
Transcriptional Regulation of the Hippo Pathway: Current Understanding and Insights from Single-Cell Technologies


Abstract
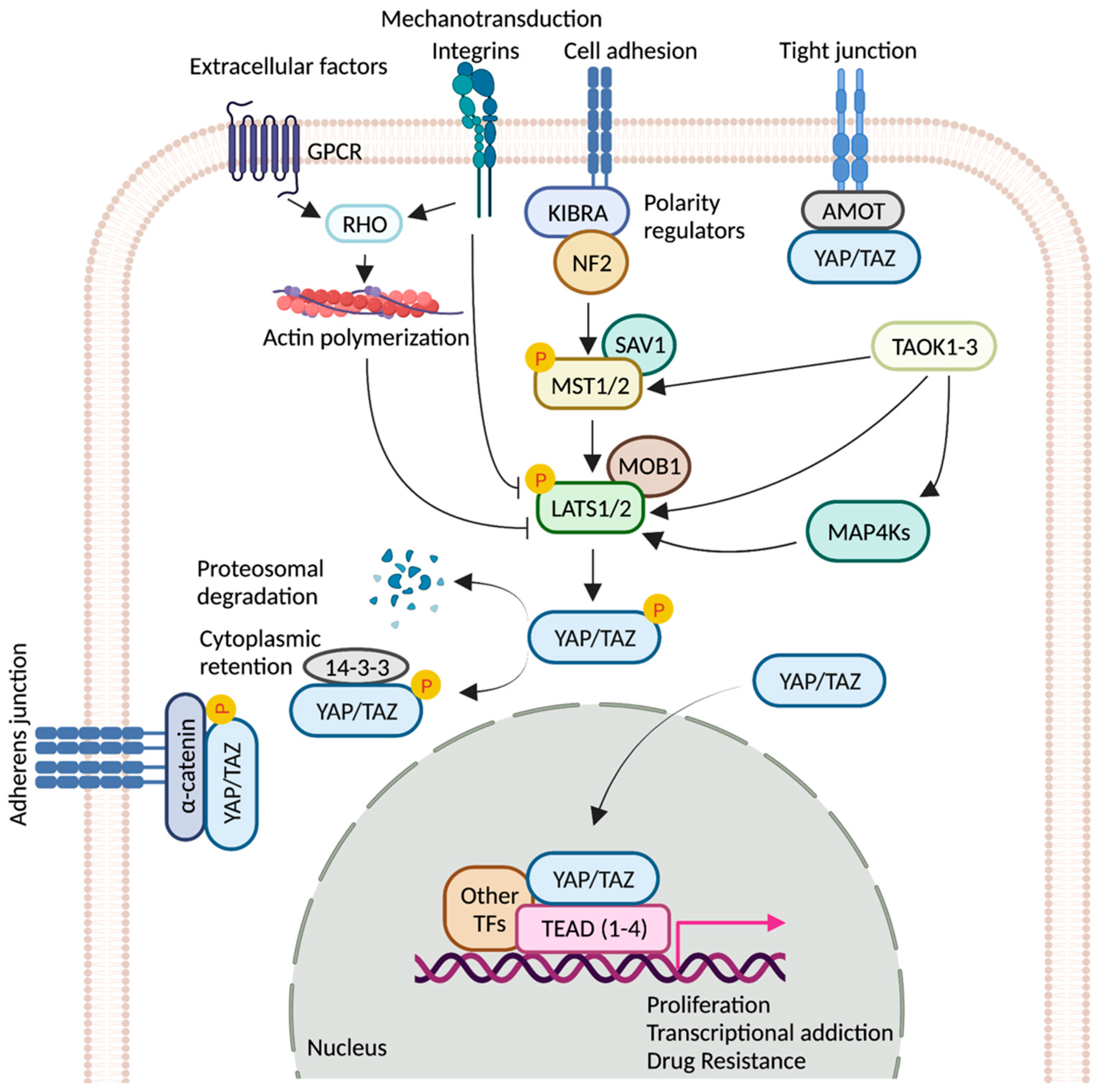
:1. Introduction
2. TEADs
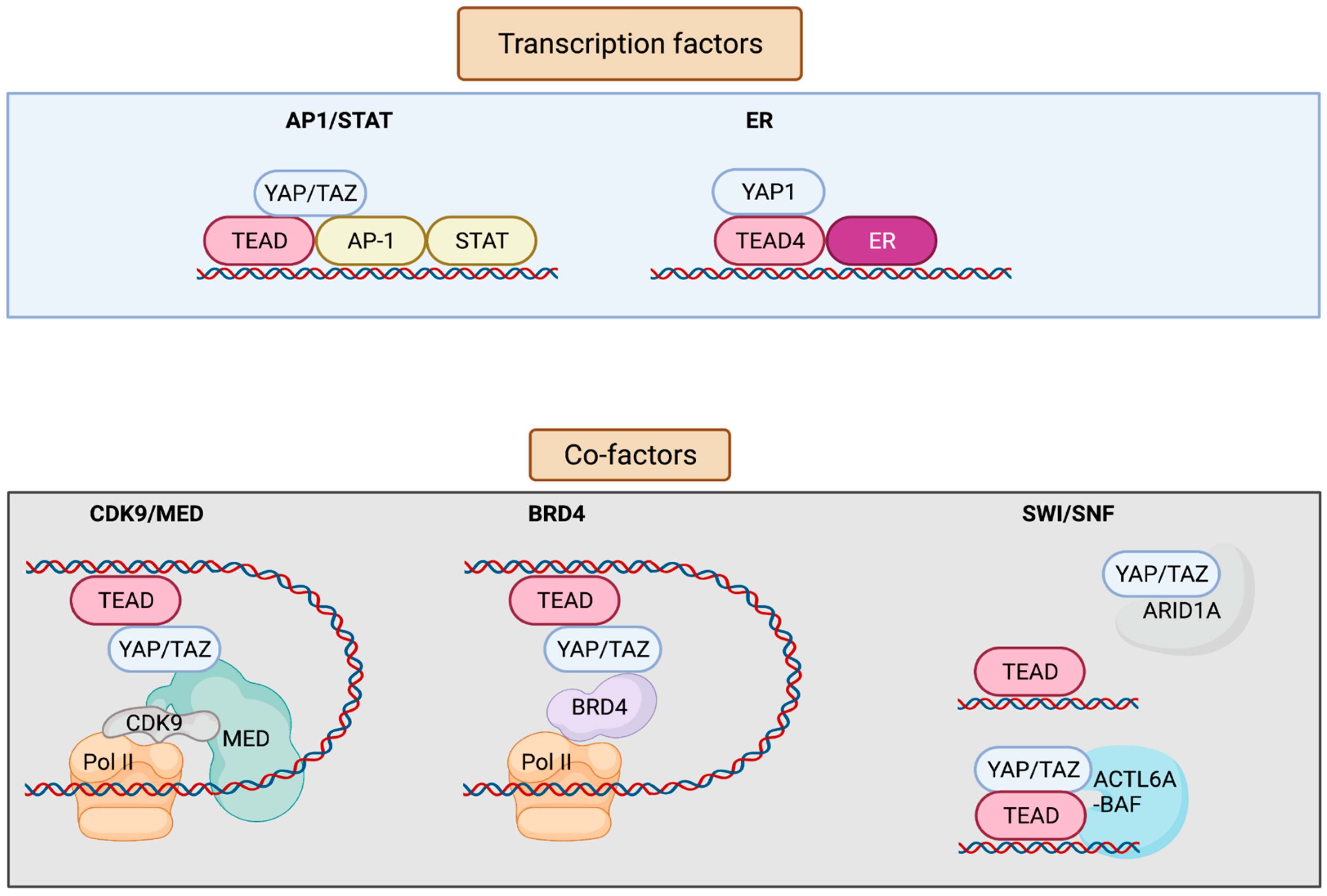
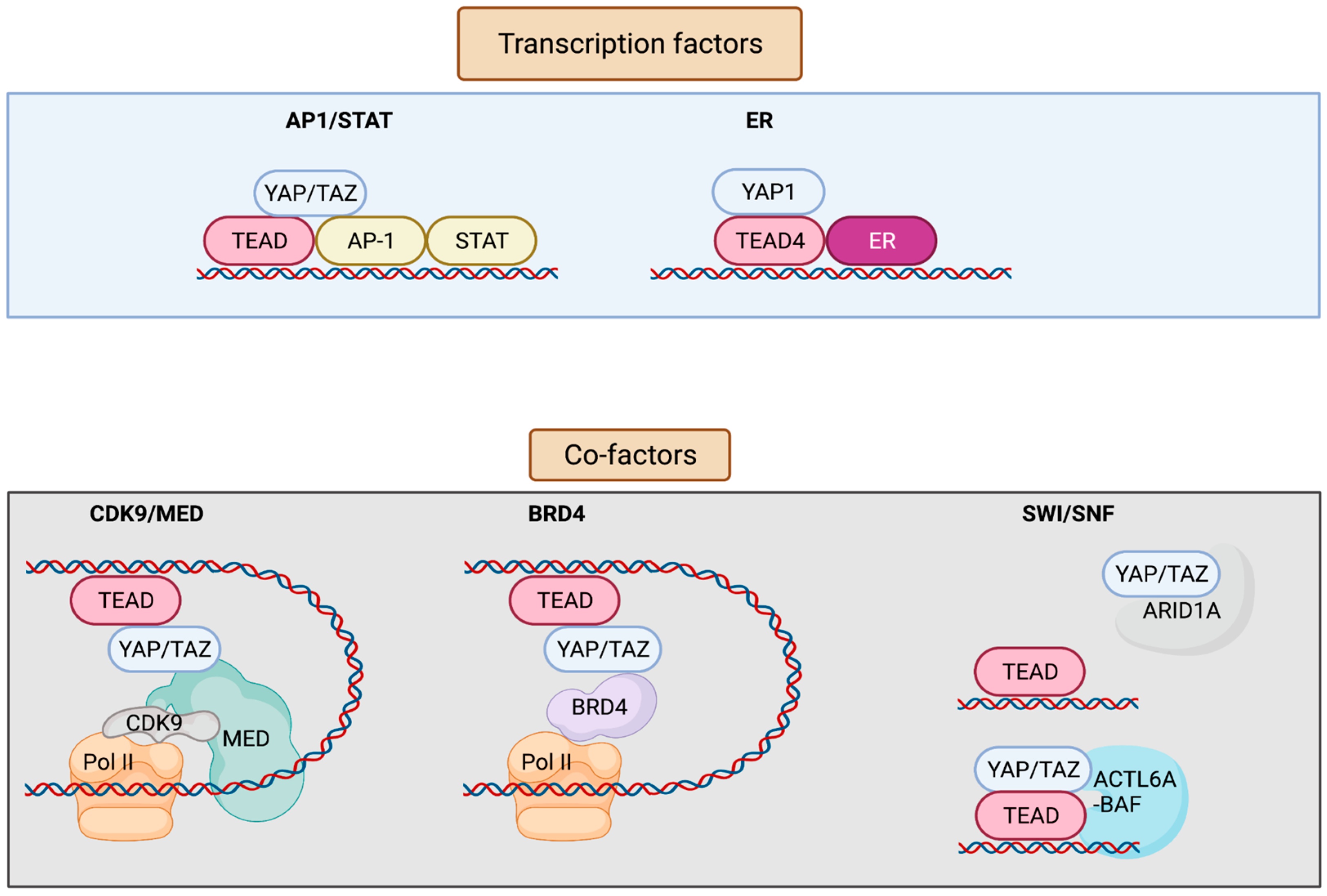
3. YAP/TAZ/TEAD Control Gene Expression from Enhancers

{kind=link}
{kind=link}
{kind=link}
| Factors | Conclusion | Tissue Origin | Reference |
|---|---|---|---|
| AP-1 and STAT | YAP/TAZ/TEAD and AP-1 transcription factors bind at the at the same genomic loci harboring TEAD and AP-1 composite sites. AP-1 enhances YAP/TAZ-induced oncogenic growth. | Breast | [14] |
| TEAD and AP-1 co-occupy the cis-regulatory region. TEAD/AP-1 engages with steroid receptor c-activators 1-3 (SRC1-3) to regulate migration and invasion. | Brain, colon, lung, endometrium | [24] | |
| Vemurafenib (small-molecule inhibitor of BRAF V600E)-induced drug resistance is partially mediated by the activity of JUN and/or AP-1 and TEAD. | Skin | [25] | |
| AP-1 drives YAP-dependent transformations. | Skin, pancreas | [26,27] | |
| AP-1 is a transcriptional target of YAP/TAZ; induced AP-1 can collaborate with YAP/TAZ to promote organ growth. | Liver | [28] | |
| FOSL1/AP-1 acts as a common node in MAPK and Hippo pathways. | Colon and lung pharynx, esophagus, cervix, ovary | [29,30] | |
| YAP/TAZ are recruited by different forms of TEAD/STAT3/AP-1 complex depending on the cis-recruiting motifs to regulate different sets of YAP/TAZ target genes. | Breast | [31] | |
| ERα/FOXA1 | YAP/TEAD act as ERα cofactors to regulate ERα-bound enhancer activation by recruiting MED1. | Breast | [32] |
| BRD4 | Enhancers occupied by YAP–TAZ show enrichment for BRD4, displaying super-enhancer-like characteristics and thus being sensitive to JQ1. | Breast | [33] |
| ARID1A sequesters YAP/TAZ from binding to TEAD to decrease YAP/TAZ activity. | Liver | [34] | |
| SWI/SNF | Pan-FGFR inhibition represses chromatin loading of BRG1, causing an epigenetic switch to promote YAP transcriptional dependency. | Breast | [35] |
| Increased ACTL6A promotes loading of TEAD-YAP binding to BAF complexes, which can enhance co-binding of each other to the chromatin through a positive feedback loop. | Pharynx, lung, esophagus (squamous cells) | [36] |
4. Role of AP-1 and STAT in YAP/TAZ/TEAD Transcriptional Regulation
5. ERα/YAP/TEAD as a Downstream Effector of Hippo Signaling
6. Role of BRD4 in Epigenetic Regulation of YAP/TAZ/TEAD-Mediated Transcription
7. Role of SWI-SNF in Epigenetic Regulation of YAP/TAZ/TEAD-Mediated Transcription
8. Phase Separation
9. Outstanding Questions
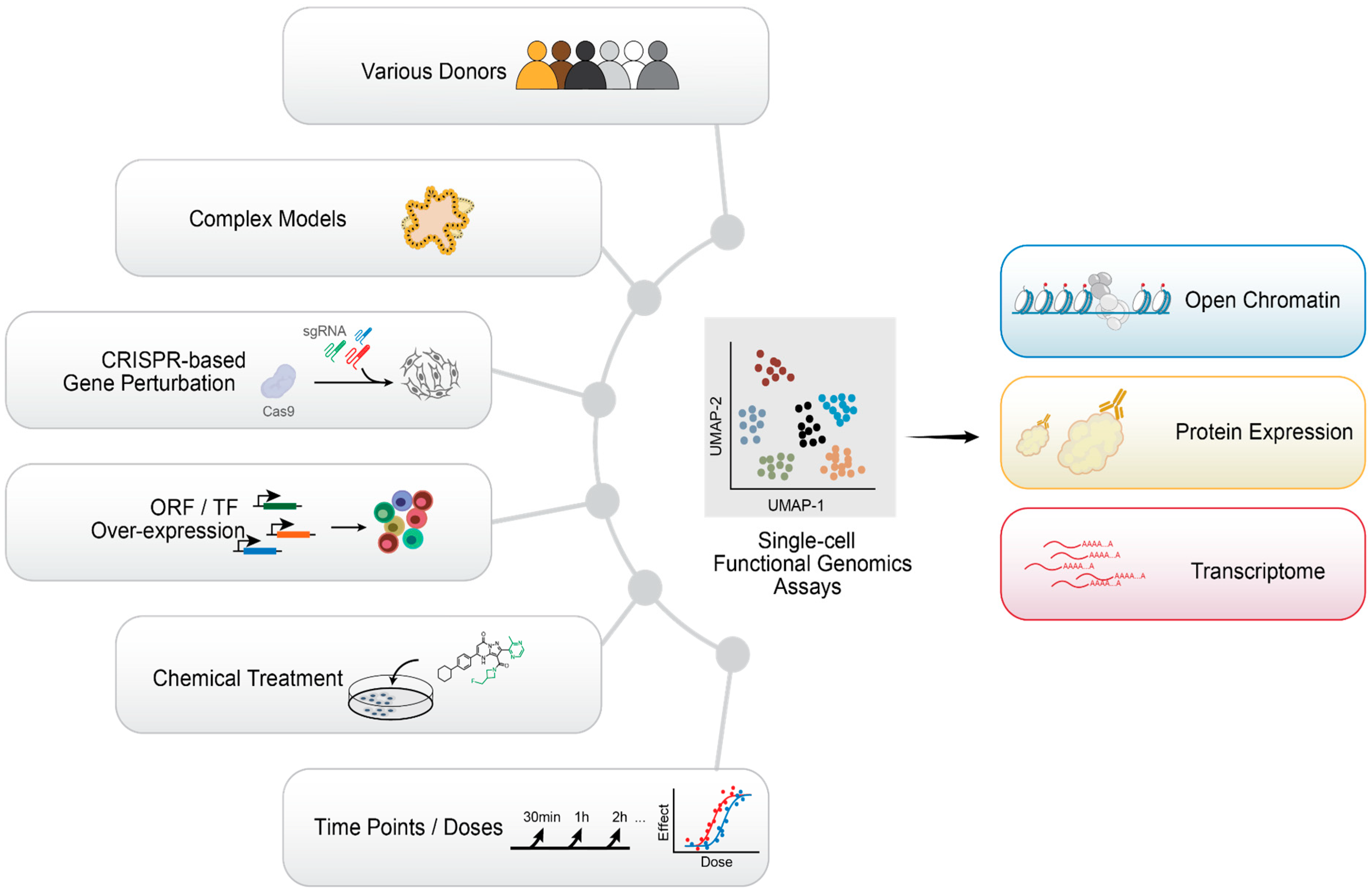
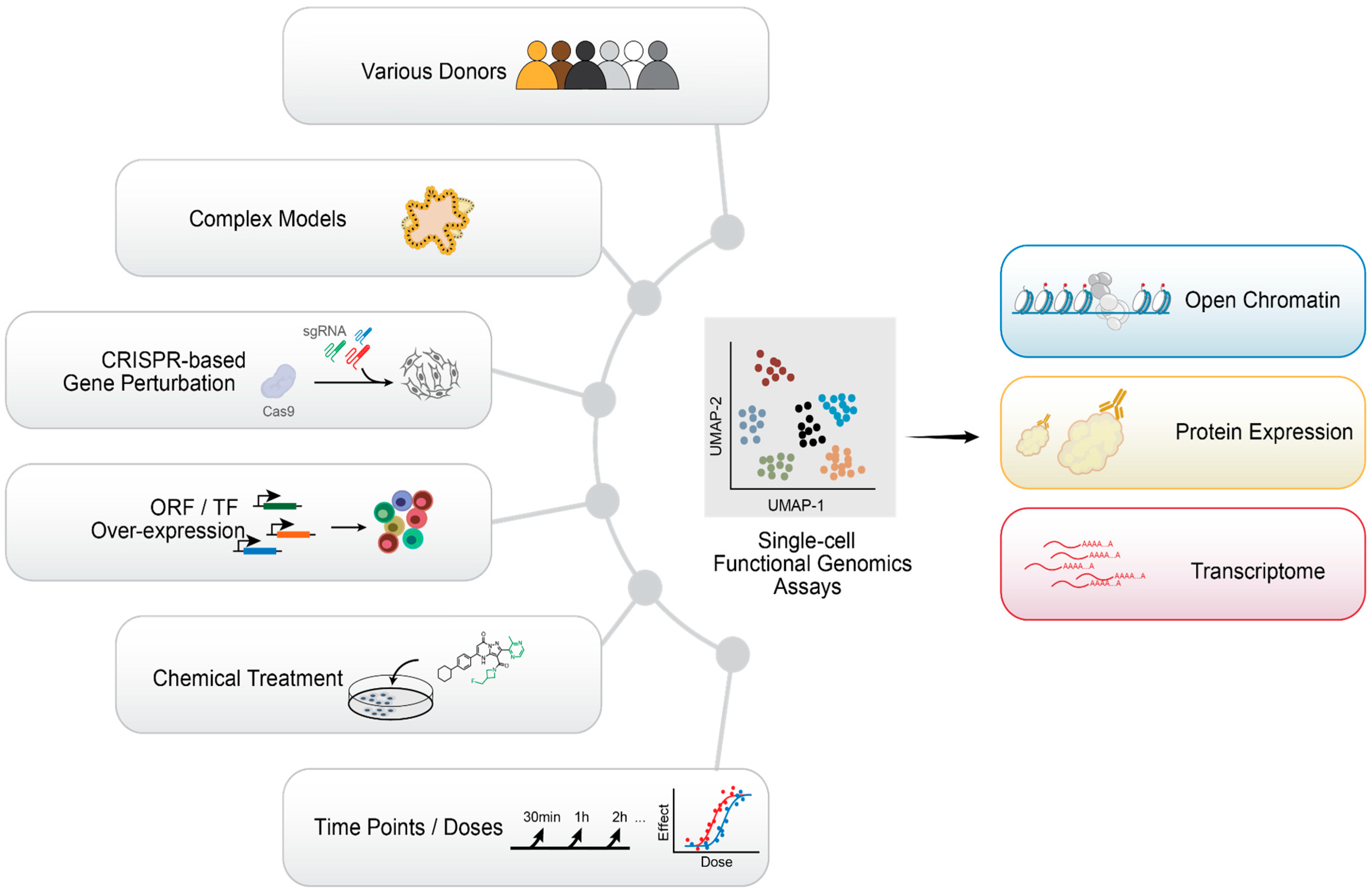
10. Emerging Single-Cell Technologies and Future Perspectives
11. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Johnson, R.; Halder, G. The Two Faces of Hippo: Targeting the Hippo Pathway for Regenerative Medicine and Cancer Treatment. Nat. Rev. Drug Discov. 2014, 13, 63–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manning, S.A.; Kroeger, B.; Harvey, K.F. The Regulation of Yorkie, YAP and TAZ: New Insights into the Hippo Pathway. Development 2020, 147, dev179069. [Google Scholar] [CrossRef]
- Oh, H.; Irvine, K.D. In Vivo Regulation of Yorkie Phosphorylation and Localization. Development 2008, 135, 1081–1088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, J.; Yu, F.-X. GPCR-Hippo Signaling in Cancer. Cells 2019, 8, 426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, X.; Wang, K.-C.; Meng, Z. Mechanoregulation of YAP and TAZ in Cellular Homeostasis and Disease Progression. Front. Cell Dev. Biol. 2021, 9, 673599. [Google Scholar] [CrossRef] [PubMed]
- Plouffe, S.W.; Meng, Z.; Lin, K.C.; Lin, B.; Hong, A.W.; Chun, J.V.; Guan, K.-L. Characterization of Hippo Pathway Components by Gene Inactivation. Mol. Cell 2016, 64, 993–1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Wang, W.; Liu, B.; Deng, H.; Uster, E.; Pan, D. Identification of Happyhour/MAP4K as Alternative Hpo/MST-like Kinases in the Hippo Kinase Cascade. Dev. Cell 2015, 34, 642–655. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.-K.; Jang, J.-W.; Bae, S.-C. DNA Binding Partners of YAP/TAZ. BMB Rep. 2018, 51, 126–133. [Google Scholar] [CrossRef] [Green Version]
- Totaro, A.; Panciera, T.; Piccolo, S. YAP/TAZ Upstream Signals and Downstream Responses. Nat. Cell Biol. 2018, 20, 888–899. [Google Scholar] [CrossRef]
- Calses, P.C.; Crawford, J.J.; Lill, J.R.; Dey, A. Hippo Pathway in Cancer: Aberrant Regulation and Therapeutic Opportunities. Trends Cancer Res. 2019, 5, 297–307. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, C.D.K.; Yi, C. YAP/TAZ Signaling and Resistance to Cancer Therapy. Trends Cancer Res. 2019, 5, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Li, X.; Huang, J.; Feng, L.; Dolinta, K.G.; Chen, J. Defining the Protein-Protein Interaction Network of the Human Hippo Pathway. Mol. Cell. Proteomics 2014, 13, 119–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Couzens, A.L.; Knight, J.D.R.; Kean, M.J.; Teo, G.; Weiss, A.; Dunham, W.H.; Lin, Z.-Y.; Bagshaw, R.D.; Sicheri, F.; Pawson, T.; et al. Protein Interaction Network of the Mammalian Hippo Pathway Reveals Mechanisms of Kinase-Phosphatase Interactions. Sci. Signal. 2013, 6, rs15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanconato, F.; Forcato, M.; Battilana, G.; Azzolin, L.; Quaranta, E.; Bodega, B.; Rosato, A.; Bicciato, S.; Cordenonsi, M.; Piccolo, S. Genome-Wide Association between YAP/TAZ/TEAD and AP-1 at Enhancers Drives Oncogenic Growth. Nat. Cell Biol. 2015, 17, 1218–1227. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Ye, X.; Yu, J.; Li, L.; Li, W.; Li, S.; Yu, J.; Lin, J.D.; Wang, C.-Y.; Chinnaiyan, A.M.; et al. TEAD Mediates YAP-Dependent Gene Induction and Growth Control. Genes Dev. 2008, 22, 1962–1971. [Google Scholar] [CrossRef] [Green Version]
- Stein, C.; Bardet, A.F.; Roma, G.; Bergling, S.; Clay, I.; Ruchti, A.; Agarinis, C.; Schmelzle, T.; Bouwmeester, T.; Schübeler, D.; et al. YAP1 Exerts Its Transcriptional Control via TEAD-Mediated Activation of Enhancers. PLoS Genet. 2015, 11, e1005465. [Google Scholar] [CrossRef] [Green Version]
- Xiao, J.H.; Davidson, I.; Matthes, H.; Garnier, J.M.; Chambon, P. Cloning, Expression, and Transcriptional Properties of the Human Enhancer Factor TEF-1. Cell 1991, 65, 551–568. [Google Scholar] [CrossRef]
- Holden, J.K.; Cunningham, C.N. Targeting the Hippo Pathway and Cancer through the TEAD Family of Transcription Factors. Cancers 2018, 10, 81. [Google Scholar] [CrossRef] [Green Version]
- Della Chiara, G.; Gervasoni, F.; Fakiola, M.; Godano, C.; D’Oria, C.; Azzolin, L.; Bonnal, R.J.P.; Moreni, G.; Drufuca, L.; Rossetti, G.; et al. Epigenomic Landscape of Human Colorectal Cancer Unveils an Aberrant Core of Pan-Cancer Enhancers Orchestrated by YAP/TAZ. Nat. Commun. 2021, 12, 2340. [Google Scholar] [CrossRef]
- Galli, G.G.; Carrara, M.; Yuan, W.-C.; Valdes-Quezada, C.; Gurung, B.; Pepe-Mooney, B.; Zhang, T.; Geeven, G.; Gray, N.S.; de Laat, W.; et al. YAP Drives Growth by Controlling Transcriptional Pause Release from Dynamic Enhancers. Mol. Cell 2015, 60, 328–337. [Google Scholar] [CrossRef] [Green Version]
- Monroe, T.O.; Hill, M.C.; Morikawa, Y.; Leach, J.P.; Heallen, T.; Cao, S.; Krijger, P.H.L.; de Laat, W.; Wehrens, X.H.T.; Rodney, G.G.; et al. YAP Partially Reprograms Chromatin Accessibility to Directly Induce Adult Cardiogenesis In Vivo. Dev. Cell 2019, 48, 765–779.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, H.; Slattery, M.; Ma, L.; Crofts, A.; White, K.P.; Mann, R.S.; Irvine, K.D. Genome-Wide Association of Yorkie with Chromatin and Chromatin-Remodeling Complexes. Cell Rep. 2013, 3, 309–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, H.; Slattery, M.; Ma, L.; White, K.P.; Mann, R.S.; Irvine, K.D. Yorkie Promotes Transcription by Recruiting a Histone Methyltransferase Complex. Cell Rep. 2014, 8, 449–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Li, H.; Rajurkar, M.; Li, Q.; Cotton, J.L.; Ou, J.; Zhu, L.J.; Goel, H.L.; Mercurio, A.M.; Park, J.-S.; et al. Tead and AP1 Coordinate Transcription and Motility. Cell Rep. 2016, 14, 1169–1180. [Google Scholar] [CrossRef] [Green Version]
- Shaffer, S.M.; Dunagin, M.C.; Torborg, S.R.; Torre, E.A.; Emert, B.; Krepler, C.; Beqiri, M.; Sproesser, K.; Brafford, P.A.; Xiao, M.; et al. Rare Cell Variability and Drug-Induced Reprogramming as a Mode of Cancer Drug Resistance. Nature 2017, 546, 431–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maglic, D.; Schlegelmilch, K.; Dost, A.F.; Panero, R.; Dill, M.T.; Calogero, R.A.; Camargo, F.D. YAP-TEAD Signaling Promotes Basal Cell Carcinoma Development via a c-JUN/AP1 Axis. EMBO J. 2018, 37, e98642. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Eisenbarth, D.; Choi, W.; Kim, H.; Choi, C.; Lee, D.; Lim, D.-S. YAP and AP-1 Cooperate to Initiate Pancreatic Cancer Development from Ductal Cells in MiceYAP and AP-1 Initiate PDAC from Ductal Cells. Cancer Res. 2020, 80, 4768–4779. [Google Scholar] [CrossRef]
- Koo, J.H.; Plouffe, S.W.; Meng, Z.; Lee, D.-H.; Yang, D.; Lim, D.-S.; Wang, C.-Y.; Guan, K.-L. Induction of AP-1 by YAP/TAZ Contributes to Cell Proliferation and Organ Growth. Genes Dev. 2020, 34, 72–86. [Google Scholar] [CrossRef]
- Shao, D.D.; Xue, W.; Krall, E.B.; Bhutkar, A.; Piccioni, F.; Wang, X.; Schinzel, A.C.; Sood, S.; Rosenbluh, J.; Kim, J.W.; et al. KRAS and YAP1 Converge to Regulate EMT and Tumor Survival. Cell 2014, 158, 171–184. [Google Scholar] [CrossRef] [Green Version]
- Pham, T.H.; Hagenbeek, T.J.; Lee, H.-J.; Li, J.; Rose, C.M.; Lin, E.; Yu, M.; Martin, S.E.; Piskol, R.; Lacap, J.A.; et al. Machine-Learning and Chemicogenomics Approach Defines and Predicts Cross-Talk of Hippo and MAPK PathwaysMachine-Learning Approach Predicts Hippo Pathway Dependency. Cancer Discov. 2021, 11, 778–793. [Google Scholar] [CrossRef]
- He, L.; Pratt, H.; Gao, M.; Wei, F.; Weng, Z.; Struhl, K. YAP and TAZ Are Transcriptional Co-Activators of AP-1 Proteins and STAT3 during Breast Cellular Transformation. Elife 2021, 10, e67312. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Li, L.; Zhang, Z.; Bi, M.; Wang, H.; Su, W.; Hernandez, K.; Liu, P.; Chen, J.; Chen, M.; et al. A Non-Canonical Role of YAP/TEAD Is Required for Activation of Estrogen-Regulated Enhancers in Breast Cancer. Mol. Cell 2019, 75, 791–806.e8. [Google Scholar] [CrossRef] [PubMed]
- Zanconato, F.; Battilana, G.; Forcato, M.; Filippi, L.; Azzolin, L.; Manfrin, A.; Quaranta, E.; Di Biagio, D.; Sigismondo, G.; Guzzardo, V.; et al. Transcriptional Addiction in Cancer Cells Is Mediated by YAP/TAZ through BRD4. Nat. Med. 2018, 24, 1599–1610. [Google Scholar] [CrossRef]
- Chang, L.; Azzolin, L.; Di Biagio, D.; Zanconato, F.; Battilana, G.; Lucon Xiccato, R.; Aragona, M.; Giulitti, S.; Panciera, T.; Gandin, A.; et al. The SWI/SNF Complex Is a Mechanoregulated Inhibitor of YAP and TAZ. Nature 2018, 563, 265–269. [Google Scholar] [CrossRef]
- Li, Y.; Qiu, X.; Wang, X.; Liu, H.; Geck, R.C.; Tewari, A.K.; Xiao, T.; Font-Tello, A.; Lim, K.; Jones, K.L.; et al. FGFR-Inhibitor-Mediated Dismissal of SWI/SNF Complexes from YAP-Dependent Enhancers Induces Adaptive Therapeutic Resistance. Nat. Cell Biol. 2021, 23, 1187–1198. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-Y.; Shipony, Z.; Lin, S.G.; Kuo, A.; Xiong, X.; Loh, K.M.; Greenleaf, W.J.; Crabtree, G.R. Increased ACTL6A Occupancy within MSWI/SNF Chromatin Remodelers Drives Human Squamous Cell Carcinoma. Mol. Cell 2021, 81, 4964–4978.e8. [Google Scholar] [CrossRef]
- Yu, F.-X.; Zhao, B.; Panupinthu, N.; Jewell, J.L.; Lian, I.; Wang, L.H.; Zhao, J.; Yuan, H.; Tumaneng, K.; Li, H.; et al. Regulation of the Hippo-YAP Pathway by G-Protein-Coupled Receptor Signaling. Cell 2012, 150, 780–791. [Google Scholar] [CrossRef] [Green Version]
- Atkins, M.; Potier, D.; Romanelli, L.; Jacobs, J.; Mach, J.; Hamaratoglu, F.; Aerts, S.; Halder, G. An Ectopic Network of Transcription Factors Regulated by Hippo Signaling Drives Growth and Invasion of a Malignant Tumor Model. Curr. Biol. 2016, 26, 2101–2113. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Wong, C.C.; Leung, K.T.; Wu, F.; Zhou, Y.; Tong, J.H.M.; Chan, R.C.K.; Li, H.; Wang, Y.; Yan, H.; et al. FGF18-FGFR2 Signaling Triggers the Activation of c-Jun-YAP1 Axis to Promote Carcinogenesis in a Subgroup of Gastric Cancer Patients and Indicates Translational Potential. Oncogene 2020, 39, 6647–6663. [Google Scholar] [CrossRef]
- Ma, S.; Wu, Z.; Yang, F.; Zhang, J.; Johnson, R.L.; Rosenfeld, M.G.; Guan, K.-L. Hippo Signalling Maintains ER Expression and ER+ Breast Cancer Growth. Nature 2021, 591, E1–E10. [Google Scholar] [CrossRef]
- Britschgi, A.; Duss, S.; Kim, S.; Couto, J.P.; Brinkhaus, H.; Koren, S.; De Silva, D.; Mertz, K.D.; Kaup, D.; Varga, Z.; et al. The Hippo Kinases LATS1 and 2 Control Human Breast Cell Fate via Crosstalk with ERα. Nature 2017, 541, 541–545. [Google Scholar] [CrossRef] [PubMed]
- Boija, A.; Klein, I.A.; Sabari, B.R.; Dall’Agnese, A.; Coffey, E.L.; Zamudio, A.V.; Li, C.H.; Shrinivas, K.; Manteiga, J.C.; Hannett, N.M.; et al. Transcription Factors Activate Genes through the Phase-Separation Capacity of Their Activation Domains. Cell 2018, 175, 1842–1855.e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhubanchaliyev, A.; Temirbekuly, A.; Kongrtay, K.; Wanshura, L.C.; Kunz, J. Targeting Mechanotransduction at the Transcriptional Level: YAP and BRD4 Are Novel Therapeutic Targets for the Reversal of Liver Fibrosis. Front. Pharmacol. 2016, 7, 462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu-Chittenden, Y.; Huang, B.; Shim, J.S.; Chen, Q.; Lee, S.-J.; Anders, R.A.; Liu, J.O.; Pan, D. Genetic and Pharmacological Disruption of the TEAD–YAP Complex Suppresses the Oncogenic Activity of YAP. Genes Dev. 2012, 26, 1300–1305. [Google Scholar] [CrossRef] [Green Version]
- Ding, N.; Hah, N.; Yu, R.T.; Sherman, M.H.; Benner, C.; Leblanc, M.; He, M.; Liddle, C.; Downes, M.; Evans, R.M. BRD4 Is a Novel Therapeutic Target for Liver Fibrosis. Proc. Natl. Acad. Sci. USA 2015, 112, 15713–15718. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; Peng, R.; Phillips, J.E.; Deguzman, J.; Ren, Y.; Apparsundaram, S.; Luo, Q.; Bauer, C.M.; Fuentes, M.E.; DeMartino, J.A.; et al. Assessment of Brd4 Inhibition in Idiopathic Pulmonary Fibrosis Lung Fibroblasts and in Vivo Models of Lung Fibrosis. Am. J. Pathol. 2013, 183, 470–479. [Google Scholar] [CrossRef]
- Gobbi, G.; Donati, B.; Do Valle, I.F.; Reggiani, F.; Torricelli, F.; Remondini, D.; Castellani, G.; Ambrosetti, D.C.; Ciarrocchi, A.; Sancisi, V. The Hippo Pathway Modulates Resistance to BET Proteins Inhibitors in Lung Cancer Cells. Oncogene 2019, 38, 6801–6817. [Google Scholar] [CrossRef]
- Song, S.; Li, Y.; Xu, Y.; Ma, L.; Pool Pizzi, M.; Jin, J.; Scott, A.W.; Huo, L.; Wang, Y.; Lee, J.H.; et al. Targeting Hippo Coactivator YAP1 through BET Bromodomain Inhibition in Esophageal Adenocarcinoma. Mol. Oncol. 2020, 14, 1410–1426. [Google Scholar] [CrossRef] [Green Version]
- Plouffe, S.W.; Lin, K.C.; Moore, J.L., 3rd; Tan, F.E.; Ma, S.; Ye, Z.; Qiu, Y.; Ren, B.; Guan, K.-L. The Hippo Pathway Effector Proteins YAP and TAZ Have Both Distinct and Overlapping Functions in the Cell. J. Biol. Chem. 2018, 293, 11230–11240. [Google Scholar] [CrossRef] [Green Version]
- Duan, Q.; Xiao, Y.; Zhu, L.; Liu, Z.; Mao, X.; Zhou, Z.; Liao, C.; Cai, J.; Huang, F.; Liu, Z.; et al. BET Bromodomain Is a Novel Regulator of TAZ and Its Activity. Biochim. Biophys. Acta 2016, 1859, 1527–1537. [Google Scholar] [CrossRef]
- Clapier, C.R.; Iwasa, J.; Cairns, B.R.; Peterson, C.L. Mechanisms of Action and Regulation of ATP-Dependent Chromatin-Remodelling Complexes. Nat. Rev. Mol. Cell Biol. 2017, 18, 407–422. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.G.; Roberts, C.W.M. SWI/SNF Nucleosome Remodellers and Cancer. Nat. Rev. Cancer 2011, 11, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Narlikar, G.J.; Fan, H.-Y.; Kingston, R.E. Cooperation between Complexes That Regulate Chromatin Structure and Transcription. Cell 2002, 108, 475–487. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Li, D.; Wang, Y.; Pei, C.; Liu, S.; Zhang, L.; Yuan, Z.; Zhang, P. Brahma Regulates the Hippo Pathway Activity through Forming Complex with Yki-Sd and Regulating the Transcription of Crumbs. Cell. Signal. 2015, 27, 606–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vierbuchen, T.; Ling, E.; Cowley, C.J.; Couch, C.H.; Wang, X.; Harmin, D.A.; Roberts, C.W.M.; Greenberg, M.E. AP-1 Transcription Factors and the BAF Complex Mediate Signal-Dependent Enhancer Selection. Mol. Cell 2017, 68, 1067–1082.e12. [Google Scholar] [CrossRef] [Green Version]
- Sabari, B.R.; Dall’Agnese, A.; Boija, A.; Klein, I.A.; Coffey, E.L.; Shrinivas, K.; Abraham, B.J.; Hannett, N.M.; Zamudio, A.V.; Manteiga, J.C.; et al. Coactivator Condensation at Super-Enhancers Links Phase Separation and Gene Control. Science 2018, 361, eaar3958. [Google Scholar] [CrossRef] [Green Version]
- Cai, D.; Feliciano, D.; Dong, P.; Flores, E.; Gruebele, M.; Porat-Shliom, N.; Sukenik, S.; Liu, Z.; Lippincott-Schwartz, J. Phase Separation of YAP Reorganizes Genome Topology for Long-Term YAP Target Gene Expression. Nat. Cell Biol. 2019, 21, 1578–1589. [Google Scholar] [CrossRef]
- Lu, Y.; Wu, T.; Gutman, O.; Lu, H.; Zhou, Q.; Henis, Y.I.; Luo, K. Phase Separation of TAZ Compartmentalizes the Transcription Machinery to Promote Gene Expression. Nat. Cell Biol. 2020, 22, 453–464. [Google Scholar] [CrossRef]
- Sigal, Y.M.; Zhou, R.; Zhuang, X. Visualizing and Discovering Cellular Structures with Super-Resolution Microscopy. Science 2018, 361, 880–887. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Tillberg, P.W.; Boyden, E.S. Optical Imaging. Expansion Microscopy. Science 2015, 347, 543–548. [Google Scholar] [CrossRef] [Green Version]
- Sawada, A.; Kiyonari, H.; Ukita, K.; Nishioka, N.; Imuta, Y.; Sasaki, H. Redundant Roles of Tead1 and Tead2 in Notochord Development and the Regulation of Cell Proliferation and Survival. Mol. Cell. Biol. 2008, 28, 3177–3189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barry, E.R.; Simov, V.; Valtingojer, I.; Venier, O. Recent Therapeutic Approaches to Modulate the Hippo Pathway in Oncology and Regenerative Medicine. Cells 2021, 10, 2715. [Google Scholar] [CrossRef] [PubMed]
- Little, D.R.; Lynch, A.M.; Yan, Y.; Akiyama, H.; Kimura, S.; Chen, J. Differential Chromatin Binding of the Lung Lineage Transcription Factor NKX2-1 Resolves Opposing Murine Alveolar Cell Fates in Vivo. Nat. Commun. 2021, 12, 2509. [Google Scholar] [CrossRef]
- Castellan, M.; Guarnieri, A.; Fujimura, A.; Zanconato, F.; Battilana, G.; Panciera, T.; Sladitschek, H.L.; Contessotto, P.; Citron, A.; Grilli, A.; et al. Single-Cell Analyses Reveal YAP/TAZ as Regulators of Stemness and Cell Plasticity in Glioblastoma. Nat Cancer 2021, 2, 174–188. [Google Scholar] [CrossRef] [PubMed]
- Stoeckius, M.; Hafemeister, C.; Stephenson, W.; Houck-Loomis, B.; Chattopadhyay, P.K.; Swerdlow, H.; Satija, R.; Smibert, P. Simultaneous Epitope and Transcriptome Measurement in Single Cells. Nat. Methods 2017, 14, 865–868. [Google Scholar] [CrossRef] [Green Version]
- Chung, H.; Parkhurst, C.N.; Magee, E.M.; Phillips, D.; Habibi, E.; Chen, F.; Yeung, B.Z.; Waldman, J.; Artis, D.; Regev, A. Joint Single-Cell Measurements of Nuclear Proteins and RNA in Vivo. Nat. Methods 2021, 18, 1204–1212. [Google Scholar] [CrossRef]
- Cao, J.; Cusanovich, D.A.; Ramani, V.; Aghamirzaie, D.; Pliner, H.A.; Hill, A.J.; Daza, R.M.; McFaline-Figueroa, J.L.; Packer, J.S.; Christiansen, L.; et al. Joint Profiling of Chromatin Accessibility and Gene Expression in Thousands of Single Cells. Science 2018, 361, 1380–1385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Lake, B.B.; Zhang, K. High-Throughput Sequencing of the Transcriptome and Chromatin Accessibility in the Same Cell. Nat. Biotechnol. 2019, 37, 1452–1457. [Google Scholar] [CrossRef]
- Ma, S.; Zhang, B.; LaFave, L.M.; Earl, A.S.; Chiang, Z.; Hu, Y.; Ding, J.; Brack, A.; Kartha, V.K.; Tay, T.; et al. Chromatin Potential Identified by Shared Single-Cell Profiling of RNA and Chromatin. Cell 2020, 183, 1103–1116.e20. [Google Scholar] [CrossRef]
- Skene, P.J.; Henikoff, S. An Efficient Targeted Nuclease Strategy for High-Resolution Mapping of DNA Binding Sites. Elife 2017, 6, e21856. [Google Scholar] [CrossRef]
- Kaya-Okur, H.S.; Wu, S.J.; Codomo, C.A.; Pledger, E.S.; Bryson, T.D.; Henikoff, J.G.; Ahmad, K.; Henikoff, S. CUT&Tag for Efficient Epigenomic Profiling of Small Samples and Single Cells. Nat. Commun. 2019, 10, 1930. [Google Scholar] [PubMed] [Green Version]
- Bartosovic, M.; Kabbe, M.; Castelo-Branco, G. Single-Cell CUT&Tag Profiles Histone Modifications and Transcription Factors in Complex Tissues. Nat. Biotechnol. 2021, 39, 825–835. [Google Scholar]
- Wu, S.J.; Furlan, S.N.; Mihalas, A.B.; Kaya-Okur, H.S.; Feroze, A.H.; Emerson, S.N.; Zheng, Y.; Carson, K.; Cimino, P.J.; Keene, C.D.; et al. Single-Cell CUT&Tag Analysis of Chromatin Modifications in Differentiation and Tumor Progression. Nat. Biotechnol. 2021, 39, 819–824. [Google Scholar] [PubMed]
- Zhu, C.; Zhang, Y.; Li, Y.E.; Lucero, J.; Behrens, M.M.; Ren, B. Joint Profiling of Histone Modifications and Transcriptome in Single Cells from Mouse Brain. Nat. Methods 2021, 18, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Srivastava, A.; Mimitou, E.; Stuart, T.; Raimondi, I.; Hao, Y.; Smibert, P.; Satija, R. Characterizing Cellular Heterogeneity in Chromatin State with ScCUT&Tag-Pro. bioRxiv 2021. [Google Scholar] [CrossRef]
- Chen, A.F.; Parks, B.; Kathiria, A.S.; Ober-Reynolds, B.; Goronzy, J.; Greenleaf, W.J. NEAT-Seq: Simultaneous Profiling of Intra-Nuclear Proteins, Chromatin Accessibility, and Gene Expression in Single Cells. bioRxiv 2021. [Google Scholar] [CrossRef]
- Swanson, E.; Lord, C.; Reading, J.; Heubeck, A.T.; Genge, P.C.; Thomson, Z.; Weiss, M.D.A.; Li, X.-J.; Savage, A.K.; Green, R.R.; et al. Simultaneous Trimodal Single-Cell Measurement of Transcripts, Epitopes, and Chromatin Accessibility Using TEA-Seq. Elife 2021, 10, e63632. [Google Scholar] [CrossRef]
- Mimitou, E.P.; Lareau, C.A.; Chen, K.Y.; Zorzetto-Fernandes, A.L.; Hao, Y.; Takeshima, Y.; Luo, W.; Huang, T.-S.; Yeung, B.Z.; Papalexi, E.; et al. Scalable, Multimodal Profiling of Chromatin Accessibility, Gene Expression and Protein Levels in Single Cells. Nat. Biotechnol. 2021, 39, 1246–1258. [Google Scholar] [CrossRef]
- Jaitin, D.A.; Weiner, A.; Yofe, I.; Lara-Astiaso, D.; Keren-Shaul, H.; David, E.; Salame, T.M.; Tanay, A.; van Oudenaarden, A.; Amit, I. Dissecting Immune Circuits by Linking CRISPR-Pooled Screens with Single-Cell RNA-Seq. Cell 2016, 167, 1883–1896.e15. [Google Scholar] [CrossRef] [Green Version]
- Adamson, B.; Norman, T.M.; Jost, M.; Cho, M.Y.; Nuñez, J.K.; Chen, Y.; Villalta, J.E.; Gilbert, L.A.; Horlbeck, M.A.; Hein, M.Y.; et al. A Multiplexed Single-Cell CRISPR Screening Platform Enables Systematic Dissection of the Unfolded Protein Response. Cell 2016, 167, 1867–1882.e21. [Google Scholar] [CrossRef] [Green Version]
- Dixit, A.; Parnas, O.; Li, B.; Chen, J.; Fulco, C.P.; Jerby-Arnon, L.; Marjanovic, N.D.; Dionne, D.; Burks, T.; Raychowdhury, R.; et al. Perturb-Seq: Dissecting Molecular Circuits with Scalable Single-Cell RNA Profiling of Pooled Genetic Screens. Cell 2016, 167, 1853–1866.e17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Datlinger, P.; Rendeiro, A.F.; Schmidl, C.; Krausgruber, T.; Traxler, P.; Klughammer, J.; Schuster, L.C.; Kuchler, A.; Alpar, D.; Bock, C. Pooled CRISPR Screening with Single-Cell Transcriptome Readout. Nat. Methods 2017, 14, 297–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, S.; Duan, J.; Li, B.; Zhou, P.; Hon, G.C. Multiplexed Engineering and Analysis of Combinatorial Enhancer Activity in Single Cells. Mol. Cell 2017, 66, 285–299.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Replogle, J.M.; Saunders, R.A.; Pogson, A.N.; Hussmann, J.A.; Lenail, A.; Guna, A.; Mascibroda, L.; Wagner, E.J.; Adelman, K.; Lithwick-Yanai, G.; et al. Mapping Information-Rich Genotype-Phenotype Landscapes with Genome-Scale Perturb-Seq. Cell 2022, 185, 2559–2575.e28. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.; Li, B.; Bhakta, M.; Xie, S.; Zhou, P.; Munshi, N.V.; Hon, G.C. Rational Reprogramming of Cellular States by Combinatorial Perturbation. Cell Rep. 2019, 27, 3486–3499.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parekh, U.; Wu, Y.; Zhao, D.; Worlikar, A.; Shah, N.; Zhang, K.; Mali, P. Mapping Cellular Reprogramming via Pooled Overexpression Screens with Paired Fitness and Single-Cell RNA-Sequencing Readout. Cell Syst 2018, 7, 548–555.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ursu, O.; Neal, J.T.; Shea, E.; Thakore, P.I.; Jerby-Arnon, L.; Nguyen, L.; Dionne, D.; Diaz, C.; Bauman, J.; Mosaad, M.M.; et al. Massively Parallel Phenotyping of Coding Variants in Cancer with Perturb-Seq. Nat. Biotechnol. 2022, 40, 896–905. [Google Scholar] [CrossRef]
- Stoeckius, M.; Zheng, S.; Houck-Loomis, B.; Hao, S.; Yeung, B.Z.; Mauck, W.M., 3rd; Smibert, P.; Satija, R. Cell Hashing with Barcoded Antibodies Enables Multiplexing and Doublet Detection for Single Cell Genomics. Genome Biol. 2018, 19, 224. [Google Scholar] [CrossRef] [Green Version]
- McGinnis, C.S.; Patterson, D.M.; Winkler, J.; Conrad, D.N.; Hein, M.Y.; Srivastava, V.; Hu, J.L.; Murrow, L.M.; Weissman, J.S.; Werb, Z.; et al. MULTI-Seq: Sample Multiplexing for Single-Cell RNA Sequencing Using Lipid-Tagged Indices. Nat. Methods 2019, 16, 619–626. [Google Scholar] [CrossRef]
- Cao, J.; Packer, J.S.; Ramani, V.; Cusanovich, D.A.; Huynh, C.; Daza, R.; Qiu, X.; Lee, C.; Furlan, S.N.; Steemers, F.J.; et al. Comprehensive Single-Cell Transcriptional Profiling of a Multicellular Organism. Science 2017, 357, 661–667. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, A.B.; Roco, C.M.; Muscat, R.A.; Kuchina, A.; Sample, P.; Yao, Z.; Graybuck, L.T.; Peeler, D.J.; Mukherjee, S.; Chen, W.; et al. Single-Cell Profiling of the Developing Mouse Brain and Spinal Cord with Split-Pool Barcoding. Science 2018, 360, 176–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McFarland, J.M.; Paolella, B.R.; Warren, A.; Geiger-Schuller, K.; Shibue, T.; Rothberg, M.; Kuksenko, O.; Colgan, W.N.; Jones, A.; Chambers, E.; et al. Multiplexed Single-Cell Transcriptional Response Profiling to Define Cancer Vulnerabilities and Therapeutic Mechanism of Action. Nat. Commun. 2020, 11, 4296. [Google Scholar] [CrossRef] [PubMed]
- Srivatsan, S.R.; McFaline-Figueroa, J.L.; Ramani, V.; Saunders, L.; Cao, J.; Packer, J.; Pliner, H.A.; Jackson, D.L.; Daza, R.M.; Christiansen, L.; et al. Massively Multiplex Chemical Transcriptomics at Single-Cell Resolution. Science 2020, 367, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Mimitou, E.P.; Cheng, A.; Montalbano, A.; Hao, S.; Stoeckius, M.; Legut, M.; Roush, T.; Herrera, A.; Papalexi, E.; Ouyang, Z.; et al. Multiplexed Detection of Proteins, Transcriptomes, Clonotypes and CRISPR Perturbations in Single Cells. Nat. Methods 2019, 16, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Frangieh, C.J.; Melms, J.C.; Thakore, P.I.; Geiger-Schuller, K.R.; Ho, P.; Luoma, A.M.; Cleary, B.; Jerby-Arnon, L.; Malu, S.; Cuoco, M.S.; et al. Multimodal Pooled Perturb-CITE-Seq Screens in Patient Models Define Mechanisms of Cancer Immune Evasion. Nat. Genet. 2021, 53, 332–341. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paul, S.; Xie, S.; Yao, X.; Dey, A. Transcriptional Regulation of the Hippo Pathway: Current Understanding and Insights from Single-Cell Technologies. Cells 2022, 11, 2225. https://doi.org/10.3390/cells11142225
Paul S, Xie S, Yao X, Dey A. Transcriptional Regulation of the Hippo Pathway: Current Understanding and Insights from Single-Cell Technologies. Cells. 2022; 11(14):2225. https://doi.org/10.3390/cells11142225
Chicago/Turabian StylePaul, Sayantanee, Shiqi Xie, Xiaosai Yao, and Anwesha Dey. 2022. "Transcriptional Regulation of the Hippo Pathway: Current Understanding and Insights from Single-Cell Technologies" Cells 11, no. 14: 2225. https://doi.org/10.3390/cells11142225
APA StylePaul, S., Xie, S., Yao, X., & Dey, A. (2022). Transcriptional Regulation of the Hippo Pathway: Current Understanding and Insights from Single-Cell Technologies. Cells, 11(14), 2225. https://doi.org/10.3390/cells11142225