibrosis (IPF): An Overview. J. Clin. Med. 2018, 7, 201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.Y.; Qiao, K.; Liu, F.; Wu, B.; Xu, X.; Jiao, G.Q.; Lu, R.G.; Li, H.X.; Zhao, J.; Huang, J.; et al. Lung transplantation as therapeutic option in acute respiratory distress syndrome for coronavirus disease 2019-related pulmonary fibrosis. Chin. Med. J. 2020, 133, 1390–1396. [Google Scholar] [CrossRef] [PubMed]
- Ley, B.; Collard, H.R.; King, T.E., Jr. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2011, 183, 431–440. [Google Scholar] [CrossRef]
- Richeldi, L.; Du Bois, R.M.; Raghu, G.; Azuma, A.; Brown, K.K.; Costabel, U.; Cottin, V.; Flaherty, K.R.; Hansell, D.M.; Inoue, Y.; et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2071–2082. [Google Scholar] [CrossRef] [Green Version]
- Noble, P.W.; Albera, C.; Bradford, W.Z.; Costabel, U.; Glassberg, M.K.; Kardatzke, D.; King, T.E., Jr.; Lancaster, L.; Sahn, S.A.; Szwarcberg, J.; et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): Two randomised trials. Lancet 2011, 377, 1760–1769. [Google Scholar] [CrossRef]
- Xu, Y.; Mizuno, T.; Sridharan, A.; Du, Y.; Guo, M.; Tang, J.; Wikenheiser-Brokamp, K.A.; Perl, A.T.; Funari, V.A.; Gokey, J.J.; et al. Single-cell RNA sequencing identifies diverse roles of epithelial cells in idiopathic pulmonary fibrosis. JCI Insight 2016, 1, e90558. [Google Scholar] [CrossRef] [Green Version]
- LaCanna, R.; Liccardo, D.; Zhang, P.; Tragesser, L.; Wang, Y.; Cao, T.; Chapman, H.A.; Morrisey, E.E.; Shen, H.; Koch, W.J.; et al. Yap/Taz regulate alveolar regeneration and resolution of lung inflammation. J. Clin. Investig. 2019, 129, 2107–2122. [Google Scholar] [CrossRef] [Green Version]
- Penkala, I.J.; Liberti, D.C.; Pankin, J.; Sivakumar, A.; Kremp, M.M.; Jayachandran, S.; Katzen, J.; Leach, J.P.; Windmueller, R.; Stolz, K.; et al. Age-dependent alveolar epithelial plasticity orchestrates lung homeostasis and regeneration. Cell Stem Cell 2021, 28, 1775–1789.e1775. [Google Scholar] [CrossRef]
- Liu, Z.; Wu, H.; Jiang, K.; Wang, Y.; Zhang, W.; Chu, Q.; Li, J.; Huang, H.; Cai, T.; Ji, H.; et al. MAPK-Mediated YAP Activation Controls Mechanical-Tension-Induced Pulmonary Alveolar Regeneration. Cell Rep. 2016, 16, 1810–1819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, C.; Sun, J.; Du, J.; Wen, D.; Lu, H.; Zhang, H.; Xue, Y.; Zhang, A.; Yang, C.; Zeng, L.; et al. The Hippo-YAP pathway regulates the proliferation of alveolar epithelial progenitors after acute lung injury. Cell Biol. Int. 2019, 43, 1174–1183. [Google Scholar] [CrossRef] [PubMed]
- Milara, J.; Navarro, R.; Juan, G.; Peiro, T.; Serrano, A.; Ramon, M.; Morcillo, E.; Cortijo, J. Sphingosine-1-phosphate is increased in patients with idiopathic pulmonary fibrosis and mediates epithelial to mesenchymal transition. Thorax 2012, 67, 147–156. [Google Scholar] [CrossRef] [Green Version]
- Zmajkovicova, K.; Bauer, Y.; Menyhart, K.; Schnoebelen, M.; Freti, D.; Boucher, M.; Renault, B.; Studer, R.; Birker-Robaczewska, M.; Klenk, A.; et al. GPCR-induced YAP activation sensitizes fibroblasts to profibrotic activity of TGFbeta1. PLoS ONE 2020, 15, e0228195. [Google Scholar] [CrossRef]
- Huang, L.S.; Sudhadevi, T.; Fu, P.; Punathil-Kannan, P.K.; Ebenezer, D.L.; Ramchandran, R.; Putherickal, V.; Cheresh, P.; Zhou, G.; Ha, A.W.; et al. Sphingosine Kinase 1/S1P Signaling Contributes to Pulmonary Fibrosis by Activating Hippo/YAP Pathway and Mitochondrial Reactive Oxygen Species in Lung Fibroblasts. Int. J. Mol. Sci. 2020, 21, 2064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aravamudhan, A.; Haak, A.J.; Choi, K.M.; Meridew, J.A.; Caporarello, N.; Jones, D.L.; Tan, Q.; Ligresti, G.; Tschumperlin, D.J. TBK1 regulates YAP/TAZ and fibrogenic fibroblast activation. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 318, L852–L863. [Google Scholar] [CrossRef]
- Santos, D.M.; Pantano, L.; Pronzati, G.; Grasberger, P.; Probst, C.K.; Black, K.E.; Spinney, J.J.; Hariri, L.P.; Nichols, R.; Lin, Y.; et al. Screening for YAP Inhibitors Identifies Statins as Modulators of Fibrosis. Am. J. Respir. Cell. Mol. Biol. 2020, 62, 479–492. [Google Scholar] [CrossRef]
- Chen, Y.; Zhao, X.; Sun, J.; Su, W.; Zhang, L.; Li, Y.; Liu, Y.; Zhang, L.; Lu, Y.; Shan, H.; et al. YAP1/Twist promotes fibroblast activation and lung fibrosis that conferred by miR-15a loss in IPF. Cell Death Differ. 2019, 26, 1832–1844. [Google Scholar] [CrossRef]
- Zhao, X.; Sun, J.; Su, W.; Shan, H.; Zhang, B.; Wang, Y.; Shabanova, A.; Shan, H.; Liang, H. Melatonin Protects against Lung Fibrosis by Regulating the Hippo/YAP Pathway. Int. J. Mol. Sci. 2018, 19, 1118. [Google Scholar] [CrossRef] [Green Version]
- Du, W.; Tang, Z.; Yang, F.; Liu, X.; Dong, J. Icariin attenuates bleomycin-induced pulmonary fibrosis by targeting Hippo/YAP pathway. Biomed. Pharmacother. 2021, 143, 112152. [Google Scholar] [CrossRef]
- Noguchi, S.; Saito, A.; Mikami, Y.; Urushiyama, H.; Horie, M.; Matsuzaki, H.; Takeshima, H.; Makita, K.; Miyashita, N.; Mitani, A.; et al. TAZ contributes to pulmonary fibrosis by activating profibrotic functions of lung fibroblasts. Sci. Rep. 2017, 7, 42595. [Google Scholar] [CrossRef] [PubMed]
- Richeldi, L.; Collard, H.R.; Jones, M.G. Idiopathic pulmonary fibrosis. Lancet 2017, 389, 1941–1952. [Google Scholar] [CrossRef]
- Russell, J.O.; Camargo, F.D. Hippo signalling in the liver: Role in development, regeneration and disease. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 297–312. [Google Scholar] [CrossRef] [PubMed]
- Pellicoro, A.; Ramachandran, P.; Iredale, J.P.; Fallowfield, J.A. Liver fibrosis and repair: Immune regulation of wound healing in a solid organ. Nat. Rev. Immunol. 2014, 14, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Allen, A.M.; Wang, Z.; Prokop, L.J.; Murad, M.H.; Loomba, R. Fibrosis progression in nonalcoholic fatty liver vs nonalcoholic steatohepatitis: A systematic review and meta-analysis of paired-biopsy studies. Clin. Gastroenterol. Hepatol. 2015, 13, 643–654. [Google Scholar] [CrossRef] [Green Version]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef]
- Zhou, D.; Conrad, C.; Xia, F.; Park, J.S.; Payer, B.; Yin, Y.; Lauwers, G.Y.; Thasler, W.; Lee, J.T.; Avruch, J.; et al. Mst1 and Mst2 maintain hepatocyte quiescence and suppress hepatocellular carcinoma development through inactivation of the Yap1 oncogene. Cancer Cell 2009, 16, 425–438. [Google Scholar] [CrossRef] [Green Version]
- Yimlamai, D.; Fowl, B.H.; Camargo, F.D. Emerging evidence on the role of the Hippo/YAP pathway in liver physiology and cancer. J. Hepatol. 2015, 63, 1491–1501. [Google Scholar] [CrossRef] [Green Version]
- Yimlamai, D.; Christodoulou, C.; Galli, G.G.; Yanger, K.; Pepe-Mooney, B.; Gurung, B.; Shrestha, K.; Cahan, P.; Stanger, B.Z.; Camargo, F.D. Hippo pathway activity influences liver cell fate. Cell 2014, 157, 1324–1338. [Google Scholar] [CrossRef] [Green Version]
- Song, H.; Mak, K.K.; Topol, L.; Yun, K.; Hu, J.; Garrett, L.; Chen, Y.; Park, O.; Chang, J.; Simpson, R.M.; et al. Mammalian Mst1 and Mst2 kinases play essential roles in organ size control and tumor suppression. Proc. Natl. Acad. Sci. USA 2010, 107, 1431–1436. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.P.; Lee, J.H.; Kim, T.S.; Kim, T.H.; Park, H.D.; Byun, J.S.; Kim, M.C.; Jeong, W.I.; Calvisi, D.F.; Kim, J.M.; et al. The Hippo-Salvador pathway restrains hepatic oval cell proliferation, liver size, and liver tumorigenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 8248–8253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, L.; Li, Y.; Kim, S.M.; Bossuyt, W.; Liu, P.; Qiu, Q.; Wang, Y.; Halder, G.; Finegold, M.J.; Lee, J.S.; et al. Hippo signaling is a potent in vivo growth and tumor suppressor pathway in the mammalian liver. Proc. Natl. Acad. Sci. USA 2010, 107, 1437–1442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pepe-Mooney, B.J.; Dill, M.T.; Alemany, A.; Ordovas-Montanes, J.; Matsushita, Y.; Rao, A.; Sen, A.; Miyazaki, M.; Anakk, S.; Dawson, P.A.; et al. Single-Cell Analysis of the Liver Epithelium Reveals Dynamic Heterogeneity and an Essential Role for YAP in Homeostasis and Regeneration. Cell Stem Cell 2019, 25, 23–38 e28. [Google Scholar] [CrossRef] [PubMed]
- Bai, H.; Zhang, N.; Xu, Y.; Chen, Q.; Khan, M.; Potter, J.J.; Nayar, S.K.; Cornish, T.; Alpini, G.; Bronk, S.; et al. Yes-associated protein regulates the hepatic response after bile duct ligation. Hepatology 2012, 56, 1097–1107. [Google Scholar] [CrossRef]
- Planas-Paz, L.; Sun, T.; Pikiolek, M.; Cochran, N.R.; Bergling, S.; Orsini, V.; Yang, Z.; Sigoillot, F.; Jetzer, J.; Syed, M.; et al. YAP, but Not RSPO-LGR4/5, Signaling in Biliary Epithelial Cells Promotes a Ductular Reaction in Response to Liver Injury. Cell Stem Cell 2019, 25, 39–53.e10. [Google Scholar] [CrossRef]
- Bangru, S.; Arif, W.; Seimetz, J.; Bhate, A.; Chen, J.; Rashan, E.H.; Carstens, R.P.; Anakk, S.; Kalsotra, A. Alternative splicing rewires Hippo signaling pathway in hepatocytes to promote liver regeneration. Nat. Struct. Mol. Biol. 2018, 25, 928–939. [Google Scholar] [CrossRef]
- Li, W.; Yang, L.; He, Q.; Hu, C.; Zhu, L.; Ma, X.; Ma, X.; Bao, S.; Li, L.; Chen, Y.; et al. A Homeostatic Arid1a-Dependent Permissive Chromatin State Licenses Hepatocyte Responsiveness to Liver-Injury-Associated YAP Signaling. Cell Stem Cell 2019, 25, 54–68 e55. [Google Scholar] [CrossRef]
- Verboven, E.; Moya, I.M.; Sansores-Garcia, L.; Xie, J.; Hillen, H.; Kowalczyk, W.; Vella, G.; Verhulst, S.; Castaldo, S.A.; Alguero-Nadal, A.; et al. Regeneration Defects in Yap and Taz Mutant Mouse Livers Are Caused by Bile Duct Disruption and Cholestasis. Gastroenterology 2021, 160, 847–862. [Google Scholar] [CrossRef]
- Machado, M.V.; Michelotti, G.A.; Pereira, T.A.; Xie, G.; Premont, R.; Cortez-Pinto, H.; Diehl, A.M. Accumulation of duct cells with activated YAP parallels fibrosis progression in non-alcoholic fatty liver disease. J. Hepatol. 2015, 63, 962–970. [Google Scholar] [CrossRef] [Green Version]
- Brenner, C.; Galluzzi, L.; Kepp, O.; Kroemer, G. Decoding cell death signals in liver inflammation. J. Hepatol. 2013, 59, 583–594. [Google Scholar] [CrossRef] [Green Version]
- Luedde, T.; Kaplowitz, N.; Schwabe, R.F. Cell death and cell death responses in liver disease: Mechanisms and clinical relevance. Gastroenterology 2014, 147, 765–783.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seki, E.; Schwabe, R.F. Hepatic inflammation and fibrosis: Functional links and key pathways. Hepatology 2015, 61, 1066–1079. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Luo, Q.; Huang, C.; Gao, Q.; Li, L.; Chen, J.; Chen, B.; Liu, W.; Zeng, W.; Chen, Z. Pathogenesis of non-alcoholic fatty liver disease mediated by YAP. Hepatol. Int. 2018, 12, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Mannaerts, I.; Leite, S.B.; Verhulst, S.; Claerhout, S.; Eysackers, N.; Thoen, L.F.; Hoorens, A.; Reynaert, H.; Halder, G.; van Grunsven, L.A. The Hippo pathway effector YAP controls mouse hepatic stellate cell activation. J. Hepatol. 2015, 63, 679–688. [Google Scholar] [CrossRef]
- Martin, K.; Pritchett, J.; Llewellyn, J.; Mullan, A.F.; Athwal, V.S.; Dobie, R.; Harvey, E.; Zeef, L.; Farrow, S.; Streuli, C.; et al. PAK proteins and YAP-1 signalling downstream of integrin beta-1 in myofibroblasts promote liver fibrosis. Nat. Commun. 2016, 7, 12502. [Google Scholar] [CrossRef]
- Konishi, T.; Schuster, R.M.; Lentsch, A.B. Proliferation of hepatic stellate cells, mediated by YAP and TAZ, contributes to liver repair and regeneration after liver ischemia-reperfusion injury. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 314, G471–G482. [Google Scholar] [CrossRef]
- Liu, Y.; Lu, T.; Zhang, C.; Xu, J.; Xue, Z.; Busuttil, R.W.; Xu, N.; Xia, Q.; Kupiec-Weglinski, J.W.; Ji, H. Activation of YAP attenuates hepatic damage and fibrosis in liver ischemia-reperfusion injury. J. Hepatol. 2019, 71, 719–730. [Google Scholar] [CrossRef]
- Zhang, K.; Chang, Y.; Shi, Z.; Han, X.; Han, Y.; Yao, Q.; Hu, Z.; Cui, H.; Zheng, L.; Han, T.; et al. Omega-3 PUFAs ameliorate liver fibrosis and inhibit hepatic stellate cells proliferation and activation by promoting YAP/TAZ degradation. Sci. Rep. 2016, 6, 30029. [Google Scholar] [CrossRef] [Green Version]
- Perumal, N.; Perumal, M.; Halagowder, D.; Sivasithamparam, N. Morin attenuates diethylnitrosamine-induced rat liver fibrosis and hepatic stellate cell activation by co-ordinated regulation of Hippo/Yap and TGF-beta1/Smad signaling. Biochimie 2017, 140, 10–19. [Google Scholar] [CrossRef]
- Qing, J.; Ren, Y.; Zhang, Y.; Yan, M.; Zhang, H.; Wu, D.; Ma, Y.; Chen, Y.; Huang, X.; Wu, Q.; et al. Dopamine receptor D2 antagonism normalizes profibrotic macrophage-endothelial crosstalk in non-alcoholic steatohepatitis. J. Hepatol. 2022, 76, 394–406. [Google Scholar] [CrossRef]
- Zhang, C.; Bian, M.; Chen, X.; Jin, H.; Zhao, S.; Yang, X.; Shao, J.; Chen, A.; Guo, Q.; Zhang, F.; et al. Oroxylin A prevents angiogenesis of LSECs in liver fibrosis via inhibition of YAP/HIF-1alpha signaling. J. Cell. Biochem. 2018, 119, 2258–2268. [Google Scholar] [CrossRef] [PubMed]
- Seo, E.; Kim, W.Y.; Hur, J.; Kim, H.; Nam, S.A.; Choi, A.; Kim, Y.M.; Park, S.H.; Chung, C.; Kim, J.; et al. The Hippo-Salvador signaling pathway regulates renal tubulointerstitial fibrosis. Sci. Rep. 2016, 6, 31931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Xu, Q.; Ren, F.; Liu, Y.; Cai, R.; Yao, Y.; Zhou, M.S. Inhibition of YAP activation attenuates renal injury and fibrosis in angiotensin II hypertensive mice. Can. J. Physiol. Pharmacol. 2021, 99, 1000–1006. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Zhang, Y.; Wang, L.; He, F.; Yan, M.; Liu, X.; Ou, Y.; Wu, Q.; Bi, T.; Wang, S.; et al. Selective Targeting of Vascular Endothelial YAP Activity Blocks EndMT and Ameliorates Unilateral Ureteral Obstruction-Induced Kidney Fibrosis. ACS Pharmacol. Transl. Sci. 2021, 4, 1066–1074. [Google Scholar] [CrossRef] [PubMed]
- Ji, C.; Zhang, J.; Zhu, Y.; Shi, H.; Yin, S.; Sun, F.; Wang, Q.; Zhang, L.; Yan, Y.; Zhang, X.; et al. Exosomes derived from hucMSC attenuate renal fibrosis through CK1delta/beta-TRCP-mediated YAP degradation. Cell Death Dis. 2020, 11, 327. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Chen, P.P.; Zheng, P.Q.; Yin, F.; Cheng, Q.; Zhou, Z.L.; Xie, H.Y.; Li, J.Y.; Ni, J.Y.; Wang, Y.Z.; et al. KLF4 initiates sustained YAP activation to promote renal fibrosis in mice after ischemia-reperfusion kidney injury. Acta Pharmacol. Sin. 2021, 42, 436–450. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Li, P.X.; Wu, J.; Gao, Y.J.; Yin, M.X.; Lin, Y.; Yang, M.; Chen, D.P.; Sun, H.P.; Liu, Z.B.; et al. Involvement of the Hippo pathway in regeneration and fibrogenesis after ischaemic acute kidney injury: YAP is the key effector. Clin. Sci. 2016, 130, 349–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.; Wang, T.; Chi, X.; Wei, X.; Xu, S.; Yu, M.; He, H.; Ma, J.; Li, X.; Du, J.; et al. Kindlin-2 Inhibits the Hippo Signaling Pathway by Promoting Degradation of MOB1. Cell Rep. 2019, 29, 3664–3677 e3665. [Google Scholar] [CrossRef] [Green Version]
- Piersma, B.; de Rond, S.; Werker, P.M.; Boo, S.; Hinz, B.; van Beuge, M.M.; Bank, R.A. YAP1 Is a Driver of Myofibroblast Differentiation in Normal and Diseased Fibroblasts. Am. J. Pathol. 2015, 185, 3326–3337. [Google Scholar] [CrossRef] [Green Version]
- Toyama, T.; Looney, A.P.; Baker, B.M.; Stawski, L.; Haines, P.; Simms, R.; Szymaniak, A.D.; Varelas, X.; Trojanowska, M. Therapeutic Targeting of TAZ and YAP by Dimethyl Fumarate in Systemic Sclerosis Fibrosis. J. Investig. Dermatol. 2018, 138, 78–88. [Google Scholar] [CrossRef]
- Scannevin, R.H.; Chollate, S.; Jung, M.Y.; Shackett, M.; Patel, H.; Bista, P.; Zeng, W.; Ryan, S.; Yamamoto, M.; Lukashev, M.; et al. Fumarates promote cytoprotection of central nervous system cells against oxidative stress via the nuclear factor (erythroid-derived 2)-like 2 pathway. J. Pharmacol. Exp. Ther. 2012, 341, 274–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, Z.; Xia, W.; Fisher, G.J.; Voorhees, J.J.; Quan, T. YAP/TAZ regulates TGF-beta/Smad3 signaling by induction of Smad7 via AP-1 in human skin dermal fibroblasts. Cell Commun. Signal. 2018, 16, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Kong, Y. YAP is essential for TGF-beta-induced retinal fibrosis in diabetic rats via promoting the fibrogenic activity of Muller cells. J. Cell. Mol. Med. 2020, 24, 12390–12400. [Google Scholar] [CrossRef]
- Futakuchi, A.; Inoue, T.; Wei, F.Y.; Inoue-Mochita, M.; Fujimoto, T.; Tomizawa, K.; Tanihara, H. YAP/TAZ Are Essential for TGF-beta2-Mediated Conjunctival Fibrosis. Investig. Ophthalmol. Vis. Sci. 2018, 59, 3069–3078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ou, W.; Xu, W.; Liu, F.; Guo, Y.; Huang, Z.; Feng, T.; Liu, C.Y.; Du, P. Increased expression of yes-associated protein/YAP and transcriptional coactivator with PDZ-binding motif/TAZ activates intestinal fibroblasts to promote intestinal obstruction in Crohn’s disease. EBioMedicine 2021, 69, 103452. [Google Scholar] [CrossRef]
- Strippoli, R.; Sandoval, P.; Moreno-Vicente, R.; Rossi, L.; Battistelli, C.; Terri, M.; Pascual-Anton, L.; Loureiro, M.; Matteini, F.; Calvo, E.; et al. Caveolin1 and YAP drive mechanically induced mesothelial to mesenchymal transition and fibrosis. Cell Death Dis. 2020, 11, 647. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mia, M.M.; Singh, M.K. New Insights into Hippo/YAP Signaling in Fibrotic Diseases. Cells 2022, 11, 2065. https://doi.org/10.3390/cells11132065
Mia MM, Singh MK. New Insights into Hippo/YAP Signaling in Fibrotic Diseases. Cells. 2022; 11(13):2065. https://doi.org/10.3390/cells11132065
Chicago/Turabian StyleMia, Masum M., and Manvendra K. Singh. 2022. "New Insights into Hippo/YAP Signaling in Fibrotic Diseases" Cells 11, no. 13: 2065. https://doi.org/10.3390/cells11132065
APA StyleMia, M. M., & Singh, M. K. (2022). New Insights into Hippo/YAP Signaling in Fibrotic Diseases. Cells, 11(13), 2065. https://doi.org/10.3390/cells11132065