
Gene Therapy in Amyotrophic Lateral Sclerosis



Abstract
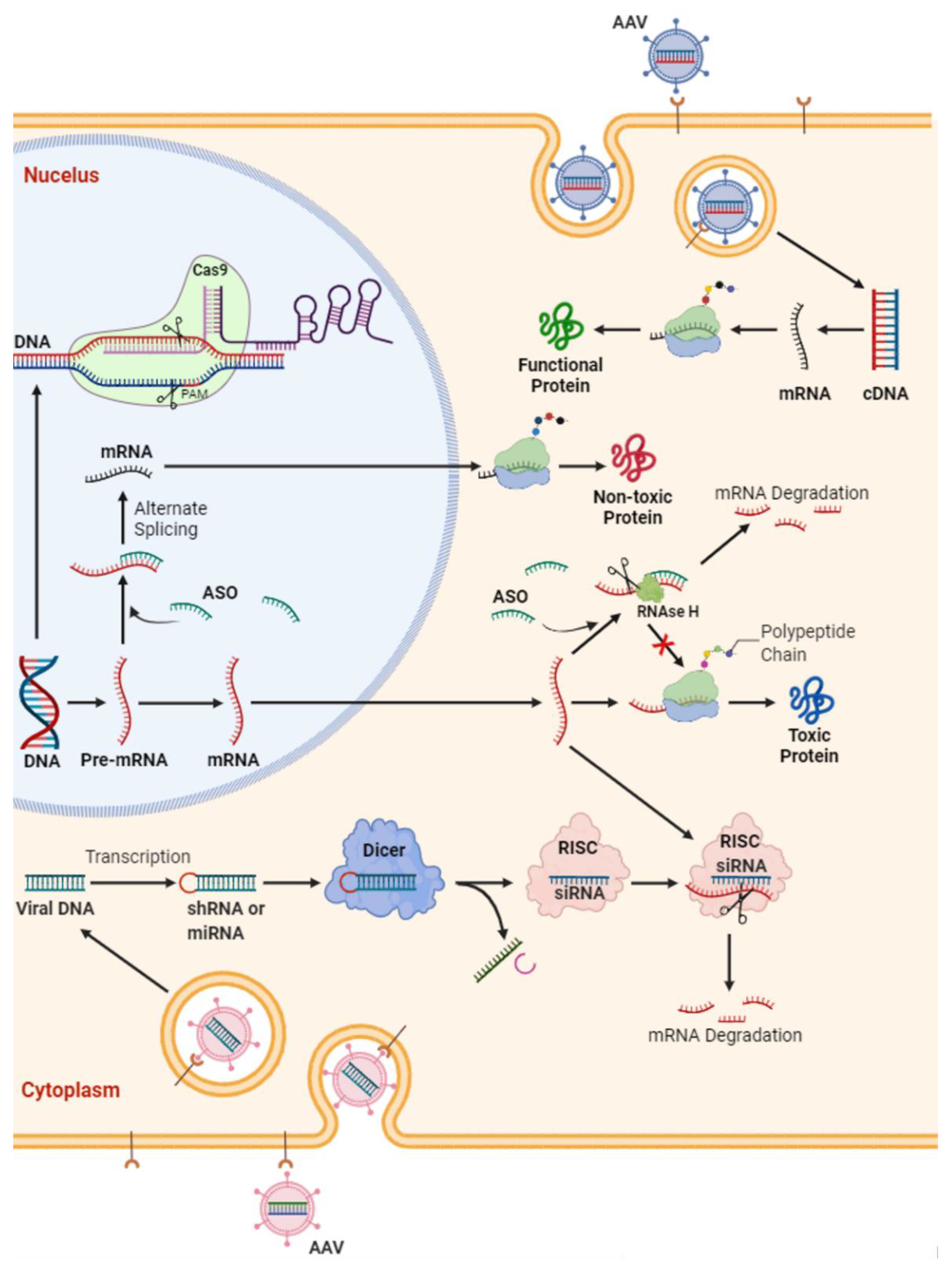
:1. Introduction
2. Strategies for Gene Therapy in ALS
- MicroRNA or antisense oligonucleotides (ASOs; complementary DNA or RNA sequences designed to pair with the target sequence and activate RNA degradation) for ablation of the RNA transcribed from the gene: Administration of ASOs, which are synthetic nucleic acids targeting/altering mRNAs, have shown promising results in treatment of other neuromuscular disorders in children, such as spinal muscle atrophy (SMA) and Duchenne muscular dystrophy (DMD). This has completely altered the original disease trajectory, which has prompted FDA approval of two ASOs, nusinersin (Spinraza) and eteplirsen (Exondys51), for respective treatment of SMA type 1 and 2 and a subset of DMD; however, these have not been tested in adult-type SMA 3 and 4, and their utility in adult disease is not yet known. Overall, ASOs either selectively degrade mRNAs through recruitment of endonuclease RNase H or prevent the interaction of RNAs with RNA binding proteins (RBPs), thereby modulating their splicing/processing without degradation [21].
- Reduction in excess mutant protein (e.g., immune-mediated reduction).
- Interference with transcriptional process with the use of small molecules.
- Somatic-cell mutagenesis, a reverse mutation of the gene back to wild-type form.
3. SOD1
3.1. ASOs
3.2. RNAi
3.3. Neurotrophins
3.4. CRISPR
4. C9orf72
4.1. Targeting C9orf72 Repeat-Expanded RNA or DNA
4.2. Targeting DPRs
4.3. Targeting Nucleocytoplasmic Transport System
5. TARDBP (TDP-43)
5.1. Targeting TARDBP
5.2. Targeting Nucleocytoplasmic Transport System
6. FUS
Targeting FUS
7. Conclusions and Perspectives
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| Abbreviation | Definition |
| AAV | Adeno-Associated Virus |
| AAV-miR-SOD1 | Adeno-Associated Virus Micro-Ribonucleic Acid against Cu/Zn Superoxide Dismutase |
| AE | Adverse Events |
| ALS | Amyotrophic Lateral Sclerosis |
| ALSFRS-R | Revised Amyotrophic Lateral Sclerosis Functional Rating Scale |
| ASO | Antisense Oligonucleotides |
| BAC | Bacterial Artificial Chromosomes |
| BDNF | Brain-Derived Neurotrophic Factor |
| Cas9 | CRISPR-Associated Protein 9 |
| cDNA | Complementary Deoxyribonucleic Acid |
| CHMP7 | Charged Multivesicular Body Protein 7 |
| CNS | Central Nervous System |
| CRISPR | Clustered Regularly Interspaced Short Palindromic Repeats |
| CSF | Cerebrospinal Fluid |
| C9ORF72 | Chromosome 9 Open Reading Frame 72 |
| DMD | Duchenne Muscular Dystrophy |
| DNA | Deoxyribonucleic Acid |
| DPK | Dependent Protein Kinase |
| DPR | Dipeptide Protein Repeats |
| fALS | Familial Amyotrophic Lateral Sclerosis |
| FTD | Frontotemporal Dementia |
| FUS | Fused in Sarcoma |
| HSPB8 | Small Heat Shock Protein B8 |
| IGF | Insulin Growth Factor |
| IMN | Induced Motor Neurons |
| iPS | Induced Pluripotent Stem Cells |
| isRIB | Integrated Stress Response Inhibitor |
| LMN | Lower Motor Neuron |
| MAPK | Mitogen-Activated Protein Kinase |
| miRNA | Micro Ribonucleic Acid |
| mRNA | Messenger Ribonucleic Acid |
| PLS | Primary Lateral Sclerosis |
| PMA | Progressive Muscular Atrophy |
| RAN | Repeat Association non-AUG |
| RBP | Ribonucleic Acid Binding Protein |
| RISC | Ribonucleic Acid-Induced Silencing Complex |
| RNA | Ribonucleic Acid |
| RNAi | Ribonucleic Acid Interference |
| saCas9 | Staphylococcus Aureus CRISPR-associated protein 9 |
| SAE | Severe Adverse Events |
| sALS | Sporadic Amyotrophic Lateral Sclerosis |
| sgRNA | Single Guide Ribonucleic Acid |
| shRNA | Short Hairpin Ribonucleic Acid |
| siRNA | Small Interfering Ribonucleic Acid |
| SMA | Spinal Muscular Atrophy |
| SOD1 | Cu/Zn Superoxide Dismutase |
| SRSF1 | Serine/Arginine-rich Splicing Factor 1 |
| TARDBP 43 | Transactive Response DNA-Binding Protein 43 |
| UMN | Upper Motor Neuron |
| VAF | Ventilation Assistance-Free Survival |
| ZFP TF | Zinc Finger Protein Transcription Factor |
References
- Peters, O.M.; Ghasemi, M.; Brown, R.H., Jr. Emerging mechanisms of molecular pathology in ALS. J. Clin. Investig. 2015, 125, 1767–1779. [Google Scholar] [CrossRef] [PubMed]
- Zucchi, E.; Ticozzi, N.; Mandrioli, J. Psychiatric Symptoms in Amyotrophic Lateral Sclerosis: Beyond a Motor Neuron Disorder. Front. Neurosci. 2019, 13, 175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghasemi, M.; Brown, R.H., Jr. Genetics of Amyotrophic Lateral Sclerosis. Cold Spring Harb. Perspect. Med. 2018, 8, a024125. [Google Scholar] [CrossRef] [PubMed]
- Al-Sarraj, S.; King, A.; Troakes, C.; Smith, B.; Maekawa, S.; Bodi, I.; Rogelj, B.; Al-Chalabi, A.; Hortobágyi, T.; Shaw, C.E. p62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72-linked FTLD and MND/ALS. Acta Neuropathol. 2011, 122, 691–702. [Google Scholar] [CrossRef] [PubMed]
- Cooper-Knock, J.; Hewitt, C.; Highley, J.R.; Brockington, A.; Milano, A.; Man, S.; Martindale, J.; Hartley, J.; Walsh, T.; Gelsthorpe, C.; et al. Clinico-pathological features in amyotrophic lateral sclerosis with expansions in C9ORF72. Brain J. Neurol. 2012, 135, 751–764. [Google Scholar] [CrossRef]
- Schipper, L.J.; Raaphorst, J.; Aronica, E.; Baas, F.; de Haan, R.; de Visser, M.; Troost, D. Prevalence of brain and spinal cord inclusions, including dipeptide repeat proteins, in patients with the C9ORF72 hexanucleotide repeat expansion: A systematic neuropathological review. Neuropathol. Appl. Neurobiol. 2016, 42, 547–560. [Google Scholar] [CrossRef] [Green Version]
- Simón-Sánchez, J.; Dopper, E.G.P.; Cohn-Hokke, P.E.; Hukema, R.K.; Nicolaou, N.; Seelaar, H.; de Graaf, J.R.A.; de Koning, I.; van Schoor, N.M.; Deeg, D.J.H.; et al. The clinical and pathological phenotype of C9ORF72 hexanucleotide repeat expansions. Brain J. Neurol. 2012, 135, 723–735. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.; Ravits, J. Pathogenic Mechanisms and Therapy Development for C9orf72 Amyotrophic Lateral Sclerosis/Frontotemporal Dementia. Neurotherapeutics 2019, 16, 1115–1132. [Google Scholar] [CrossRef]
- Kang, S.H.; Li, Y.; Fukaya, M.; Lorenzini, I.; Cleveland, D.W.; Ostrow, L.W.; Rothstein, J.D.; Bergles, D.E. Degeneration and impaired regeneration of gray matter oligodendrocytes in amyotrophic lateral sclerosis. Nat. Neurosci. 2013, 16, 571–579. [Google Scholar] [CrossRef] [Green Version]
- Filipi, T.; Hermanova, Z.; Tureckova, J.; Vanatko, O.; Anderova, A.M. Glial Cells-The Strategic Targets in Amyotrophic Lateral Sclerosis Treatment. J. Clin. Med. 2020, 9, 261. [Google Scholar] [CrossRef] [Green Version]
- Andersen, P.M.; Al-Chalabi, A. Clinical genetics of amyotrophic lateral sclerosis: What do we really know? Nat. Rev. Neurol. 2011, 7, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.X.; Chen, W.; Hong, S.T.; Boycott, K.M.; Gorrie, G.H.; Siddique, N.; Yang, Y.; Fecto, F.; Shi, Y.; Zhai, H.; et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 2011, 477, 211–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.H.; Al-Chalabi, A. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2017, 377, 162–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dejesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC Hexanucleotide Repeat in Noncoding Region of C9ORF72 Causes Chromosome 9p-Linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [Green Version]
- Renton, A.E.; Majounie, E.; Waite, A.; Simon-Sanchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A Hexanucleotide Repeat Expansion in C9ORF72 Is the Cause of Chromosome 9p21-Linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef] [Green Version]
- Gijselinck, I.; Van Langenhove, T.; van der Zee, J.; Sleegers, K.; Philtjens, S.; Kleinberger, G.; Janssens, J.; Bettens, K.; Van Cauwenberghe, C.; Pereson, S.; et al. A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: A gene identification study. Lancet Neurol. 2012, 11, 54–65. [Google Scholar] [CrossRef]
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E.; et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef]
- Kwiatkowski, T.J., Jr.; Bosco, D.A.; Leclerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T.; et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef] [Green Version]
- Vance, C.; Rogelj, B.; Hortobagyi, T.; De Vos, K.J.; Nishimura, A.L.; Sreedharan, J.; Hu, X.; Smith, B.; Ruddy, D.; Wright, P.; et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009, 323, 1208–1211. [Google Scholar] [CrossRef] [Green Version]
- Bennett, C.F.; Krainer, A.R.; Cleveland, D.W. Antisense Oligonucleotide Therapies for Neurodegenerative Diseases. Annu. Rev. Neurosci. 2019, 42, 385–406. [Google Scholar] [CrossRef]
- Pickles, S.; Velde, C.V. Misfolded SOD1 and ALS: Zeroing in on mitochondria. Amyotroph. Lateral. Scler. 2012, 13, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Sayana, P.; Zhang, X.; Le, W. Genetics of amyotrophic lateral sclerosis: An update. Mol. Neurodegener. 2013, 8, 28. [Google Scholar] [CrossRef] [Green Version]
- Bunton-Stasyshyn, R.K.; Saccon, R.A.; Fratta, P.; Fisher, E.M. SOD1 Function and Its Implications for Amyotrophic Lateral Sclerosis Pathology: New and Renascent Themes. Neurosci. Rev. J. Bringing Neurobiol. Neurol. Psychiatry 2015, 21, 519–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowling, A.C.; Schulz, J.B.; Brown, R.H., Jr.; Beal, M.F. Superoxide dismutase activity, oxidative damage, and mitochondrial energy metabolism in familial and sporadic amyotrophic lateral sclerosis. J. Neurochem. 1993, 61, 2322–2325. [Google Scholar] [CrossRef] [PubMed]
- Abel, O.; Powell, J.F.; Andersen, P.M.; Al-Chalabi, A. ALSoD: A user-friendly online bioinformatics tool for amyotrophic lateral sclerosis genetics. Hum. Mutat. 2012, 33, 1345–1351. [Google Scholar] [CrossRef] [PubMed]
- Penco, S.; Lunetta, C.; Mosca, L.; Maestri, E.; Avemaria, F.; Tarlarini, C.; Patrosso, M.C.; Marocchi, A.; Corbo, M. Phenotypic heterogeneity in a SOD1 G93D Italian ALS family: An example of human model to study a complex disease. J. Mol. Neurosci. 2011, 44, 25–30. [Google Scholar] [CrossRef]
- Zhao, X.; Feng, X.; Li, X.; Mou, J.; Liu, H.; Chen, J.; Wu, J. The G41D mutation in SOD1-related amyotrophic lateral sclerosis exhibits phenotypic heterogeneity among individuals: A case report and literature review. Medicine 2022, 101, e28771. [Google Scholar] [CrossRef]
- Cudkowicz, M.E.; McKenna-Yasek, D.; Sapp, P.E.; Chin, W.; Geller, B.; Hayden, D.L.; Schoenfeld, D.A.; Hosler, B.A.; Horvitz, H.R.; Brown, R.H. Epidemiology of mutations in superoxide dismutase in amyotrophic lateal sclerosis. Ann. Neurol. 1997, 41, 210–221. [Google Scholar] [CrossRef]
- Liang, X.-H.; Sun, H.; Nichols, J.G.; Crooke, S.T. RNase H1-Dependent Antisense Oligonucleotides Are Robustly Active in Directing RNA Cleavage in Both the Cytoplasm and the Nucleus. Mol. Ther. 2017, 25, 2075–2092. [Google Scholar] [CrossRef] [Green Version]
- Finkel, R.S.; Mercuri, E.; Darras, B.T.; Connolly, A.M.; Kuntz, N.L.; Kirschner, J.; Chiriboga, C.A.; Saito, K.; Servais, L.; Tizzano, E.; et al. Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy. N. Engl. J. Med. 2017, 377, 1723–1732. [Google Scholar] [CrossRef] [Green Version]
- Smith, R.A.; Miller, T.M.; Yamanaka, K.; Monia, B.P.; Condon, T.P.; Hung, G.; Lobsiger, C.S.; Ward, C.M.; McAlonis-Downes, M.; Wei, H.; et al. Antisense oligonucleotide therapy for neurodegenerative disease. J. Clin. Investig. 2006, 116, 2290–2296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, T.M.; Pestronk, A.; David, W.; Rothstein, J.; Simpson, E.; Appel, S.H.; Andres, P.L.; Mahoney, K.; Allred, P.; Alexander, K.; et al. An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: A phase 1, randomised, first-in-man study. Lancet Neurol. 2013, 12, 435–442. [Google Scholar] [CrossRef] [Green Version]
- Safety, Tolerability, and Activity Study of ISIS SOD1Rx to Treat Familial Amyotrophic Lateral Sclerosis (ALS) Caused by SOD1 Gene Mutations (SOD-1). ClinicalTrials.gov Identifier: NCT01041222. 2012. Available online: https://clinicaltrials.gov/ct2/show/NCT01041222 (accessed on 20 June 2022).
- Miller, T.; Cudkowicz, M.; Shaw, P.J.; Andersen, P.M.; Atassi, N.; Bucelli, R.C.; Genge, A.; Glass, J.; Ladha, S.; Ludolph, A.L.; et al. Phase 1-2 Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N. Engl. J. Med. 2020, 383, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Biogen. Biogen Announces Topline Results from the Tofersen Phase 3 Study and Its Open-Label Extension in SOD1-ALS. 2021. Available online: https://investors.biogen.com/news-releases/news-release-details/biogen-announces-topline-results-tofersen-phase-3-study-and-its#:~:text=%E2%80%9CData%20from%20the%20tofersen%20Phase,Research%20and%20Development%20at%20Biogen. (accessed on 20 June 2022).
- Mueller, C.; Berry, J.D.; McKenna-Yasek, D.M.; Gernoux, G.; Owegi, M.A.; Pothier, L.M.; Douthwright, C.L.; Gelevski, D.; Luppino, S.D.; Blackwood, M.; et al. SOD1 Suppression with Adeno-Associated Virus and MicroRNA in Familial ALS. N. Engl. J. Med. 2020, 383, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Scarrott, J.M.; Herranz-Martín, S.; Alrafiah, A.R.; Shaw, P.J.; Azzouz, M. Current developments in gene therapy for amyotrophic lateral sclerosis. Expert Opin. Biol. Ther. 2015, 15, 935–947. [Google Scholar] [CrossRef] [Green Version]
- Castanotto, D.; Rossi, J.J. The promises and pitfalls of RNA-interference-based therapeutics. Nature 2009, 457, 426–433. [Google Scholar] [CrossRef] [Green Version]
- Foust, K.D.; Salazar, D.L.; Likhite, S.; Ferraiuolo, L.; Ditsworth, D.; Ilieva, H.; Meyer, K.; Schmelzer, L.; Braun, L.; Cleveland, D.W.; et al. Therapeutic AAV9-mediated suppression of mutant SOD1 slows disease progression and extends survival in models of inherited ALS. Mol. Ther. J. Am. Soc. Gene Ther. 2013, 21, 2148–2159. [Google Scholar] [CrossRef] [Green Version]
- Thomsen, G.M.; Gowing, G. Delayed disease onset and extended survival in the SOD1G93A rat model of amyotrophic lateral sclerosis after suppression of mutant SOD1 in the motor cortex. J. Neurosci. 2014, 34, 15587–15600. [Google Scholar] [CrossRef] [Green Version]
- Stoica, L.; Todeasa, S.H.; Cabrera, G.T.; Salameh, J.S.; ElMallah, M.K.; Mueller, C.; Brown, R.H., Jr.; Sena-Esteves, M. Adeno-associated virus-delivered artificial microRNA extends survival and delays paralysis in an amyotrophic lateral sclerosis mouse model. Ann. Neurol. 2016, 79, 687–700. [Google Scholar] [CrossRef]
- Huang, E.J.; Reichardt, L.F. Neurotrophins: Roles in neuronal development and function. Annu. Rev. Neurosci. 2001, 24, 677–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, H.; Hu, H.; Duan, W.; Liu, Y.; Tan, G.; Li, Z.; Liu, Y.; Deng, B.; Song, X.; Wang, W.; et al. Intramuscular Delivery of scAAV9-hIGF1 Prolongs Survival in the hSOD1(G93A) ALS Mouse Model via Upregulation of D-Amino Acid Oxidase. Mol. Neurobiol. 2018, 55, 682–695. [Google Scholar] [CrossRef] [PubMed]
- Allodi, I.; Comley, L.; Nichterwitz, S.; Nizzardo, M.; Simone, C.; Benitez, J.A.; Cao, M.; Corti, S.; Hedlund, E. Differential neuronal vulnerability identifies IGF-2 as a protective factor in ALS. Sci. Rep. 2016, 6, 25960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Duan, W.; Wang, W.; Di, W.; Liu, Y.; Liu, Y.; Li, Z.; Hu, H.; Lin, H.; Cui, C.; et al. scAAV9-VEGF prolongs the survival of transgenic ALS mice by promoting activation of M2 microglia and the PI3K/Akt pathway. Brain Res. 2016, 1648, 1–10. [Google Scholar] [CrossRef]
- Dodge, J.C.; Treleaven, C.M.; Fidler, J.A.; Hester, M.; Haidet, A.; Handy, C.; Rao, M.; Eagle, A.; Matthews, J.C.; Taksir, T.V.; et al. AAV4-mediated expression of IGF-1 and VEGF within cellular components of the ventricular system improves survival outcome in familial ALS mice. Mol. Ther. J. Am. Soc. Gene Ther. 2010, 18, 2075–2084. [Google Scholar] [CrossRef]
- Heidenreich, M.; Zhang, F. Applications of CRISPR-Cas systems in neuroscience. Nat. Rev. Neurosci. 2016, 17, 36–44. [Google Scholar] [CrossRef] [Green Version]
- Gaj, T.; Ojala, D.S.; Ekman, F.K.; Byrne, L.C.; Limsirichai, P.; Schaffer, D.V. In vivo genome editing improves motor function and extends survival in a mouse model of ALS. Sci. Adv. 2017, 3, eaar3952. [Google Scholar] [CrossRef] [Green Version]
- Duan, W.; Guo, M.; Yi, L.; Liu, Y.; Li, Z.; Ma, Y.; Zhang, G.; Liu, Y.; Bu, H.; Song, X.; et al. The deletion of mutant SOD1 via CRISPR/Cas9/sgRNA prolongs survival in an amyotrophic lateral sclerosis mouse model. Gene Ther. 2020, 27, 157–169. [Google Scholar] [CrossRef]
- Lim, C.K.W.; Gapinske, M.; Brooks, A.K.; Woods, W.S.; Powell, J.E.; Zeballos, C.M.; Winter, J.; Perez-Pinera, P.; Gaj, T. Treatment of a Mouse Model of ALS by In Vivo Base Editing. Mol. Ther. J. Am. Soc. Gene Ther. 2020, 28, 1177–1189. [Google Scholar] [CrossRef]
- Brooks, B.R.; Miller, R.G.; Swash, M.; Munsat, T.L. El Escorial revisited: Revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Mot. Neuron Disord. 2000, 1, 293–299. [Google Scholar] [CrossRef]
- Nguyen, H.P.; Van Broeckhoven, C.; van der Zee, J. ALS Genes in the Genomic Era and their Implications for FTD. Trends Genet. 2018, 34, 404–423. [Google Scholar] [CrossRef] [Green Version]
- Ghasemi, M.; Keyhanian, K.; Douthwright, C. Glial Cell Dysfunction in C9orf72-Related Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Cells 2021, 10, 249. [Google Scholar] [CrossRef] [PubMed]
- Haeusler, A.R.; Donnelly, C.J.; Rothstein, J.D. The expanding biology of the C9orf72 nucleotide repeat expansion in neurodegenerative disease. Nat. Rev. Neurosci. 2016, 17, 383–395. [Google Scholar] [CrossRef] [PubMed]
- Iacoangeli, A.; Al Khleifat, A.; Jones, A.R.; Sproviero, W.; Shatunov, A.; Opie-Martin, S.; Morrison, K.E.; Shaw, P.J.; Shaw, C.E.; Fogh, I.; et al. C9orf72 intermediate expansions of 24–30 repeats are associated with ALS. Acta Neuropathol. Commun. 2019, 7, 115. [Google Scholar] [CrossRef] [Green Version]
- Cammack, A.J.; Atassi, N.; Hyman, T.; van den Berg, L.H.; Harms, M.; Baloh, R.H.; Brown, R.H.; van Es, M.A.; Veldink, J.H.; de Vries, B.S.; et al. Prospective natural history study of C9orf72 ALS clinical characteristics and biomarkers. Neurology 2019, 93, e1605–e1617. [Google Scholar] [CrossRef] [PubMed]
- Byrne, S.; Elamin, M.; Bede, P.; Shatunov, A.; Walsh, C.; Corr, B.; Heverin, M.; Jordan, N.; Kenna, K.; Lynch, C.; et al. Cognitive and clinical characteristics of patients with amyotrophic lateral sclerosis carrying a C9orf72 repeat expansion: A population-based cohort study. Lancet Neurol. 2012, 11, 232–240. [Google Scholar] [CrossRef] [Green Version]
- Irwin, D.J.; McMillan, C.T.; Brettschneider, J.; Libon, D.J.; Powers, J.; Rascovsky, K.; Toledo, J.B.; Boller, A.; Bekisz, J.; Chandrasekaran, K.; et al. Cognitive decline and reduced survival in C9orf72 expansion frontotemporal degeneration and amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2013, 84, 163–169. [Google Scholar] [CrossRef] [Green Version]
- Millecamps, S.; Boillée, S.; Le Ber, I.; Seilhean, D.; Teyssou, E.; Giraudeau, M.; Moigneu, C.; Vandenberghe, N.; Danel-Brunaud, V.; Corcia, P.; et al. Phenotype difference between ALS patients with expanded repeats in C9ORF72 and patients with mutations in other ALS-related genes. J. Med. Genet. 2012, 49, 258–263. [Google Scholar] [CrossRef]
- Chiò, A.; Borghero, G.; Restagno, G.; Mora, G.; Drepper, C.; Traynor, B.J.; Sendtner, M.; Brunetti, M.; Ossola, I.; Calvo, A.; et al. Clinical characteristics of patients with familial amyotrophic lateral sclerosis carrying the pathogenic GGGGCC hexanucleotide repeat expansion of C9ORF72. Brain J. Neurol. 2012, 135, 784–793. [Google Scholar] [CrossRef]
- Zhu, Q.; Jiang, J.; Gendron, T.F.; McAlonis-Downes, M.; Jiang, L.; Taylor, A.; Diaz Garcia, S.; Ghosh Dastidar, S.; Rodriguez, M.J.; King, P.; et al. Reduced C9ORF72 function exacerbates gain of toxicity from ALS/FTD-causing repeat expansion in C9orf72. Nat. Neurosci. 2020, 23, 615–624. [Google Scholar] [CrossRef]
- Shi, Y.; Lin, S.; Staats, K.A.; Li, Y.; Chang, W.-H.; Hung, S.-T.; Hendricks, E.; Linares, G.R.; Wang, Y.; Son, E.Y.; et al. Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat. Med. 2018, 24, 313–325. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, C.J.; Zhang, P.W.; Pham, J.T.; Haeusler, A.R.; Mistry, N.A.; Vidensky, S.; Daley, E.L.; Poth, E.M.; Hoover, B.; Fines, D.M.; et al. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 2013, 80, 415–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagier-Tourenne, C.; Baughn, M.; Rigo, F.; Sun, S.; Liu, P.; Li, H.R.; Jiang, J.; Watt, A.T.; Chun, S.; Katz, M.; et al. Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc. Natl. Acad. Sci. USA 2013, 110, E4530–E4539. [Google Scholar] [CrossRef] [Green Version]
- Gendron, T.F.; Chew, J.; Stankowski, J.N.; Hayes, L.R.; Zhang, Y.-J.; Prudencio, M.; Carlomagno, Y.; Daughrity, L.M.; Jansen-West, K.; Perkerson, E.A.; et al. Poly(GP) proteins are a useful pharmacodynamic marker for C9ORF72-associated amyotrophic lateral sclerosis. Sci. Transl. Med. 2017, 9, eaai7866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sareen, D.; O’Rourke, J.G.; Meera, P.; Muhammad, A.K.; Grant, S.; Simpkinson, M.; Bell, S.; Carmona, S.; Ornelas, L.; Sahabian, A.; et al. Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci. Transl. Med. 2013, 5, 208ra149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, K.; Donnelly, C.J.; Haeusler, A.R.; Grima, J.C.; Machamer, J.B.; Steinwald, P.; Daley, E.L.; Miller, S.J.; Cunningham, K.M.; Vidensky, S.; et al. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 2015, 525, 56–61. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.; Zhu, Q.; Gendron, T.F.; Saberi, S.; McAlonis-Downes, M.; Seelman, A.; Stauffer, J.E.; Jafar-Nejad, P.; Drenner, K.; Schulte, D.; et al. Gain of Toxicity from ALS/FTD-Linked Repeat Expansions in C9ORF72 Is Alleviated by Antisense Oligonucleotides Targeting GGGGCC-Containing RNAs. Neuron 2016, 90, 535–550. [Google Scholar] [CrossRef] [Green Version]
- Tran, H.; Moazami, M.P.; Yang, H.; McKenna-Yasek, D.; Douthwright, C.L.; Pinto, C.; Metterville, J.; Shin, M.; Sanil, N.; Dooley, C.; et al. Suppression of mutant C9orf72 expression by a potent mixed backbone antisense oligonucleotide. Nat. Med. 2022, 28, 117–124. [Google Scholar] [CrossRef]
- Biogen. Biogen and Ionis Announce Topline Phase 1 Study Results of Investigational Drug in C9orf72 Amyotrophic Lateral Sclerosis. 2022. Available online: https://investors.biogen.com/news-releases/news-release-details/biogen-and-ionis-announce-topline-phase-1-study-results (accessed on 20 June 2022).
- Hu, J.; Rigo, F.; Prakash, T.P.; Corey, D.R. Recognition of c9orf72 Mutant RNA by Single-Stranded Silencing RNAs. Nucleic Acid Ther. 2017, 27, 87–94. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Liu, J.; Li, L.; Gagnon, K.T.; Corey, D.R. Engineering Duplex RNAs for Challenging Targets: Recognition of GGGGCC/CCCCGG Repeats at the ALS/FTD C9orf72 Locus. Chem. Biol. 2015, 22, 1505–1511. [Google Scholar] [CrossRef] [Green Version]
- Drouet, V.; Perrin, V.; Hassig, R.; Dufour, N.; Auregan, G.; Alves, S.; Bonvento, G.; Brouillet, E.; Luthi-Carter, R.; Hantraye, P.; et al. Sustained effects of nonallele-specific Huntingtin silencing. Ann. Neurol. 2009, 65, 276–285. [Google Scholar] [CrossRef]
- Stanek, L.M.; Sardi, S.P.; Mastis, B.; Richards, A.R.; Treleaven, C.M.; Taksir, T.; Misra, K.; Cheng, S.H.; Shihabuddin, L.S. Silencing mutant huntingtin by adeno-associated virus-mediated RNA interference ameliorates disease manifestations in the YAC128 mouse model of Huntington’s disease. Hum. Gene Ther. 2014, 25, 461–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martier, R.; Liefhebber, J.M.; García-Osta, A.; Miniarikova, J.; Cuadrado-Tejedor, M.; Espelosin, M.; Ursua, S.; Petry, H.; van Deventer, S.J.; Evers, M.M.; et al. Targeting RNA-Mediated Toxicity in C9orf72 ALS and/or FTD by RNAi-Based Gene Therapy. Mol. Ther. Nucleic Acids 2019, 16, 26–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martier, R.; Liefhebber, J.M.; Miniarikova, J.; van der Zon, T.; Snapper, J.; Kolder, I.; Petry, H.; van Deventer, S.J.; Evers, M.M.; Konstantinova, P. Artificial MicroRNAs Targeting C9orf72 Can Reduce Accumulation of Intra-nuclear Transcripts in ALS and FTD Patients. Mol. Ther. Nucleic Acids 2019, 14, 593–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, Z.; Zhang, Y.; Gendron, T.F.; Bauer, P.O.; Chew, J.; Yang, W.Y.; Fostvedt, E.; Jansen-West, K.; Belzil, V.V.; Desaro, P.; et al. Discovery of a biomarker and lead small molecules to target r(GGGGCC)-associated defects in c9FTD/ALS. Neuron 2014, 83, 1043–1050. [Google Scholar] [CrossRef] [Green Version]
- Zamiri, B.; Reddy, K.; Macgregor, R.B., Jr.; Pearson, C.E. TMPyP4 porphyrin distorts RNA G-quadruplex structures of the disease-associated r(GGGGCC)n repeat of the C9orf72 gene and blocks interaction of RNA-binding proteins. J. Biol. Chem. 2014, 289, 4653–4659. [Google Scholar] [CrossRef] [Green Version]
- Simone, R.; Balendra, R.; Moens, T.G.; Preza, E.; Wilson, K.M.; Heslegrave, A.; Woodling, N.S.; Niccoli, T.; Gilbert-Jaramillo, J.; Abdelkarim, S.; et al. G-quadruplex-binding small molecules ameliorate C9orf72 FTD/ALS pathology in vitro and in vivo. EMBO Mol. Med. 2018, 10, 22–31. [Google Scholar] [CrossRef]
- Alniss, H.; Zamiri, B.; Khalaj, M.; Pearson, C.E.; Macgregor, R.B., Jr. Thermodynamic and spectroscopic investigations of TMPyP4 association with guanine- and cytosine-rich DNA and RNA repeats of C9orf72. Biochem. Biophys. Res. Commun. 2018, 495, 2410–2417. [Google Scholar] [CrossRef]
- Wang, Z.-F.; Ursu, A.; Childs-Disney, J.L.; Guertler, R.; Yang, W.-Y.; Bernat, V.; Rzuczek, S.G.; Fuerst, R.; Zhang, Y.-J.; Gendron, T.F.; et al. The Hairpin Form of r(G(4)C(2))(exp) in c9ALS/FTD Is Repeat-Associated Non-ATG Translated and a Target for Bioactive Small Molecules. Cell Chem. Biol. 2019, 26, 179–190e12. [Google Scholar] [CrossRef]
- Skourti-Stathaki, K.; Proudfoot, N.J.; Gromak, N. Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2-dependent termination. Mol. Cell 2011, 42, 794–805. [Google Scholar] [CrossRef]
- Yuce, O.; West, S.C. Senataxin, defective in the neurodegenerative disorder ataxia with oculomotor apraxia 2, lies at the interface of transcription and the DNA damage response. Mol. Cell. Biol. 2013, 33, 406–417. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.Z.; Bennett, C.L.; Huynh, H.M.; Blair, I.P.; Puls, I.; Irobi, J.; Dierick, I.; Abel, A.; Kennerson, M.L.; Rabin, B.A.; et al. DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4). Am. J. Hum. Genet. 2004, 74, 1128–1135. [Google Scholar] [CrossRef] [Green Version]
- Walker, C.; Herranz-Martin, S.; Karyka, E.; Liao, C.; Lewis, K.; Elsayed, W.; Lukashchuk, V.; Chiang, S.C.; Ray, S.; Mulcahy, P.J.; et al. C9orf72 expansion disrupts ATM-mediated chromosomal break repair. Nat. Neurosci. 2017, 20, 1225–1235. [Google Scholar] [CrossRef]
- Hirtreiter, A.; Damsma, G.E.; Cheung, A.C.M.; Klose, D.; Grohmann, D.; Vojnic, E.; Martin, A.C.R.; Cramer, P.; Werner, F. Spt4/5 stimulates transcription elongation through the RNA polymerase clamp coiled-coil motif. Nucleic Acids Res. 2010, 38, 4040–4051. [Google Scholar] [CrossRef] [PubMed]
- Rondón, A.G.; García-Rubio, M.; González-Barrera, S.; Aguilera, A. Molecular evidence for a positive role of Spt4 in transcription elongation. EMBO J. 2003, 22, 612–620. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.-R.; Chang, C.-R.; Chern, Y.; Wang, T.-H.; Hsieh, W.-C.; Shen, W.-C.; Chang, C.-Y.; Chu, I.C.; Deng, N.; Cohen, S.N.; et al. Spt4 is selectively required for transcription of extended trinucleotide repeats. Cell 2012, 148, 690–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kramer, N.J.; Carlomagno, Y.; Zhang, Y.-J.; Almeida, S.; Cook, C.N.; Gendron, T.F.; Prudencio, M.; Van Blitterswijk, M.; Belzil, V.; Couthouis, J.; et al. Spt4 selectively regulates the expression of C9orf72 sense and antisense mutant transcripts. Science 2016, 353, 708–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodman, L.D.; Prudencio, M.; Kramer, N.J.; Martinez-Ramirez, L.F.; Srinivasan, A.R.; Lan, M.; Parisi, M.J.; Zhu, Y.; Chew, J.; Cook, C.N.; et al. Toxic expanded GGGGCC repeat transcription is mediated by the PAF1 complex in C9orf72-associated FTD. Nat. Neurosci. 2019, 22, 863–874. [Google Scholar] [CrossRef]
- Pinto, B.S.; Saxena, T.; Oliveira, R.; Méndez-Gómez, H.R.; Cleary, J.D.; Denes, L.T.; McConnell, O.; Arboleda, J.; Xia, G.; Swanson, M.S.; et al. Impeding Transcription of Expanded Microsatellite Repeats by Deactivated Cas9. Mol. Cell 2017, 68, 479–490e5. [Google Scholar] [CrossRef] [Green Version]
- Batra, R.; Nelles, D.A.; Pirie, E.; Blue, S.M.; Marina, R.J.; Wang, H.; Chaim, I.A.; Thomas, J.D.; Zhang, N.; Nguyen, V.; et al. Elimination of Toxic Microsatellite Repeat Expansion RNA by RNA-Targeting Cas9. Cell 2017, 170, 899–912e10. [Google Scholar] [CrossRef]
- Paganoni, S.; Macklin, E.A.; Lee, A.; Murphy, A.; Chang, J.; Zipf, A.; Cudkowicz, M.; Atassi, N. Diagnostic timelines and delays in diagnosing amyotrophic lateral sclerosis (ALS). Amyotroph. Lateral Scler. Front. Degener. 2014, 15, 453–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallardo, G.; Holtzman, D.M. Antibody Therapeutics Targeting Aβ and Tau. Cold Spring Harb. Perspect. Med. 2017, 7, a024331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masliah, E.; Rockenstein, E.; Adame, A.; Alford, M.; Crews, L.; Hashimoto, M.; Seubert, P.; Lee, M.; Goldstein, J.; Chilcote, T.; et al. Effects of alpha-synuclein immunization in a mouse model of Parkinson’s disease. Neuron 2005, 46, 857–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Q.; Lehmer, C.; Michaelsen, M.; Mori, K.; Alterauge, D.; Baumjohann, D.; Schludi, M.H.; Greiling, J.; Farny, D.; Flatley, A.; et al. Antibodies inhibit transmission and aggregation of C9orf72 poly-GA dipeptide repeat proteins. EMBO Mol. Med. 2017, 9, 687–702. [Google Scholar] [CrossRef]
- Cristofani, R.; Crippa, V.; Vezzoli, G.; Rusmini, P.; Galbiati, M.; Cicardi, M.E.; Meroni, M.; Ferrari, V.; Tedesco, B.; Piccolella, M.; et al. The small heat shock protein B8 (HSPB8) efficiently removes aggregating species of dipeptides produced in C9ORF72-related neurodegenerative diseases. Cell Stress Chaperones 2018, 23, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Licata, N.V.; Cristofani, R.; Salomonsson, S.; Wilson, K.M.; Kempthorne, L.; Vaizoglu, D.; D’Agostino, V.G.; Pollini, D.; Loffredo, R.; Pancher, M.; et al. C9orf72 ALS/FTD dipeptide repeat protein levels are reduced by small molecules that inhibit PKA or enhance protein degradation. EMBO J. 2022, 41, e105026. [Google Scholar] [CrossRef]
- Shi, Y.; Hung, S.T.; Rocha, G.; Lin, S.; Linares, G.R.; Staats, K.A.; Seah, C.; Wang, Y.; Chickering, M.; Lai, J.; et al. Identification and therapeutic rescue of autophagosome and glutamate receptor defects in C9ORF72 and sporadic ALS neurons. JCI Insight 2019, 5, e127736. [Google Scholar] [CrossRef]
- Cheng, W.; Wang, S.; Zhang, Z.; Morgens, D.W.; Hayes, L.R.; Lee, S.; Portz, B.; Xie, Y.; Nguyen, B.V.; Haney, M.S.; et al. CRISPR-Cas9 Screens Identify the RNA Helicase DDX3X as a Repressor of C9ORF72 (GGGGCC)n Repeat-Associated Non-AUG Translation. Neuron 2019, 104, 885–898. [Google Scholar] [CrossRef]
- Cheng, W.; Wang, S.; Mestre, A.A.; Fu, C.; Makarem, A.; Xian, F.; Hayes, L.R.; Lopez-Gonzalez, R.; Drenner, K.; Jiang, J.; et al. C9ORF72 GGGGCC repeat-associated non-AUG translation is upregulated by stress through eIF2alpha phosphorylation. Nat. Commun. 2018, 9, 51. [Google Scholar] [CrossRef]
- Hautbergue, G.M.; Castelli, L.M.; Ferraiuolo, L.; Sanchez-Martinez, A.; Cooper-Knock, J.; Higginbottom, A.; Lin, Y.-H.; Bauer, C.S.; Dodd, J.E.; Myszczynska, M.A.; et al. SRSF1-dependent nuclear export inhibition of C9ORF72 repeat transcripts prevents neurodegeneration and associated motor deficits. Nat. Commun. 2017, 8, 16063. [Google Scholar] [CrossRef]
- Zhang, K.; Daigle, J.G.; Cunningham, K.M.; Coyne, A.N.; Ruan, K.; Grima, J.C.; Bowen, K.E.; Wadhwa, H.; Yang, P.; Rigo, F.; et al. Stress Granule Assembly Disrupts Nucleocytoplasmic Transport. Cell 2018, 173, 958–971.e17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arai, T.; Hasegawa, M.; Akiyama, H.; Ikeda, K.; Nonaka, T.; Mori, H.; Mann, D.; Tsuchiya, K.; Yoshida, M.; Hashizume, Y.; et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 2006, 351, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [Green Version]
- Gitcho, M.A.; Baloh, R.H.; Chakraverty, S.; Mayo, K.; Norton, J.B.; Levitch, D.; Hatanpaa, K.J.; White, C.L., 3rd; Bigio, E.H.; Caselli, R.; et al. TDP-43 A315T mutation in familial motor neuron disease. Ann. Neurol. 2008, 63, 535–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabashi, E.; Valdmanis, P.N.; Dion, P.; Spiegelman, D.; McConkey, B.J.; Vande Velde, C.; Bouchard, J.P.; Lacomblez, L.; Pochigaeva, K.; Salachas, F.; et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet. 2008, 40, 572–574. [Google Scholar] [CrossRef] [PubMed]
- Van Deerlin, V.M.; Leverenz, J.B.; Bekris, L.M.; Bird, T.D.; Yuan, W.; Elman, L.B.; Clay, D.; Wood, E.M.; Chen-Plotkin, A.S.; Martinez-Lage, M.; et al. TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: A genetic and histopathological analysis. Lancet Neurol. 2008, 7, 409–416. [Google Scholar] [CrossRef] [Green Version]
- Lattante, S.; Rouleau, G.A.; Kabashi, E. TARDBP and FUS mutations associated with amyotrophic lateral sclerosis: Summary and update. Hum. Mutat. 2013, 34, 812–826. [Google Scholar] [CrossRef]
- Giordana, M.T.; Piccinini, M.; Grifoni, S.; De Marco, G.; Vercellino, M.; Magistrello, M.; Pellerino, A.; Buccinna, B.; Lupino, E.; Rinaudo, M.T. TDP-43 redistribution is an early event in sporadic amyotrophic lateral sclerosis. Brain Pathol. 2010, 20, 351–360. [Google Scholar] [CrossRef] [Green Version]
- Suk, T.R.; Rousseaux, M.W.C. The role of TDP-43 mislocalization in amyotrophic lateral sclerosis. Mol. Neurodegener. 2020, 15, 45. [Google Scholar] [CrossRef]
- Takeuchi, R.; Tada, M.; Shiga, A.; Toyoshima, Y.; Konno, T.; Sato, T.; Nozaki, H.; Kato, T.; Horie, M.; Shimizu, H.; et al. Heterogeneity of cerebral TDP-43 pathology in sporadic amyotrophic lateral sclerosis: Evidence for clinico-pathologic subtypes. Acta Neuropathol. Commun. 2016, 4, 61. [Google Scholar] [CrossRef] [Green Version]
- Neumann, M.; Kwong, L.K.; Sampathu, D.M.; Trojanowski, J.Q.; Lee, V.M. TDP-43 proteinopathy in frontotemporal lobar degeneration and amyotrophic lateral sclerosis: Protein misfolding diseases without amyloidosis. Arch. Neurol. 2007, 64, 1388–1394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buratti, E.; Baralle, F.E. The multiple roles of TDP-43 in pre-mRNA processing and gene expression regulation. RNA Biol. 2010, 7, 420–429. [Google Scholar] [CrossRef] [Green Version]
- Ratti, A.; Buratti, E. Physiological functions and pathobiology of TDP-43 and FUS/TLS proteins. J. Neurochem. 2016, 138 (Suppl. 1), 95–111. [Google Scholar] [CrossRef] [PubMed]
- Ayala, Y.M.; Zago, P.; D’Ambrogio, A.; Xu, Y.F.; Petrucelli, L.; Buratti, E.; Baralle, F.E. Structural determinants of the cellular localization and shuttling of TDP-43. J. Cell Sci. 2008, 121, 3778–3785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraemer, B.C.; Schuck, T.; Wheeler, J.M.; Robinson, L.C.; Trojanowski, J.Q.; Lee, V.M.; Schellenberg, G.D. Loss of murine TDP-43 disrupts motor function and plays an essential role in embryogenesis. Acta Neuropathol. 2010, 119, 409–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ash, P.E.; Zhang, Y.J.; Roberts, C.M.; Saldi, T.; Hutter, H.; Buratti, E.; Petrucelli, L.; Link, C.D. Neurotoxic effects of TDP-43 overexpression in C. elegans. Hum. Mol. Genet. 2010, 19, 3206–3218. [Google Scholar] [CrossRef]
- Kabashi, E.; Lin, L.; Tradewell, M.L.; Dion, P.A.; Bercier, V.; Bourgouin, P.; Rochefort, D.; Bel Hadj, S.; Durham, H.D.; Vande Velde, C.; et al. Gain and loss of function of ALS-related mutations of TARDBP (TDP-43) cause motor deficits in vivo. Hum. Mol. Genet. 2010, 19, 671–683. [Google Scholar] [CrossRef] [Green Version]
- Wils, H.; Kleinberger, G.; Janssens, J.; Pereson, S.; Joris, G.; Cuijt, I.; Smits, V.; Ceuterick-de Groote, C.; Van Broeckhoven, C.; Kumar-Singh, S. TDP-43 transgenic mice develop spastic paralysis and neuronal inclusions characteristic of ALS and frontotemporal lobar degeneration. Proc. Natl. Acad. Sci. USA 2010, 107, 3858–3863. [Google Scholar] [CrossRef] [Green Version]
- Buratti, E. TDP-43 post-translational modifications in health and disease. Expert. Opin. Ther. Targets 2018, 22, 279–293. [Google Scholar] [CrossRef]
- Hasegawa, M.; Arai, T.; Nonaka, T.; Kametani, F.; Yoshida, M.; Hashizume, Y.; Beach, T.G.; Buratti, E.; Baralle, F.; Morita, M.; et al. Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann. Neurol. 2008, 64, 60–70. [Google Scholar] [CrossRef] [Green Version]
- Becker, L.A.; Huang, B.; Bieri, G.; Ma, R.; Knowles, D.A.; Jafar-Nejad, P.; Messing, J.; Kim, H.J.; Soriano, A.; Auburger, G.; et al. Therapeutic reduction of ataxin-2 extends lifespan and reduces pathology in TDP-43 mice. Nature 2017, 544, 367–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coyne Alyssa, N.; Baskerville, V.; Zaepfel Benjamin, L.; Dickson Dennis, W.; Rigo, F.; Bennett, F.; Lusk, C.P.; Rothstein Jeffrey, D. Nuclear accumulation of CHMP7 initiates nuclear pore complex injury and subsequent TDP-43 dysfunction in sporadic and familial ALS. Sci. Transl. Med. 2021, 13, eabe1923. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, A.L.; Shum, C.; Scotter, E.L.; Abdelgany, A.; Sardone, V.; Wright, J.; Lee, Y.B.; Chen, H.J.; Bilican, B.; Carrasco, M.; et al. Allele-specific knockdown of ALS-associated mutant TDP-43 in neural stem cells derived from induced pluripotent stem cells. PLoS ONE 2014, 9, e91269. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.Y.; Ieraci, A.; Teng, H.; Dall, H.; Meng, C.X.; Herrera, D.G.; Nykjaer, A.; Hempstead, B.L.; Lee, F.S. Sortilin controls intracellular sorting of brain-derived neurotrophic factor to the regulated secretory pathway. J. Neurosci. 2005, 25, 6156–6166. [Google Scholar] [CrossRef] [PubMed]
- Tann, J.Y.; Wong, L.W.; Sajikumar, S.; Ibanez, C.F. Abnormal TDP-43 function impairs activity-dependent BDNF secretion, synaptic plasticity, and cognitive behavior through altered Sortilin splicing. EMBO J. 2019, 38, e100989. [Google Scholar] [CrossRef]
- Archbold, H.C.; Jackson, K.L.; Arora, A.; Weskamp, K.; Tank, E.M.; Li, X.; Miguez, R.; Dayton, R.D.; Tamir, S.; Klein, R.L.; et al. TDP43 nuclear export and neurodegeneration in models of amyotrophic lateral sclerosis and frontotemporal dementia. Sci. Rep. 2018, 8, 4606. [Google Scholar] [CrossRef] [Green Version]
- Deng, H.; Gao, K.; Jankovic, J. The role of FUS gene variants in neurodegenerative diseases. Nat. Rev. Neurol. 2014, 10, 337–348. [Google Scholar] [CrossRef]
- Mackenzie, I.R.; Rademakers, R.; Neumann, M. TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol. 2010, 9, 995–1007. [Google Scholar] [CrossRef]
- Baumer, D.; Hilton, D.; Paine, S.M.; Turner, M.R.; Lowe, J.; Talbot, K.; Ansorge, O. Juvenile ALS with basophilic inclusions is a FUS proteinopathy with FUS mutations. Neurology 2010, 75, 611–618. [Google Scholar] [CrossRef] [Green Version]
- Waibel, S.; Neumann, M.; Rosenbohm, A.; Birve, A.; Volk, A.E.; Weishaupt, J.H.; Meyer, T.; Muller, U.; Andersen, P.M.; Ludolph, A.C. Truncating mutations in FUS/TLS give rise to a more aggressive ALS-phenotype than missense mutations: A clinico-genetic study in Germany. Eur. J. Neurol. 2013, 20, 540–546. [Google Scholar] [CrossRef]
- Ravits, J.; Appel, S.; Baloh, R.H.; Barohn, R.; Brooks, B.R.; Elman, L.; Floeter, M.K.; Henderson, C.; Lomen-Hoerth, C.; Macklis, J.D.; et al. Deciphering amyotrophic lateral sclerosis: What phenotype, neuropathology and genetics are telling us about pathogenesis. Amyotroph. Lateral Scler. Front. Degener. 2013, 14 (Suppl. 1), 5–18. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Rademakers, R.; Roeber, S.; Baker, M.; Kretzschmar, H.A.; Mackenzie, I.R. A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain J. Neurol. 2009, 132, 2922–2931. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.Y.; Pan, L.; Su, S.C.; Quinn, E.J.; Sasaki, M.; Jimenez, J.C.; Mackenzie, I.R.; Huang, E.J.; Tsai, L.H. Interaction of FUS and HDAC1 regulates DNA damage response and repair in neurons. Nat. Neurosci. 2013, 16, 1383–1391. [Google Scholar] [CrossRef] [PubMed]
- Hennig, S.; Kong, G.; Mannen, T.; Sadowska, A.; Kobelke, S.; Blythe, A.; Knott, G.J.; Iyer, K.S.; Ho, D.; Newcombe, E.A.; et al. Prion-like domains in RNA binding proteins are essential for building subnuclear paraspeckles. J. Cell Biol. 2015, 210, 529–539. [Google Scholar] [CrossRef]
- Vance, C.; Scotter, E.L.; Nishimura, A.L.; Troakes, C.; Mitchell, J.C.; Kathe, C.; Urwin, H.; Manser, C.; Miller, C.C.; Hortobagyi, T.; et al. ALS mutant FUS disrupts nuclear localization and sequesters wild-type FUS within cytoplasmic stress granules. Hum. Mol. Genet. 2013, 22, 2676–2688. [Google Scholar] [CrossRef] [PubMed]
- Sasayama, H.; Shimamura, M.; Tokuda, T.; Azuma, Y.; Yoshida, T.; Mizuno, T.; Nakagawa, M.; Fujikake, N.; Nagai, Y.; Yamaguchi, M. Knockdown of the Drosophila fused in sarcoma (FUS) homologue causes deficient locomotive behavior and shortening of motoneuron terminal branches. PLoS ONE 2012, 7, e39483. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, J.C.; McGoldrick, P.; Vance, C.; Hortobagyi, T.; Sreedharan, J.; Rogelj, B.; Tudor, E.L.; Smith, B.N.; Klasen, C.; Miller, C.C.; et al. Overexpression of human wild-type FUS causes progressive motor neuron degeneration in an age- and dose-dependent fashion. Acta Neuropathol. 2013, 125, 273–288. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Yi, F.; Fu, L.; Yang, J.; Wang, S.; Wang, Z.; Suzuki, K.; Sun, L.; Xu, X.; Yu, Y.; et al. CRISPR/Cas9-mediated targeted gene correction in amyotrophic lateral sclerosis patient iPSCs. Protein Cell 2017, 8, 365–378. [Google Scholar] [CrossRef]
- Bhinge, A.; Namboori, S.C.; Zhang, X.; VanDongen, A.M.J.; Stanton, L.W. Genetic Correction of SOD1 Mutant iPSCs Reveals ERK and JNK Activated AP1 as a Driver of Neurodegeneration in Amyotrophic Lateral Sclerosis. Stem Cell Rep. 2017, 8, 856–869. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Guo, W.; Mitra, J.; Hegde, P.M.; Vandoorne, T.; Eckelmann, B.J.; Mitra, S.; Tomkinson, A.E.; Van Den Bosch, L.; Hegde, M.L. Mutant FUS causes DNA ligation defects to inhibit oxidative damage repair in Amyotrophic Lateral Sclerosis. Nat. Commun. 2018, 9, 3683. [Google Scholar] [CrossRef] [Green Version]
- Vandoorne, T.; Veys, K.; Guo, W.; Sicart, A.; Vints, K.; Swijsen, A.; Moisse, M.; Eelen, G.; Gounko, N.V.; Fumagalli, L.; et al. Differentiation but not ALS mutations in FUS rewires motor neuron metabolism. Nat. Commun. 2019, 10, 4147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, W.; Naujock, M.; Fumagalli, L.; Vandoorne, T.; Baatsen, P.; Boon, R.; Ordovas, L.; Patel, A.; Welters, M.; Vanwelden, T.; et al. HDAC6 inhibition reverses axonal transport defects in motor neurons derived from FUS-ALS patients. Nat. Commun. 2017, 8, 861. [Google Scholar] [CrossRef] [PubMed]
- Benatar, M.; Wuu, J.; Andersen, P.M.; Bucelli, R.C.; Andrews, J.A.; Otto, M.; Farahany, N.A.; Harrington, E.A.; Chen, W.; Mitchell, A.A.; et al. Design of a Randomized, Placebo-Controlled, Phase 3 Trial of Tofersen Initiated in Clinically Presymptomatic SOD1 Variant Carriers: The ATLAS Study. Neurotherapeutics 2022. Online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Benatar, M.; Wuu, J. Presymptomatic studies in ALS: Rationale, challenges, and approach. Neurology 2012, 79, 1732–1739. [Google Scholar] [CrossRef] [Green Version]
- Benatar, M.; Wuu, J.; Lombardi, V.; Jeromin, A.; Bowser, R.; Andersen, P.M.; Malaspina, A. Neurofilaments in pre-symptomatic ALS and the impact of genotype. Amyotroph. Lateral Scler. Front. Degener. 2019, 20, 538–548. [Google Scholar] [CrossRef]
- Benatar, M.; Wuu, J.; Andersen, P.M.; Lombardi, V.; Malaspina, A. Neurofilament light: A candidate biomarker of presymptomatic amyotrophic lateral sclerosis and phenoconversion. Ann. Neurol. 2018, 84, 130–139. [Google Scholar] [CrossRef]

{kind=link}
| Agent | Mechanism of Action | Primary Measure Outcomes | Trial Design | N | Sites of Study | Status | CTI | Primary Outcome |
|---|---|---|---|---|---|---|---|---|
| BIIB067 or Tofersen (VALOR Trial) | ASO against SOD1 mRNA | Safety, tolerability, pharmacokinetics, biomarkers, ALSFRS-R change at 28 weeks | Phase 3, randomized, quadruple-blinded, placebo-controlled | 183 | USA, Canada, Europe | Complete | NCT-02623699 | N/A |
| AE and SAE up to 248 weeks | Extension of Phase 3, placebo-cotrolled, open label | 183 | USA, Canada, Europe | Active | NCT-03070119 | N/A | ||
| ISIS 333611 [33,34] | ASO against SOD1 mRNA | Safety, tolerability, and pharmacokinetics at unknown time | Phase 1, quadruple-blinded, randomized, placebo-controlled | 33 | USA | Complete | NCT-01041222 | No AE, Well tolerated, dose-dependent CSF and plasma concentrations |
| [37] | AAV-miR-SOD1 | Safety, tolerability, and pharmacokinetics | Open-label | 2 | USA | Complete | N/A | Meningoradiculitis in case 1, but not in case 2 with immunosuppressive therapy; Transient improvement in muscle sctregnth in case 1; |
| BIIB078 | ASO against C9orf72 mRNA | Safety at 323 days | Phase 1, quadruple-blinded, randomized, placebo-controlled | 90 | USA, Canada, Europe | Complete | NCT-03626012 | N/A |
| SB-509 [38] | Plasmid encoding a zinc finger DNA-binding protein transcription factor (ZFP TF(TM)) designed to up-regulate the expression of the gene encoding vascular endothelial growth factor (VEGF-A) | Change in ALSFRS-R at 11 months | Phase 2, open label | 45 | USA | Complete | NCT-00748501 | Safe, delayed deterioration in ankle and toe strength in 40% of treated subjects |
| ION363 (Jacifusen) | ASO against FUS mRNA | Change in ALSFRS-R and Ventilation Assistance-free survival (VAFS) at 505 days | Phase 1–3, double-blinded, randomized, placebo-controlled | 77 | USA, Canada, Belgium, UK | Active | NCT-04768972 | N/A |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fang, T.; Je, G.; Pacut, P.; Keyhanian, K.; Gao, J.; Ghasemi, M. Gene Therapy in Amyotrophic Lateral Sclerosis. Cells 2022, 11, 2066. https://doi.org/10.3390/cells11132066
Fang T, Je G, Pacut P, Keyhanian K, Gao J, Ghasemi M. Gene Therapy in Amyotrophic Lateral Sclerosis. Cells. 2022; 11(13):2066. https://doi.org/10.3390/cells11132066
Chicago/Turabian StyleFang, Ton, Goun Je, Peter Pacut, Kiandokht Keyhanian, Jeff Gao, and Mehdi Ghasemi. 2022. "Gene Therapy in Amyotrophic Lateral Sclerosis" Cells 11, no. 13: 2066. https://doi.org/10.3390/cells11132066
APA StyleFang, T., Je, G., Pacut, P., Keyhanian, K., Gao, J., & Ghasemi, M. (2022). Gene Therapy in Amyotrophic Lateral Sclerosis. Cells, 11(13), 2066. https://doi.org/10.3390/cells11132066