
Differences in Antioxidant and Lipid Handling Protein Expression Influence How Cells Expressing Distinct Mutant TP53 Subtypes Maintain Iron Homeostasis

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
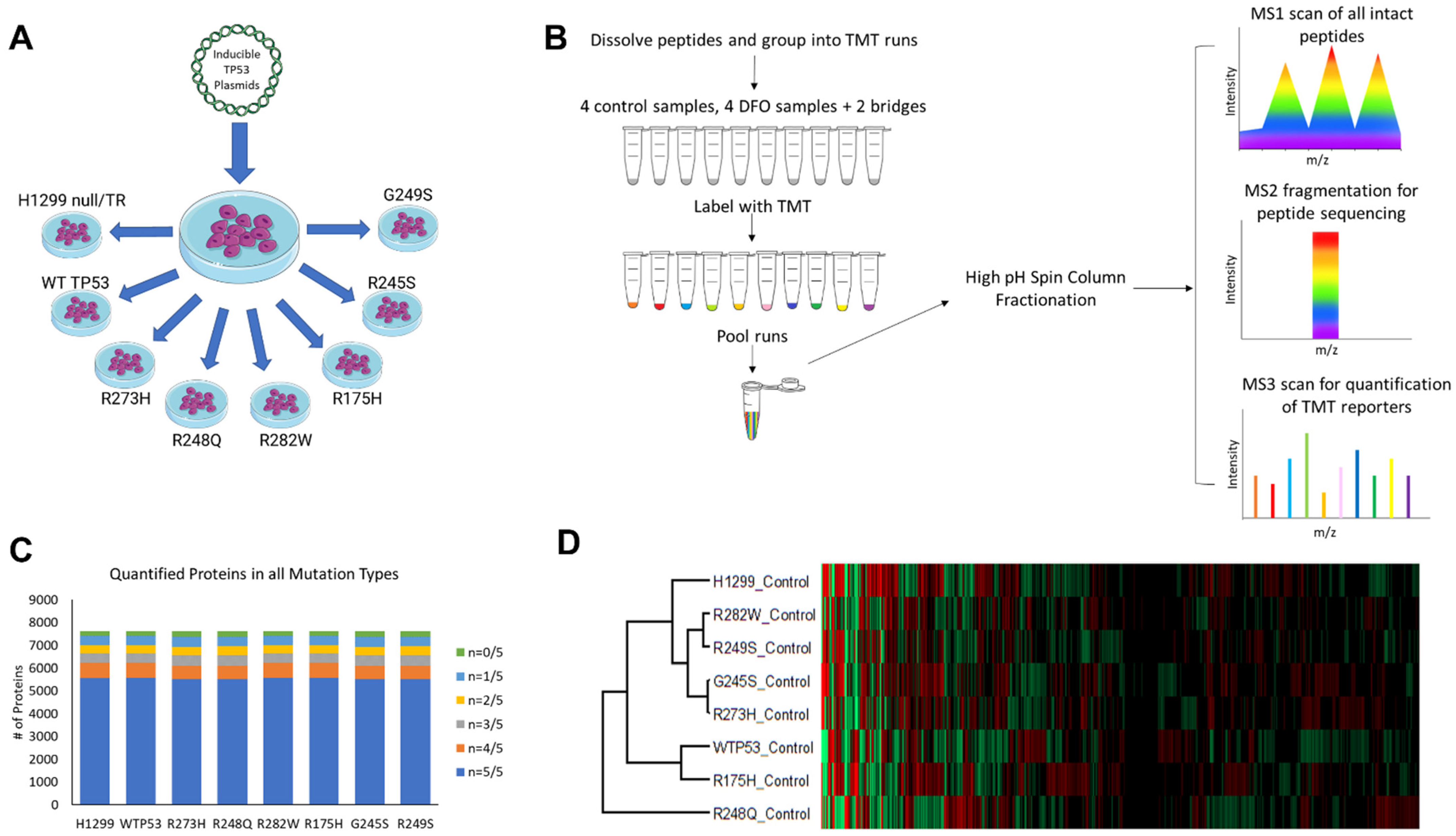
2.1. Cell Line Construction and Care
2.2. Treatment and Collection for Total Iron Quantitation and Proteomic Analyses
2.3. Tandem Mass Tag Proteomics
2.4. Bioinformatics
2.5. Enzymatic Assays
2.6. RNA Isolation and Real-Time qPCR
2.7. Viability Assays
2.8. Statistical Analyses
3. Results
3.1. In-Depth Acquisition of the Mutant TP53 Proteome
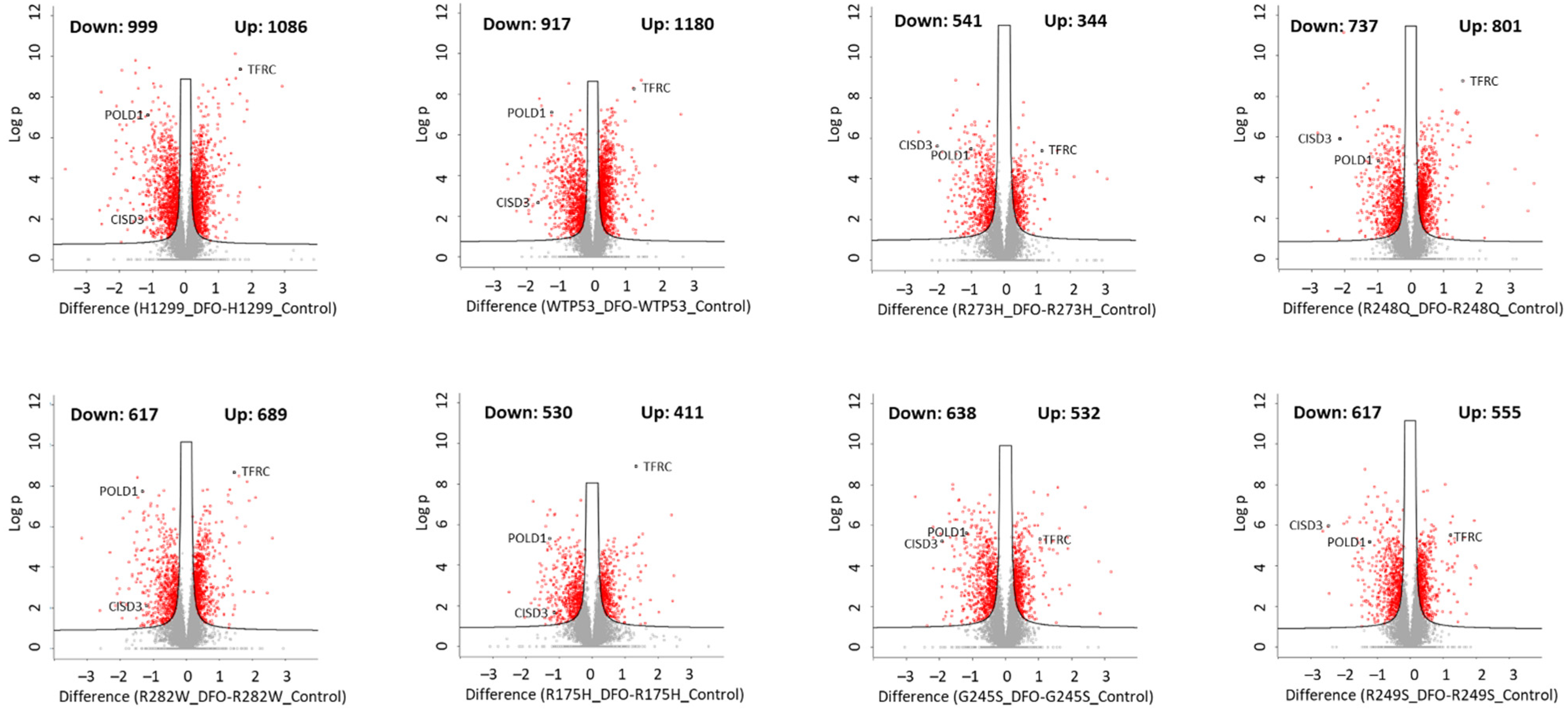
3.2. Iron Chelation Elicits Widespread Changes in Total Protein Expression in Wild-Type and Mutant TP53-Expressing Cells
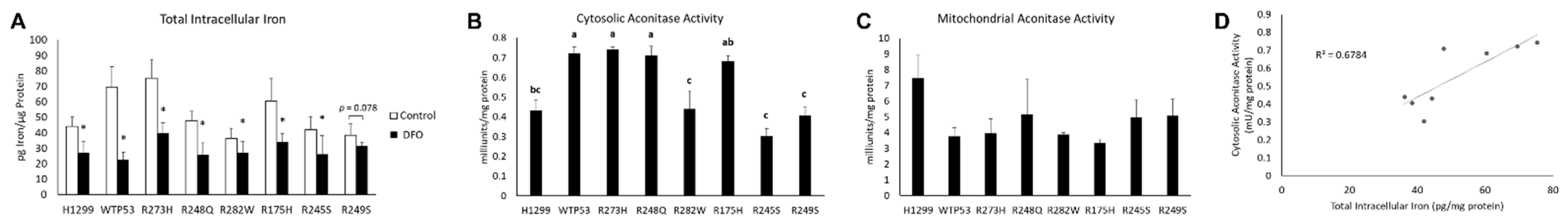
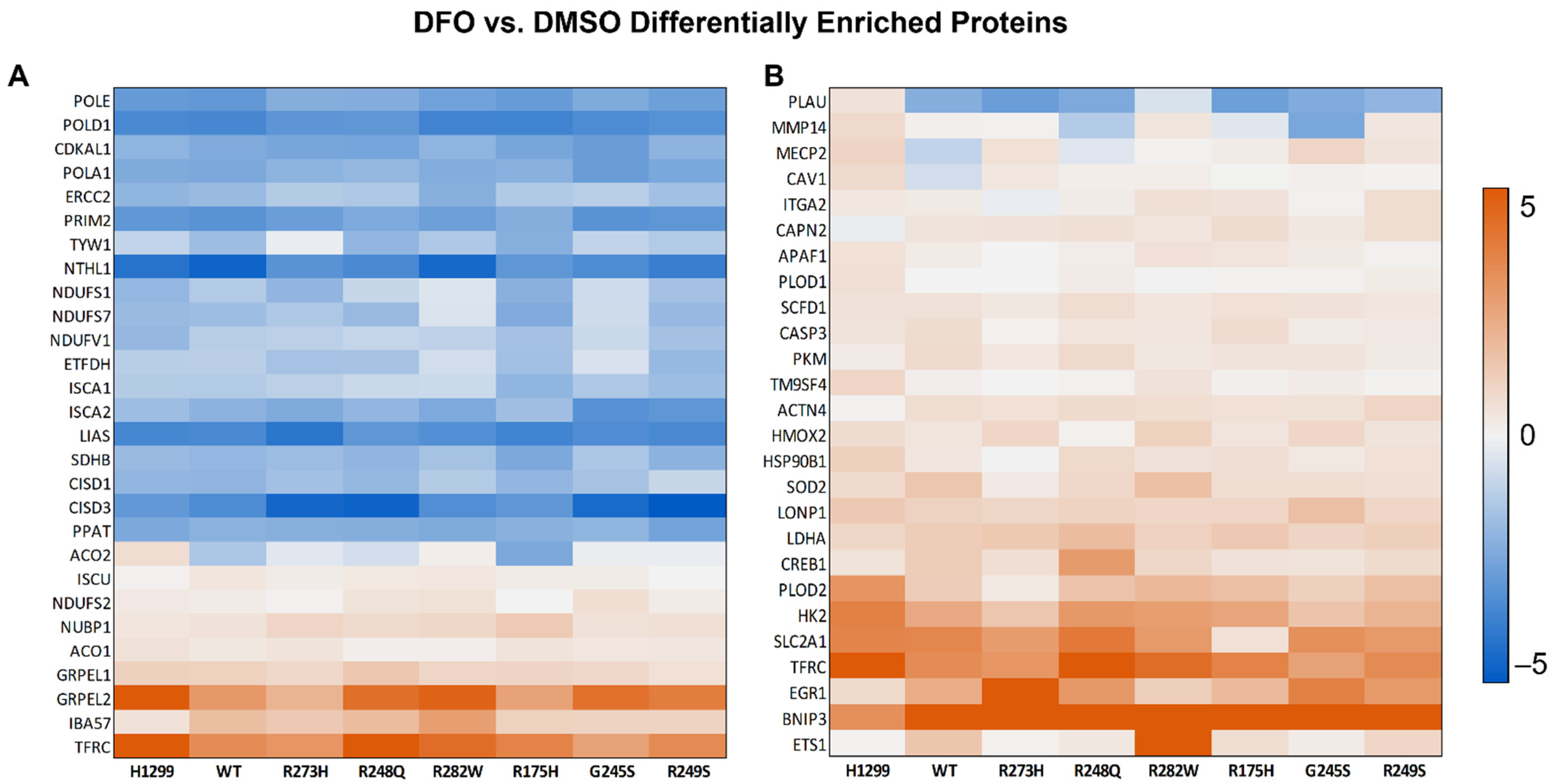
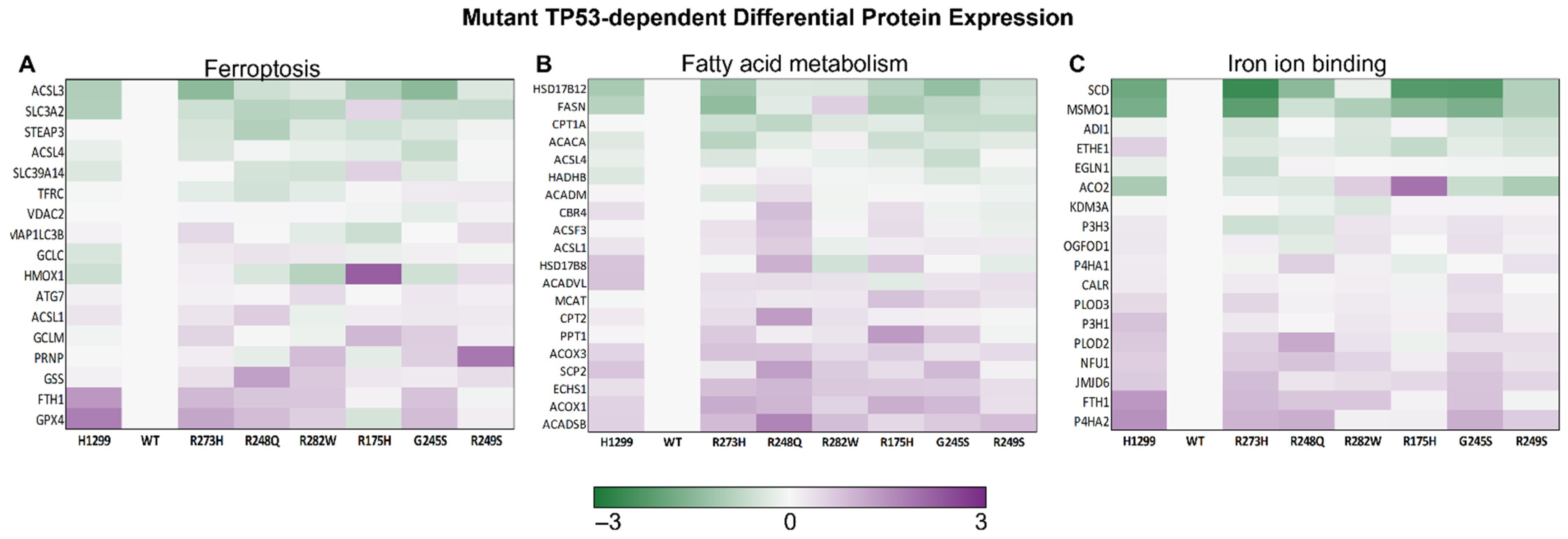
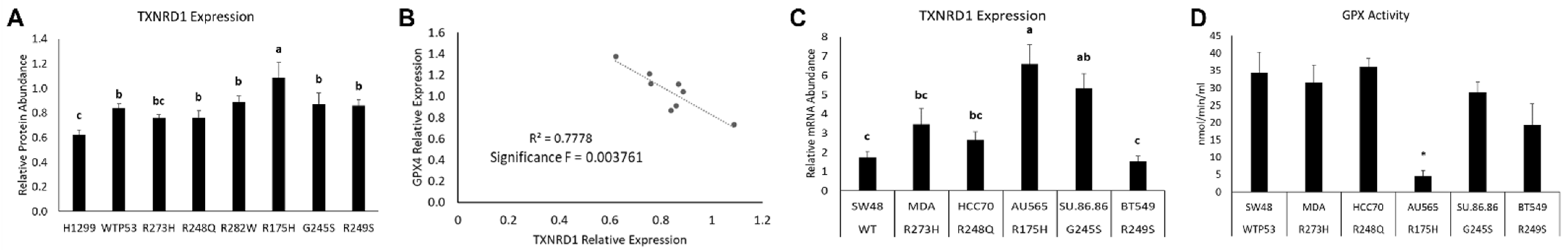
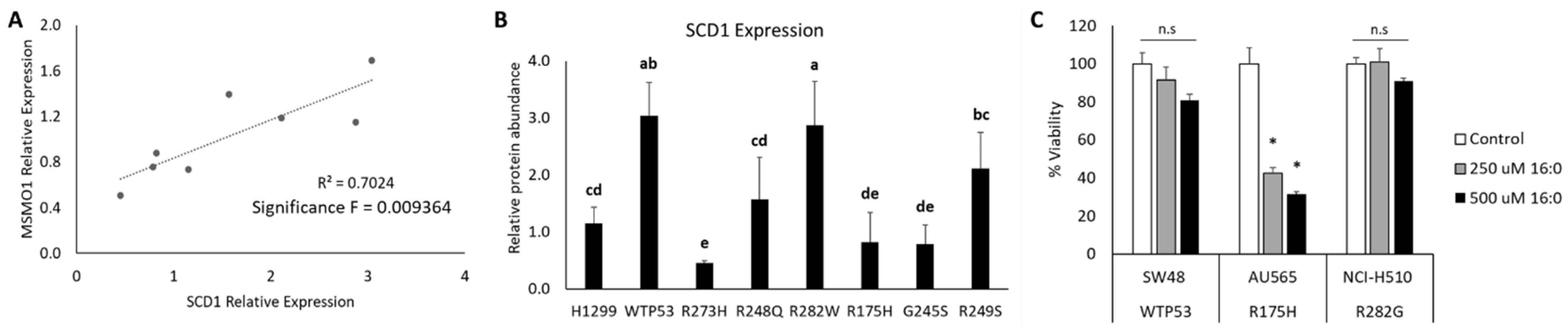
3.3. Distinct Mutant TP53 Protein Expression Patterns Influence Antioxidant and Lipid Handling Capacity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dixon, S.J.; Stockwell, B.R. The Hallmarks of Ferroptosis. Annu. Rev. Cancer Biol. 2019, 3, 35–54. [Google Scholar] [CrossRef]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 5, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef] [Green Version]
- Tarangelo, A.; Magtanong, L.; Bieging-Rolett, K.T.; Li, Y.; Ye, J.; Attardi, L.D.; Dixon, S.J. p53 Suppresses Metabolic Stress-Induced Ferroptosis in Cancer Cells. Cell Rep. 2018, 22, 569–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.J.; Li, D.; Ou, Y.; Jiang, L.; Chen, Y.; Zhao, Y.; Gu, W. Acetylation Is Crucial for p53-Mediated Ferroptosis and Tumor Suppression. Cell Rep. 2016, 17, 366–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Y.; Zhu, S.; Song, X.; Sun, X.; Fan, Y.; Liu, J.; Zhong, M.; Yuan, H.; Zhang, L.; Billiar, T.R.; et al. The Tumor Suppressor p53 Limits Ferroptosis by Blocking DPP4 Activity. Cell Rep. 2017, 20, 1692–1704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gnanapradeepan, K.; Basu, S.; Barnoud, T.; Budina-Kolomets, A.; Kung, C.P.; Murphy, M.E. The p53 Tumor Suppressor in the Control of Metabolism and Ferroptosis. Front Endocrinol. 2018, 9, 124. [Google Scholar] [CrossRef] [Green Version]
- Thompson, L.R.; Oliveira, T.G.; Hermann, E.R.; Chowanadisai, W.; Clarke, S.L.; Montgomery, M.R. Distinct TP53 Mutation Types Exhibit Increased Sensitivity to Ferroptosis Independently of Changes in Iron Regulatory Protein Activity. Int. J. Mol. Sci. 2020, 21, 6751. [Google Scholar] [CrossRef]
- Clarke, S.L.; Thompson, L.R.; Dandekar, E.; Srinivasan, A.; Montgomery, M.R. Distinct TP53 Mutation Subtypes Differentially Influence Cellular Iron Metabolism. Nutrients 2019, 11, 2144. [Google Scholar] [CrossRef] [Green Version]
- Zecha, J.; Satpathy, S.; Kanashova, T.; Avanessian, S.C.; Kane, M.H.; Clauser, K.R.; Mertins, P.; Carr, S.A.; Kuster, B. TMT Labeling for the Masses: A Robust and Cost-efficient, In-solution Labeling Approach. Mol. Cell Proteom. 2019, 18, 1468–1478. [Google Scholar] [CrossRef] [Green Version]
- McAlister, G.C.; Nusinow, D.P.; Jedrychowski, M.P.; Wuhr, M.; Huttlin, E.L.; Erickson, B.K.; Rad, R.; Haas, W.; Gygi, S.P. MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal. Chem. 2014, 86, 7150–7158. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Tusher, V.G.; Tibshirani, R.; Chu, G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. USA 2001, 98, 5116–5121. [Google Scholar] [CrossRef] [Green Version]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W. DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 2022, 10, gkac194. [Google Scholar] [CrossRef]
- Safran, M.; Rosen, N.; Twik, M.; BarShir, R.; Iny Stein, T.; Dahary, D.; Fishilevich, S.; Lancet, D. The GeneCards Suite Chapter. In Practical Guide to Life Science Databases; Springer: Singapore, 2022; pp. 27–56. [Google Scholar]
- Javvadi, P.; Hertan, L.; Kosoff, R.; Datta, T.; Kolev, J.; Mick, R.; Tuttle, S.W.; Koumenis, C. Thioredoxin reductase-1 mediates curcumin-induced radiosensitization of squamous carcinoma cells. Cancer Res. 2010, 70, 1941–1950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cousin, S.P.; Hugl, S.R.; Wrede, C.E.; Kajio, H.; Myers, M.G., Jr.; Rhodes, C.J. Free fatty acid-induced inhibition of glucose and insulin-like growth factor I-induced deoxyribonucleic acid synthesis in the pancreatic beta-cell line INS-1. Endocrinology 2001, 142, 229–240. [Google Scholar] [CrossRef]
- Altman, D.G.; Bland, J.M. Standard deviations and standard errors. BMJ 2005, 331, 903. [Google Scholar] [CrossRef] [Green Version]
- Gilar, M.; Olivova, P.; Daly, A.E.; Gebler, J.C. Orthogonality of separation in two-dimensional liquid chromatography. Anal. Chem. 2005, 77, 6426–6434. [Google Scholar] [CrossRef]
- Yang, F.; Shen, Y.; Camp, D.G., 2nd; Smith, R.D. High-pH reversed-phase chromatography with fraction concatenation for 2D proteomic analysis. Expert Rev. Proteom. 2012, 9, 129–134. [Google Scholar] [CrossRef] [Green Version]
- Magtanong, L.; Ko, P.J.; To, M.; Cao, J.Y.; Forcina, G.C.; Tarangelo, A.; Ward, C.C.; Cho, K.; Patti, G.J.; Nomura, D.K.; et al. Exogenous Monounsaturated Fatty Acids Promote a Ferroptosis-Resistant Cell State. Cell Chem. Biol. 2019, 26, 420–432.e9. [Google Scholar] [CrossRef]
- Wang, C.; Shi, M.; Ji, J.; Cai, Q.; Zhao, Q.; Jiang, J.; Liu, J.; Zhang, H.; Zhu, Z.; Zhang, J. Stearoyl-CoA desaturase 1 (SCD1) facilitates the growth and anti-ferroptosis of gastric cancer cells and predicts poor prognosis of gastric cancer. Aging 2020, 12, 15374–15391. [Google Scholar] [CrossRef] [PubMed]
- Man, W.C.; Miyazaki, M.; Chu, K.; Ntambi, J.M. Membrane topology of mouse stearoyl-CoA desaturase 1. J. Biol. Chem. 2006, 281, 1251–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, X.; Zhao, Y.; Xu, Y.; Zheng, M.; Feng, Z.; Hu, W. Mutant p53 in Cancer: Accumulation, Gain-of-Function, and Therapy. J. Mol. Biol. 2017, 429, 1595–1606. [Google Scholar] [CrossRef] [PubMed]
- Freed-Pastor, W.A.; Prives, C. Mutant p53: One name, many proteins. Genes Dev. 2012, 26, 1268–1286. [Google Scholar] [CrossRef] [Green Version]
- Funauchi, Y.; Tanikawa, C.; Yi Lo, P.H.; Mori, J.; Daigo, Y.; Takano, A.; Miyagi, Y.; Okawa, A.; Nakamura, Y.; Matsuda, K. Regulation of iron homeostasis by the p53-ISCU pathway. Sci. Rep. 2015, 5, 16497. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Wang, W.; Tsuji, Y.; Torti, S.V.; Torti, F.M. Post-transcriptional modulation of iron homeostasis during p53-dependent growth arrest. J. Biol. Chem. 2008, 283, 33911–33918. [Google Scholar] [CrossRef] [Green Version]
- Sheftel, A.; Stehling, O.; Lill, R. Iron-sulfur proteins in health and disease. Trends Endocrinol. Metab. 2010, 21, 302–314. [Google Scholar] [CrossRef]
- Davis, M.R.; Shawron, K.M.; Rendina, E.; Peterson, S.K.; Lucas, E.A.; Smith, B.J.; Clarke, S.L. Hypoxia inducible factor-2 alpha is translationally repressed in response to dietary iron deficiency in Sprague-Dawley rats. J. Nutr. 2011, 141, 1590–1596. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, M.; Galy, B.; Muckenthaler, M.U.; Hentze, M.W. Iron-regulatory proteins limit hypoxia-inducible factor-2alpha expression in iron deficiency. Nat. Struct. Mol. Biol. 2007, 14, 420–426. [Google Scholar] [CrossRef]
- Torti, S.V.; Manz, D.H.; Paul, B.T.; Blanchette-Farra, N.; Torti, F.M. Iron and Cancer. Annu. Rev. Nutr. 2018, 38, 97–125. [Google Scholar] [CrossRef]
- Tesfay, L.; Paul, B.T.; Konstorum, A.; Deng, Z.; Cox, A.O.; Lee, J.; Furdui, C.M.; Hegde, P.; Torti, F.M.; Torti, S.V. Stearoyl-CoA Desaturase 1 Protects Ovarian Cancer Cells from Ferroptotic Cell Death. Cancer Res. 2019, 79, 5355–5366. [Google Scholar] [CrossRef] [PubMed]
- Mirza, A.; Wu, Q.; Wang, L.; McClanahan, T.; Bishop, W.R.; Gheyas, F.; Ding, W.; Hutchins, B.; Hockenberry, T.; Kirschmeier, P.; et al. Global transcriptional program of p53 target genes during the process of apoptosis and cell cycle progression. Oncogene 2003, 22, 3645–3654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, L.L.; Ruberto, R.A.; Ryan, M.J.; Eaton, J.K.; Schreiber, S.L.; Viswanathan, V.S. Modulation of ferroptosis sensitivity by TXNRD1 in pancreatic cancer cells. BioRxiv 2020. [Google Scholar] [CrossRef]
- Freire Boullosa, L.; Van Loenhout, J.; Flieswasser, T.; De Waele, J.; Hermans, C.; Lambrechts, H.; Cuypers, B.; Laukens, K.; Bartholomeus, E.; Siozopoulou, V.; et al. Auranofin reveals therapeutic anticancer potential by triggering distinct molecular cell death mechanisms and innate immunity in mutant p53 non-small cell lung cancer. Redox Biol 2021, 42, 101949. [Google Scholar] [CrossRef]
- Adedoyin, O.; Boddu, R.; Traylor, A.; Lever, J.M.; Bolisetty, S.; George, J.F.; Agarwal, A. Heme oxygenase-1 mitigates ferroptosis in renal proximal tubule cells. Am. J. Physiol. Ren. Physiol. 2018, 314, F702–F714. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cardona, C.J.; Hermann, E.R.; Kouplen, K.N.; Hartson, S.D.; Montgomery, M.R. Differences in Antioxidant and Lipid Handling Protein Expression Influence How Cells Expressing Distinct Mutant TP53 Subtypes Maintain Iron Homeostasis. Cells 2022, 11, 2064. https://doi.org/10.3390/cells11132064
Cardona CJ, Hermann ER, Kouplen KN, Hartson SD, Montgomery MR. Differences in Antioxidant and Lipid Handling Protein Expression Influence How Cells Expressing Distinct Mutant TP53 Subtypes Maintain Iron Homeostasis. Cells. 2022; 11(13):2064. https://doi.org/10.3390/cells11132064
Chicago/Turabian StyleCardona, Cameron J., Evan R. Hermann, Kate N. Kouplen, Steven D. Hartson, and McKale R. Montgomery. 2022. "Differences in Antioxidant and Lipid Handling Protein Expression Influence How Cells Expressing Distinct Mutant TP53 Subtypes Maintain Iron Homeostasis" Cells 11, no. 13: 2064. https://doi.org/10.3390/cells11132064
APA StyleCardona, C. J., Hermann, E. R., Kouplen, K. N., Hartson, S. D., & Montgomery, M. R. (2022). Differences in Antioxidant and Lipid Handling Protein Expression Influence How Cells Expressing Distinct Mutant TP53 Subtypes Maintain Iron Homeostasis. Cells, 11(13), 2064. https://doi.org/10.3390/cells11132064