
CAR T Cell Locomotion in Solid Tumor Microenvironment

 ,
,  , , ,
, , ,
Abstract
1. Introduction
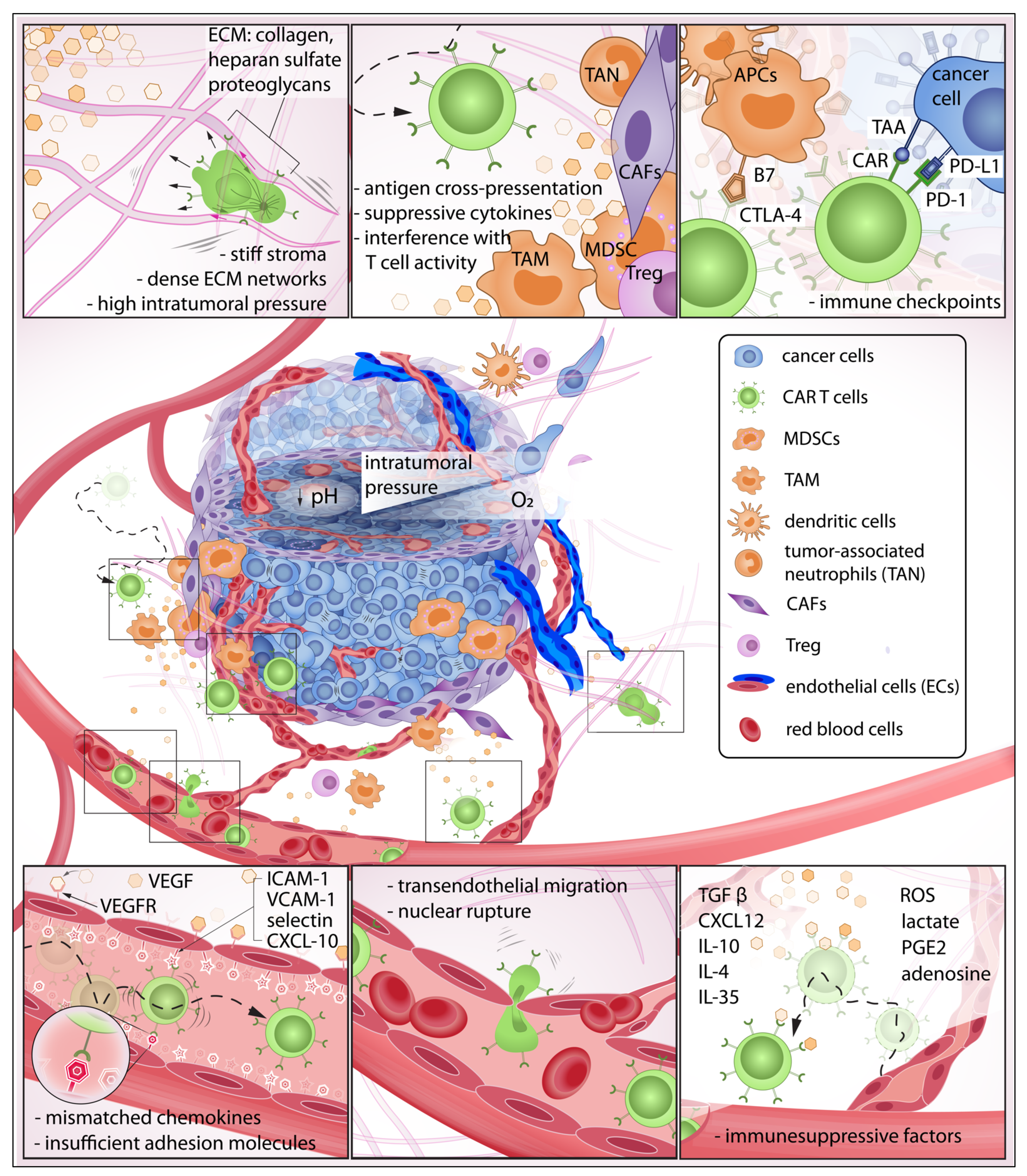
2. CAR T Cell Trafficking and Infiltration
2.1. The Immunosuppressive Tumor Microenvironment and Physical Barriers Favorable to Resilient Tumor Growth
2.2. Immunosuppressive Chemokines: The Invisible Barrier against CAR T Infiltration
2.3. Pro-Tumor Stromal and Immune Cells Are Active Contributors to Immune Suppression
2.4. Physical Confinement and Mechanical Properties of Cell Nucleus Modulate T Cell Locomotion
3. Preclinical Models for Trafficking and Infiltration
3.1. In Vivo Models
3.2. Three-Dimensional (3D) Supports and In Vitro Tumor Models
3.3. Common Three-Dimensional Support Materials for In Vitro Assays
3.4. Tumor Spheroids
3.5. Tumor Organoids
3.6. Tumor Explants
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Melenhorst, J.J.; Castillo, P.; Hanley, P.J.; Keller, M.D.; Krance, R.A.; Margolin, J.; Leen, A.M.; Heslop, H.E.; Barrett, A.J.; Rooney, C.M.; et al. Graft versus leukemia response without graft-versus-host disease elicited by adoptively transferred multivirus-specific T-cells. Mol. Ther. 2015, 23, 179–183. [Google Scholar] [CrossRef]
- Castillo, P.; Wright, K.E.; Kontoyiannis, D.P.; Walsh, T.; Patel, S.; Chorvinsky, E.; Bose, S.; Hazrat, Y.; Omer, B.; Albert, N.; et al. A New Method for Reactivating and Expanding T Cells Specific for Rhizopus oryzae. Mol. Ther.-Methods Clin. Dev. 2018, 9, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Sayour, E.J.; Grippin, A.; De Leon, G.; Stover, B.; Rahman, M.; Karachi, A.; Wummer, B.; Moore, G.; Castillo-Caro, P.; Fredenburg, K.; et al. Personalized Tumor RNA Loaded Lipid-Nanoparticles Prime the Systemic and Intratumoral Milieu for Response to Cancer Immunotherapy. Nano Lett. 2018, 18, 6195–6206. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Restifo, N.P.; Yang, J.C.; Morgan, R.A.; Dudley, M.E. Adoptive cell transfer: A clinical path to effective cancer immunotherapy. Nat. Rev. Cancer 2008, 8, 299–308. [Google Scholar] [CrossRef]
- Maude, S.L.; Teachey, D.T.; Porter, D.L.; Grupp, S.A. CD19-targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Blood 2015, 125, 4017–4023. [Google Scholar] [CrossRef]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric Antigen Receptor T Cells for Sustained Remissions in Leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Zhang, W.; Moore, L.; Ji, P. Mouse models for cancer research. Chin. J. Cancer 2007, 30, 149–152. [Google Scholar] [CrossRef]
- Convery, N.; Gadegaard, N. 30 Years of Microfluidics. Micro Nano Eng. 2019, 2, 76–91. [Google Scholar] [CrossRef]
- Park, D.; Son, K.; Hwang, Y.; Ko, J.; Lee, Y.; Doh, J.; Jeon, N.L. High-throughput microfluidic 3D cytotoxicity assay for cancer immunotherapy (CACI-ImpacT platform). Front. Immunol. 2019, 10, 1133. [Google Scholar] [CrossRef] [PubMed]
- Haeseleer, F.; Eichholz, K.; Tareen, S.U.; Iwamoto, N.; Roederer, M.; Kirchhoff, F.; Park, H.; Okoye, A.A.; Corey, L. Real-Time Killing Assays to Assess the Potency of a New Anti-Simian Immunodeficiency Virus Chimeric Antigen Receptor T Cell. AIDS Res. Hum. Retrovir. 2020, 36, 998–1009. [Google Scholar] [CrossRef] [PubMed]
- Bellone, M.; Calcinotto, A. Ways to enhance lymphocyte trafficking into tumors and fitness of tumor infiltrating lymphocytes. Front. Oncol. 2013, 3, 231. [Google Scholar] [CrossRef] [PubMed]
- Chinnasamy, D.; Yu, Z.; Theoret, M.R.; Zhao, Y.; Shrimali R, R.K.; Morgan, R.A.; Feldman, S.A.; Restifo, N.P.; Rosenberg, S.A. Gene therapy using genetically modified lymphocytes targeting VEGFR-2 inhibits the growth of vascularized syngenic tumors in mice. J. Clin. Investig. 2010, 120, 3953–3968. [Google Scholar] [CrossRef] [PubMed]
- Englisch, A.; Altvater, B.; Kailayangiri, S.; Hartmann, W.; Rossig, C. VEGFR2 as a target for CAR T cell therapy of Ewing sarcoma. Pediatr. Blood Cancer 2020, 67, e28313. [Google Scholar] [CrossRef]
- Chinnasamy, D.; Yu, Z.; Kerkar, S.P.; Zhang, L.; Morgan, R.A.; Restifo, N.P.; Rosenberg, S.A. Local delivery of interleukin-12 using T cells targeting VEGF receptor-2 eradicates multiple vascularized tumors in mice. Clin. Cancer Res. 2012, 18, 1672–1683. [Google Scholar] [CrossRef]
- Chinnasamy, D.; Tran, E.; Yu, Z.; Morgan, R.A.; Restifo, N.P.; Rosenberg, S.A. Simultaneous targeting of tumor antigens and the tumor vasculature using t lymphocyte transfer synergize to induce regression of established tumors in mice. Cancer Res. 2013, 73, 3371–3380. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Kai, F.B.; Drain, A.P.; Weaver, V.M. The Extracellular Matrix Modulates the Metastatic Journey. Dev. Cell 2019, 49, 332–346. [Google Scholar] [CrossRef]
- Napoli, M.; Flores, E.R. Beware of thy neighbor: Senescent cancer cells feast on adjacent ells to persist. J. Cell Biol. 2019, 218, 3535–3536. [Google Scholar] [CrossRef] [PubMed]
- Frantz, C.; Stewart, K.M.; Weaver, V.M. The extracellular matrix at a glance. J. Cell Sci. 2010, 123, 4195–4200. [Google Scholar] [CrossRef]
- Cox, T.R.; Erler, J.T. Remodeling and homeostasis of the extracellular matrix: Implications for fibrotic diseases and cancer. Dis. Model. Mech. 2011, 4, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef] [PubMed]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix Metalloproteinases: Regulators of the Tumor Microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef] [PubMed]
- Moon, E.K.; Wang, L.C.; Dolfi, D.V.; Wilson, C.B.; Ranganathan, R.; Sun, J.; Kapoor, V.; Scholler, J.; Puré, E.; Milone, M.C.; et al. Multifactorial T-cell hypofunction that is reversible can limit the efficacy of chimeric antigen receptor-transduced human T cells in solid tumors. Clin. Cancer Res. 2014, 20, 4262–4273. [Google Scholar] [CrossRef] [PubMed]
- Gustave, I.; Albini, A. Contribution to Tumor Angiogenesis From innate immune Cells within the Tumor Microenvironment: Implications for immunotherapy. Front. Immunol. 2018, 9, 527. [Google Scholar] [CrossRef]
- Van Helvert, S.; Friedl, P. Strain Stiffening of Fibrillar Collagen during Individual and Collective Cell Migration Identified by AFM Nanoindentation. ACS Appl. Mater. Interfaces 2016, 8, 21946–21955. [Google Scholar] [CrossRef] [PubMed]
- Ahmadzadeh, H.; Webster, M.R.; Behera, R.; Jimenez, A.M.; Wirtz, D. Modeling the two-way feedback between contractility and matrix realignment reveals a nonlinear mode of cancer cell invasion. Proc. Natl. Acad. Sci. USA 2017, 114, E1617–E1626. [Google Scholar] [CrossRef]
- Butcher, D.T.; Alliston, T.; Weaver, V.M. A tense situation: Forcing tumour progression. Nat. Rev. Cancer 2009, 9, 108–122. [Google Scholar] [CrossRef]
- D’Aloia, M.M.; Zizzari, I.G.; Sacchetti, B.; Pierelli, L.; Alimandi, M. CAR-T cells: The long and winding road to solid tumors review-article. Cell Death Dis. 2018, 9, 282. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, W.; Huang, K.; Zhang, Y.; Kupfer, G.; Zhao, Q. Chimeric antigen receptor T cell (CAR-T) immunotherapy for solid tumors: Lessons learned and strategies for moving forward. J. Hematol. Oncol. 2018, 11, 22. [Google Scholar] [CrossRef] [PubMed]
- Yong, C.S.M.; Dardalhon, V.; Devaud, C.; Taylor, N.; Darcy, P.K.; Kershaw, M.H. CAR T-cell therapy of solid tumors. Immunol. Cell Biol. 2017, 95, 356–363. [Google Scholar] [CrossRef]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef] [PubMed]
- Levental, K.R.; Yu, H.; Kass, L.; Lakins, J.N.; Egeblad, M.; Erler, J.T.; Fong, S.F.T.; Csiszar, K.; Giaccia, A.; Weninger, W.; et al. Matrix Crosslinking Forces Tumor Progression by Enhancing Integrin Signaling. Cell 2009, 139, 891–906. [Google Scholar] [CrossRef] [PubMed]
- Coste, B.; Xiao, B.; Santos, J.S.; Syeda, R.; Grandl, J.; Spencer, K.S.; Kim, S.E.; Schmidt, M.; Mathur, J.; Dubin, A.E.; et al. Piezo proteins are pore-forming subunits of mechanically activated channels. Nature 2012, 483, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP/TAZ at the Roots of Cancer. Cancer Cell 2016, 29, 783–803. [Google Scholar] [CrossRef]
- Chin, L.K.; Xia, Y.; Discher, D.E.; Janmey, P.A. Mechanotransduction in cancer. Curr. Opin. Chem. Eng. 2016, 11, 77–84. [Google Scholar] [CrossRef]
- Feitelson, M.A.; Arzumanyan, A.; Kulathinal, R.J.; Blain, S.W.; Holcombe, R.F.; Mahajna, J.; Marino, M.; Martinez-Chantar, M.L.; Nawroth, R.; Sanchez-Garcia, I.; et al. Sustained proliferation in cancer: Mechanisms and novel therapeutic targets. Semin. Cancer Biol. 2015, 35, S25–S54. [Google Scholar] [CrossRef] [PubMed]
- Wei, B.; Zhou, X.; Liang, C.; Zheng, X.; Lei, P.; Fang, J.; Han, X.; Wang, L.; Qi, C.; Wei, H. Human colorectal cancer progression correlates with LOX-induced ECM stiffening. Int. J. Biol. Sci. 2017, 13, 1450–1457. [Google Scholar] [CrossRef]
- Su, G.; Meyer, K.; Nandini, C.D.; Qiao, D.; Salamat, S.; Friedl, A. Glypican-1 is frequently overexpressed in human gliomas and enhances FGF-2 signaling in glioma cells. Am. J. Pathol. 2006, 168, 2014–2026. [Google Scholar] [CrossRef] [PubMed]
- Iozzo, R.V.; Sanderson, R.D. Proteoglycans in cancer biology, tumour microenvironment and angiogenesis. J. Cell. Mol. Med. 2011, 15, 1013–1031. [Google Scholar] [CrossRef] [PubMed]
- Bordeleau, F.; Mason, B.N.; Lollis, E.M.; Mazzola, M.; Zanotelli, M.R.; Somasegar, S.; Califano, J.P.; Montague, C.; LaValley, D.J.; Huynh, J.; et al. Matrix stiffening promotes a tumor vasculature phenotype. Proc. Natl. Acad. Sci. USA 2017, 114, 492–497. [Google Scholar] [CrossRef] [PubMed]
- Mcmahon, M.; Ye, S.; Pedrina, J.; Dlugolenski, D.; Stambas, J. Extracellular Matrix Enzymes and Immune Cell Biology. Front. Mol. Biosci. 2021, 8, 703868. [Google Scholar] [CrossRef]
- Mondal, S.; Adhikari, N.; Banerjee, S.; Amin, S.A.; Jha, T. Matrix metalloproteinase-9 ( MMP-9 ) and its inhibitors in cancer: A minireview. Eur. J. Med. Chem. 2020, 194, 112260. [Google Scholar] [CrossRef]
- Kumar, S.; Das, A.; Barai, A.; Sen, S. MMP Secretion Rate and Inter-invadopodia Spacing Collectively Govern Cancer Invasiveness. Biophysj 2018, 114, 650–662. [Google Scholar] [CrossRef]
- Caruana, I.; Savoldo, B.; Hoyos, V.; Weber, G.; Liu, H.; Kim, E.S.; Ittmann, M.M.; Marchetti, D.; Dotti, G. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat. Med. 2015, 21, 524–529. [Google Scholar] [CrossRef] [PubMed]
- Jabłońska-Trypuć, A.; Matejczyk, M.; Rosochacki, S. Matrix metalloproteinases (MMPs), the main extracellular matrix (ECM) enzymes in collagen degradation, as a target for anticancer drugs. J. Enzyme Inhib. Med. Chem. 2016, 31, 177–183. [Google Scholar] [CrossRef]
- Renkawitz, J.; Kopf, A.; Stopp, J.; de Vries, I.; Driscoll, M.K.; Merrin, J.; Hauschild, R.; Welf, E.S.; Danuser, G.; Fiolka, R.; et al. Nuclear positioning facilitates amoeboid migration along the path of least resistance. Nature 2019, 568, 546–550. [Google Scholar] [CrossRef]
- Krummel, M.F.; Friedman, R.S.; Jacobelli, J. Modes and mechanisms of T cell motility: Roles for confinement and Myosin-IIA. Curr. Opin. Cell Biol. 2014, 30, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Tabdanov, E.D.; Rodríguez-Merced, N.J.; Cartagena-Rivera, A.X.; Puram, V.V.; Callaway, M.K.; Ensminger, E.A.; Pomeroy, E.J.; Yamamoto, K.; Lahr, W.S.; Webber, B.R.; et al. Engineering T cells to enhance 3D migration through structurally and mechanically complex tumor microenvironments. Nat. Commun. 2021, 12, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Salmon, H.; Franciszkiewicz, K.; Damotte, D.; Dieu-Nosjean, M.C.; Validire, P.; Trautmann, A.; Mami-Chouaib, F.; Donnadieu, E. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J. Clin. Investig. 2012, 122, 899–910. [Google Scholar] [CrossRef] [PubMed]
- Fanfone, D.; Wu, Z.; Mammi, J.; Berthenet, K.; Neves, D.; Weber, K.; Halaburkova, A.; Virard, F.; Bunel, F.; Jamard, C.; et al. Confined migration promotes cancer metastasis through resistance to anoikis and increased invasiveness. Elife 2022, 11, e73150. [Google Scholar] [CrossRef] [PubMed]
- De Cicco, P.; Ercolano, G.; Ianaro, A. The New Era of Cancer Immunotherapy: Targeting Myeloid-Derived Suppressor Cells to Overcome Immune Evasion. Front. Immunol. 2020, 11, 1680. [Google Scholar] [CrossRef] [PubMed]
- Rianna, C.; Radmacher, M.; Kumar, S.; Discher, D. Direct evidence that tumor cells soften when navigating confined spaces. Mol. Biol. Cell 2020, 31, 1726–1734. [Google Scholar] [CrossRef] [PubMed]
- Condeelis, J.; Pollard, J.W. Macrophages: Obligate partners for tumor cell migration, invasion, and metastasis. Cell 2006, 124, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Bufi, N.; Saitakis, M.; Dogniaux, S.; Buschinger, O.; Bohineust, A.; Richert, A.; Maurin, M.; Hivroz, C.; Asnacios, A. Human primary immune cells exhibit distinct mechanical properties that are modified by inflammation. Biophys. J. 2015, 108, 2181–2190. [Google Scholar] [CrossRef] [PubMed]
- Wullkopf, L.; West, A.K.V.; Leijnse, N.; Cox, T.R.; Madsen, C.D.; Oddershede, L.B.; Erler, J.T. Cancer cells’ ability to mechanically adjust to extracellular matrix stiffness correlates with their invasive potential. Mol. Biol. Cell 2018, 29, 2378–2385. [Google Scholar] [CrossRef]
- Dhar, M.; Lam, J.N.; Walser, T.; Dubinett, S.M.; Rettig, M.B.; Di Carlo, D. Functional profiling of circulating tumor cells with an integrated vortex capture and single-cell protease activity assay. Proc. Natl. Acad. Sci. USA 2018, 115, 9986–9991. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef]
- Xiang, H.; Ramil, C.P.; Hai, J.; Zhang, C.; Wang, H.; Watkins, A.A.; Afshar, R.; Georgiev, P.; Sze, M.A.; Song, X.S.; et al. Cancer-associated fibroblasts promote immunosuppression by inducing ROS-generating monocytic MDSCs in lung squamous cell carcinoma. Cancer Immunol. Res. 2020, 8, 436–450. [Google Scholar] [CrossRef] [PubMed]
- Tamura, R.; Tanaka, T.; Akasaki, Y.; Murayama, Y.; Yoshida, K.; Sasaki, H. The role of vascular endothelial growth factor in the hypoxic and immunosuppressive tumor microenvironment: Perspectives for therapeutic implications. Med. Oncol. 2020, 37, 2. [Google Scholar] [CrossRef] [PubMed]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Cañellas, A.; Hernando-Momblona, X.; et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef] [PubMed]
- Deleon-pennell, K.Y.; Barker, T.H.; Lindsey, M.L. Fibroblasts: The arbiters of extracellular matrix remodeling. Matrix Biol. 2020, 91–92, 1–7. [Google Scholar] [CrossRef]
- Tracy, L.E.; Minasian, R.A.; Caterson, E.J. Extracellular Matrix and Dermal Fibroblast Function in the Healing Wound. Adv. Wound Care 2016, 5, 119–136. [Google Scholar] [CrossRef]
- Gubin, M.M.; Zhang, X.; Schuster, H.; Caron, E.; Ward, J.P.; Noguchi, T.; Ivanova, Y.; Hundal, J.; Arthur, C.D.; Krebber, W.J.; et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 2014, 515, 577–581. [Google Scholar] [CrossRef]
- Jang, I.; Beningo, K.A. Integrins, CAFs and mechanical forces in the progression of cancer. Cancers 2019, 11, 721. [Google Scholar] [CrossRef]
- Paszek, M.J.; Zahir, N.; Johnson, K.R.; Lakins, J.N.; Rozenberg, G.I.; Gefen, A.; Reinhart-King, C.A.; Margulies, S.S.; Dembo, M.; Boettiger, D.; et al. Tensional homeostasis and the malignant phenotype. Cancer Cell 2005, 8, 241–254. [Google Scholar] [CrossRef]
- Kim, J.; Feng, J.; Jones, C.A.R.; Mao, X.; Sander, L.M.; Levine, H.; Sun, B. Stress-induced plasticity of dynamic collagen networks. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef]
- Swift, J.; Discher, D.E. The nuclear lamina is mechano-responsive to ECM elasticity in mature tissue. J. Cell Sci. 2014, 127, 3005–3015. [Google Scholar] [CrossRef]
- Northcott, J.M.; Dean, I.S.; Mouw, J.K.; Weaver, V.M. Feeling Stress: The Mechanics of Cancer Progression and Aggression. Front. Cell Dev. Biol. 2018, 6, 17. [Google Scholar] [CrossRef] [PubMed]
- Stylianopoulos, T.; Martin, J.D.; Snuderl, M.; Mpekris, F.; Jain, S.R.; Jain, R.K. Co-evolution of solid stress and interstitial fluid pressure in tumors during progression: Implications for vascular collapse. Cancer Res. 2013, 73, 3833–3841. [Google Scholar] [CrossRef] [PubMed]
- Goetz, J.G.; Minguet, S.; Navarro-Lérida, I.; Lazcano, J.J.; Samaniego, R.; Calvo, E.; Tello, M.; Osteso-Ibáñez, T.; Pellinen, T.; Echarri, A.; et al. Biomechanical remodeling of the microenvironment by stromal caveolin-1 favors tumor invasion and metastasis. Cell 2011, 146, 148–163. [Google Scholar] [CrossRef] [PubMed]
- Lugano, R.; Ramachandran, M.; Dimberg, A. Tumor angiogenesis: Causes, consequences, challenges and opportunities. Cell. Mol. Life Sci. 2020, 77, 1745–1770. [Google Scholar] [CrossRef]
- Zuazo-gaztelu, I.; Casanovas, O. Unraveling the Role of Angiogenesis in Cancer ecosystems. Front. Oncol. 2018, 8, 248. [Google Scholar] [CrossRef]
- Forster, J.C.; Harriss-phillips, W.M.; Douglass, M.J.J.; Bezak, E. A review of the development of tumor vasculature and its effects on the tumor microenvironment. Hypoxia 2017, 5, 21–32. [Google Scholar] [CrossRef]
- Nishida, N.; Yano, H.; Nishida, T.; Kamura, T.; Kojiro, M. Angiogenesis in cancer. Vasc. Health Risk Manag. 2006, 2, 213–219. [Google Scholar] [CrossRef]
- Cheng, G.; Tse, J.; Jain, R.K.; Munn, L.L. Micro-environmental mechanical stress controls tumor spheroid size and morphology by suppressing proliferation and inducing apoptosis in cancer cells. PLoS ONE 2009, 4, e4632. [Google Scholar] [CrossRef]
- Nia, H.T.; Liu, H.; Seano, G.; Datta, M.; Jones, D.; Rahbari, N.; Incio, J.; Chauhan, V.P.; Jung, K.; Martin, J.D.; et al. Solid stress and elastic energy as measures of tumour mechanopathology. Nat. Biomed. Eng. 2016, 1, 0004. [Google Scholar] [CrossRef]
- Hofmann, M.; Guschel, M.; Bernd, A.; Bereiter-Hahn, J.; Kaufmann, R.; Tandi, C.; Helge, W.; Kippenberger, S. Lowering of tumor interstitial fluid pressure reduces tumor cell proliferation in a xenograft tumor model. Neoplasia 2006, 8, 89–95. [Google Scholar] [CrossRef]
- Rofstad, E.K.; Galappathi, K.; Mathiesen, B.S. Tumor Interstitial Fluid Pressure-A Link between Tumor Hypoxia, Microvascular Density, and Lymph Node Metastasis. Neoplasia 2014, 16, 586–594. [Google Scholar] [CrossRef] [PubMed]
- Rens, E.G.; Merks, R.M.H. Cell Contractility Facilitates Alignment of Cells and Tissues to Static Uniaxial Stretch. Biophys. J. 2017, 112, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, P.P.; Eliceiri, K.W.; Campbell, J.M.; Inman, D.R.; White, J.G.; Keely, P.J. Collagen reorganization at the tumor-stromal interface facilitates local invasion. BMC Med. 2006, 4, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Kuczek, D.E.; Larsen, A.M.H.; Thorseth, M.L.; Carretta, M.; Kalvisa, A.; Siersbæk, M.S.; Simões, A.M.C.; Roslind, A.; Engelholm, L.H.; Noessner, E.; et al. Collagen density regulates the activity of tumor-infiltrating T cells. J. Immunother. Cancer 2019, 7, 68. [Google Scholar] [CrossRef]
- Riching, K.M.; Cox, B.L.; Salick, M.R.; Pehlke, C.; Riching, A.S.; Ponik, S.M.; Bass, B.R.; Crone, W.C.; Jiang, Y.; Weaver, A.M.; et al. 3D collagen alignment limits protrusions to enhance breast cancer cell persistence. Biophys. J. 2015, 107, 2546–2558. [Google Scholar] [CrossRef]
- Gocheva, V.; Wang, H.W.; Gadea, B.B.; Shree, T.; Hunter, K.E.; Garfall, A.L.; Berman, T.; Joyce, J.A. IL-4 induces cathepsin protease activity in tumor-associated macrophages to promote cancer growth and invasion. Genes Dev. 2010, 24, 241–255. [Google Scholar] [CrossRef]
- Erdogan, B.; Ao, M.; White, L.M.; Means, A.L.; Brewer, B.M.; Yang, L.; Washington, M.K.; Shi, C.; Franco, O.E.; Weaver, A.M.; et al. Cancer-associated fibroblasts promote directional cancer cell migration by aligning fibronectin. J. Cell Biol. 2017, 216, 3799–3816. [Google Scholar] [CrossRef]
- Koorman, T.; Jansen, K.A.; Khalil, A.; Haughton, P.D.; Visser, D.; Rätze, M.A.K.; Haakma, W.E.; Sakalauskaitè, G.; van Diest, P.J.; de Rooij, J.; et al. Spatial collagen stiffening promotes collective breast cancer cell invasion by reinforcing extracellular matrix alignment. Oncogene 2022. [Google Scholar] [CrossRef]
- Jain, R.K. Delivery of molecular and cellular medicine to solid tumors. Adv. Drug Deliv. Rev. 2012, 64, 353–365. [Google Scholar] [CrossRef]
- Heldin, C.H.; Rubin, K.; Pietras, K.; Östman, A. High interstitial fluid pressure—An obstacle in cancer therapy. Nat. Rev. Cancer 2004, 4, 806–813. [Google Scholar] [CrossRef]
- Provenzano, P.P.; Hingorani, S.R. Hyaluronan, fluid pressure, and stromal resistance in pancreas cancer. Br. J. Cancer 2013, 108, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Libutti, S.K.; Tamarkin, L.; Nilubol, N. Targeting the invincible barrier for drug delivery in solid cancers: Interstitial fluid pressure. Oncotarget 2018, 9, 35723–35725. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.C.; Wu, P.C.; Wang, C.Y.; Kuo, P.L. Evaluation of cytotoxic T lymphocyte-mediated anticancer response against tumor interstitium-simulating physical barriers. Sci. Rep. 2020, 10, 13662. [Google Scholar] [CrossRef] [PubMed]
- Helmlinger, G.; Netti, P.A.; Lichtenbeld, H.C.; Melder, R.J.; Jain, R.K. Solid stress inhibits the growth of multicellular tumor spheroids. Nat. Biotechnol. 1997, 15, 778–783. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Sceneay, J.; Gödde, N.; Kinwel, T.; Ham, S.; Thompson, E.W.; Humbert, P.O.; Möller, A. Intermittent hypoxia induces a metastatic phenotype in breast cancer. Oncogene 2018, 37, 4214–4225. [Google Scholar] [CrossRef] [PubMed]
- Lewis, C.; Murdoch, C. Macrophage responses to hypoxia: Implications for tumor progression and anti-cancer therapies. Am. J. Pathol. 2005, 167, 627–635. [Google Scholar] [CrossRef]
- Winkler, J.; Werb, Z.; Metcalf, K.J. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat. Commun. 2020, 11, 5120. [Google Scholar] [CrossRef]
- Pickup, M.; Novitskiy, S.; Moses, H.L. The roles of TGFβ in the tumour microenvironment. Nat. Rev. Cancer 2013, 13, 788–799. [Google Scholar] [CrossRef]
- Martinez, M.; Moon, E.K. CAR T cells for solid tumors: New strategies for finding, infiltrating, and surviving in the tumor microenvironment. Front. Immunol. 2019, 10, 1–21. [Google Scholar] [CrossRef]
- Gargalionis, A.N.; Basdra, E.K.; Papavassiliou, A.G. Mechanosignalling in tumour progression. J. Cell. Mol. Med. 2018, 22, 704–705. [Google Scholar] [CrossRef]
- Chaudhuri, O.; Koshy, S.T.; Branco Da Cunha, C.; Shin, J.W.; Verbeke, C.S.; Allison, K.H.; Mooney, D.J. Extracellular matrix stiffness and composition jointly regulate the induction of malignant phenotypes in mammary epithelium. Nat. Mater. 2014, 13, 970–978. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Weaver, V.M.; Werb, Z. The extracellular matrix: A dynamic niche in cancer progression. J. Cell Biol. 2012, 196, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Crotti, S.; Piccoli, M.; Rizzolio, F.; Giordano, A.; Nitti, D.; Agostini, M. Extracellular Matrix and Colorectal Cancer: How Surrounding Microenvironment Affects Cancer Cell Behavior? J. Cell. Physiol. 2017, 232, 967–975. [Google Scholar] [CrossRef]
- Moriggi, M.; Giussani, M.; Torretta, E.; Capitanio, D.; Sandri, M.; Leone, R.; De Palma, S.; Vasso, M.; Vozzi, G.; Tagliabue, E.; et al. ECM Remodeling in Breast Cancer with Different Grade: Contribution of 2D-DIGE Proteomics. Proteomics 2018, 18, e1800278. [Google Scholar] [CrossRef] [PubMed]
- Ingber, D.E. Mechanical signaling and the cellular response to extracellular matrix in angiogenesis and cardiovascular physiology. Circ. Res. 2002, 91, 877–887. [Google Scholar] [CrossRef]
- Tuxhorn, J.A.; Mcalhany, S.J.; Dang, T.D.; Ayala, G.E.; Rowley, D.R. Stromal Cells Promote Angiogenesis and Growth of Human Prostate Tumors in a Differential Reactive Stroma ( DRS ) Xenograft Model 1. Cancer Res. 2002, 62, 3298–3307. [Google Scholar]
- Gupta, M.K.; Qin, R. Mechanism and its regulation of tumor-induced angiogenesis. World J. Gastroenterol. WJG 2003, 9, 1144–1155. [Google Scholar] [CrossRef]
- Friedl, P.; Alexander, S. Cancer invasion and the microenvironment: Plasticity and reciprocity. Cell 2011, 147, 992–1009. [Google Scholar] [CrossRef]
- Kim, J.; Zheng, Y.; Alobaidi, A.A.; Nan, H.; Tian, J.; Jiao, Y.; Sun, B. Collective ECM remodeling organizes 3D collective cancer invasion. arXiv 2019, arXiv:1903.03290. [Google Scholar]
- Chaudhuri, P.K.; Low, B.C.; Lim, C.T. Mechanobiology of Tumor Growth. Chem. Rev. 2018, 118, 6499–6515. [Google Scholar] [CrossRef]
- Henke, E.; Nandigama, R.; Ergün, S. Extracellular Matrix in the Tumor Microenvironment and Its Impact on Cancer Therapy. Front. Mol. Biosci. 2020, 6, 160. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Yuan, J.; Righi, E.; Kamoun, W.S.; Ancukiewicz, M.; Nezivar, J.; Santosuosso, M.; Martin, J.D.; Martin, M.R.; Vianello, F.; et al. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc. Natl. Acad. Sci. USA 2012, 109, 17561–17566. [Google Scholar] [CrossRef] [PubMed]
- Whatcott, C.J.; Han, H.; Posner, R.G.; Hostetter, G.; Von Hoff, D.D. Targeting the tumor microenvironment in cancer: Why hyaluronidase deserves a second look. Cancer Discov. 2011, 1, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Venning, F.A.; Wullkopf, L.; Erler, J.T. Targeting ECM disrupts cancer progression. Front. Oncol. 2015, 5, 224. [Google Scholar] [CrossRef]
- Barry-Hamilton, V.; Spangler, R.; Marshall, D.; McCauley, S.; Rodriguez, H.M.; Oyasu, M.; Mikels, A.; Vaysberg, M.; Ghermazien, H.; Wai, C.; et al. Allosteric inhibition of lysyl oxidase-like-2 impedes the development of a pathologic microenvironment. Nat. Med. 2010, 16, 1009–1017. [Google Scholar] [CrossRef]
- Lanitis, E.; Irving, M.; Coukos, G. Targeting the tumor vasculature to enhance T cell activity. Curr. Opin. Immunol. 2015, 33, 55–63. [Google Scholar] [CrossRef]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.B.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217. [Google Scholar] [CrossRef]
- Zboralski, D.; Hoehlig, K.; Eulberg, D.; Frömming, A.; Vater, A. Increasing tumor-infiltrating T cells through inhibition of CXCL12 with NOX-A12 synergizes with PD-1 blockade. Cancer Immunol. Res. 2017, 5, 950–956. [Google Scholar] [CrossRef]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E.; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef]
- Ito, T.; Hanabuchi, S.; Wang, Y.H.; Park, W.R.; Arima, K.; Bover, L.; Qin, F.X.F.; Gilliet, M.; Liu, Y.J. Two Functional Subsets of FOXP3+ Regulatory T Cells in Human Thymus and Periphery. Immunity 2008, 28, 870–880. [Google Scholar] [CrossRef]
- Hombach, A.A.; Heiders, J.; Foppe, M.; Chmielewski, M.; Abken, H. OX40 costimulation by a chimeric antigen receptor abrogates CD28 and IL-2 induced IL-10 secretion by redirected CD4+ T cells. Oncoimmunology 2012, 1, 458–466. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Terai, M.; Tamura, Y.; Alexeev, V.; Mastrangelo, M.J.; Selvan, S.R. Interleukin 10 in the tumor microenvironment: A target for anticancer immunotherapy. Immunol. Res. 2011, 51, 170–182. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jiang, H.; Luo, H.; Sun, Y.; Shi, B.; Sun, R.; Li, Z. An IL-4/21 Inverted Cytokine Receptor Improving CAR-T Cell Potency in Immunosuppressive Solid-Tumor Microenvironment. Front. Immunol. 2019, 10, 1691. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Huang, A.; Nie, J.; Tan, J.; Xing, S.; Qu, Y.; Jiang, K. IL-35 Regulates the Function of Immune Cells in Tumor Microenvironment. Front. Immunol. 2021, 12, 683332. [Google Scholar] [CrossRef]
- Heim, L.; Kachler, K.; Siegmund, R.; Trufa, D.I.; Mittler, S.; Geppert, C.I.; Friedrich, J.; Rieker, R.J.; Sirbu, H.; Finotto, S. Increased expression of the immunosuppressive interleukin-35 in patients with non-small cell lung cancer. Br. J. Cancer 2019, 120, 903–912. [Google Scholar] [CrossRef]
- Morrot, A.; da Fonseca, L.M.; Salustiano, E.J.; Gentile, L.B.; Conde, L.; Filardy, A.A.; Franklim, T.N.; da Costa, K.M.; Freire-de-Lima, C.G.; Freire-de-Lima, L. Metabolic symbiosis and immunomodulation: How tumor cell-derived lactate may disturb innate and adaptive immune responses. Front. Oncol. 2018, 8, 81. [Google Scholar] [CrossRef]
- Vigano, S.; Alatzoglou, D.; Irving, M.; Ménétrier-Caux, C.; Caux, C.; Romero, P.; Coukos, G. Targeting adenosine in cancer immunotherapy to enhance T-Cell function. Front. Immunol. 2019, 10, 925. [Google Scholar] [CrossRef]
- Moon, E.K.; Wang, L.C.S.; Bekdache, K.; Lynn, R.C.; Lo, A.; Thorne, S.H.; Albelda, S.M. Intra-tumoral delivery of CXCL11 via a vaccinia virus, but not by modified T cells, enhances the efficacy of adoptive T cell therapy and vaccines. Oncoimmunology 2018, 7, e1395997. [Google Scholar] [CrossRef]
- Nishio, N.; Diaconu, I.; Liu, H.; Cerullo, V.; Caruana, I.; Hoyos, V.; Bouchier-Hayes, L.; Savoldo, B.; Dotti, G. Armed oncolytic virus enhances immune functions of chimeric antigen receptor-modified T cells in solid tumors. Cancer Res. 2014, 74, 5195–5205. [Google Scholar] [CrossRef]
- McGrath, K.; Dotti, G. Combining oncolytic viruses with chimeric antigen receptor T cell therapy. Hum. Gene Ther. 2021, 32, 150–157. [Google Scholar] [CrossRef]
- Koneru, M.; Purdon, T.J.; Spriggs, D.; Koneru, S.; Brentjens, R.J. IL-12 secreting tumor-targeted chimeric antigen receptor T cells eradicate ovarian tumors in vivo. Oncoimmunology 2015, 4, e994446. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Ren, J.; Luo, Y.; Keith, B.; Young, R.M.; Scholler, J.; Zhao, Y.; June, C.H. Augmentation of Antitumor Immunity by Human and Mouse CAR T Cells Secreting IL-18. Cell Rep. 2017, 20, 3025–3033. [Google Scholar] [CrossRef] [PubMed]
- Shum, T.; Omer, B.; Tashiro, H.; Kruse, R.L.; Wagner, D.L.; Parikh, K.; Yi, Z.; Sauer, T.; Liu, D.; Parihar, R.; et al. Constitutive signaling from an engineered IL7 receptor promotes durable tumor elimination by tumor-redirected T cells. Cancer Discov. 2017, 7, 1238–1247. [Google Scholar] [CrossRef] [PubMed]
- Adachi, K.; Kano, Y.; Nagai, T.; Okuyama, N.; Sakoda, Y.; Tamada, K. IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nat. Biotechnol. 2018, 36, 346–351. [Google Scholar] [CrossRef]
- Kershaw, M.H.; Wang, G.; Westwood, J.A.; Pachynski, R.K.; Tiffany, H.L.; Marincola, F.M.; Wang, E.; Young, H.A.; Murphy, P.M.; Hwu, P. Redirecting migration of T cells to chemokine secreted from tumors by genetic modification with CXCR2. Hum. Gene Ther. 2002, 13, 1971–1980. [Google Scholar] [CrossRef]
- Craddock, J.A.; Lu, A.; Bear, A.; Pule, M.; Brenner, M.K.; Rooney, C.M.; Foster, A.E. Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J. Immunother. 2010, 33, 780–788. [Google Scholar] [CrossRef]
- Moon, E.K.; Carpenito, C.; Sun, J.; Wang, L.C.S.; Kapoor, V.; Predina, J.; Powell, D.J.; Riley, J.L.; June, C.H.; Albelda, S.M. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin. Cancer Res. 2011, 17, 4719–4730. [Google Scholar] [CrossRef]
- Jin, L.; Tao, H.; Karachi, A.; Long, Y.; Hou, A.Y.; Na, M.; Dyson, K.A.; Grippin, A.J.; Deleyrolle, L.P.; Zhang, W.; et al. CXCR1- or CXCR2-modi fi ed CAR T cells co-opt IL- 8 for maximal antitumor ef fi cacy in solid tumors. Nat. Commun. 2019, 10, 4016. [Google Scholar] [CrossRef]
- Jin, L.; Ge, H.; Long, Y.; Yang, C.; Chang, Y.E.; Mu, L.; Sayour, E.J.; De Leon, G.; Wang, Q.J.; Yang, J.C.; et al. CD70, a novel target of CAR T-cell therapy for gliomas. Neuro. Oncol. 2018, 20, 55–65. [Google Scholar] [CrossRef]
- Zhang, L.; Yu, Z.; Muranski, P.; Palmer, D.C.; Restifo, N.P.; Rosenberg, S.A.; Morgan, R.A. Inhibition of TGF-β signaling in genetically engineered tumor antigen-reactive T cells significantly enhances tumor treatment efficacy. Gene Ther. 2013, 20, 575–580. [Google Scholar] [CrossRef]
- Chen, N.; Morello, A.; Tano, Z.; Adusumilli, P.S. CAR T-cell intrinsic PD-1 checkpoint blockade: A two-in-one approach for solid tumor immunotherapy. Oncoimmunology 2017, 6, e1273302. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, S.; Sukumaran, S.; Bajgain, P.; Watanabe, N.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K.; Fisher, W.E.; Leen, A.M.; Vera, J.F. Improving Chimeric Antigen Receptor-Modified T Cell Function by Reversing the Immunosuppressive Tumor Microenvironment of Pancreatic Cancer. Mol. Ther. 2017, 25, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Bajgain, P.; Tawinwung, S.; D’Elia, L.; Sukumaran, S.; Watanabe, N.; Hoyos, V.; Lulla, P.; Brenner, M.K.; Leen, A.M.; Vera, J.F. CAR T cell therapy for breast cancer: Harnessing the tumor milieu to drive T cell activation. J. Immunother. Cancer 2018, 6, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Han, C.; Wang, S.; Fang, P.; Ma, Z.; Xu, L.; Yin, R. Cancer-associated fibroblasts: An emerging target of anti-cancer immunotherapy. J. Hematol. Oncol. 2019, 12, 86. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhou, L.; Li, D.; Andl, T.; Zhang, Y. Cancer-associated fibroblasts build and secure the tumor microenvironment. Front. Cell Dev. Biol. 2019, 7, 60. [Google Scholar] [CrossRef]
- Harryvan, T.J.; Verdegaal, E.M.E.; Hardwick, J.C.H.; Hawinkels, L.J.A.C.; van der Burg, S.H. Targeting of the cancer-associated fibroblast—t-cell axis in solid malignancies. J. Clin. Med. 2019, 8, 1989. [Google Scholar] [CrossRef]
- Lakins, M.A.; Ghorani, E.; Munir, H.; Martins, C.P.; Shields, J.D. Cancer-associated fibroblasts induce antigen-specific deletion of CD8 + T Cells to protect tumour cells. Nat. Commun. 2018, 9, 948. [Google Scholar] [CrossRef]
- Lo, A.; Wang, L.C.S.; Scholler, J.; Monslow, J.; Avery, D.; Newick, K.; O’Brien, S.; Evans, R.A.; Bajor, D.J.; Clendenin, C.; et al. Tumor-promoting desmoplasia is disrupted by depleting FAP-expressing stromal cells. Cancer Res. 2015, 75, 2800–2810. [Google Scholar] [CrossRef]
- Kakarla, S.; Chow, K.K.H.; Mata, M.; Shaffer, D.R.; Song, X.T.; Wu, M.F.; Liu, H.; Wang, L.L.; Rowley, D.R.; Pfizenmaier, K.; et al. Antitumor effects of chimeric receptor engineered human T cells directed to tumor stroma. Mol. Ther. 2013, 21, 1611–1620. [Google Scholar] [CrossRef]
- Özdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef]
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the immune system in cancer: From tumor initiation to metastatic progression. Genes Dev. 2018, 32, 1267–1284. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage biology in development, homeostasis and disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Law, A.M.K.; Valdes-mora, F.; Gallego-ortega, D. Myeloid-Derived Suppressor Cells as a Therapeutic. Cells 2020, 27, 561. [Google Scholar] [CrossRef] [PubMed]
- Pollard, J.W. Trophic macrophages in development and disease. Nat. Rev. Immunol. 2009, 9, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.-Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef]
- Sica, A.; Saccani, A.; Bottazzi, B.; Polentarutti, N.; Vecchi, A.; Van Damme, J.; Mantovani, A. Autocrine Production of IL-10 Mediates Defective IL-12 Production and NF-κB Activation in Tumor-Associated Macrophages. J. Immunol. 2000, 164, 762–767. [Google Scholar] [CrossRef]
- Rodriguez-Garcia, A.; Lynn, R.C.; Poussin, M.; Eiva, M.A.; Shaw, L.C.; O’Connor, R.S.; Minutolo, N.G.; Casado-Medrano, V.; Lopez, G.; Matsuyama, T.; et al. CAR-T cell-mediated depletion of immunosuppressive tumor-associated macrophages promotes endogenous antitumor immunity and augments adoptive immunotherapy. Nat. Commun. 2021, 12, 877. [Google Scholar] [CrossRef]
- Wang, G.; Lu, X.; Dey, P.; Deng, P.; Wu, C.C.; Jiang, S.; Fang, Z.; Zhao, K.; Konaparthi, R.; Hua, S.; et al. Targeting YAP-dependent MDSC infiltration impairs tumor progression. Cancer Discov. 2016, 6, 80–95. [Google Scholar] [CrossRef]
- Alban, T.J.; Bayik, D.; Otvos, B.; Rabljenovic, A.; Leng, L.; Jia-Shiun, L.; Roversi, G.; Lauko, A.; Momin, A.A.; Mohammadi, A.M.; et al. Glioblastoma Myeloid-Derived Suppressor Cell Subsets Express Differential Macrophage Migration Inhibitory Factor Receptor Profiles That Can Be Targeted to Reduce Immune Suppression. Front. Immunol. 2020, 11, 1191. [Google Scholar] [CrossRef]
- Nalawade, S.A.; Shafer, P.; Bajgain, P.; McKenna, M.K.; Ali, A.; Kelly, L.; Joubert, J.; Gottschalk, S.; Watanabe, N.; Leen, A.; et al. Selectively targeting myeloid-derived suppressor cells through TRAIL receptor 2 to enhance the efficacy of CAR T cell therapy for treatment of breast cancer. J. Immunother. Cancer 2021, 9, e003237. [Google Scholar] [CrossRef] [PubMed]
- Shaul, M.E.; Fridlender, Z.G. Tumour-associated neutrophils in patients with cancer. Nat. Rev. Clin. Oncol. 2019, 16, 601–620. [Google Scholar] [CrossRef] [PubMed]
- Togashi, Y.; Shitara, K.; Nishikawa, H. Regulatory T cells in cancer immunosuppression—Implications for anticancer therapy. Nat. Rev. Clin. Oncol. 2019, 16, 356–371. [Google Scholar] [CrossRef]
- Amoozgar, Z.; Kloepper, J.; Ren, J.; Tay, R.E.; Kazer, S.W.; Kiner, E.; Krishnan, S.; Posada, J.M.; Ghosh, M.; Mamessier, E.; et al. Targeting Treg cells with GITR activation alleviates resistance to immunotherapy in murine glioblastomas. Nat. Commun. 2021, 12, 2582. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Reinhard, K.; Rengstl, B.; Oehm, P.; Michel, K.; Billmeier, A.; Hayduk, N.; Klein, O.; Kuna, K.; Ouchan, Y.; Wöll, S.; et al. An RNA vaccine drives expansion and efficacy of claudin-CAR-T cells against solid tumors. Science 2020, 367, 446–453. [Google Scholar] [CrossRef]
- Sadjadi, Z.; Zhao, R.; Hoth, M.; Qu, B.; Rieger, H. Migration of Cytotoxic T Lymphocytes in 3D Collagen Matrices. Biophys. J. 2020, 119, 2141–2152. [Google Scholar] [CrossRef]
- Wolf, K.; Müller, R.; Borgmann, S.; Bröcker, E.B.; Friedl, P. Amoeboid shape change and contact guidance: T-lymphocyte crawling through fibrillar collagen is independent of matrix remodeling by MMPs and other proteases. Blood 2003, 102, 3262–3269. [Google Scholar] [CrossRef]
- Jacobelli, J.; Friedman, R.S.; Conti, M.A.; Lennon-Dumenil, A.M.; Piel, M.; Sorensen, C.M.; Adelstein, R.S.; Krummel, M.F. Confinement-optimized three-dimensional T cell amoeboid motility is modulated via myosin IIA-regulated adhesions. Nat. Immunol. 2010, 11, 953–961. [Google Scholar] [CrossRef]
- Kawauchi, T. Cell adhesion and its endocytic regulation in cell migration during neural development and cancer metastasis. Int. J. Mol. Sci. 2012, 13, 4564–4590. [Google Scholar] [CrossRef]
- Petrie, R.J.; Harlin, H.M.; Korsak, L.I.T.; Yamada, K.M. Activating the nuclear piston mechanism of 3D migration in tumor cells. J. Cell Biol. 2017, 216, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Ruprecht, V.; Wieser, S.; Callan-Jones, A.; Smutny, M.; Morita, H.; Sako, K.; Barone, V.; Ritsch-Marte, M.; Sixt, M.; Voituriez, R.; et al. Cortical contractility triggers a stochastic switch to fast amoeboid cell motility. Cell 2015, 160, 673–685. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.M.; Sixt, M. Mechanisms of 3D cell migration. Nat. Rev. Mol. Cell Biol. 2019, 20, 738–752. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.J.; Wei, S.H.; Parker, I.; Cahalan, M.D. Two-photon imaging of lymphocyte motility and antigen response in intact lymph node. Science 2002, 296, 1869–1873. [Google Scholar] [CrossRef]
- Friedl, P.; Entschladen, F.; Conrad, C.; Niggemann, B.; Zänker, K.S. CD4+ T lymphocytes migrating in three-dimensional collagen lattices lack focal adhesions and utilize β1 integrin-independent strategies for polarization, interaction with collagen fibers and locomotion. Eur. J. Immunol. 1998, 28, 2331–2343. [Google Scholar] [CrossRef]
- Paluch, E.K.; Aspalter, I.M.; Sixt, M. Focal Adhesion-Independent Cell Migration. Annu. Rev. Cell Dev. Biol. 2016, 32, 469–490. [Google Scholar] [CrossRef]
- Lämmermann, T.; Bader, B.L.; Monkley, S.J.; Worbs, T.; Wedlich-Söldner, R.; Hirsch, K.; Keller, M.; Förster, R.; Critchley, D.R.; Fässler, R.; et al. Rapid leukocyte migration by integrin-independent flowing and squeezing. Nature 2008, 453, 51–55. [Google Scholar] [CrossRef]
- Goudarzi, M.; Banisch, T.U.; Mobin, M.B.; Maghelli, N.; Tarbashevich, K.; Strate, I.; van den Berg, J.; Blaser, H.; Bandemer, S.; Paluch, E.; et al. Identification and Regulation of a Molecular Module for Bleb-Based Cell Motility. Dev. Cell 2012, 23, 210–218. [Google Scholar] [CrossRef]
- Wu, S.K.; Priya, R. Spatio-temporal regulation of RhoGTPases signaling by myosin II. Front. Cell Dev. Biol. 2019, 7, 90. [Google Scholar] [CrossRef]
- Wolf, K.; te Lindert, M.; Krause, M.; Alexander, S.; te Riet, J.; Willis, A.L.; Hoffman, R.M.; Figdor, C.G.; Weiss, S.J.; Friedl, P. Physical limits of cell migration: Control by ECM space and nuclear deformation and tuning by proteolysis and traction force. J. Cell Biol. 2013, 201, 1069–1084. [Google Scholar] [CrossRef]
- Lele, T.P.; Dickinson, R.B.; Gundersen, G.G. Mechanical principles of nuclear shaping and positioning. J. Cell Biol. 2018, 217, 3330–3342. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Pfeifer, C.R.; Discher, D.E. Nuclear mechanics during and after constricted migration. Acta Mech. Sin. Xuebao 2019, 35, 299–308. [Google Scholar] [CrossRef]
- Swift, J.; Ivanovska, I.L.; Buxboim, A.; Harada, T.; Dingal, P.C.D.P.; Pinter, J.; Pajerowski, J.D.; Spinler, K.R.; Shin, J.W.; Tewari, M.; et al. Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science 2013, 341, 1240104. [Google Scholar] [CrossRef] [PubMed]
- Harada, T.; Swift, J.; Irianto, J.; Shin, J.W.; Spinler, K.R.; Athirasala, A.; Diegmiller, R.; Dingal, P.C.D.P.; Ivanovska, I.L.; Discher, D.E. Nuclear lamin stiffness is a barrier to 3D migration, but softness can limit survival. J. Cell Biol. 2014, 204, 669–682. [Google Scholar] [CrossRef]
- Stecklein, S.R.; Kumaraswamy, E.; Behbod, F.; Wang, W.; Chaguturu, V.; Harlan-Williams, L.M.; Jensen, R.A. BRCA1 and HSP90 cooperate in homologous and non-homologous DNA double-strand-break repair and G2/M checkpoint activation. Proc. Natl. Acad. Sci. USA 2012, 109, 13650–13655. [Google Scholar] [CrossRef]
- Stephens, A.D.; Banigan, E.J.; Adam, S.A.; Goldman, R.D.; Marko, J.F. Chromatin and lamin A determine two different mechanical response regimes of the cell nucleus. Mol. Biol. Cell 2017, 28, 1984–1996. [Google Scholar] [CrossRef]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef]
- Lambert, L.H.; Goebrecht, G.K.E.; De Leo, S.E.; O’Connor, R.S.; Nunez-Cruz, S.; De Li, T.; Yuan, J.; Milone, M.C.; Kam, L.C. Improving T Cell Expansion with a Soft Touch. Nano Lett. 2017, 17, 821–826. [Google Scholar] [CrossRef]
- Bashoura, K.T.; Gondarenko, A.; Chen, H.; Shen, K.; Liu, X.; Huse, M.; Hone, J.C.; Kam, L.C. CD28 and CD3 have complementary roles in T-cell traction forces. Proc. Natl. Acad. Sci. USA 2014, 111, 2241–2246. [Google Scholar] [CrossRef]
- Kim, S.T.; Takeuchi, K.; Sun, Z.Y.J.; Touma, M.; Castro, C.E.; Fahmy, A.; Lang, M.J.; Wagner, G.; Reinherz, E.L. The αβ T cell receptor is an anisotropic mechanosensor. J. Biol. Chem. 2009, 284, 31028–31037. [Google Scholar] [CrossRef]
- Basu, R.; Whitlock, B.M.; Husson, J.; Le Floc’h, A.; Jin, W.; Oyler-Yaniv, A.; Dotiwala, F.; Giannone, G.; Hivroz, C.; Biais, N.; et al. Cytotoxic T cells use mechanical force to potentiate target cell killing. Cell 2016, 165, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Buxboim, A.; Swift, J.; Irianto, J.; Spinler, K.R.; Dingal, P.C.D.P.; Athirasala, A.; Kao, Y.R.C.; Cho, S.; Harada, T.; Shin, J.W.; et al. Matrix elasticity regulates lamin-A,C phosphorylation and turnover with feedback to actomyosin. Curr. Biol. 2014, 24, 1909–1917. [Google Scholar] [CrossRef] [PubMed]
- Judokusumo, E.; Tabdanov, E.; Kumari, S.; Dustin, M.L.; Kam, L.C. Mechanosensing in T lymphocyte activation. Biophys. J. 2012, 102, L5–L7. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.; Tamzalit, F.; Chaudhuri, P.K.; Black, C.T.; Huse, M.; Kam, L.C. T cell activation and immune synapse organization respond to the microscale mechanics of structured surfaces. Proc. Natl. Acad. Sci. USA 2019, 116, 19835–19840. [Google Scholar] [CrossRef]
- Pruitt, H.C.; Lewis, D.; Ciccaglione, M.; Connor, S.; Smith, Q.; Hickey, J.W.; Schneck, J.P.; Gerecht, S. Collagen fiber structure guides 3D motility of cytotoxic T lymphocytes. Matrix Biol. 2020, 85–86, 147–159. [Google Scholar] [CrossRef]
- Lanitis, E.; Kosti, P.; Ronet, C.; Cribioli, E.; Rota, G.; Spill, A.; Reichenbach, P.; Zoete, V.; Dangaj Laniti, D.; Coukos, G.; et al. VEGFR-2 redirected CAR-T cells are functionally impaired by soluble VEGF-A competition for receptor binding. J. Immunother. Cancer 2021, 9, e002151. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, L.; Su, H.F.; Liu, Q.; Shen, J.; Dai, H.; Zheng, W.; Lu, Y.; Zhang, W.; Bei, Y.; et al. Chimeric antigen receptor macrophage therapy for breast tumours mediated by targeting the tumour extracellular matrix. Br. J. Cancer 2019, 121, 837–845. [Google Scholar] [CrossRef]
- Tran, E.; Chinnasamy, D.; Yu, Z.; Morgan, R.A.; Lee, C.C.R.; Restifo, N.P.; Rosenberg, S.A. Immune targeting of fibroblast activation protein triggers recognition of multipotent bone marrow stromal cells and cachexia. J. Exp. Med. 2013, 210, 1065–1068. [Google Scholar] [CrossRef]
- Liu, G.; Rui, W.; Zheng, H.; Huang, D.; Yu, F.; Zhang, Y.; Dong, J.; Zhao, X.; Lin, X. CXCR2-modified CAR-T cells have enhanced trafficking ability that improves treatment of hepatocellular carcinoma. Eur. J. Immunol. 2020, 50, 712–724. [Google Scholar] [CrossRef]
- Berahovich, R.; Liu, X.; Zhou, H.; Tsadik, E.; Xu, S.; Golubovskaya, V.; Wu, L. Hypoxia selectively impairs CAR-T cells in vitro. Cancers 2019, 11, 602. [Google Scholar] [CrossRef]
- Liao, Q.; He, H.; Mao, Y.; Ding, X.; Zhang, X.; Xu, J. Engineering T cells with hypoxia-inducible chimeric antigen receptor (HiCAR) for selective tumor killing. Biomark. Res. 2020, 8, 56. [Google Scholar] [CrossRef] [PubMed]
- Kersten, K.; Visser, K.E.; Miltenburg, M.H.; Jonkers, J. Genetically engineered mouse models in oncology research and cancer medicine. EMBO Mol. Med. 2017, 9, 137–153. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Yu, X.Z. Modelling CAR-T therapy in humanized mice. EBioMedicine 2019, 40, 25–26. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.H.; Xia, J.; Rafiq, S.; Huang, X.; Hu, Z.; Zhou, X.; Brentjens, R.J.; Yang, Y.G. Modeling anti-CD19 CAR T cell therapy in humanized mice with human immunity and autologous leukemia. EBioMedicine 2019, 39, 173–181. [Google Scholar] [CrossRef]
- Drake, A.C.; Chen, Q.; Chen, J. Engineering humanized mice for improved hematopoietic reconstitution. Cell. Mol. Immunol. 2012, 9, 215–224. [Google Scholar] [CrossRef]
- Holzapfel, B.M.; Wagner, F.; Thibaudeau, L.; Levesque, J.P.; Hutmacher, D.W. Concise review: Humanized models of tumor immunology in the 21st century: Convergence of cancer research and tissue engineering. Stem Cells 2015, 33, 1696–1704. [Google Scholar] [CrossRef]
- Wang, M.; Yao, L.C.; Cheng, M.; Cai, D.; Martinek, J.; Pan, C.X.; Shi, W.; Ma, A.H.; De Vere White, R.W.; Airhart, S.; et al. Humanized mice in studying efficacy and mechanisms of PD-1-targeted cancer immunotherapy. FASEB J. 2018, 32, 1537–1549. [Google Scholar] [CrossRef]
- Gitto, S.B.; Kim, H.; Rafail, S.; Omran, D.K.; Medvedev, S.; Kinose, Y.; Rodriguez-Garcia, A.; Flowers, A.J.; Xu, H.; Schwartz, L.E.; et al. An autologous humanized patient-derived-xenograft platform to evaluate immunotherapy in ovarian cancer. Gynecol. Oncol. 2020, 156, 222–232. [Google Scholar] [CrossRef]
- Bareham, B.; Georgakopoulos, N.; Matas-Céspedes, A.; Curran, M.; Saeb-Parsy, K. Modeling human tumor-immune environments in vivo for the preclinical assessment of immunotherapies. Cancer Immunol. Immunother. 2021, 70, 2737–2750. [Google Scholar] [CrossRef]
- Lang, G.A.; Iwakuma, T.; Suh, Y.A.; Liu, G.; Rao, V.A.; Parant, J.M.; Valentin-Vega, Y.A.; Terzian, T.; Caldwell, L.C.; Strong, L.C.; et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell 2004, 119, 861–872. [Google Scholar] [CrossRef]
- Olive, K.P.; Tuveson, D.A.; Ruhe, Z.C.; Yin, B.; Willis, N.A.; Bronson, R.T.; Crowley, D.; Jacks, T. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell 2004, 119, 847–860. [Google Scholar] [CrossRef] [PubMed]
- Drost, R.; Bouwman, P.; Rottenberg, S.; Boon, U.; Schut, E.; Klarenbeek, S.; Klijn, C.; van der Heijden, I.; van der Gulden, H.; Wientjens, E.; et al. BRCA1 RING function is essential for tumor suppression but dispensable for therapy resistance. Cancer Cell 2011, 20, 797–809. [Google Scholar] [CrossRef] [PubMed]
- Drost, R.; Dhillon, K.K.; Van Der Gulden, H.; Van Der Heijden, I.; Brandsma, I.; Cruz, C.; Chondronasiou, D.; Castroviejo-Bermejo, M.; Boon, U.; Schut, E.; et al. BRCA1185delAG tumors may acquire therapy resistance through expression of RING-less BRCA1. J. Clin. Investig. 2016, 126, 2903–2918. [Google Scholar] [CrossRef] [PubMed]
- Willimsky, G.; Blankenstein, T. Sporadic immunogenic tumours avoid destruction by inducing T-cell tolerance. Nature 2005, 437, 141–146. [Google Scholar] [CrossRef]
- Garbe, A.I.; Vermeer, B.; Gamrekelashvili, J.; Von Wasielewski, R.; Greten, F.R.; Westendorf, A.M.; Buer, J.; Schmid, R.M.; Manns, M.P.; Korangy, F.; et al. Genetically induced pancreatic adenocarcinoma is highly immunogenic and causes spontaneous tumor-specific immune responses. Cancer Res. 2006, 66, 508–516. [Google Scholar] [CrossRef]
- DuPage, M.; Cheung, A.F.; Mazumdar, C.; Winslow, M.M.; Bronson, R.; Schmidt, L.M.; Crowley, D.; Chen, J.; Jacks, T. Endogenous T cell responses to antigens expressed in lung adenocarcinomas delay malignant tumor progression. Cancer Cell 2011, 19, 72–85. [Google Scholar] [CrossRef]
- Rivera-Rodriguez, A.; Hoang-Minh, L.B.; Chiu-Lam, A.; Sarna, N.; Marrero-Morales, L.; Mitchell, D.A.; Rinaldi-Ramos, C.M. Tracking adoptive t cell immunotherapy using magnetic particle imaging. Nanotheranostics 2021, 5, 431–444. [Google Scholar] [CrossRef]
- Cazaux, M.; Grandjean, C.L.; Lemaître, F.; Garcia, Z.; Beck, R.J.; Milo, I.; Postat, J.; Beltman, J.B.; Cheadle, E.J.; Bousso, P. Single-cell imaging of CAR T cell activity in vivo reveals extensive functional and anatomical heterogeneity. J. Exp. Med. 2019, 216, 1038–1049. [Google Scholar] [CrossRef]
- Zhang, S.; Black, R.G.; Kohli, K.; Hayes, B.J.; Miller, C.; Koehne, A.; Schroeder, B.A.; Abrams, K.; Schulte, B.C.; Alexiev, B.A.; et al. B7-H3 Specific CAR T Cells for the Naturally Occurring, Spontaneous Canine Sarcoma Model. Mol. Cancer Ther. 2022, 21, 999–1009. [Google Scholar] [CrossRef]
- Schachtschneider, K.M.; Schwind, R.M.; Newson, J.; Kinachtchouk, N.; Rizko, M.; Mendoza-Elias, N.; Grippo, P.; Principe, D.R.; Park, A.; Overgaard, N.H.; et al. The oncopig cancer model: An innovative large animal translational oncology platform. Front. Oncol. 2017, 7, 190. [Google Scholar] [CrossRef]
- Schook, L.B.; Collares, T.V.; Hu, W.; Liang, Y.; Rodrigues, F.M.; Rund, L.A.; Schachtschneider, K.M.; Seixas, F.K.; Singh, K.; Wells, K.D.; et al. A genetic porcine model of cancer. PLoS ONE 2015, 10, e0128864. [Google Scholar] [CrossRef] [PubMed]
- Overgaard, N.H.; Frøsig, T.M.; Welner, S.; Rasmussen, M.; Ilsøe, M.; Sørensen, M.R.; Andersen, M.H.; Buus, S.; Jungersen, G. Establishing the pig as a large animal model for vaccine development against human cancer. Front. Genet. 2015, 6, 286. [Google Scholar] [CrossRef] [PubMed]
- Mair, K.H.; Sedlak, C.; Käser, T.; Pasternak, A.; Levast, B.; Gerner, W.; Saalmüller, A.; Summerfield, A.; Gerdts, V.; Wilson, H.L.; et al. The porcine innate immune system: An update. Dev. Comp. Immunol. 2014, 45, 321–343. [Google Scholar] [CrossRef] [PubMed]
- Darfour-Oduro, K.A.; Megens, H.J.; Roca, A.L.; Groenen, M.A.M.; Schook, L.B. Adaptive evolution of Toll-like receptors (TLRs) in the family Suidae. PLoS ONE 2015, 10, e0124069. [Google Scholar] [CrossRef] [PubMed]
- Darfour-Oduro, K.A.; Megens, H.J.; Roca, A.L.; Groenen, M.A.M.; Schook, L.B. Evolutionary patterns of Toll-like receptor signaling pathway genes in the Suidae. BMC Evol. Biol. 2016, 16, 33. [Google Scholar] [CrossRef]
- Darfour-Oduro, K.A.; Megens, H.J.; Roca, A.; Groenen, M.A.M.; Schook, L.B. Evidence for adaptation of porcine Toll-like receptors. Immunogenetics 2016, 68, 179–189. [Google Scholar] [CrossRef][Green Version]
- Ogando-Rivas, E.; Castillo, P.; Jones, N.; Trivedi, V.; Drake, J.; Dechkovskaia, A.; Candelario, K.M.; Yang, C.; Mitchell, D.A. Effects of immune checkpoint blockade on antigen-specific CD8+ T cells for use in adoptive cellular therapy. Microbiol. Immunol. 2022, 66, 201–211. [Google Scholar] [CrossRef]
- Anton, D.; Burckel, H.; Josset, E.; Noel, G. Three-dimensional cell culture: A breakthrough in vivo. Int. J. Mol. Sci. 2015, 16, 5517–5527. [Google Scholar] [CrossRef]
- Kaczmarczyk, J.A.; Roberts, R.R.; Luke, B.T.; Chan, K.C.; van Wagoner, C.M.; Felder, R.A.; Saul, R.G.; Simona, C.; Blonder, J. Comparative microsomal proteomics of a model lung cancer cell line NCI-H23 reveals distinct differences between molecular profiles of 3D and 2D cultured cells. Oncotarget 2021, 12, 2022–2038. [Google Scholar] [CrossRef]
- Tagle, D.A. The NIH microphysiological systems program: Developing in vitro tools for safety and efficacy in drug development. Curr. Opin. Pharmacol. 2019, 48, 146–154. [Google Scholar] [CrossRef]
- Zips, D.; Kunz-schughart, L.A.; Cordes, N. EGFR / JIP-4 / JNK2 Signaling Attenuates Cetuximab- Mediated Radiosensitization of Squamous Cell Carcinoma. Cancer Res. 2021, 73, 297–307. [Google Scholar] [CrossRef]
- Jensen, C.; Teng, Y. Is It Time to Start Transitioning From 2D to 3D Cell Culture? Front. Mol. Biosci. 2020, 7, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Longati, P.; Jia, X.; Eimer, J.; Wagman, A.; Witt, M.R.; Rehnmark, S.; Verbeke, C.; Toftgård, R.; Löhr, M.; Heuchel, R.L. 3D pancreatic carcinoma spheroids induce a matrix-rich, chemoresistant phenotype offering a better model for drug testing. BMC Cancer 2013, 13, 95. [Google Scholar] [CrossRef] [PubMed]
- Enzerink, A.; Salmenperä, P.; Kankuri, E.; Vaheri, A. Clustering of fibroblasts induces proinflammatory chemokine secretion promoting leukocyte migration. Mol. Immunol. 2009, 46, 1787–1795. [Google Scholar] [CrossRef]
- Sureban, S.M.; Berahovich, R.; Zhou, H.; Xu, S.; Wu, L.; Ding, K.; May, R.; Qu, D.; Bannerman-menson, E.; Golubovskaya, V.; et al. DCLK1 Monoclonal Antibody-Based CAR-T Cells as a Novel Treatment Strategy against Human Colorectal Cancers. Cancers 2020, 12, 54. [Google Scholar] [CrossRef]
- Dillard, P.; Köksal, H.; Inderberg, E.M.; Wälchli, S. A spheroid killing assay by CAR T cells. J. Vis. Exp. 2018, 2018, 1–7. [Google Scholar] [CrossRef]
- Wallstabe, L.; Göttlich, C.; Nelke, L.C.; Kühnemundt, J.; Schwarz, T.; Nerreter, T.; Einsele, H.; Walles, H.; Dandekar, G.; Nietzer, S.L.; et al. ROR1-CAR T cells are effective against lung and breast cancer in advanced microphysiologic 3D tumor models. JCI Insight 2019, 4, 1–14. [Google Scholar] [CrossRef]
- Ando, Y.; Siegler, E.L.; Ta, H.P.; Cinay, G.E.; Zhou, H.; Gorrell, K.A.; Au, H.; Jarvis, B.M.; Wang, P.; Shen, K. Evaluating CAR-T Cell Therapy in a Hypoxic 3D Tumor Model. Adv. Healthc. Mater. 2019, 8, 1–15. [Google Scholar] [CrossRef]
- Schnalzger, T.E.; Groot, M.H.; Zhang, C.; Mosa, M.H.; Michels, B.E.; Röder, J.; Darvishi, T.; Wels, W.S.; Farin, H.F. 3D model for CAR -mediated cytotoxicity using patient-derived colorectal cancer organoids. EMBO J. 2019, 38, e100928. [Google Scholar] [CrossRef]
- Jacob, F.; Ming, G.-L.; Song, H. Generation and biobanking of patient-derived glioblastoma organoids and their application in CAR T cell testing. Nat. Protoc. 2020, 15, 4000–4033. [Google Scholar] [CrossRef]
- Powley, I.R.; Patel, M.; Miles, G.; Pringle, H.; Howells, L.; Thomas, A.; Kettleborough, C.; Bryans, J.; Hammonds, T.; MacFarlane, M.; et al. Patient-derived explants (PDEs) as a powerful preclinical platform for anti-cancer drug and biomarker discovery. Br. J. Cancer 2020, 122, 735–744. [Google Scholar] [CrossRef] [PubMed]
- Kleinman, H.K.; Martin, G.R. Matrigel: Basement membrane matrix with biological activity. Semin. Cancer Biol. 2005, 15, 378–386. [Google Scholar] [CrossRef] [PubMed]
- Aisenbrey, E.A.; Murphy, W.L. Synthetic alternatives to Matrigel. Nat. Rev. Mater. 2020, 5, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Caliari, S.R.; Burdick, J.A. A practical guide to hydrogels for cell culture. Nat. Methods 2016, 13, 405–414. [Google Scholar] [CrossRef]
- Mourran, A.; Wu, Y.; Gumerov, R.A.; Rudov, A.A.; Potemkin, I.I.; Pich, A.; Möller, M. When Colloidal Particles Become Polymer Coils. Langmuir 2016, 32, 723–730. [Google Scholar] [CrossRef]
- Plamper, F.A.; Richtering, W. Functional Microgels and Microgel Systems. Acc. Chem. Res. 2017, 50, 131–140. [Google Scholar] [CrossRef]
- Agrawal, G.; Agrawal, R. Stimuli-responsive microgels and microgel-based systems: Advances in the exploitation of microgel colloidal properties and their interfacial activity. Polymers 2018, 10, 418. [Google Scholar] [CrossRef]
- Lee, J.; Kim, S.E.; Moon, D.; Doh, J. A multilayered blood vessel/tumor tissue chip to investigate T cell infiltration into solid tumor tissues. Lab Chip 2021, 21, 2142–2152. [Google Scholar] [CrossRef]
- Hughes, C.S.; Postovit, L.M.; Lajoie, G.A. Matrigel: A complex protein mixture required for optimal growth of cell culture. Proteomics 2010, 10, 1886–1890. [Google Scholar] [CrossRef]
- Shin, J.-W.; Discher, D.E. Soft gels select tumorigenic cells. Nat. Mater. 2012, 11, 662–663. [Google Scholar] [CrossRef]
- Janmey, P.A.; Winer, J.P.; Weisel, J.W. Fibrin gels and their clinical and bioengineering applications. J. R. Soc. Interface 2009, 6, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.Y.; Mooney, D.J. Alginate: Properties and biomedical applications. Prog. Polym. Sci. 2012, 37, 106–126. [Google Scholar] [CrossRef] [PubMed]
- Vining, K.H.; Mooney, D.J. Mechanical forces direct stem cell behaviour in development and regeneration. Nat. Rev. Mol. Cell Biol. 2017, 18, 728–742. [Google Scholar] [CrossRef] [PubMed]
- Pelham, R.J.; Wang, Y.L. Cell locomotion and focal adhesions are regulated by substrate flexibility. Proc. Natl. Acad. Sci. USA 1997, 94, 13661–13665. [Google Scholar] [CrossRef] [PubMed]
- Burdick, J.A.; Anseth, K.S. Photoencapsulation of osteoblasts in injectable RGD-modified PEG hydrogels for bone tissue engineering. Biomaterials 2002, 23, 4315–4323. [Google Scholar] [CrossRef]
- Acerbi, I.; Cassereau, L.; Dean, I.; Shi, Q.; Au, A.; Park, C.; Chen, Y.Y.; Liphardt, J.; Hwang, E.S.; Weaver, V.M. Human breast cancer invasion and aggression correlates with ECM stiffening and immune cell infiltration. Integr. Biol. 2015, 7, 1120–1134. [Google Scholar] [CrossRef]
- Pedro, D.I.; Nguyen, D.T.; Trachsel, L.; Rosa, J.G.; Chu, B.; Eikenberry, S.; Sumerlin, B.S.; Sawyer, W.G. Superficial Modulus, Water-Content, and Mesh-Size at Hydrogel Surfaces. Tribol. Lett. 2021, 69, 1–9. [Google Scholar] [CrossRef]
- Appel, E.A.; del Barrio, J.; Loh, X.J.; Scherman, O.A. Supramolecular polymeric hydrogels. Chem. Soc. Rev. 2012, 41, 6195–6214. [Google Scholar] [CrossRef]
- Lin, C.C.; Anseth, K.S. PEG hydrogels for the controlled release of biomolecules in regenerative medicine. Pharm. Res. 2009, 26, 631–643. [Google Scholar] [CrossRef]
- Kozlowski, M.T.; Crook, C.J.; Ku, H.T. Towards organoid culture without Matrigel. Commun. Biol. 2021, 4, 1387. [Google Scholar] [CrossRef]
- Vukicevic, S.; Kleinman, H.K.; Luyten, F.P.; Roberts, A.B.; Roche, N.S.; Reddi, A.H. Identification of multiple active growth factors in basement membrane matrigel suggests caution in interpretation of cellular activity related to extracellular matrix components. Exp. Cell Res. 1992, 202, 1–8. [Google Scholar] [CrossRef]
- Talbot, N.C.; Caperna, T.J. Proteome array identification of bioactive soluble proteins/peptides in Matrigel: Relevance to stem cell responses. Cytotechnology 2015, 67, 873–883. [Google Scholar] [CrossRef] [PubMed]
- Tse, J.R.; Engler, A.J. Preparation of hydrogel substrates with tunable mechanical properties. Curr. Protoc. Cell Biol. 2010, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Griffin, D.R.; Weaver, W.M.; Scumpia, P.; Di Carlo, D.; Segura, T. Scaffolds Assembled From Annealed Building Blocks. Nat. Mater. 2015, 14, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Lutolf, M.P.; Lauer-Fields, J.L.; Schmoekel, H.G.; Metters, A.T.; Weber, F.E.; Fields, G.B.; Hubbell, J.A. Synthetic matrix metalloproteinase-sensitive hydrogels for the conduction of tissue regeneration: Engineering cell-invasion characteristics. Proc. Natl. Acad. Sci. USA 2003, 100, 5413–5418. [Google Scholar] [CrossRef] [PubMed]
- Girardo, S.; Träber, N.; Wagner, K.; Cojoc, G.; Herold, C.; Goswami, R.; Schlüßler, R.; Abuhattum, S.; Taubenberger, A.; Reichel, F.; et al. Standardized microgel beads as elastic cell mechanical probes. J. Mater. Chem. B 2018, 6, 6245–6261. [Google Scholar] [CrossRef]
- Sideris, E.; Griffin, D.R.; Ding, Y.; Li, S.; Weaver, W.M.; Di Carlo, D.; Hsiai, T.; Segura, T. Particle Hydrogels Based on Hyaluronic Acid Building Blocks. ACS Biomater. Sci. Eng. 2016, 2, 2034–2041. [Google Scholar] [CrossRef]
- Sheikhi, A.; de Rutte, J.; Haghniaz, R.; Akouissi, O.; Sohrabi, A.; Di Carlo, D.; Khademhosseini, A. Microfluidic-enabled bottom-up hydrogels from annealable naturally-derived protein microbeads. Biomaterials 2019, 192, 560–568. [Google Scholar] [CrossRef]
- Malmsten, M.; Bysell, H.; Hansson, P. Biomacromolecules in microgels—Opportunities and challenges for drug delivery. Curr. Opin. Colloid Interface Sci. 2010, 15, 435–444. [Google Scholar] [CrossRef]
- Oh, J.K.; Drumright, R.; Siegwart, D.J.; Matyjaszewski, K. The development of microgels/nanogels for drug delivery applications. Prog. Polym. Sci. 2008, 33, 448–477. [Google Scholar] [CrossRef]
- Wagner, K.; Girardo, S.; Goswami, R.; Rosso, G.; Ulbricht, E.; Müller, P.; Soteriou, D.; Träber, N.; Guck, J. Colloidal crystals of compliant microgel beads to study cell migration and mechanosensitivity in 3D. Soft Matter 2019, 15, 9776–9787. [Google Scholar] [CrossRef] [PubMed]
- Foty, R. A simple hanging drop cell culture protocol for generation of 3D spheroids. J. Vis. Exp. 2011, 20, 4–7. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, S.; Mehta, P.; Horst, E.N.; Ward, M.R.; Rowley, K.R.; Mehta, G. Comparative analysis of tumor spheroid generation techniques for differential in vitro drug toxicity. Oncotarget 2016, 7, 16948–16961. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.Z.; Chang, H.Y. Recent advances in three-dimensional multicellular spheroid culture for biomedical research. Biotechnol. J. 2008, 3, 1172–1184. [Google Scholar] [CrossRef]
- Grunewald, L.; Lam, T.; Andersch, L.; Klaus, A.; Schwiebert, S.; Winkler, A.; Gauert, A.; Heeren-Hagemann, A.I.; Astrahantseff, K.; Klironomos, F.; et al. A Reproducible Bioprinted 3D Tumor Model Serves as a Preselection Tool for CAR T Cell Therapy Optimization. Front. Immunol. 2021, 12, 689697. [Google Scholar] [CrossRef]
- Gunti, S.; Hoke, A.T.K.; Vu, K.P.; London, N.R. Organoid and spheroid tumor models: Techniques and applications. Cancers 2021, 13, 874. [Google Scholar] [CrossRef]
- Clevers, H.; Tuveson, D.A. Organoid models for cancer research. Annu. Rev. Cancer Biol. 2019, 3, 223–234. [Google Scholar] [CrossRef]
- Freeman, A.E.; Hoffman, R.M. In vivo-like growth of human tumors in vitro. Proc. Natl. Acad. Sci. USA 1986, 83, 2694–2698. [Google Scholar] [CrossRef]
- Nguyen, D.T.; Famiglietti, J.E.; Smolchek, R.A.; Dupee, Z.; Diodati, N.; Pedro, D.I.; Urueña, J.M.; Schaller, M.A.; Sawyer, W.G. 3D In Vitro Platform for Cell and Explant Culture in Liquid-like Solids. Cells 2022, 11, 967. [Google Scholar] [CrossRef]
- He, C.; Xu, K.; Zhu, X.; Dunphy, P.S.; Gudenas, B.; Lin, W.; Twarog, N.; Hover, L.D.; Kwon, C.H.; Kasper, L.H.; et al. Patient-derived models recapitulate heterogeneity of molecular signatures and drug response in pediatric high-grade glioma. Nat. Commun. 2021, 12, 4089. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Reference | Tumor | TME | Appproach | Outcome(s) |
|---|---|---|---|---|
| [196] | B16 melanoma | Vasculature | VEFGR-2 CAR T cells in combination with anti-VEFG-A ligand antibody | Combination of anti-VEFG-A ligand to overcome competition enhances CAR T cell activity |
| [197] | Murine breast carcinoma | ECM | Engineered macrophages to HER2 CAR T cells and production of metalloproteases as byproduct of activation | In addition to controlling HER2+ tumors, this approach decreased collagen deposition and facilitated T cell infiltration |
| [47] | Stroma-rich solid tumors | ECM | CAR T cells expressing enzyme heparanase (HPSE) | The expression of HPSE improves CAR T cell infiltration through degradation of the ECM |
| [198] | Breast, colon, melanoma, fibrosarcoma | ECM | Fibroblast activated protein targeted CAR T cells (FAP-CAR T cells) | Toxicity and off-target effects in bone marrow killed mouse models and therapy showed limited antitumor results |
| [148] | Carcinomas | ECM | FAP-CAR T cells | Decreased tumor density and reduced autochthonous pancreatic cancer growth |
| [149] | Non-small cell lung carcinoma | ECM | FAP-CAR T cells | Reduction in FAP positive stroma and improved antitumor killing alone or when paired with anti-EpHA2 CAR T cells |
| [199] | Hepatocellular carcinoma (HCC) | Migration | CXCR2-expressing CAR T cells | Improved T cell migration and accumulation at the tumor site compared to control |
| [200] | Solid tumors | Hypoxemia | CD19 and B cell maturation antigen (BCMA) CAR T cells at normoxic and hypoxic oxygen levels | Hypoxic conditions attenuated CAR T cell expansion, differentiation to CD8 T cells, and cytokine production |
| [201] | Solid tumor | Hypoxemia | CAR T cells were transcriptionally paired to hypoxia response elements (HREs) including hypoxia-inducible-1 factor-alpha (HIF1α) | Hypoxia-induced CAR T cells showed improved antitumor activity in hypoxia compared with normoxia |
| [166] | Solid tumor | Immune cells | Delivery of developmental antigen-encoding RNA via nanoparticles | RNA loaded nanoparticles enhanced expansion and activity of CAR T cells in claudin-expression solid tumors |
| NCT | Title | Phase | Author(s) | Disease(s) | Approach | Status |
|---|---|---|---|---|---|---|
| NCT04185038 | Study of B7-H3-Specific CAR T Cell Locoregional Immunotherapy for Diffuse Intrinsic Pontine Glioma/Diffuse Midline Glioma and Recurrent or Refractory Pediatric Central Nervous System Tumors | I | Vitanza et al. | DIPG/recurrent pediatric CNS tumors | Locoregional delivery targeting B7-H3 | Recruiting |
| NCT03696030 | HER2-CAR T Cells in Treating Patients with Recurrent Brain or Leptomeningeal Metastases | I | Pornow et al. | CNS metastasis | Intraventricular delivery of HER2 CAR T cells for CNS metastasis from HER2+ tumors. | Recruiting |
| NCT03638167 | EGFR806-specific CAR T Cell Locoregional Immunotherapy for EGFR-positive Recurrent or Refractory Pediatric CNS Tumors | I | Gust et al. | EGFR-positive recurrent or refractory pediatric CNS tumors | Locoregional delivery targeting EGFR806 | Recruiting |
| NCT03283631 | Intracerebral EGFR-vIII CAR-T Cells for Recurrent GBM (INTERCEPT) | I | Landi et al. | Recurrent GBM | Intracerebral EGFR-vIII CAR T cells | Terminated. Patient enrollment was halted. |
| NCT04153799 | Study of CXCR5 Modified EGFE Chimeric Antigen Receptor Autologous T cells in EGFR-Positive Patients with Advanced Non-Small Cell Lung Cancer | I | Zhang et al. | Advanced non-small-cell lung cancer | CXCR5 modified CAR T cells targeting EGFR | Recruiting |
| NCT03602157 | Study of CAR-T Cells Expressing CD30 and CCR4 for r/r CD30+ HL and CTCL | I | Grover et al. | Relapsed and recurrent Hodgkin lymphoma and cutaneous T cell lymphoma | CCR4 modified CAR T cells targeting CD30 | Recruiting |
| NCT05081479 | A Study of N-Acetylcysteine (N-AC) in People Receiving CAR T cell Therapy for Lymphoma | I | Batlevi et al. | B cell lymphoma | Reduce tumor reactive oxygen species to condition lymphoma TME for CD19 CAR T cells | Recruiting |
| NCT04976218 | TGFβR-KO CAR-EGFR T Cells in Previously Treated Advanced EGFR-positive Solid Tumors | I | Han et al. | EGFR-positive solid tumors | CAR T cells resistant to TGFβ receptor | Recruiting |
| NCT03740256 | Binary Oncolytic Adenovirus in Combination with HER2-Specific Autologous CAR VST, Advanced HER2 Positive Solid Tumors (VISTA) | I | Wang et al. | Advanced HER2+ solid tumors | Oncolytic adenovirus delivers PDL1 blocking mini antibody to enhance CAR T cell killing | Recruiting |
| NCT04381741 | CD19 CAR-T Expressing IL7 and CCL19 Combined with PD1 mAb for Relapsed or Refractory Diffuse Large B Cell Lymphoma (CICPD) | I | Qian and Liu et al. | Recurrent or relapsed diffuse large B cell lymphoma | CAR T cells are potentiated with co-expression of IL-7 and CCL19 to better migrate into the TME and enhanced T cell fitness. This approach is also combined with PD1 blocking antibody. | Recruiting |
| NCT03545815 | Study of CRISPR-Cas9 Mediated PD-1 and TCR Gene-Knocked Out Mesothelin-Directed CAR-T Cells in Patients with Mesothelin Positive Multiple Solid Tumors | I | Han et al. | Mesothelin-directed CAR T cells | CAR T cells have PD1 TCR receptor knocked out to enhance their activity | Recruiting |
| NCT02706405 | JCAR014 and Durvalumab in Treating Patients with Relapsed or Refractory B-Cell Non-Hodgkin Lymphoma | 1b | Gauthier et al. | B cell non-Hodgkin lymphoma | PDL1 blocking antibody to improved CD19 CAR T cells | Terminated due to slow accrual |
| NCT03070327 | BCMA Targeted CAR T Cells with or without Lenalidomide for the Treatment of Multiple Myeloma | I | Mailankody et al. | Multiple myeloma | Lenalidomide has been shown to inhibit regulatory T cells and activate CD8 T cells | Active, not recruiting |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, D.T.; Ogando-Rivas, E.; Liu, R.; Wang, T.; Rubin, J.; Jin, L.; Tao, H.; Sawyer, W.W.; Mendez-Gomez, H.R.; Cascio, M.; et al. CAR T Cell Locomotion in Solid Tumor Microenvironment. Cells 2022, 11, 1974. https://doi.org/10.3390/cells11121974
Nguyen DT, Ogando-Rivas E, Liu R, Wang T, Rubin J, Jin L, Tao H, Sawyer WW, Mendez-Gomez HR, Cascio M, et al. CAR T Cell Locomotion in Solid Tumor Microenvironment. Cells. 2022; 11(12):1974. https://doi.org/10.3390/cells11121974
Chicago/Turabian StyleNguyen, Duy T., Elizabeth Ogando-Rivas, Ruixuan Liu, Theodore Wang, Jacob Rubin, Linchun Jin, Haipeng Tao, William W. Sawyer, Hector R. Mendez-Gomez, Matthew Cascio, and et al. 2022. "CAR T Cell Locomotion in Solid Tumor Microenvironment" Cells 11, no. 12: 1974. https://doi.org/10.3390/cells11121974
APA StyleNguyen, D. T., Ogando-Rivas, E., Liu, R., Wang, T., Rubin, J., Jin, L., Tao, H., Sawyer, W. W., Mendez-Gomez, H. R., Cascio, M., Mitchell, D. A., Huang, J., Sawyer, W. G., Sayour, E. J., & Castillo, P. (2022). CAR T Cell Locomotion in Solid Tumor Microenvironment. Cells, 11(12), 1974. https://doi.org/10.3390/cells11121974