
Lethal and Non-Lethal Functions of Caspases in the DNA Damage Response


Abstract
1. Introduction
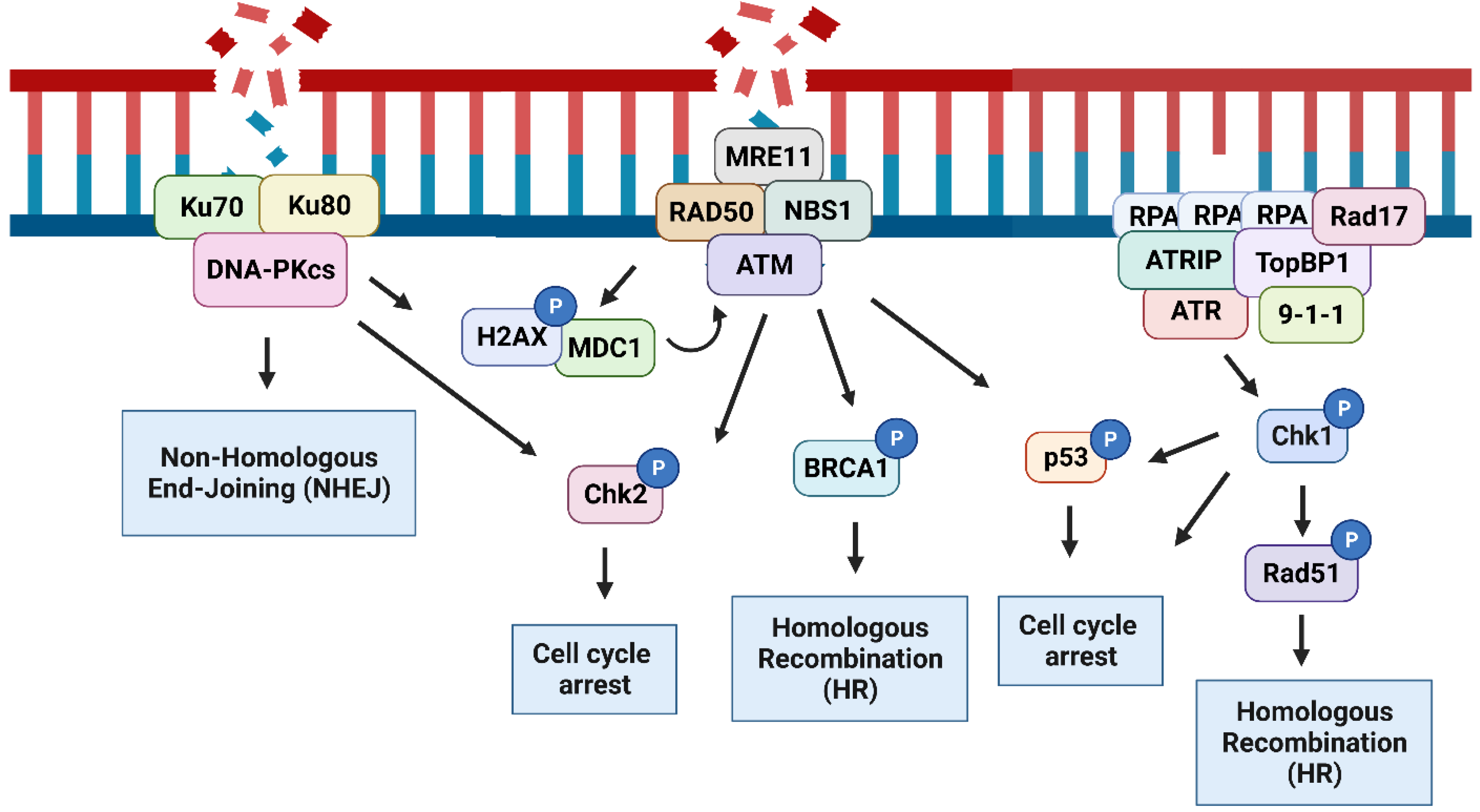
2. The DNA Damage Response
3. DNA Damage-Induced Apoptosis
4. DNA Damage Induced by Caspases without Apoptosis
5. Caspase Function in DNA Repair
6. Crosstalk between Inflammation, Caspases, and DNA Damage
7. Caspases in Cancer
8. Closing Remarks
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef]
- Lindahl, T.; Barnes, D.E. Repair of endogenous DNA damage. Cold Spring Harb. Symp. Quant. Biol. 2000, 65, 127–133. [Google Scholar] [CrossRef]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef]
- Boice, A.; Bouchier-Hayes, L. Targeting apoptotic caspases in cancer. Biochim. Biophys. Acta 2020, 1867, 118688. [Google Scholar] [CrossRef] [PubMed]
- Cheng, B.; Pan, W.; Xing, Y.; Xiao, Y.; Chen, J.; Xu, Z. Recent advances in DDR (DNA damage response) inhibitors for cancer therapy. Eur. J. Med. Chem. 2022, 230, 114109. [Google Scholar] [CrossRef]
- Ali, J.Y.H.; Fitieh, A.M.; Ismail, I.H. The Role of DNA Repair in Genomic Instability of Multiple Myeloma. Int. J. Mol. Sci. 2022, 23, 5688. [Google Scholar] [CrossRef]
- Srinivas, U.S.; Tan, B.W.Q.; Vellayappan, B.A.; Jeyasekharan, A.D. ROS and the DNA damage response in cancer. Redox Biol. 2019, 25, 101084. [Google Scholar] [CrossRef]
- Van de Kamp, G.; Heemskerk, T.; Kanaar, R.; Essers, J. DNA Double Strand Break Repair Pathways in Response to Different Types of Ionizing Radiation. Front. Genet. 2021, 12, 738230. [Google Scholar] [CrossRef]
- Arya, R.; Bassing, C.H. V(D)J Recombination Exploits DNA Damage Responses to Promote Immunity. Trends Genet. 2017, 33, 479–489. [Google Scholar] [CrossRef]
- Nicolas, L.; Cols, M.; Choi, J.E.; Chaudhuri, J.; Vuong, B. Generating and repairing genetically programmed DNA breaks during immunoglobulin class switch recombination. F1000Research 2018, 7, 458. [Google Scholar] [CrossRef]
- Ebel, K.; Bald, I. Low-Energy (5–20 eV) Electron-Induced Single and Double Strand Breaks in Well-Defined DNA Sequences. J. Phys. Chem. Lett. 2022, 13, 4871–4876. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Kim, Y.; Ra, J.S.; Wie, M.W.; Kang, M.-S.; Kang, S.; Myung, K.; Lee, K.-Y. Timely termination of repair DNA synthesis by ATAD5 is important in oxidative DNA damage-induced single-strand break repair. Nucleic Acids Res. 2021, 49, 11746–11764. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, A.R.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef]
- Tewari, M.; Quan, L.T.; O’Rourke, K.; Desnoyers, S.; Zeng, Z.; Beidler, D.R.; Poirier, G.G.; Salvesen, G.S.; Dixit, V.M. Yama/CPP32 beta, a mammalian homolog of CED-3, is a CrmA-inhibitable protease that cleaves the death substrate poly(ADP-ribose) polymerase. Cell 1995, 81, 801–809. [Google Scholar] [CrossRef]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2013, 16, 2–9. [Google Scholar] [CrossRef]
- Maréchal, A.; Zou, L. RPA-coated single-stranded DNA as a platform for post-translational modifications in the DNA damage response. Cell Res. 2014, 25, 9–23. [Google Scholar] [CrossRef]
- Lin, Y.; Bai, L.; Cupello, S.; Hossain, A.; Deem, B.; McLeod, M.; Raj, J.; Yan, S. APE2 promotes DNA damage response pathway from a single-strand break. Nucleic Acids Res. 2018, 46, 2479–2494. [Google Scholar] [CrossRef]
- Kumagai, A.; Lee, J.; Yoo, H.Y.; Dunphy, W.G. TopBP1 Activates the ATR-ATRIP Complex. Cell 2006, 124, 943–955. [Google Scholar] [CrossRef]
- Acevedo, J.; Yan, S.; Michael, W.M. Direct Binding to Replication Protein A (RPA)-coated Single-stranded DNA Allows Recruitment of the ATR Activator TopBP1 to Sites of DNA Damage. J. Biol. Chem. 2016, 291, 13124–13131. [Google Scholar] [CrossRef]
- Mailand, N.; Falck, J.; Lukas, C.; Syljuåsen, R.G.; Welcker, M.; Bartek, J.; Lukas, J. Rapid Destruction of Human Cdc25A in Response to DNA Damage. Science 2000, 288, 1425–1429. [Google Scholar] [CrossRef]
- Sanchez, Y.; Wong, C.; Thoma, R.S.; Richman, R.; Wu, Z.; Piwnica-Worms, H.; Elledge, S.J. Conservation of the Chk1 Checkpoint Pathway in Mammals: Linkage of DNA Damage to Cdk Regulation Through Cdc25. Science 1997, 277, 1497–1501. [Google Scholar] [CrossRef] [PubMed]
- Peng, B.; Shi, R.; Bian, J.; Li, Y.; Wag, P.; Wang, H.; Liao, J.; Zhu, W.-G.; Xu, X. PARP1 and CHK1 coordinate PLK1 enzymatic activity during the DNA damage response to promote homologous recombination-mediated repair. Nucleic Acids Res. 2021, 49, 7554–7570. [Google Scholar] [CrossRef] [PubMed]
- Ishiai, M.; Kitao, H.; Smogorzewska, A.; Tomida, J.; Kinomura, A.; Uchida, E.; Saberi, A.; Kinoshita, E.; Kinoshita-Kikuta, E.; Koike, T.; et al. FANCI phosphorylation functions as a molecular switch to turn on the Fanconi anemia pathway. Nat. Struct. Mol. Biol. 2008, 15, 1138–1146. [Google Scholar] [CrossRef] [PubMed]
- Ho, G.; Margossian, S.; Taniguchi, T.; D’Andrea, A.D. Phosphorylation of FANCD2 on Two Novel Sites Is Required for Mitomycin C Resistance. Mol. Cell. Biol. 2006, 26, 7005–7015. [Google Scholar] [CrossRef]
- Wang, X.; Kennedy, R.D.; Ray, K.; Stuckert, P.; Ellenberger, T.; D’Andrea, A.D. Chk1-Mediated Phosphorylation of FANCE Is Required for the Fanconi Anemia/BRCA Pathway. Mol. Cell. Biol. 2007, 27, 3098–3108. [Google Scholar] [CrossRef]
- Hastak, K.; Paul, R.K.; Agarwal, M.K.; Thakur, V.S.; Amin, A.R.M.R.; Agrawal, S.; Sramkoski, R.M.; Jacobberger, J.W.; Jackson, M.W.; Stark, G.R.; et al. DNA synthesis from unbalanced nucleotide pools causes limited DNA damage that triggers ATR-CHK1-dependent p53 activation. Proc. Natl. Acad. Sci. USA 2008, 105, 6314–6319. [Google Scholar] [CrossRef]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef]
- Saintigny, Y.; Delacôte, F.; Vares, G.; Petitot, F.; Lambert, S.; Averbeck, D.; Lopez, B.S. Characterization of homologous recombination induced by replication inhibition in mammalian cells. EMBO J. 2001, 20, 3861–3870. [Google Scholar] [CrossRef]
- Karanam, K.; Kafri, R.; Loewer, A.; Lahav, G. Quantitative Live Cell Imaging Reveals a Gradual Shift between DNA Repair Mechanisms and a Maximal Use of HR in Mid S Phase. Mol. Cell 2012, 47, 320–329. [Google Scholar] [CrossRef]
- Warren, C.; Pavletich, N.P. Structure of the human ATM kinase and mechanism of Nbs1 binding. eLife 2022, 11, e74218. [Google Scholar] [CrossRef]
- Stiff, T.; O’Driscoll, M.; Rief, N.; Iwabuchi, K.; Löbrich, M.; Jeggo, P.A. ATM and DNA-PK Function Redundantly to Phosphorylate H2AX after Exposure to Ionizing Radiation. Cancer Res. 2004, 64, 2390–2396. [Google Scholar] [CrossRef] [PubMed]
- Lou, Z.; Minter-Dykhouse, K.; Franco, S.; Gostissa, M.; Rivera, M.A.; Celeste, A.; Manis, J.P.; van Deursen, J.; Nussenzweig, A.; Paull, T.T.; et al. MDC1 Maintains Genomic Stability by Participating in the Amplification of ATM-Dependent DNA Damage Signals. Mol. Cell 2006, 21, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Stucki, M.; Clapperton, J.A.; Mohammad, D.; Yaffe, M.B.; Smerdon, S.J.; Jackson, S.P. MDC1 Directly Binds Phosphorylated Histone H2AX to Regulate Cellular Responses to DNA Double-Strand Breaks. Cell 2005, 123, 1213–1226. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, S.; Rotman, G.; Ogawa, A.; Shiloh, Y.; Tamai, K.; Elledge, S.J. Ataxia telangiectasia-mutated phosphorylates Chk2 in vivo and in vitro. Proc. Natl. Acad. Sci. USA 2000, 97, 10389–10394. [Google Scholar] [CrossRef] [PubMed]
- Falck, J.; Mailand, N.; Syljuåsen, R.G.; Bartek, J.; Lukas, J. The ATM–Chk2–Cdc25A checkpoint pathway guards against radioresistant DNA synthesis. Nature 2001, 410, 842–847. [Google Scholar] [CrossRef]
- Matsuoka, S.; Huang, M.; Elledge, S.J. Linkage of ATM to Cell Cycle Regulation by the Chk2 Protein Kinase. Science 1998, 282, 1893–1897. [Google Scholar] [CrossRef]
- Chao, C.; Herr, D.; Chun, J.; Xu, Y. Ser18 and 23 phosphorylation is required for p53-dependent apoptosis and tumor suppression. EMBO J. 2006, 25, 2615–2622. [Google Scholar] [CrossRef]
- Foo, T.K.; Vincelli, G.; Huselid, E.; Her, J.; Zheng, H.; Simhadri, S.; Wang, M.; Huo, Y.; Li, T.; Yu, X.; et al. ATR/ATM-Mediated Phosphorylation of BRCA1 T1394 Promotes Homologous Recombinational Repair and G2–M Checkpoint Maintenance. Cancer Res. 2021, 81, 4676–4684. [Google Scholar] [CrossRef]
- Chaplin, A.K.; Hardwick, S.W.; Liang, S.; Kefala Stavridi, A.; Hnizda, A.; Cooper, L.R.; De Oliveira, T.M.; Chirgadze, D.Y.; Blundell, T.L. Dimers of DNA-PK create a stage for DNA double-strand break repair. Nat. Struct Mol. Biol. 2021, 28, 13–19. [Google Scholar] [CrossRef]
- Betermier, M.; Bertrand, P.; Lopez, B.S. Is non-homologous end-joining really an inherently error-prone process? PLoS Genet. 2014, 10, e1004086. [Google Scholar] [CrossRef]
- Bigot, N.; Day, M.; A Baldock, R.; Watts, F.Z.; Oliver, A.W.; Pearl, L.H. Phosphorylation-mediated interactions with TOPBP1 couple 53BP1 and 9-1-1 to control the G1 DNA damage checkpoint. eLife 2019, 8, e44353. [Google Scholar] [CrossRef] [PubMed]
- Stiff, T.; A Walker, S.; Cerosaletti, K.; A Goodarzi, A.; Petermann, E.; Concannon, P.; O’Driscoll, M.; A Jeggo, P. ATR-dependent phosphorylation and activation of ATM in response to UV treatment or replication fork stalling. EMBO J. 2006, 25, 5775–5782. [Google Scholar] [CrossRef] [PubMed]
- Leiter, L.M.; Chen, J.; Marathe, T.; Tanaka, M.; Dutta, A. Loss of transactivation and transrepression function, and not RPA binding, alters growth suppression by p53. Oncogene 1996, 12, 2661–2668. [Google Scholar] [PubMed]
- Serrano, M.A.; Li, Z.; Dangeti, M.; Musich, P.; Patrick, S.; Roginskaya, M.; Cartwright, B.; Zou, Y. DNA-PK, ATM and ATR collaboratively regulate p53–RPA interaction to facilitate homologous recombination DNA repair. Oncogene 2013, 32, 2452–2462. [Google Scholar] [CrossRef]
- Shang, Z.-F.; Huang, B.; Xu, Q.-Z.; Zhang, S.-M.; Fan, R.; Liu, X.-D.; Wang, Y.; Zhou, P.-K. Inactivation of DNA-Dependent Protein Kinase Leads to Spindle Disruption and Mitotic Catastrophe with Attenuated Checkpoint Protein 2 Phosphorylation in Response to DNA Damage. Cancer Res. 2010, 70, 3657–3666. [Google Scholar] [CrossRef]
- Tomimatsu, N.; Mukherjee, B.; Burma, S. Distinct roles of ATR and DNA-PKcs in triggering DNA damage responses in ATM-deficient cells. EMBO Rep. 2009, 10, 629–635. [Google Scholar] [CrossRef]
- Boatright, K.M.; Salvesen, G.S. Mechanisms of caspase activation. Curr. Opin. Cell Biol. 2003, 15, 725–731. [Google Scholar] [CrossRef]
- Martin, S.J.; Green, D.R. Protease activation during apoptosis: Death by a thousand cuts? Cell 1995, 82, 349–352. [Google Scholar] [CrossRef]
- Boatright, K.M.; Renatus, M.; Scott, F.L.; Sperandio, S.; Shin, H.; Pedersen, I.M.; Ricci, J.E.; A Edris, W.; Sutherlin, D.P.; Green, D.; et al. A Unified Model for Apical Caspase Activation. Mol. Cell 2003, 11, 529–541. [Google Scholar] [CrossRef]
- Baliga, B.C.; Read, S.H.; Kumar, S. The biochemical mechanism of caspase-2 activation. Cell Death Differ. 2004, 11, 1234–1241. [Google Scholar] [CrossRef]
- Oberst, A.; Pop, C.; Tremblay, A.G.; Blais, V.; Denault, J.-B.; Salvesen, G.S.; Green, D.R. Inducible Dimerization and Inducible Cleavage Reveal a Requirement for Both Processes in Caspase-8 Activation. J. Biol. Chem. 2010, 285, 16632–16642. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Srinivasula, S.M.; Druilhe, A.; Fernandes-Alnemri, T.; Alnemri, E.S. Caspase-2 Induces Apoptosis by Releasing Proapoptotic Proteins from Mitochondria. J. Biol. Chem. 2002, 277, 13430–13437. [Google Scholar] [CrossRef] [PubMed]
- Heilig, R.; Dilucca, M.; Boucher, D.; Chen, K.W.; Hancz, D.; Demarco, B.; Shkarina, K.; Broz, P. Caspase-1 cleaves Bid to release mitochondrial SMAC and drive secondary necrosis in the absence of GSDMD. Life Sci. Alliance 2020, 3, e202000735. [Google Scholar] [CrossRef] [PubMed]
- Rogers, C.; Erkes, D.A.; Nardone, A.; Aplin, A.E.; Fernandes-Alnemri, T.; Alnemri, E.S. Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation. Nat. Commun. 2019, 10, 1689. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, K.; Nakajima, S.; Hosojima, S.; Nguyen, D.T.; Hattori, T.; Le, T.M.; Hori, O.; Mahib, M.R.; Yamaguchi, Y.; Miura, M.; et al. Caspase-1 initiates apoptosis in the absence of gasdermin D. Nat. Commun. 2019, 10, 2091. [Google Scholar] [CrossRef]
- Hu, B.; Elinav, E.; Huber, S.; Booth, C.J.; Strowig, T.; Jin, C.; Eisenbarth, S.C.; Flavell, R.A. Inflammation-induced tumorigenesis in the colon is regulated by caspase-1 and NLRC4. Proc. Natl. Acad. Sci. USA 2010, 107, 21635–21640. [Google Scholar] [CrossRef]
- Boice, A.G.; Lopez, K.E.; Pandita, R.K.; Parsons, M.J.; Charendoff, C.I.; Charaka, V.; Carisey, A.F.; Pandita, T.K.; Bouchier-Hayes, L. Caspase-2 regulates S-phase cell cycle events to protect from DNA damage accumulation independent of apoptosis. Oncogene 2021, 41, 204–219. [Google Scholar] [CrossRef]
- Fava, L.L.; Schuler, F.; Sladky, V.; Haschka, M.D.; Soratroi, C.; Eiterer, L.; Demetz, E.; Weiss, G.; Geley, S.; Nigg, E.A.; et al. The PIDDosome activates p53 in response to supernumerary centrosomes. Genes Dev. 2017, 31, 34–45. [Google Scholar] [CrossRef]
- Oliver, T.J.; Meylan, E.; Chang, G.P.; Xue, W.; Burke, J.R.; Humpton, T.J.; Hubbard, D.; Bhutkar, A.; Jacks, T. Caspase-2-Mediated Cleavage of Mdm2 Creates a p53-Induced Positive Feedback Loop. Mol. Cell 2011, 43, 57–71. [Google Scholar] [CrossRef]
- Chipuk, J.E.; Kuwana, T.; Bouchier-Hayes, L.; Droin, N.M.; Newmeyer, D.D.; Schuler, M.; Green, D.R. Direct Activation of Bax by p53 Mediates Mitochondrial Membrane Permeabilization and Apoptosis. Science 2004, 303, 1010–1014. [Google Scholar] [CrossRef]
- Muller, M.; Wilder, S.; Bannasch, D.; Israeli, D.; Lehlbach, K.; Li-Weber, M.; Friedman, S.L.; Galle, P.R.; Stremmel, W.; Oren, M.; et al. p53 activates the CD95 (APO-1/Fas) gene in response to DNA damage by anticancer drugs. J. Exp. Med. 1998, 188, 2033–2045. [Google Scholar] [CrossRef] [PubMed]
- Hill, L.L.; Ouhtit, A.; Loughlin, S.M.; Kripke, M.L.; Ananthaswamy, H.N.; Owen-Schaub, L.B. Fas Ligand: A Sensor for DNA Damage Critical in Skin Cancer Etiology. Science 1999, 285, 898–900. [Google Scholar] [CrossRef] [PubMed]
- Ichim, G.; Lopez, J.; Ahmed, S.U.; Muthalagu, N.; Giampazolias, E.; Delgado, M.E.; Haller, M.; Riley, J.; Mason, S.; Athineos, D.; et al. Limited Mitochondrial Permeabilization Causes DNA Damage and Genomic Instability in the Absence of Cell Death. Mol. Cell 2015, 57, 860–872. [Google Scholar] [CrossRef] [PubMed]
- Rongvaux, A.; Jackson, R.; Harman, C.C.; Li, T.; West, A.P.; De Zoete, M.R.; Wu, Y.; Yordy, B.; Lakhani, S.; Kuan, C.-Y.; et al. Apoptotic Caspases Prevent the Induction of Type I Interferons by Mitochondrial DNA. Cell 2014, 159, 1563–1577. [Google Scholar] [CrossRef]
- Enari, M.; Sakahira, H.; Yokoyama, H.; Okawa, K.; Iwamatsu, A.; Nagata, S. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature 1998, 391, 43–50. [Google Scholar] [CrossRef]
- Dawar, S.; Lim, Y.; Puccini, J.; White, M.; Thomas, P.; Bouchier-Hayes, L.; Green, D.R.; Dorstyn, L.; Kumar, S. Caspase-2-mediated cell death is required for deleting aneuploid cells. Oncogene 2016, 36, 2704–2714. [Google Scholar] [CrossRef]
- López-García, C.; Sansregret, L.; Domingo, E.; McGranahan, N.; Hobor, S.; Birkbak, N.J.; Horswell, S.; Grönroos, E.; Favero, F.; Rowan, A.J.; et al. BCL9L Dysfunction Impairs Caspase-2 Expression Permitting Aneuploidy Tolerance in Colorectal Cancer. Cancer Cell 2017, 31, 79–93. [Google Scholar] [CrossRef]
- Koganti, S.; Burgula, S.; Bhaduri-McIntosh, S. STAT3 activates the anti-apoptotic form of caspase 9 in oncovirus-infected B lymphocytes. Virology 2019, 540, 160–164. [Google Scholar] [CrossRef]
- Cory, S.; Adams, J.M. The Bcl2 family: Regulators of the cellular life-or-death switch. Nat. Rev. Cancer 2002, 2, 647–656. [Google Scholar] [CrossRef]
- Chipuk, J.E.; Bouchier-Hayes, L.; Green, D.R. Mitochondrial outer membrane permeabilization during apoptosis: The innocent bystander scenario. Cell Death Differ. 2006, 13, 1396–1402. [Google Scholar] [CrossRef]
- Li, P.; Nijhawan, D.; Budihardjo, I.; Srinivasula, S.M.; Ahmad, M.; Alnemri, E.S.; Wang, X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 1997, 91, 479–489. [Google Scholar] [CrossRef]
- Tinel, A.; Tschopp, J. The PIDDosome, a Protein Complex Implicated in Activation of Caspase-2 in Response to Genotoxic Stress. Science 2004, 304, 843–846. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Ma, W.; Benchimol, S. Pidd, a new death-domain–containing protein, is induced by p53 and promotes apoptosis. Nat. Genet. 2000, 26, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Bradley, G.; Tremblay, S.; Irish, J.; Macmillan, C.; Baker, G.; Gullane, P.; Benchimol, S. The expression of p53-induced protein with death domain (Pidd) and apoptosis in oral squamous cell carcinoma. Br. J. Cancer 2007, 96, 1425–1432. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sidi, S.; Sanda, T.; Kennedy, R.D.; Hagen, A.T.; Jette, C.A.; Hoffmans, R.; Pascual, J.; Imamura, S.; Kishi, S.; Amatruda, J.F.; et al. Chk1 Suppresses a Caspase-2 Apoptotic Response to DNA Damage that Bypasses p53, Bcl-2, and Caspase-3. Cell 2008, 133, 864–877. [Google Scholar] [CrossRef]
- Ando, K.; Kernan, J.L.; Liu, P.; Sanda, T.; Logette, E.; Tschopp, J.; Look, A.T.; Wang, J.; Bouchier-Hayes, L.; Sidi, S. PIDD Death-Domain Phosphorylation by ATM Controls Prodeath versus Prosurvival PIDDosome Signaling. Mol. Cell 2012, 47, 681–693. [Google Scholar] [CrossRef]
- Shah, R.B.; Kernan, J.L.; van Hoogstraten, A.; Ando, K.; Li, Y.; Belcher, A.L.; Mininger, I.; Bussenault, A.M.; Raman, R.; Ramanagoudr-Bhojappa, R.; et al. FANCI functions as a repair/apoptosis switch in response to DNA crosslinks. Dev. Cell 2021, 56, 2207–2222. [Google Scholar] [CrossRef]
- Ando, K.; Parsons, M.J.; Shah, R.B.; Charendoff, C.I.; Paris, S.L.; Liu, P.H.; Fassio, S.R.; Rohrman, B.A.; Thompson, R.; Oberst, A.; et al. NPM1 directs PIDDosome-dependent caspase-2 activation in the nucleolus. J. Cell Biol. 2017, 216, 1795–1810. [Google Scholar] [CrossRef]
- Robeson, A.C.; Lindblom, K.R.; Wojton, J.; Kornbluth, S.; Matsuura, K. Dimer-specific immunoprecipitation of active caspase-2 identifies TRAF proteins as novel activators. EMBO J. 2018, 37, e97072. [Google Scholar] [CrossRef]
- Ho, L.H.; Taylor, R.; Dorstyn, L.; Cakouros, D.; Bouillet, P.; Kumar, S. A tumor suppressor function for caspase-2. Proc. Natl. Acad. Sci. USA 2009, 106, 5336–5341. [Google Scholar] [CrossRef]
- Kim, H.; Rafiuddin-Shah, M.; Tu, H.-C.; Jeffers, J.R.; Zambetti, G.P.; Hsieh, J.J.-D.; Cheng, E.H.-Y. Hierarchical regulation of mitochondrion-dependent apoptosis by BCL-2 subfamilies. Nat. Cell Biol. 2006, 8, 1348–1358. [Google Scholar] [CrossRef] [PubMed]
- Letai, A.; Bassik, M.C.; Walensky, L.D.; Sorcinelli, M.D.; Weiler, S.; Korsmeyer, S.J. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2002, 2, 183–192. [Google Scholar] [CrossRef]
- Kischkel, F.C.; Hellbardt, S.; Behrmann, I.; Germer, M.; Pawlita, M.; Krammer, P.H.; E Peter, M. Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. EMBO J. 1995, 14, 5579–5588. [Google Scholar] [CrossRef] [PubMed]
- Takimoto, R.; El-Deiry, W.S. Wild-type p53 transactivates the KILLER/DR5 gene through an intronic sequence-specific DNA-binding site. Oncogene 2000, 19, 1735–1743. [Google Scholar] [CrossRef]
- Micheau, O.; Tschopp, J. Induction of TNF Receptor I-Mediated Apoptosis via Two Sequential Signaling Complexes. Cell 2003, 114, 181–190. [Google Scholar] [CrossRef]
- Kaiser, W.J.; Upton, J.W.; Long, A.B.; Livingston-Rosanoff, D.; Daley-Bauer, L.P.; Hakem, R.; Caspary, T.; Mocarski, E.S. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature 2011, 471, 368–372. [Google Scholar] [CrossRef]
- Cai, Z.; Jitkaew, S.; Zhao, J.; Chiang, H.-C.; Choksi, S.; Liu, J.; Ward, Y.; Wu, L.-G.; Liu, Z.-G. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat. Cell Biol. 2013, 16, 55–65. [Google Scholar] [CrossRef]
- Tenev, T.; Bianchi, K.; Darding, M.; Broemer, M.; Langlais, C.; Wallberg, F.; Zachariou, A.; Lopez, J.; MacFarlane, M.; Cain, K.; et al. The Ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol. Cell 2011, 43, 432–448. [Google Scholar] [CrossRef]
- Orth, J.D.; Loewer, A.; Lahav, G.; Mitchison, T.J. Prolonged mitotic arrest triggers partial activation of apoptosis, resulting in DNA damage and p53 induction. Mol. Biol. Cell 2012, 23, 567–576. [Google Scholar] [CrossRef]
- Solier, S.; Pommier, Y. MDC1 Cleavage by Caspase-3: A Novel Mechanism for Inactivating the DNA Damage Response during Apoptosis. Cancer Res. 2010, 71, 906–913. [Google Scholar] [CrossRef]
- Van Loo, G.; Schotte, P.; Van Gurp, M.; Demol, H.; Hoorelbeke, B.; Gevaert, K.; Rodriguez, I.; Ruiz-Carrillo, A.; Vandekerckhove, J.; Declercq, W.; et al. Endonuclease G: A mitochondrial protein released in apoptosis and involved in caspase-independent DNA degradation. Cell Death Differ. 2001, 8, 1136–1142. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; He, Y.; Li, F.; Huang, Q.; Kato, T.A.; Hall, R.P.; Li, C.-Y. Caspase-3 Promotes Genetic Instability and Carcinogenesis. Mol. Cell 2015, 58, 284–296. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.L.; Mak, K.H.; Hu, S.; Wang, S.S.; Wong, K.M.; Wong, C.S.T.; Wu, H.Y.; Law, H.T.; Liu, K.; Talbot, C.C.; et al. Cell survival, DNA damage, and oncogenic transformation after a transient and reversible apoptotic response. Mol. Biol. Cell 2012, 23, 2240–2252. [Google Scholar] [CrossRef] [PubMed]
- Fernald, K.; Kurokawa, M. Evading apoptosis in cancer. Trends Cell Biol. 2013, 23, 620–633. [Google Scholar] [CrossRef] [PubMed]
- Miles, M.; Hawkins, C.J. Executioner caspases and CAD are essential for mutagenesis induced by TRAIL or vincristine. Cell Death Dis. 2017, 8, e3062. [Google Scholar] [CrossRef]
- Lovric, M.M.; Hawkins, C.J. TRAIL treatment provokes mutations in surviving cells. Oncogene 2010, 29, 5048–5060. [Google Scholar] [CrossRef]
- Muller, I.; Strozyk, E.; Schindler, S.; Beissert, S.; Oo, H.Z.; Sauter, T.; Lucarelli, P.; Raeth, S.; Hausser, A.; Al Nakouzi, N.; et al. Cancer Cells Employ Nuclear Caspase-8 to Overcome the p53-Dependent G2/M Checkpoint through Cleavage of USP28. Mol. Cell 2020, 77, 970–984. [Google Scholar] [CrossRef]
- Srinivasula, S.M.; Ahmad, M.; Guo, Y.; Zhan, Y.; Lazebnik, Y.; Fernandes-Alnemri, T.; Alnemri, E.S. Identification of an endogenous dominant-negative short isoform of caspase-9 that can regulate apoptosis. Cancer Res. 1999, 59, 999–1002. [Google Scholar]
- McStay, G.P.; Salvesen, G.S.; Green, D.R. Overlapping cleavage motif selectivity of caspases: Implications for analysis of apoptotic pathways. Cell Death Differ. 2007, 15, 322–331. [Google Scholar] [CrossRef]
- Seol, D.-W.; Billiar, T.R. A Caspase-9 Variant Missing the Catalytic Site Is an Endogenous Inhibitor of Apoptosis. J. Biol. Chem. 1999, 274, 2072–2076. [Google Scholar] [CrossRef]
- Pop, C.; Oberst, A.; Drag, M.; Van Raam, B.J.; Riedl, S.J.; Green, D.R.; Salvesen, G.S. FLIP(L) induces caspase 8 activity in the absence of interdomain caspase 8 cleavage and alters substrate specificity. Biochem. J. 2011, 433, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Clarke, C.A.L.; Bennett, L.N.; Clarke, P.R. Cleavage of Claspin by Caspase-7 during Apoptosis Inhibits the Chk1 Pathway. J. Biol. Chem. 2005, 280, 35337–35345. [Google Scholar] [CrossRef] [PubMed]
- Goehe, R.W.; Shultz, J.C.; Murudkar, C.; Usanovic, S.; Lamour, N.F.; Massey, D.H.; Zhang, L.; Camidge, D.R.; Shay, J.W.; Minna, J.D.; et al. Chalfant, hnRNP L regulates the tumorigenic capacity of lung cancer xenografts in mice via caspase-9 pre-mRNA processing. J. Clin. Invest. 2010, 120, 3923–3939. [Google Scholar] [CrossRef] [PubMed]
- Dorstyn, L.; Puccini, J.; Wilson, C.H.; Shalini, S.; Nicola, M.; Moore, S.; Kumar, S. Caspase-2 deficiency promotes aberrant DNA-damage response and genetic instability. Cell Death Differ. 2012, 19, 1288–1298. [Google Scholar] [CrossRef]
- Parsons, M.J.; McCormick, L.; Janke, L.; Howard, A.; Bouchier-Hayes, L.; Green, D.R. Genetic deletion of caspase-2 accelerates MMTV/c-neu-driven mammary carcinogenesis in mice. Cell Death Differ. 2013, 20, 1174–1182. [Google Scholar] [CrossRef]
- Puccini, J.; Shalini, S.; Voss, A.K.; Gatei, M.; Wilson, C.H.; Hiwase, D.K.; Lavin, M.F.; Dorstyn, L.; Kumar, S. Loss of caspase-2 augments lymphomagenesis and enhances genomic instability in Atm -deficient mice. Proc. Natl. Acad. Sci. USA 2013, 110, 19920–19925. [Google Scholar] [CrossRef]
- Lim, Y.; De Bellis, D.; Sandow, J.J.; Capalbo, L.; D’Avino, P.P.; Murphy, J.M.; Webb, A.I.; Dorstyn, L.; Kumar, S. Phosphorylation by Aurora B kinase regulates caspase-2 activity and function. Cell Death Differ. 2021, 28, 349–366. [Google Scholar] [CrossRef]
- Andersen, J.L.; E Johnson, C.; Freel, C.D.; Parrish, A.B.; Day, J.L.; Buchakjian, M.; Nutt, L.K.; Thompson, J.W.; Moseley, M.A.; Kornbluth, S. Restraint of apoptosis during mitosis through interdomain phosphorylation of caspase-2. EMBO J. 2009, 28, 3216–3227. [Google Scholar] [CrossRef]
- Zamaraev, A.V.; Volik, P.I.; Nilov, D.K.; Turkina, M.V.; Egorshina, A.Y.; Gorbunova, A.S.; Iarovenko, S.I.; Zhivotovsky, B.; Kopeina, G.S. Requirement for Serine-384 in Caspase-2 processing and activity. Cell Death Dis. 2020, 11, 825. [Google Scholar] [CrossRef]
- Beisner, D.R.; Ch’En, I.L.; Kolla, R.V.; Hoffmann, A.; Hedrick, S. Cutting Edge: Innate Immunity Conferred by B Cells Is Regulated by Caspase-8. J. Immunol. 2005, 175, 3469–3473. [Google Scholar] [CrossRef]
- Salmena, L.; Lemmers, B.; Hakem, A.; Matysiak-Zablocki, E.; Murakami, K.; Au, P.B.; Berry, D.M.; Tamblyn, L.; Shehabeldin, A.; Migon, E.; et al. Essential role for caspase 8 in T-cell homeostasis and T-cell-mediated immunity. Genes Dev. 2003, 17, 883–895. [Google Scholar] [CrossRef] [PubMed]
- Hakem, A.; El Ghamrasni, S.; Maire, G.; Lemmers, B.; Karaskova, J.; Jurisicova, A.; Sanchez, O.; Squire, J.; Hakem, R. Caspase-8 is essential for maintaining chromosomal stability and suppressing B-cell lymphomagenesis. Blood 2012, 119, 3495–3502. [Google Scholar] [CrossRef] [PubMed]
- Boege, Y.; Malehmir, M.; Healy, M.; Bettermann, K.; Lorentzen, A.; Vucur, M.; Ahuja, A.K.; Böhm, F.; Mertens, J.C.; Shimizu, Y.; et al. A Dual Role of Caspase-8 in Triggering and Sensing Proliferation-Associated DNA Damage, a Key Determinant of Liver Cancer Development. Cancer Cell 2017, 32, 342–359. [Google Scholar] [CrossRef] [PubMed]
- Liccardi, G.; Garcia, L.R.; Tenev, T.; Annibaldi, A.; Legrand, A.; Robertson, D.; Feltham, R.; Anderton, H.; Darding, M.; Peltzer, N.; et al. RIPK1 and Caspase-8 Ensure Chromosome Stability Independently of Their Role in Cell Death and Inflammation. Mol. Cell 2018, 73, 413–428. [Google Scholar] [CrossRef]
- Petronczki, M.; Lénárt, P.; Peters, J.-M. Polo on the Rise—from Mitotic Entry to Cytokinesis with Plk1. Dev. Cell 2008, 14, 646–659. [Google Scholar] [CrossRef]
- Elowe, S. Bub1 and BubR1: At the Interface between Chromosome Attachment and the Spindle Checkpoint. Mol. Cell. Biol. 2011, 31, 3085–3093. [Google Scholar] [CrossRef]
- Bolívar, B.; Vogel, T.P.; Bouchier-Hayes, L. Inflammatory caspase regulation: Maintaining balance between inflammation and cell death in health and disease. FEBS J. 2019, 286, 2628–2644. [Google Scholar] [CrossRef]
- Hu, B.; Jin, C.; Li, H.-B.; Tong, J.; Ouyang, X.; Cetinbas, N.M.; Zhu, S.; Strowig, T.; Lam, F.C.; Zhao, C.; et al. The DNA-sensing AIM2 inflammasome controls radiation-induced cell death and tissue injury. Science 2016, 354, 765–768. [Google Scholar] [CrossRef]
- Stoecklein, V.M.; Osuka, A.; Ishikawa, S.; Lederer, M.R.; Wanke-Jellinek, L.; Lederer, J.A. Radiation Exposure Induces Inflammasome Pathway Activation in Immune Cells. J. Immunol. 2014, 194, 1178–1189. [Google Scholar] [CrossRef]
- Zanoni, I.; Tan, Y.; Di Gioia, M.; Broggi, A.; Ruan, J.; Shi, J.; Donado, C.A.; Shao, F.; Wu, H.; Springstead, J.R.; et al. An endogenous caspase-11 ligand elicits interleukin-1 release from living dendritic cells. Science 2016, 352, 1232–1236. [Google Scholar] [CrossRef]
- Wolf, A.J.; Reyes, C.N.; Liang, W.; Becker, C.; Shimada, K.; Wheeler, M.L.; Cho, H.C.; Popescu, N.; Coggeshall, K.M.; Arditi, M.; et al. Hexokinase Is an Innate Immune Receptor for the Detection of Bacterial Peptidoglycan. Cell 2016, 166, 624–636. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.W.; Gross, C.J.; Sotomayor, F.V.; Stacey, K.J.; Tschopp, J.; Sweet, M.J.; Schroder, K. The neutrophil NLRC4 inflammasome selectively promotes IL-1beta maturation without pyroptosis during acute Salmonella challenge. Cell Rep. 2014, 8, 570–582. [Google Scholar] [CrossRef] [PubMed]
- Gaidt, M.M.; Ebert, T.S.; Chauhan, D.; Schmidt, T.; Schmid-Burgk, J.; Rapino, F.; Robertson, A.; Cooper, M.; Graf, T.; Hornung, V. Human Monocytes Engage an Alternative Inflammasome Pathway. Immunity 2016, 44, 833–846. [Google Scholar] [CrossRef] [PubMed]
- Evavold, C.; Ruan, J.; Tan, Y.; Xia, S.; Wu, H.; Kagan, J.C. The Pore-Forming Protein Gasdermin D Regulates Interleukin-1 Secretion from Living Macrophages. Immunity 2017, 48, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhang, Y.; Xia, S.; Kong, Q.; Li, S.; Liu, X.; Junqueira, C.; Meza-Sosa, K.F.; Mok, T.M.Y.; Ansara, J.; et al. Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature 2020, 579, 415–420. [Google Scholar] [CrossRef]
- Riley, J.S.; Quarato, G.; Cloix, C.; Lopez, J.; O’Prey, J.; Pearson, M.; Chapman, J.; Sesaki, H.; Carlin, L.M.; Passos, J.F.; et al. Mitochondrial inner membrane permeabilisation enables mtDNA release during apoptosis. EMBO J. 2018, 37, e99238. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, H.; Wu, X.; Ma, D.; Wu, J.; Wang, L.; Jiang, Y.; Fei, Y.; Zhu, C.; Tan, R.; et al. Nuclear cGAS suppresses DNA repair and promotes tumorigenesis. Nature 2018, 563, 131–136. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Terry, M.R.; Arya, R.; Mukhopadhyay, A.; Berrett, K.C.; Clair, P.M.; Witt, B.; E Salama, M.; Bhutkar, A.; Oliver, T.G. Caspase-2 impacts lung tumorigenesis and chemotherapy response in vivo. Cell Death Differ. 2014, 22, 719–730. [Google Scholar] [CrossRef]
- Hackl, H.; Astanina, K.; Wieser, R. Molecular and genetic alterations associated with therapy resistance and relapse of acute myeloid leukemia. J. Hematol. Oncol. 2017, 10, 51. [Google Scholar] [CrossRef]
- Kim, M.S.; Kim, H.S.; Jeong, E.G.; Soung, Y.H.; Yoo, N.J.; Lee, S.H. Somatic mutations of caspase-2 gene in gastric and colorectal cancers. Pathol. Res. Pract. 2011, 207, 640–644. [Google Scholar] [CrossRef] [PubMed]
- Hopkins-Donaldson, S.; Ziegler, A.; Kurtz, S.; Bigosch, C.; Kandioler, D.; Ludwig, C.; Zangemeister-Wittke, U.; Stahel, R. Silencing of death receptor and caspase-8 expression in small cell lung carcinoma cell lines and tumors by DNA methylation. Cell Death Differ. 2003, 10, 356–364. [Google Scholar] [CrossRef] [PubMed]
- Teitz, T.; Wei, T.; Valentine, M.B.; Vanin, E.F.; Grenet, J.; Valentine, V.A.; Behm, F.G.; Look, A.T.; Lahti, J.M.; Kidd, V.J. Caspase 8 is deleted or silenced preferentially in childhood neuroblastomas with amplification of MYCN. Nat. Med. 2000, 6, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Soung, Y.H.; Lee, J.W.; Kim, S.Y.; Sung, Y.J.; Park, W.S.; Nam, S.W.; Kim, S.H.; Lee, J.Y.; Yoo, N.J.; Lee, S.H. Caspase-8 gene is frequently inactivated by the frameshift somatic mutation 1225_1226delTG in hepatocellular carcinomas. Oncogene 2005, 24, 141–147. [Google Scholar] [CrossRef]
- Dorstyn, L.; Puccini, J.; A Nikolic, A.; Shalini, S.; Wilson, C.H.; Norris, M.D.; Haber, M.; Kumar, S. An unexpected role for caspase-2 in neuroblastoma. Cell Death Dis. 2014, 5, e1383. [Google Scholar] [CrossRef]
- Helfer, B.; Boswell, B.C.; Finlay, D.; Cipres, A.; Vuori, K.; Kang, T.B.; Wallach, D.; Dorfleutner, A.; Lahti, J.M.; Flynn, D.C.; et al. Caspase-8 Promotes Cell Motility and Calpain Activity under Nonapoptotic Conditions. Cancer Res. 2006, 66, 4273–4278. [Google Scholar] [CrossRef]
- Devarajan, E.; A Sahin, A.; Chen, J.S.; Krishnamurthy, R.R.; Aggarwal, N.; Brun, A.-M.; Sapino, A.; Zhang, F.; Sharma, D.; Yang, X.-H.; et al. Down-regulation of caspase 3 in breast cancer: A possible mechanism for chemoresistance. Oncogene 2002, 21, 8843–8851. [Google Scholar] [CrossRef]
- O’Donovan, N.; Crown, J.; Stunell, H.; Hill, A.D.K.; McDermott, E.; O’Higgins, N.; Duffy, M.J. Caspase 3 in breast cancer. Clin. Cancer Res. 2003, 9, 738–742. [Google Scholar]
- Li, H.; Zhu, H.; Xu, C.-J.; Yuan, J. Cleavage of BID by Caspase 8 Mediates the Mitochondrial Damage in the Fas Pathway of Apoptosis. Cell 1998, 94, 491–501. [Google Scholar] [CrossRef]
- Green, D.R. A BH3 Mimetic for Killing Cancer Cells. Cell 2016, 165, 1560. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Caspase | Lethal Role | Non-Lethal Role |
|---|---|---|
| Caspase-1/4/5/11 | GSDMD and pro-IL-1β cleavage. | Potential sublethal activation of caspase-3 [53,54,55]; suppression of proliferation in colorectal cancer [56] |
| Caspase-2 | Bid cleavage; intrinsic pathway activation | DNA replication fork protection; ploidy-induced cell cycle arrest; MDM2 cleavage [57,58,59]. |
| Caspase-3 | Execution of apoptosis | ICAD cleavage; DNA damage; EndoG release [60,61,62,63]; inhibition of cGAS/STING [64] |
| Caspase-8 | Extrinsic pathway initiation; caspase-3 and -7 activation; Bid cleavage | Cleavage of USP28; RIPoptosome formation; PLK1 cleavage [65,66,67] |
| Caspase-9 | Intrinsic pathway initiation; caspase-3 and -7 activation. | Reduced Chk1 activation by caspase-9b [68] |
| Caspase | Role in Human Cancers |
|---|---|
| Caspase-1 | Tumor suppressor in a colon cancer model [56] |
| Caspase-2 | Tumor suppressor in hematological malignancies and epithelial cancers [80,106]; promotes tumorigenesis in TH-MYC neuroblastoma [135]. |
| Caspase-3 | Promotion and prevention of tumorigenesis in breast cancer [137,138]. |
| Caspase-8 | Low expression in hepatocellular carcinomas, small cell lung carcinoma, and neuroblastomas with N-MYC amplification associated with tumorigenesis [132,133,134]; high expression in breast and pancreatic cancer associated with tumorigenesis [136]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lopez, K.E.; Bouchier-Hayes, L. Lethal and Non-Lethal Functions of Caspases in the DNA Damage Response. Cells 2022, 11, 1887. https://doi.org/10.3390/cells11121887
Lopez KE, Bouchier-Hayes L. Lethal and Non-Lethal Functions of Caspases in the DNA Damage Response. Cells. 2022; 11(12):1887. https://doi.org/10.3390/cells11121887
Chicago/Turabian StyleLopez, Karla E., and Lisa Bouchier-Hayes. 2022. "Lethal and Non-Lethal Functions of Caspases in the DNA Damage Response" Cells 11, no. 12: 1887. https://doi.org/10.3390/cells11121887
APA StyleLopez, K. E., & Bouchier-Hayes, L. (2022). Lethal and Non-Lethal Functions of Caspases in the DNA Damage Response. Cells, 11(12), 1887. https://doi.org/10.3390/cells11121887