
Neutrophils Actively Contribute to Obesity-Associated Inflammation and Pathological Complications


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Obesity
3. Microbiota and Obesity
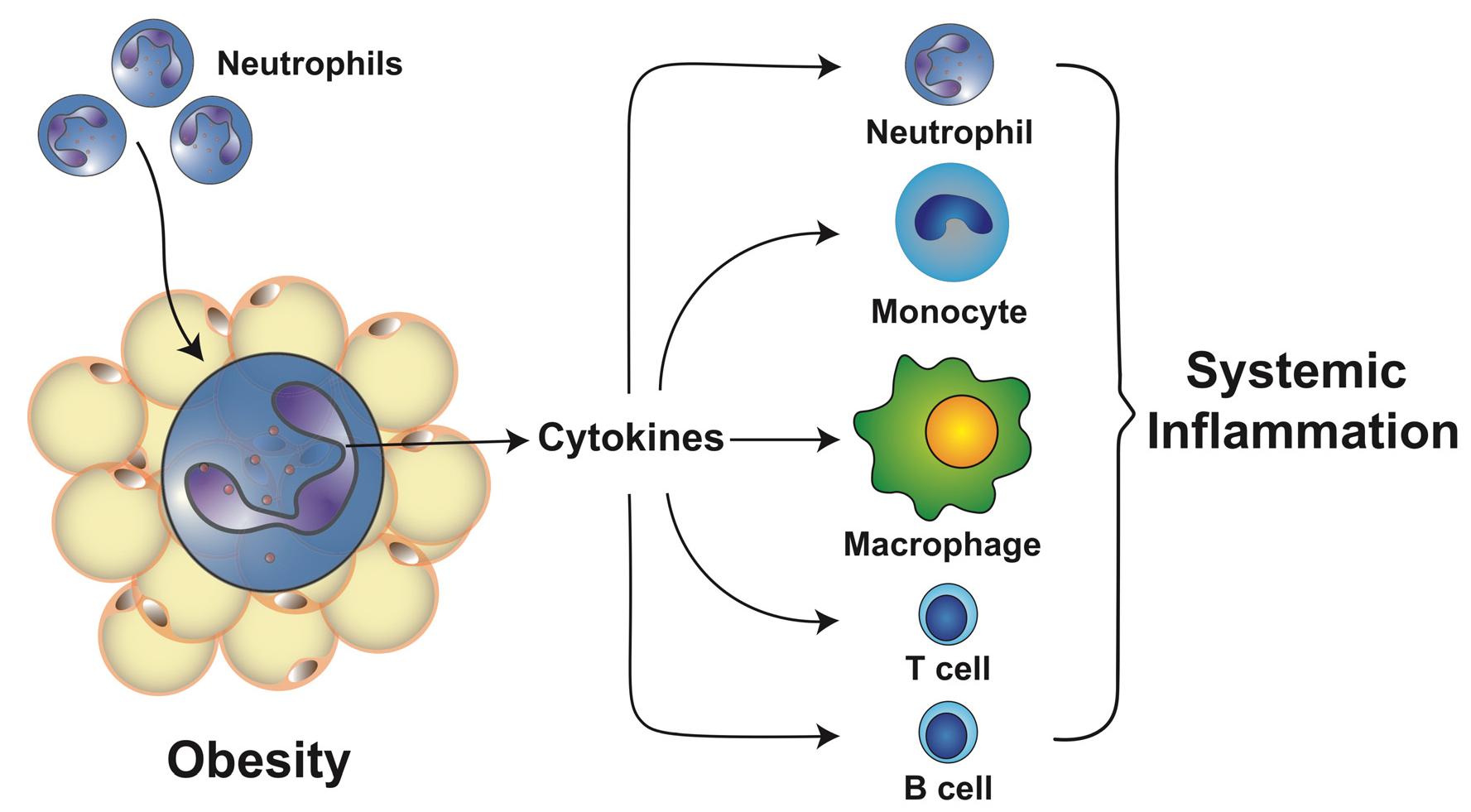
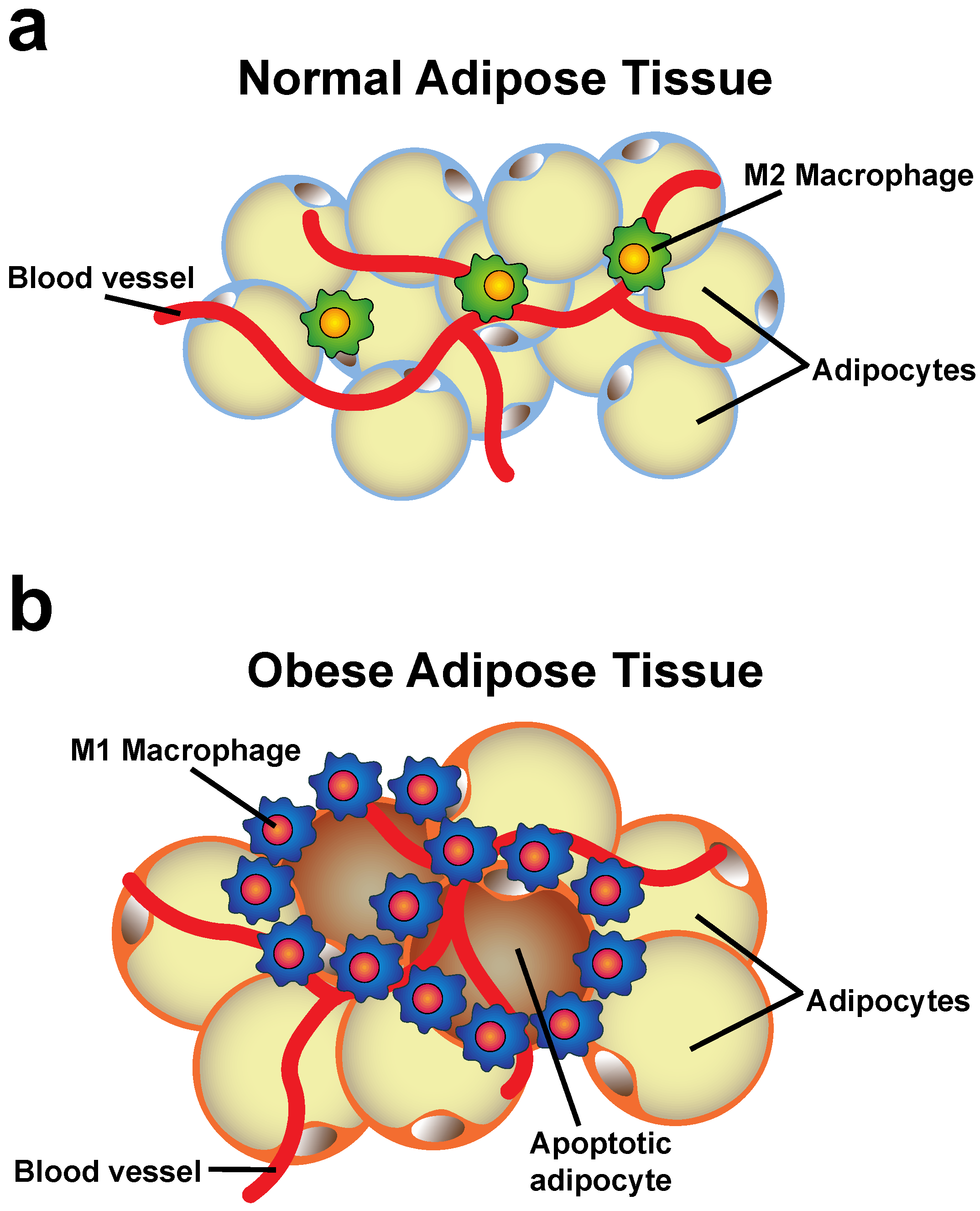
4. Changes of the Adipose Tissue during Obesity
5. Neutrophils in Obesity
5.1. Circulating Neutrophils Increase in Obesity
5.2. Neutrophil-to-Lymphocyte Ratio (NLR)
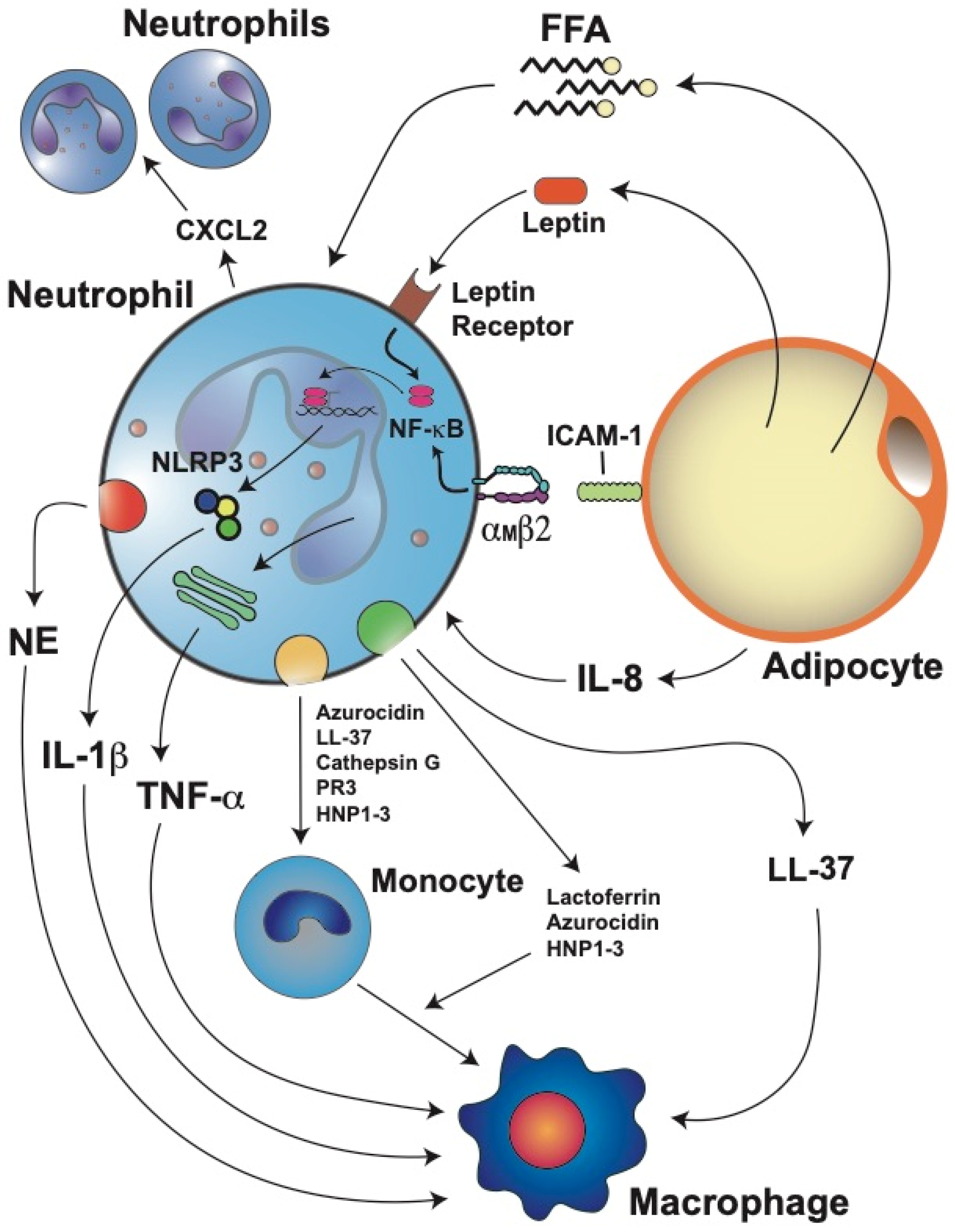
5.3. Neutrophil Infiltration into Adipose Tissue
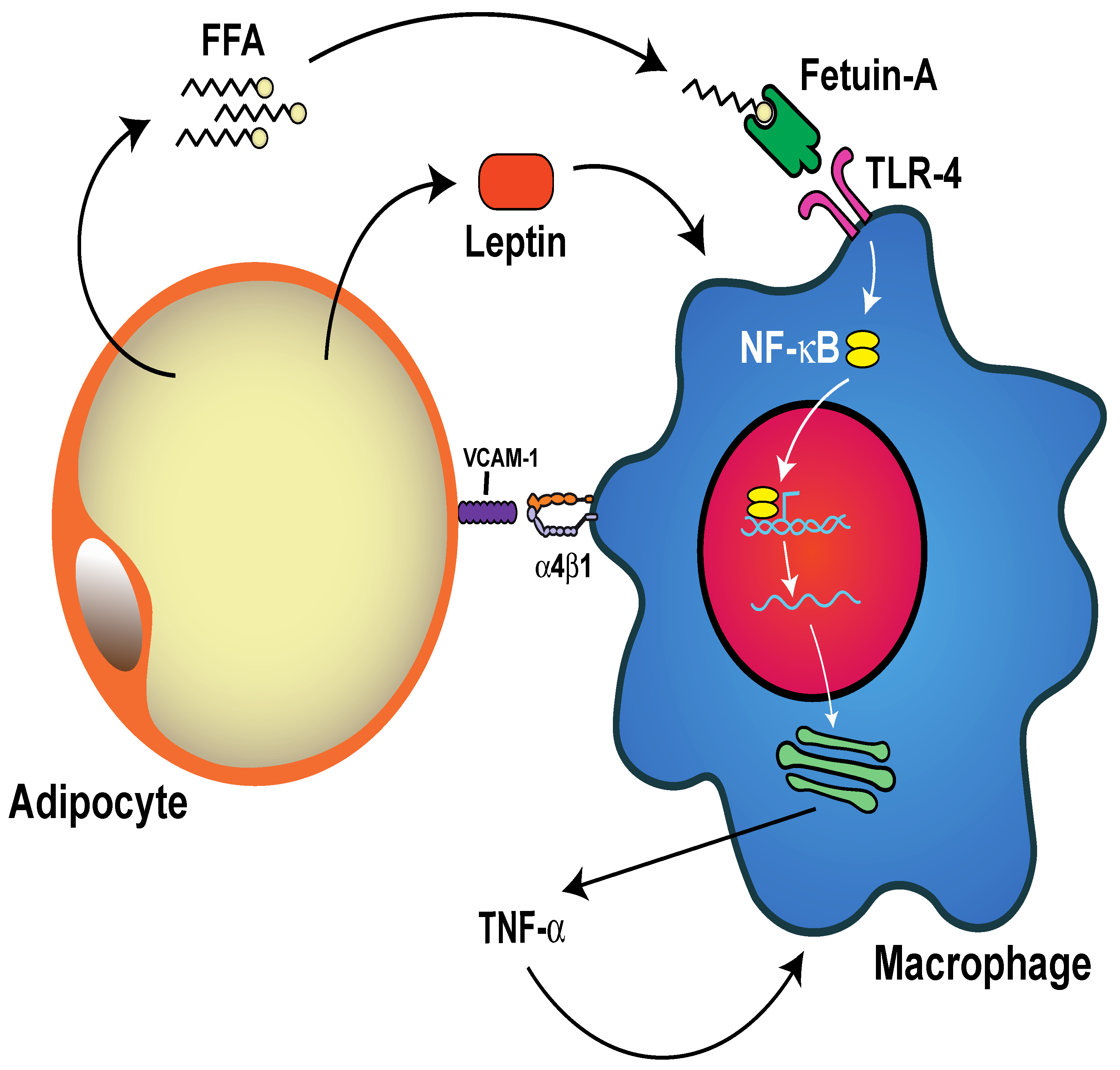
5.4. Neutrophil Activation and Inflammation
5.5. Neutrophil Extracellular Traps (NETs)
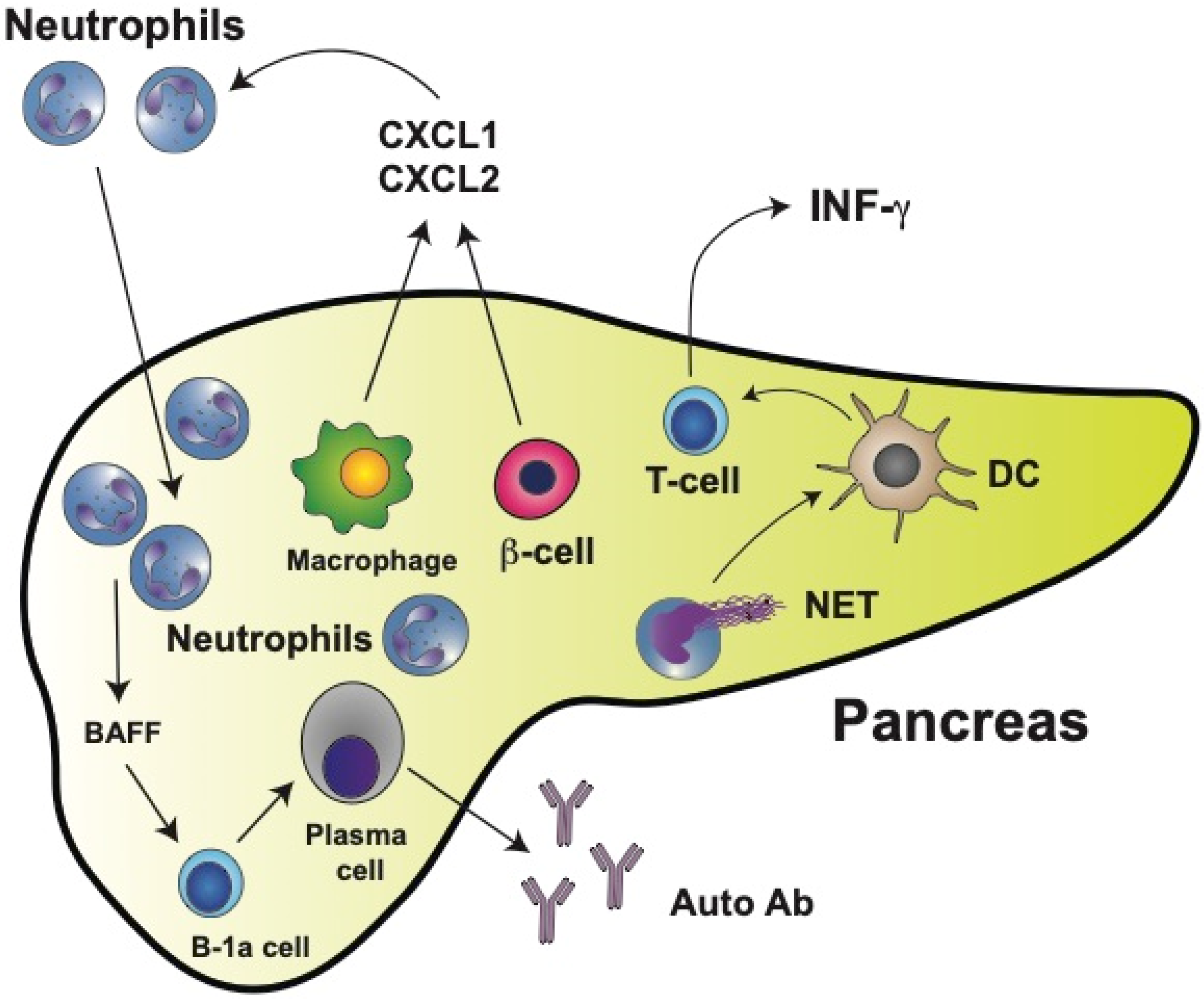
6. Neutrophils in Type 1 Diabetes (T1D)
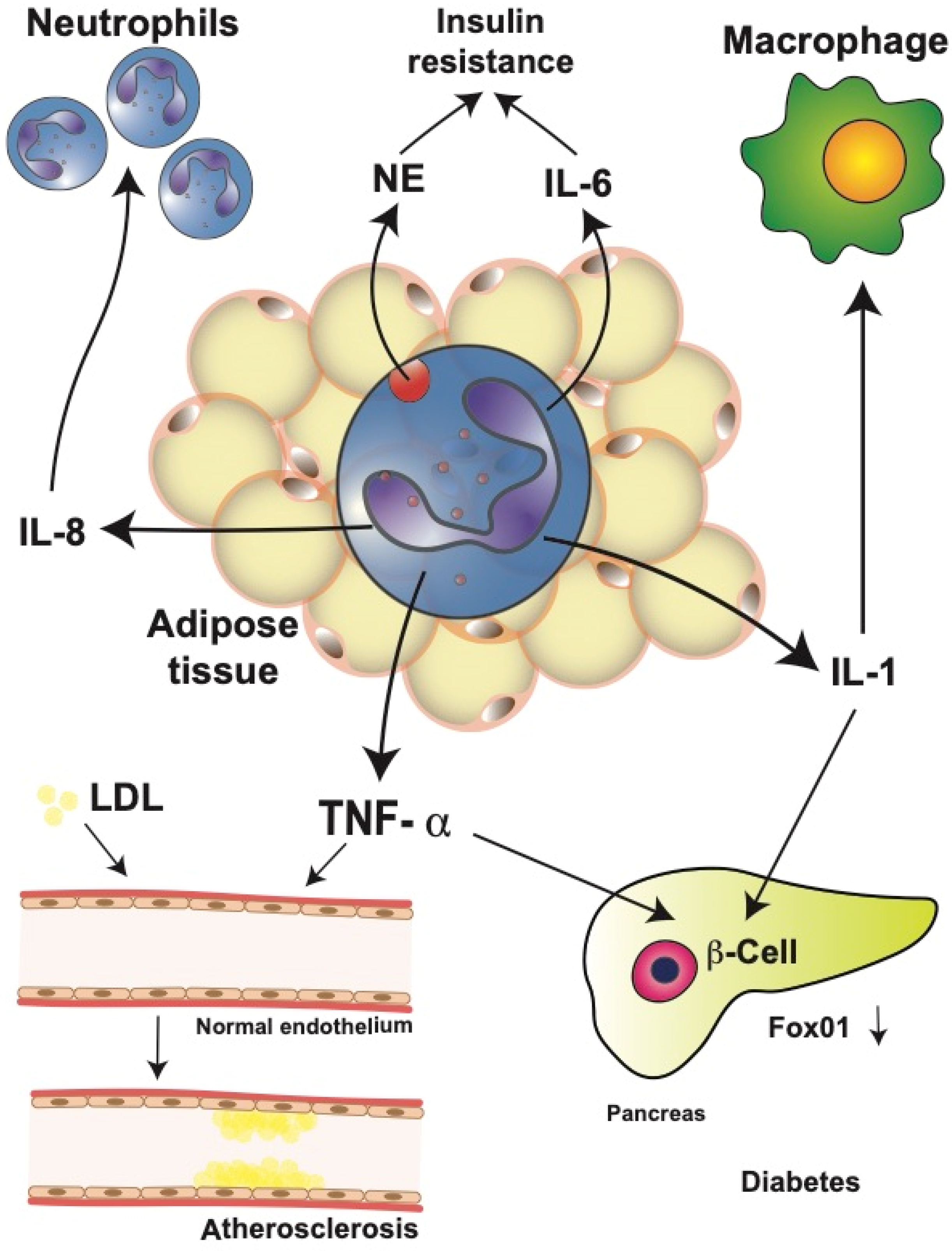
7. Neutrophils in Type 2 Diabetes (T2D)
8. Concluding Ideas
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Engin, A. The definition and prevalence of obesity and metabolic syndrome. Adv. Exp. Med. Biol. 2017, 960, 1–17. [Google Scholar] [CrossRef]
- González-Muniesa, P.; Mártinez-González, M.A.; Hu, F.B.; Després, J.P.; Matsuzawa, Y.; Loos, R.J.F.; Moreno, L.A.; Bray, G.A.; Martinez, J.A. Obesity. Nat. Rev. Dis. Primers 2017, 3, 17034. [Google Scholar] [CrossRef]
- Williams, E.P.; Mesidor, M.; Winters, K.; Dubbert, P.M.; Wyatt, S.B. Overweight and obesity: Prevalence, consequences, and causes of a growing public health problem. Curr. Obes. Rep. 2015, 4, 363–370. [Google Scholar] [CrossRef]
- McAllister, E.J.; Dhurandhar, N.V.; Keith, S.W.; Aronne, L.J.; Barger, J.; Baskin, M.; Benca, R.M.; Biggio, J.; Boggiano, M.M.; Eisenmann, J.C.; et al. Ten putative contributors to the obesity epidemic. Crit. Rev. Food Sci. Nutr. 2009, 49, 868–913. [Google Scholar] [CrossRef]
- Ng, M.; Fleming, T.; Robinson, M.; Thomson, B.; Graetz, N.; Margono, C.; Mullany, E.C.; Biryukov, S.; Abbafati, C.; Abera, S.F.; et al. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980-2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet 2014, 384, 766–781. [Google Scholar] [CrossRef]
- NCD Risk Factor Collaboration (NCD-RisC). Trends in adult body-mass index in 200 countries from 1975 to 2014: A pooled analysis of 1698 population-based measurement studies with 19·2 million participants. Lancet 2016, 397, 1998. [Google Scholar] [CrossRef]
- Blüher, M. Obesity: Global epidemiology and pathogenesis. Nat. Rev. Endocrinol. 2019, 15, 288–298. [Google Scholar] [CrossRef]
- Chooi, Y.C.; Ding, C.; Magkos, F. The epidemiology of obesity. Metab. Clin. Exp. 2019, 92, 6–10. [Google Scholar] [CrossRef]
- Artemniak-Wojtowicz, D.; Kucharska, A.M.; Pyrżak, B. Obesity and chronic inflammation crosslinking. Cent.-Eur. J. Immunol. 2020, 45, 461–468. [Google Scholar] [CrossRef]
- Zatterale, F.; Longo, M.; Naderi, J.; Raciti, G.A.; Desiderio, A.; Miele, C.; Beguinot, F. Chronic adipose tissue inflammation linking obesity to insulin resistance and type 2 diabetes. Front. Physiol. 2020, 10, 1607. [Google Scholar] [CrossRef]
- Zhou, Y.; Chi, J.; Lv, W.; Wang, Y. Obesity and diabetes as high-risk factors for severe coronavirus disease 2019 (COVID-19). Diabetes/Metab. Res. Rev. 2021, 37, e3377. [Google Scholar] [CrossRef]
- Ghaben, A.L.; Scherer, P.E. Adipogenesis and metabolic health. Nat. Rev. Mol. Cell Biol. 2019, 20, 242–258. [Google Scholar] [CrossRef]
- Gregor, M.F.; Hotamisligil, G.S. Inflammatory mechanisms in obesity. Annu. Rev. Immunol. 2011, 29, 415–445. [Google Scholar] [CrossRef]
- Illán-Gómez, F.; Gonzálvez-Ortega, M.; Orea-Soler, I.; Alcaraz-Tafalla, M.S.; Aragón-Alonso, A.; Pascual-Díaz, M.; Pérez-Paredes, M.; Lozano-Almela, M.L. Obesity and inflammation: Change in adiponectin, C-reactive protein, tumour necrosis factor-alpha and interleukin-6 after bariatric surgery. Obes. Surg. 2012, 22, 950–955. [Google Scholar] [CrossRef]
- Shoelson, S.E.; Herrero, L.; Naaz, A. Obesity, inflammation, and insulin resistance. Gastroenterology 2007, 132, 2169–2180. [Google Scholar] [CrossRef]
- Chung, K.J.; Nati, M.; Chavakis, T.; Chatzigeorgiou, A. Innate immune cells in the adipose tissue. Rev. Endocr. Metab. Disord. 2018, 19, 283–292. [Google Scholar] [CrossRef]
- Michailidou, Z.; Gomez-Salazar, M.; Alexaki, V.I. Innate immune cells in the adipose tissue in health and metabolic disease. J. Innate Immun. 2022, 14, 4–30. [Google Scholar] [CrossRef]
- Kawai, T.; Autieri, M.V.; Scalia, R. Adipose tissue inflammation and metabolic dysfunction in obesity. Am. J. Physiol. Cell Physiol. 2021, 320, C375–C391. [Google Scholar] [CrossRef]
- Blaszczak, A.M.; Jalilvand, A.; Hsueh, W.A. Adipocytes, innate Immunity and obesity: A mini-review. Front. Immunol. 2021, 12, 650768. [Google Scholar] [CrossRef]
- Maurizi, G.; Della Guardia, L.; Maurizi, A.; Poloni, A. Adipocytes properties and crosstalk with immune system in obesity-related inflammation. J. Cell. Physiol. 2018, 233, 88–97. [Google Scholar] [CrossRef]
- Harman-Boehm, I.; Blüher, M.; Redel, H.; Sion-Vardy, N.; Ovadia, S.; Avinoach, E.; Shai, I.; Klöting, N.; Stumvoll, M.; Bashan, N.; et al. Macrophage infiltration into omental versus subcutaneous fat across different populations: Effect of regional adiposity and the comorbidities of obesity. J. Clin. Endocrinol. Metab. 2007, 92, 2240–2247. [Google Scholar] [CrossRef]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W., Jr. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef]
- Cinti, S.; Mitchell, G.; Barbatelli, G.; Murano, I.; Ceresi, E.; Faloia, E.; Wang, S.; Fortier, M.; Greenberg, A.S.; Obin, M.S. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J. Lipid Res. 2005, 46, 2347–2355. [Google Scholar] [CrossRef]
- Haase, J.; Weyer, U.; Immig, K.; Klöting, N.; Blüher, M.; Eilers, J.; Bechmann, I.; Gericke, M. Local proliferation of macrophages in adipose tissue during obesity-induced inflammation. Diabetologia 2014, 57, 562–571. [Google Scholar] [CrossRef]
- Elgazar-Carmon, V.; Rudich, A.; Hadad, N.; Levy, R. Neutrophils transiently infiltrate intra-abdominal fat early in the course of high-fat feeding. J. Lipid Res. 2008, 49, 1894–1903. [Google Scholar] [CrossRef]
- Nijhuis, J.; Rensen, S.S.; Slaats, Y.; van Dielen, F.M.; Buurman, W.A.; Greve, J.W. Neutrophil activation in morbid obesity, chronic activation of acute inflammation. Obesity 2009, 17, 2014–2018. [Google Scholar] [CrossRef]
- Shah, T.J.; Leik, C.E.; Walsh, S.W. Neutrophil infiltration and systemic vascular inflammation in obese women. Reprod. Sci. 2010, 17, 116–124. [Google Scholar] [CrossRef]
- Xu, X.; Su, S.; Wang, X.; Barnes, V.; De Miguel, C.; Ownby, D.; Pollock, J.; Snieder, H.; Chen, W.; Wang, X. Obesity is associated with more activated neutrophils in African American male youth. Int. J. Obes. 2015, 39, 26–32. [Google Scholar] [CrossRef]
- Herrero-Cervera, A.; Soehnlein, O.; Kenne, E. Neutrophils in chronic inflammatory diseases. Cell. Mol. Immunol. 2022, 19, 177–191. [Google Scholar] [CrossRef]
- Watanabe, Y.; Nagai, Y.; Honda, H.; Okamoto, N.; Yanagibashi, T.; Ogasawara, M.; Yamamoto, S.; Imamura, R.; Takasaki, I.; Hara, H.; et al. Bidirectional crosstalk between neutrophils and adipocytes promotes adipose tissue inflammation. FASEB J. 2019, 33, 11821–11835. [Google Scholar] [CrossRef]
- World Health Organization. Obesity and Overweight. Available online: https://www.who.int/en/news-room/fact-sheets/detail/obesity-and-overweight (accessed on 10 January 2022).
- Cerhan, J.R.; Moore, S.C.; Jacobs, E.J.; Kitahara, C.M.; Rosenberg, P.S.; Adami, H.O.; Ebbert, J.O.; English, D.R.; Gapstur, S.M.; Giles, G.G.; et al. A pooled analysis of waist circumference and mortality in 650,000 adults. Mayo Clin. Proc. 2014, 89, 335–345. [Google Scholar] [CrossRef]
- Alberti, K.G.; Eckel, R.H.; Grundy, S.M.; Zimmet, P.Z.; Cleeman, J.I.; Donato, K.A.; Fruchart, J.C.; James, W.P.; Loria, C.M.; Smith, S.C., Jr.; et al. Harmonizing the metabolic syndrome: A joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation 2009, 120, 1640–1645. [Google Scholar] [CrossRef]
- Favieri, F.; Forte, G.; Casagrande, M. The executive functions in overweight and obesity: A systematic review of neuropsychological cross-sectional and longitudinal studies. Front. Psychol. 2019, 10, 2126. [Google Scholar] [CrossRef]
- Jauch-Chara, K.; Oltmanns, K.M. Obesity–A neuropsychological disease? Systematic review and neuropsychological model. Prog. Neurobiol. 2014, 114, 84–101. [Google Scholar] [CrossRef]
- Tilg, H.; Zmora, N.; Adolph, T.E.; Elinav, E. The intestinal microbiota fuelling metabolic inflammation. Nat. Rev. Immunol. 2020, 20, 40–54. [Google Scholar] [CrossRef]
- Gill, S.R.; Pop, M.; Deboy, R.T.; Eckburg, P.B.; Turnbaugh, P.J.; Samuel, B.S.; Gordon, J.I.; Relman, D.A.; Fraser-Liggett, C.M.; Nelson, K.E. Metagenomic analysis of the human distal gut microbiome. Science 2006, 312, 1355–1359. [Google Scholar] [CrossRef]
- Ursell, L.K.; Haiser, H.J.; Van Treuren, W.; Garg, N.; Reddivari, L.; Vanamala, J.; Dorrestein, P.C.; Turnbaugh, P.J.; Knight, R. The intestinal metabolome: An intersection between microbiota and host. Gastroenterology 2014, 146, 1470–1476. [Google Scholar] [CrossRef]
- Cani, P.D.; Van Hul, M. Gut microbiota and obesity: Causally linked? Expert Rev. Gastroenterol. Hepatol. 2020, 14, 401–403. [Google Scholar] [CrossRef]
- Crovesy, L.; Masterson, D.; Rosado, E.L. Profile of the gut microbiota of adults with obesity: A systematic review. Eur. J. Clin. Nutr. 2020, 74, 1251–1262. [Google Scholar] [CrossRef]
- Cuevas-Sierra, A.; Ramos-Lopez, O.; Riezu-Boj, J.I.; Milagro, F.I.; Martinez, J.A. Diet, gut microbiota, and obesity: Links with host genetics and epigenetics and potential applications. Adv. Nutr. 2019, 10, S17–S30. [Google Scholar] [CrossRef]
- Cunningham, A.L.; Stephens, J.W.; Harris, D.A. A review on gut microbiota: A central factor in the pathophysiology of obesity. Lipids Health Dis. 2021, 20, 65. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.J.; Sears, C.L.; Maruthur, N. Gut microbiome and its role in obesity and insulin resistance. Ann. N. Y. Acad. Sci. 2020, 1461, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Bäckhed, F.; Ding, H.; Wang, T.; Hooper, L.V.; Koh, G.Y.; Nagy, A.; Semenkovich, C.F.; Gordon, J.I. The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl. Acad. Sci. USA 2004, 101, 15718–15723. [Google Scholar] [CrossRef] [PubMed]
- Bäckhed, F.; Manchester, J.K.; Semenkovich, C.F.; Gordon, J.I. Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc. Natl. Acad. Sci. USA 2007, 104, 979–984. [Google Scholar] [CrossRef]
- Ley, R.E.; Bäckhed, F.; Turnbaugh, P.; Lozupone, C.A.; Knight, R.D.; Gordon, J.I. Obesity alters gut microbial ecology. Proc. Natl. Acad. Sci. USA 2005, 102, 11070–11075. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef]
- Cani, P.D. Human gut microbiome: Hopes, threats and promises. Gut 2018, 67, 1716–1725. [Google Scholar] [CrossRef]
- Liu, B.N.; Liu, X.T.; Liang, Z.H.; Wang, J.H. Gut microbiota in obesity. World J. Gastroenterol. 2021, 27, 3837–3850. [Google Scholar] [CrossRef]
- Wei, Y.X.; Zheng, K.Y.; Wang, Y.G. Gut microbiota-derived metabolites as key mucosal barrier modulators in obesity. World J. Gastroenterol. 2021, 27, 5555–5565. [Google Scholar] [CrossRef]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef]
- Cani, P.D.; Bibiloni, R.; Knauf, C.; Waget, A.; Neyrinck, A.M.; Delzenne, N.M.; Burcelin, R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 2008, 57, 1470–1481. [Google Scholar] [CrossRef] [PubMed]
- Amar, J.; Burcelin, R.; Ruidavets, J.B.; Cani, P.D.; Fauvel, J.; Alessi, M.C.; Chamontin, B.; Ferriéres, J. Energy intake is associated with endotoxemia in apparently healthy men. Am. J. Clin. Nutr. 2008, 87, 1219–1223. [Google Scholar] [CrossRef] [PubMed]
- Boulangé, C.L.; Neves, A.L.; Chilloux, J.; Nicholson, J.K.; Dumas, M.E. Impact of the gut microbiota on inflammation, obesity, and metabolic disease. Genome Med. 2016, 8, 42. [Google Scholar] [CrossRef]
- Janssen, A.W.; Kersten, S. Potential mediators linking gut bacteria to metabolic health: A critical view. J. Physiol. 2017, 595, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Maslowski, K.M.; Vieira, A.T.; Ng, A.; Kranich, J.; Sierro, F.; Yu, D.; Schilter, H.C.; Rolph, M.S.; Mackay, F.; Artis, D.; et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 2009, 461, 1282–1286. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.V.; Frassetto, A.; Kowalik, E.J., Jr.; Nawrocki, A.R.; Lu, M.M.; Kosinski, J.R.; Hubert, J.A.; Szeto, D.; Yao, X.; Forrest, G.; et al. Butyrate and propionate protect against diet-induced obesity and regulate gut hormones via free fatty acid receptor 3-independent mechanisms. PLoS ONE 2012, 7, e35240. [Google Scholar] [CrossRef]
- Murphy, K.G.; Bloom, S.R. Gut hormones and the regulation of energy homeostasis. Nature 2006, 444, 854–859. [Google Scholar] [CrossRef]
- Kimura, I.; Ichimura, A.; Ohue-Kitano, R.; Igarashi, M. Free fatty acid receptors in health and disease. Physiol. Rev. 2020, 100, 171–210. [Google Scholar] [CrossRef]
- Overby, H.B.; Ferguson, J.F. Gut microbiota-derived short-chain fatty acids facilitate microbiota:host cross talk and modulate obesity and hypertension. Curr. Hypertens. Rep. 2021, 23, 8. [Google Scholar] [CrossRef]
- Vinolo, M.A.; Rodrigues, H.G.; Nachbar, R.T.; Curi, R. Regulation of inflammation by short chain fatty acids. Nutrients 2011, 3, 858–876. [Google Scholar] [CrossRef]
- Aoyama, M.; Kotani, J.; Usami, M. Butyrate and propionate induced activated or non-activated neutrophil apoptosis via HDAC inhibitor activity but without activating GPR-41/GPR-43 pathways. Nutrition 2010, 26, 653–661. [Google Scholar] [CrossRef] [PubMed]
- da Silva, T.F.; Casarotti, S.N.; de Oliveira, G.L.V.; Penna, A.L.B. The impact of probiotics, prebiotics, and synbiotics on the biochemical, clinical, and immunological markers, as well as on the gut microbiota of obese hosts. Crit. Rev. Food Sci. Nutr. 2021, 61, 337–355. [Google Scholar] [CrossRef] [PubMed]
- Green, M.; Arora, K.; Prakash, S. Microbial medicine: Prebiotic and probiotic functional foods to target obesity and metabolic syndrome. Int. J. Mol. Sci. 2020, 21, 2890. [Google Scholar] [CrossRef]
- Hijova, E. Probiotics and prebiotics, targeting obesity with functional foods. Bratisl. Lek. Listy 2021, 122, 647–652. [Google Scholar] [CrossRef] [PubMed]
- Fuhri Snethlage, C.M.; Nieuwdorp, M.; Hanssen, N.M.J. Faecal microbiota transplantation in endocrine diseases and obesity. Best Pract. Res. Clin. Endocrinol. Metab. 2021, 35, 101483. [Google Scholar] [CrossRef]
- Pitocco, D.; Di Leo, M.; Tartaglione, L.; De Leva, F.; Petruzziello, C.; Saviano, A.; Pontecorvi, A.; Ojetti, V. The role of gut microbiota in mediating obesity and diabetes mellitus. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 1548–1562. [Google Scholar] [CrossRef]
- Boscaini, S.; Leigh, S.J.; Lavelle, A.; García-Cabrerizo, R.; Lipuma, T.; Clarke, G.; Schellekens, H.; Cryan, J.F. Microbiota and body weight control: Weight watchers within? Mol. Metab. 2022, 57, 101427. [Google Scholar] [CrossRef]
- Giralt, M.; Villarroya, F. White, brown, beige/brite: Different adipose cells for different functions? Endocrinology 2013, 154, 2992–3000. [Google Scholar] [CrossRef]
- Sacks, H.; Symonds, M.E. Anatomical locations of human brown adipose tissue: Functional relevance and implications in obesity and type 2 diabetes. Diabetes 2013, 62, 1783–1790. [Google Scholar] [CrossRef]
- Chen, Y.; Pan, R.; Pfeifer, A. Fat tissues, the brite and the dark sides. Pflug. Arch. Eur. J. Physiol. 2016, 468, 1803–1807. [Google Scholar] [CrossRef]
- Matsuzawa, Y. Adipocytokines and metabolic syndrome. Semin. Vasc. Med. 2005, 5, 34–39. [Google Scholar] [CrossRef]
- Fasshauer, M.; Blüher, M. Adipokines in health and disease. Trends Pharmacol. Sci. 2015, 36, 461–470. [Google Scholar] [CrossRef]
- Ibrahim, M.M. Subcutaneous and visceral adipose tissue: Structural and functional differences. Obes. Rev. 2010, 11, 11–18. [Google Scholar] [CrossRef]
- Després, J.P.; Lemieux, I. Abdominal obesity and metabolic syndrome. Nature 2006, 444, 881–887. [Google Scholar] [CrossRef]
- Hill, J.H.; Solt, C.; Foster, M.T. Obesity associated disease risk: The role of inherent differences and location of adipose depots. Horm. Mol. Biol. Clin. Investig. 2018, 33, 20180012. [Google Scholar] [CrossRef]
- Karpe, F.; Pinnick, K.E. Biology of upper-body and lower-body adipose tissue–link to whole-body phenotypes. Nat. Rev. Endocrinol. 2015, 11, 90–100. [Google Scholar] [CrossRef]
- Piché, M.E.; Tchernof, A.; Després, J.P. Obesity phenotypes, diabetes, and cardiovascular diseases. Circ. Res. 2020, 127, e107. [Google Scholar] [CrossRef]
- Cotillard, A.; Poitou, C.; Torcivia, A.; Bouillot, J.L.; Dietrich, A.; Klöting, N.; Grégoire, C.; Lolmede, K.; Blüher, M.; Clément, K. Adipocyte size threshold matters: Link with risk of type 2 diabetes and improved insulin resistance after gastric bypass. J. Clin. Endocrinol. Metab. 2014, 99, E1466–E1470. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 2010, 140, 900–917. [Google Scholar] [CrossRef]
- Boden, G.; Duan, X.; Homko, C.; Molina, E.J.; Song, W.; Perez, O.; Cheung, P.; Merali, S. Increase in endoplasmic reticulum stress-related proteins and genes in adipose tissue of obese, insulin-resistant individuals. Diabetes 2008, 57, 2438–2444. [Google Scholar] [CrossRef]
- Michailidou, Z. Fundamental roles for hypoxia signalling in adipose tissue metabolism and inflammation in obesity. Curr. Opin. Physiol. 2019, 12, 39–43. [Google Scholar] [CrossRef]
- La Cava, A. Leptin in inflammation and autoimmunity. Cytokine 2017, 98, 51–58. [Google Scholar] [CrossRef]
- Inouye, K.E.; Shi, H.; Howard, J.K.; Daly, C.H.; Lord, G.M.; Rollins, B.J.; Flier, J.S. Absence of CC chemokine ligand 2 does not limit obesity-associated infiltration of macrophages into adipose tissue. Diabetes 2007, 56, 2242–2250. [Google Scholar] [CrossRef]
- Chung, K.J.; Chatzigeorgiou, A.; Economopoulou, M.; Garcia-Martin, R.; Alexaki, V.I.; Mitroulis, I.; Nati, M.; Gebler, J.; Ziemssen, T.; Goelz, S.E.; et al. A self-sustained loop of inflammation-driven inhibition of beige adipogenesis in obesity. Nat. Immunol. 2017, 18, 654–664. [Google Scholar] [CrossRef]
- Braune, J.; Weyer, U.; Hobusch, C.; Mauer, J.; Brüning, J.C.; Bechmann, I.; Gericke, M. IL-6 regulates M2 polarization and local proliferation of adipose tissue macrophages in obesity. J. Immunol. 2017, 198, 2927–2934. [Google Scholar] [CrossRef]
- Lumeng, C.N.; Bodzin, J.L.; Saltiel, A.R. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Investig. 2007, 117, 175–184. [Google Scholar] [CrossRef]
- Shaul, M.E.; Bennett, G.; Strissel, K.J.; Greenberg, A.S.; Obin, M.S. Dynamic, M2-like remodeling phenotypes of CD11c+ adipose tissue macrophages during high-fat diet–induced obesity in mice. Diabetes 2010, 59, 1171–1181. [Google Scholar] [CrossRef]
- Hill, A.A.; Reid Bolus, W.; Hasty, A.H. A decade of progress in adipose tissue macrophage biology. Immunol. Rev. 2014, 262, 134–152. [Google Scholar] [CrossRef]
- Suganami, T.; Tanimoto-Koyama, K.; Nishida, J.; Itoh, M.; Yuan, X.; Mizuarai, S.; Kotani, H.; Yamaoka, S.; Miyake, K.; Aoe, S.; et al. Role of the Toll-like receptor 4/NF-kappaB pathway in saturated fatty acid-induced inflammatory changes in the interaction between adipocytes and macrophages. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 84–91. [Google Scholar] [CrossRef]
- Suganami, T.; Nishida, J.; Ogawa, Y. A paracrine loop between adipocytes and macrophages aggravates inflammatory changes: Role of free fatty acids and tumor necrosis factor alpha. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2062–2068. [Google Scholar] [CrossRef]
- Erridge, C.; Samani, N.J. Saturated fatty acids do not directly stimulate Toll-like receptor signaling. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1944–1949. [Google Scholar] [CrossRef] [PubMed]
- Pal, D.; Dasgupta, S.; Kundu, R.; Maitra, S.; Das, G.; Mukhopadhyay, S.; Ray, S.; Majumdar, S.S.; Bhattacharya, S. Fetuin-A acts as an endogenous ligand of TLR4 to promote lipid-induced insulin resistance. Nat. Med. 2012, 18, 1279–1285. [Google Scholar] [CrossRef] [PubMed]
- Engin, A. The pathogenesis of obesity-associated adipose tissue inflammation. Adv. Exp. Med. Biol. 2017, 960, 221–245. [Google Scholar] [CrossRef] [PubMed]
- Russo, L.; Lumeng, C.N. Properties and functions of adipose tissue macrophages in obesity. Immunology 2018, 155, 407–417. [Google Scholar] [CrossRef]
- Lee, B.C.; Lee, J. Cellular and molecular players in adipose tissue inflammation in the development of obesity-induced insulin resistance. Biochim. Biophys. Acta 2014, 1842, 446–462. [Google Scholar] [CrossRef]
- Suganami, T.; Ogawa, Y. Adipose tissue macrophages: Their role in adipose tissue remodeling. J. Leukoc. Biol. 2010, 88, 33–39. [Google Scholar] [CrossRef]
- Asghar, A.; Sheikh, N. Role of immune cells in obesity induced low grade inflammation and insulin resistance. Cell. Immunol. 2017, 315, 18–26. [Google Scholar] [CrossRef]
- Liew, P.X.; Kubes, P. The neutrophil’s role during health and disease. Physiol. Rev. 2019, 99, 1223–1248. [Google Scholar] [CrossRef]
- Fine, N.; Tasevski, N.; McCulloch, C.A.; Tenenbaum, H.C.; Glogauer, M. The neutrophil: Constant defender and first responder. Front. Immunol. 2020, 11, 571085. [Google Scholar] [CrossRef]
- Uribe-Querol, E.; Rosales, C. Phagocytosis: Our current understading of a universal biological process. Front. Immunol. 2020, 11, 1066. [Google Scholar] [CrossRef]
- Rosales, C.; Lowell, C.A.; Schnoor, M.; Uribe-Querol, E. Neutrophils: Their role in innate and adaptive immunity 2017. J. Immunol. Res. 2017, 2017, 9748345. [Google Scholar] [CrossRef] [PubMed]
- Rosales, C.; Uribe-Querol, E. Phagocytosis: A fundamental process in immunity. BioMed Res. Int. 2017, 2017, 9042851. [Google Scholar] [CrossRef] [PubMed]
- Tecchio, C.; Cassatella, M.A. Neutrophil-derived chemokines on the road to immunity. Semin. Immunol. 2016, 28, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Rosales, C. Neutrophils at the crossroads of innate and adaptive immunity. J. Leukoc. Biol. 2020, 108, 377–396. [Google Scholar] [CrossRef]
- Tsai, C.Y.; Hsieh, S.C.; Liu, C.W.; Lu, C.S.; Wu, C.H.; Liao, H.T.; Chen, M.H.; Li, K.J.; Shen, C.Y.; Kuo, Y.M.; et al. Cross-talk among polymorphonuclear neutrophils, immune, and non-Immune cells via released cytokines, granule proteins, microvesicles, and neutrophil extracellular trap formation: A novel concept of biology and pathobiology for neutrophils. Int. J. Mol. Sci. 2021, 22, 3119. [Google Scholar] [CrossRef]
- Herishanu, Y.; Rogowski, O.; Polliack, A.; Marilus, R. Leukocytosis in obese individuals: Possible link in patients with unexplained persistent neutrophilia. Eur. J. Haematol. 2006, 76, 516–520. [Google Scholar] [CrossRef]
- Kim, J.A.; Park, H.S. White blood cell count and abdominal fat distribution in female obese adolescents. Metab. Clin. Exp. 2008, 57, 1375–1379. [Google Scholar] [CrossRef]
- Weir, A.B.; Lewis, J.B., Jr.; Arteta-Bulos, R. Chronic idiopathic neutrophilia: Experience and recommendations. South. Med. J. 2011, 104, 499–504. [Google Scholar] [CrossRef]
- Rhee, H.; Love, T.; Harrington, D. Blood neutrophil count is associated with body mass index in adolescents with asthma. JSM Allergy Asthma 2018, 3, 1019. [Google Scholar]
- Gállego-Suárez, C.; Bulan, A.; Hirschfeld, E.; Wachowiak, P.; Abrishami, S.; Griffin, C.; Sturza, J.; Tzau, A.; Hayes, T.; Woolford, S.J.; et al. Enhanced myeloid leukocytes in obese children and adolescents at risk for metabolic impairment. Front. Endocrinol. 2020, 11, 327. [Google Scholar] [CrossRef]
- Zernecke, A.; Bot, I.; Djalali-Talab, Y.; Shagdarsuren, E.; Bidzhekov, K.; Meiler, S.; Krohn, R.; Schober, A.; Sperandio, M.; Soehnlein, O.; et al. Protective role of CXC receptor 4/CXC ligand 12 unveils the importance of neutrophils in atherosclerosis. Circ. Res. 2008, 102, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.; Jasmin, S.; Fariduddin, M.; Alam, S.M.K.; Arslan, M.I.; Biswas, S.K. Neutrophil elastase and myeloperoxidase mRNA expression in overweight and obese subjects. Mol. Biol. Rep. 2018, 45, 1245–1252. [Google Scholar] [CrossRef] [PubMed]
- Roberts, H.M.; Grant, M.M.; Hubber, N.; Super, P.; Singhal, R.; Chapple, I.L.C. Impact of bariatric surgical intervention on peripheral blood neutrophil (PBN) function in obesity. Obes. Surg. 2018, 28, 1611–1621. [Google Scholar] [CrossRef] [PubMed]
- Dixon, J.B.; O’Brien, P.E. Obesity and the white blood cell count: Changes with sustained weight loss. Obes. Surg. 2006, 16, 251–257. [Google Scholar] [CrossRef]
- Festa, A.; D’Agostino, R., Jr.; Williams, K.; Karter, A.J.; Mayer-Davis, E.J.; Tracy, R.P.; Haffner, S.M. The relation of body fat mass and distribution to markers of chronic inflammation. Int. J. Obes. Relat. Metab. Disord. 2001, 25, 1407–1415. [Google Scholar] [CrossRef]
- Bruun, J.M.; Pedersen, S.B.; Richelsen, B. Regulation of interleukin 8 production and gene expression in human adipose tissue in vitro. J. Clin. Endocrinol. Metab. 2001, 86, 1267–1273. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.; Arner, P.; Caro, J.F.; Atkinson, R.L.; Spiegelman, B.M. Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J. Clin. Investig. 1995, 95, 2409–2415. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.; Shargill, N.S.; Spiegelman, B.M. Adipose expression of tumor necrosis factor-alpha: Direct role in obesity-linked insulin resistance. Science 1993, 259, 87–91. [Google Scholar] [CrossRef]
- Kern, P.A.; Saghizadeh, M.; Ong, J.M.; Bosch, R.J.; Deem, R.; Simsolo, R.B. The expression of tumor necrosis factor in human adipose tissue. Regulation by obesity, weight loss, and relationship to lipoprotein lipase. J. Clin. Investig. 1995, 95, 2111–2119. [Google Scholar] [CrossRef]
- Mohamed-Ali, V.; Goodrick, S.; Rawesh, A.; Katz, D.R.; Miles, J.M.; Yudkin, J.S.; Klein, S.; Coppack, S.W. Subcutaneous adipose tissue releases interleukin-6, but not tumor necrosis factor-alpha, in vivo. J. Clin. Endocrinol. Metab. 1997, 82, 4196–4200. [Google Scholar] [CrossRef]
- Zhang, H.H.; Kumar, S.; Barnett, A.H.; Eggo, M.C. Dexamethasone inhibits tumor necrosis factor-alpha-induced apoptosis and interleukin-1 beta release in human subcutaneous adipocytes and preadipocytes. J. Clin. Endocrinol. Metab. 2001, 86, 2817–2825. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rusten, L.S.; Jacobsen, F.W.; Lesslauer, W.; Loetscher, H.; Smeland, E.B.; Jacobsen, S.E. Bifunctional effects of tumor necrosis factor alpha (TNF alpha) on the growth of mature and primitive human hematopoietic progenitor cells: Involvement of p55 and p75 TNF receptors. Blood 1994, 83, 3152–3159. [Google Scholar] [CrossRef] [PubMed]
- Sieff, C.A.; Niemeyer, C.M.; Mentzer, S.J.; Faller, D.V. Interleukin-1, tumor necrosis factor, and the production of colony-stimulating factors by cultured mesenchymal cells. Blood 1988, 72, 1316–1323. [Google Scholar] [CrossRef]
- Veltri, S.; Smith, J.W., 2nd. Interleukin 1 trials in cancer patients: A review of the toxicity, antitumor and hematopoietic effects. Stem Cells 1996, 14, 164–176. [Google Scholar] [CrossRef] [PubMed]
- Suwa, T.; Hogg, J.C.; English, D.; Van Eeden, S.F. Interleukin-6 induces demargination of intravascular neutrophils and shortens their transit in marrow. Am. J. Physiol. Heart Circ. Physiol. 2000, 279, H2954–H2960. [Google Scholar] [CrossRef] [PubMed]
- Bruno, A.; Conus, S.; Schmid, I.; Simon, H.U. Apoptotic pathways are inhibited by leptin receptor activation in neutrophils. J. Immunol. 2005, 174, 8090–8096. [Google Scholar] [CrossRef]
- Claycombe, K.; King, L.E.; Fraker, P.J. A role for leptin in sustaining lymphopoiesis and myelopoiesis. Proc. Natl. Acad. Sci. USA 2008, 105, 2017–2021. [Google Scholar] [CrossRef]
- Laharrague, P.; Oppert, J.M.; Brousset, P.; Charlet, J.P.; Campfield, A.; Fontanilles, A.M.; Guy-Grand, B.; Corberand, J.X.; Pénicaud, L.; Casteilla, L. High concentration of leptin stimulates myeloid differentiation from human bone marrow CD34+ progenitors: Potential involvement in leukocytosis of obese subjects. Int. J. Obes. Relat. Metab. Disord. 2000, 24, 1212–1216. [Google Scholar] [CrossRef]
- Zahorec, R. Neutrophil-to-lymphocyte ratio, past, present and future perspectives. Bratisl. Lek. Listy 2021, 122, 474–488. [Google Scholar] [CrossRef]
- Keskin Kurt, R.; Okyay, A.G.; Hakverdi, A.U.; Gungoren, A.; Dolapcioglu, K.S.; Karateke, A.; Dogan, M.O. The effect of obesity on inflammatory markers in patients with PCOS: A BMI-matched case-control study. Arch. Gynecol. Obstet. 2014, 290, 315–319. [Google Scholar] [CrossRef]
- Aydin, M.; Yilmaz, A.; Donma, M.M.; Tulubas, F.; Demirkol, M.; Erdogan, M.; Gurel, A. Neutrophil/lymphocyte ratio in obese adolescents. North. Clin. Istanb. 2015, 2, 87–91. [Google Scholar] [CrossRef][Green Version]
- Fang, Q.; Tong, Y.W.; Wang, G.; Zhang, N.; Chen, W.G.; Li, Y.F.; Shen, K.W.; Wu, B.W.; Chen, X.S. Neutrophil-to-lymphocyte ratio, obesity, and breast cancer risk in Chinese population. Medicine 2018, 97, e11692. [Google Scholar] [CrossRef]
- Kain, V.; Van Der Pol, W.; Mariappan, N.; Ahmad, A.; Eipers, P.; Gibson, D.L.; Gladine, C.; Vigor, C.; Durand, T.; Morrow, C.; et al. Obesogenic diet in aging mice disrupts gut microbe composition and alters neutrophil:lymphocyte ratio, leading to inflamed milieu in acute heart failure. FASEB J. 2019, 33, 6456–6469. [Google Scholar] [CrossRef]
- Țaranu, I.; Lazea, C.; Creț, V.; Răcătăianu, N.; Iancu, M.; Bolboacă, S.D. Inflammation-related markers and thyroid function measures in pediatric patients: Is the grade of obesity relevant? Diagnostics 2021, 11, 485. [Google Scholar] [CrossRef]
- Eren, C.; Cecen, S. The relationship between childhood obesity with inflammatory mediator. J. Pak. Med. Assoc. 2020, 70, 1737–1741. [Google Scholar] [CrossRef]
- Mărginean, C.O.; Meliţ, L.E.; Huţanu, A.; Ghiga, D.V.; Săsăran, M.O. The gap between overweight and obesity status in children—(STROBE-compliant article). Medicine 2021, 100, e24520. [Google Scholar] [CrossRef]
- Orlandini, L.F.; Pimentel, F.F.; Andrade, J.M.; Reis, F.J.C.D.; Mattos-Arruda, L.; Tiezzi, D.G. Obesity and high neutrophil-to-lymphocyte ratio are prognostic factors in non-metastatic breast cancer patients. Braz. J. Med. Biol. Res. 2021, 54, e11409. [Google Scholar] [CrossRef]
- Trellakis, S.; Rydleuskaya, A.; Fischer, C.; Canbay, A.; Tagay, S.; Scherag, A.; Bruderek, K.; Schuler, P.J.; Brandau, S. Low adiponectin, high levels of apoptosis and increased peripheral blood neutrophil activity in healthy obese subjects. Obes. Facts 2012, 5, 305–318. [Google Scholar] [CrossRef]
- Osadnik, T.; Bujak, K.; Osadnik, K.; Czarnecka, H.; Pawlas, N.; Reguła, R.; Fronczek, M.; Lejawa, M.; Gawlita, M.; Gonera, M.; et al. Novel inflammatory biomarkers may reflect subclinical inflammation in young healthy adults with obesity. Endokrynol. Pol. 2019, 70, 135–142. [Google Scholar] [CrossRef]
- Rodríguez-Rodríguez, E.; López-Sobaler, A.M.; Ortega, R.M.; Delgado-Losada, M.L.; López-Parra, A.M.; Aparicio, A. Association between neutrophil-to-lymphocyte ratio with abdominal obesity and healthy eating index in a representative older Spanish population. Nutrients 2020, 12, 855. [Google Scholar] [CrossRef]
- Suárez-Cuenca, J.A.; Ruíz-Hernández, A.S.; Mendoza-Castañeda, A.A.; Domínguez-Pérez, G.A.; Hernández-Patricio, A.; Vera-Gómez, E.; De la Peña-Sosa, G.; Banderas-Lares, D.Z.; Montoya-Ramírez, J.; Blas-Azotla, R.; et al. Neutrophil-to-lymphocyte ratio and its relation with pro-inflammatory mediators, visceral adiposity and carotid intima-media thickness in population with obesity. Eur. J. Clin. Investig. 2019, 49, e13085. [Google Scholar] [CrossRef]
- Yilmaz, H.; Ucan, B.; Sayki, M.; Unsal, I.; Sahin, M.; Ozbek, M.; Delibasi, T. Usefulness of the neutrophil-to-lymphocyte ratio to prediction of type 2 diabetes mellitus in morbid obesity. Diabetes Metab. Syndr. 2015, 9, 299–304. [Google Scholar] [CrossRef]
- Ferrante, A.W., Jr. The immune cells in adipose tissue. Diabetes Obes. Metab. 2013, 15, 34–38. [Google Scholar] [CrossRef]
- Skinner, A.C.; Steiner, M.J.; Henderson, F.W.; Perrin, E.M. Multiple markers of inflammation and weight status: Cross-sectional analyses throughout childhood. Pediatrics 2010, 125, e801–e809. [Google Scholar] [CrossRef]
- Mantovani, A.; Cassatella, M.A.; Constantini, C.; Jaillon, S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat. Rev. Immunol. 2011, 11, 519–531. [Google Scholar] [CrossRef]
- Talukdar, S.; Oh, D.Y.; Bandyopadhyay, G.; Li, D.; Xu, J.; McNelis, J.; Lu, M.; Li, P.; Yan, Q.; Zhu, Y.; et al. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat. Med. 2012, 18, 1407–1412. [Google Scholar] [CrossRef]
- Girbl, T.; Lenn, T.; Perez, L.; Rolas, L.; Barkaway, A.; Thiriot, A.; Del Fresno, C.; Lynam, E.; Hub, E.; Thelen, M.; et al. Distinct compartmentalization of the chemokines CXCL1 and CXCL2 and the atypical receptor ACKR1 determine discrete stages of neutrophil diapedesis. Immunity 2018, 49, 1062–1076. [Google Scholar] [CrossRef]
- Tynan, G.A.; Hearnden, C.H.; Oleszycka, E.; Lyons, C.L.; Coutts, G.; O’Connell, J.; Corrigan, M.A.; Lynch, L.; Campbell, M.; Callanan, J.J.; et al. Endogenous oils derived from human adipocytes are potent adjuvants that promote IL-1α-dependent inflammation. Diabetes 2014, 63, 2037–2050. [Google Scholar] [CrossRef]
- Dam, V.; Sikder, T.; Santosa, S. From neutrophils to macrophages: Differences in regional adipose tissue depots. Obes. Rev. 2016, 17, 1–17. [Google Scholar] [CrossRef]
- Trim, W.; Turner, J.E.; Thompson, D. Parallels in immunometabolic adipose tissue dysfunction with ageing and obesity. Front. Immunol. 2018, 9, 169. [Google Scholar] [CrossRef]
- Mansuy-Aubert, V.; Zhou, Q.L.; Xie, X.; Gong, Z.; Huang, J.Y.; Khan, A.R.; Aubert, G.; Candelaria, K.; Thomas, S.; Shin, D.J.; et al. Imbalance between neutrophil elastase and its inhibitor α1-antitrypsin in obesity alters insulin sensitivity, inflammation, and energy expenditure. Cell Metab. 2013, 17, 534–548. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Foundations of immunometabolism and implications for metabolic health and disease. Immunity 2017, 47, 406–420. [Google Scholar] [CrossRef] [PubMed]
- Vandanmagsar, B.; Youm, Y.H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 2011, 17, 179–188. [Google Scholar] [CrossRef]
- Wiersma, J.J.; Meuwese, M.C.; van Miert, J.N.; Kastelein, A.; Tijssen, J.G.; Piek, J.J.; Trip, M.D. Diabetes mellitus type 2 is associated with higher levels of myeloperoxidase. Med. Sci. Monit. 2008, 14, CR406–CR410. [Google Scholar]
- Sun, Z.; Dragon, S.; Becker, A.; Gounni, A.S. Leptin inhibits neutrophil apoptosis in children via ERK/NF-κB-dependent pathways. PLoS ONE 2013, 8, e55249. [Google Scholar] [CrossRef]
- Bosco, A.M.; Almeida, B.F.M.; Valadares, T.C.; Baptistiolli, L.; Hoffmann, D.J.; Pereira, A.A.F.; Lima, V.M.F.; Ciarlini, P.C. Preactivation of neutrophils and systemic oxidative stress in dogs with hyperleptinemia. Vet. Immunol. Immunopathol. 2018, 202, 18–24. [Google Scholar] [CrossRef]
- Salinas, C.; Espinosa, G.; Morales, N.; Henríquez, C.; Morán, G.; Gajardo, G.; Uberti, B. Assessment of peripheral blood neutrophil respiratory burst, phagocytosis and apoptosis in obese non-insulin dysregulated horses. Res. Vet. Sci. 2020, 132, 127–132. [Google Scholar] [CrossRef]
- Wilson, R.M.; Reeves, W.G. Neutrophil phagocytosis and killing in insulin-dependent diabetes. Clin. Exp. Immunol. 1986, 63, 478–484. [Google Scholar]
- Mancuso, P.; Curtis, J.L.; Weitzel, A.M.; Griffin, C.A.; Bouchard, B.; Freeman, C.M.; Bridges, D.; Singer, K. Diet-induced obesity in mice impairs host defense against Klebsiella pneumonia in vivo and glucose transport and bactericidal functions in neutrophils in vitro. Am. J. Physiol. Lung Cell. Mol. Physiol. 2022, 322, L116–L128. [Google Scholar] [CrossRef]
- Dobner, J.; Kaser, S. Body mass index and the risk of infection—From underweight to obesity. Clin. Microbiol. Infect. 2018, 24, 24–28. [Google Scholar] [CrossRef]
- Frydrych, L.M.; Bian, G.; O’Lone, D.E.; Ward, P.A.; Delano, M.J. Obesity and type 2 diabetes mellitus drive immune dysfunction, infection development, and sepsis mortality. J. Leukoc. Biol. 2018, 104, 525–534. [Google Scholar] [CrossRef]
- Amulic, B.; Cazalet, C.; Hayes, G.L.; Metzler, K.D.; Zychlinsky, A. Neutrophil function: From mechanisms to disease. Annu. Rev. Immunol. 2012, 30, 459–489. [Google Scholar] [CrossRef]
- Soehnlein, O.; Zernecke, A.; Eriksson, E.E.; Rothfuchs, A.G.; Pham, C.T.; Herwald, H.; Bidzhekov, K.; Rottenberg, M.E.; Weber, C.; Lindbom, L. Neutrophil secretion products pave the way for inflammatory monocytes. Blood 2008, 112, 1461–1471. [Google Scholar] [CrossRef]
- Soehnlein, O.; Lindbom, L.; Weber, C. Mechanisms underlying neutrophil-mediated monocyte recruitment. Blood 2008, 114, 4613–4623. [Google Scholar] [CrossRef]
- Gao, C.H.; Dong, H.L.; Tai, L.; Gao, X.M. Lactoferrin-containing immunocomplexes drive the conversion of human macrophages from M2- into M1-like phenotype. Front. Immunol. 2018, 9, 37. [Google Scholar] [CrossRef]
- Xing, L.; Zhongqian, L.; Chunmei, S.; Pingfa, C.; Lei, H.; Qin, J.; Genhua, M.; Yijun, D. Activation of M1 macrophages in sepsis-induced acute kidney injury in response to heparin-binding protein. PLoS ONE 2018, 13, e0196423. [Google Scholar] [CrossRef]
- Soehnlein, O.; Kai-Larsen, Y.; Frithiof, R.; Sorensen, O.E.; Kenne, E.; Scharffetter-Kochanek, K.; Eriksson, E.E.; Herwald, H.; Agerberth, B.; Lindbom, L. Neutrophil primary granule proteins HBP and HNP1-3 boost bacterial phagocytosis by human and murine macrophages. J. Clin. Investig. 2008, 118, 3491–3502. [Google Scholar] [CrossRef]
- Ribon, M.; Seninet, S.; Mussard, J.; Sebbag, M.; Clavel, C.; Serre, G.; Boissier, M.C.; Semerano, L.; Decker, P. Neutrophil extracellular traps exert both pro- and anti-inflammatory actions in rheumatoid arthritis that are modulated by C1q and LL-37. J. Autoimmun. 2019, 98, 122–131. [Google Scholar] [CrossRef]
- van den Bosch, M.H.; Blom, A.B.; Schelbergen, R.F.; Koenders, M.I.; van de Loo, F.A.; van den Berg, W.B.; Vogl, T.; Roth, J.; van der Kraan, P.M.; van Lent, P.L. Alarmin S100A9 induces proinflammatory and catabolic effects predominantly in the M1 macrophages of human osteoarthritic synovium. J. Rheumatol. 2016, 43, 1874–1884. [Google Scholar] [CrossRef]
- Castanheira, F.V.S.; Kubes, P. Neutrophils and NETs in modulating acute and chronic inflammation. Blood 2019, 133, 2178–2185. [Google Scholar] [CrossRef]
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2018, 18, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, Q.; Venugopal, J.; Wang, J.; Kleiman, K.; Guo, C.; Eitzman, D.T. Obesity-induced endothelial dysfunction is prevented by neutrophil extracellular trap inhibition. Sci. Rep. 2018, 8, 4881. [Google Scholar] [CrossRef] [PubMed]
- D’Abbondanza, M.; Martorelli, E.E.; Ricci, M.A.; De Vuono, S.; Migliola, E.N.; Godino, C.; Corradetti, S.; Siepi, D.; Paganelli, M.T.; Maugeri, N.; et al. Increased plasmatic NETs by-products in patients in severe obesity. Sci. Rep. 2019, 9, 14678. [Google Scholar] [CrossRef] [PubMed]
- Valeria Oliveira de Sousa, B.; de Freitas, D.F.; Monteiro-Junior, R.S.; Mendes, I.H.R.; Sousa, J.N.; Guimarães, V.H.D.; Santos, S.H.S. Physical exercise, obesity, inflammation and neutrophil extracellular traps (NETs): A review with bioinformatics analysis. Mol. Biol. Rep. 2021, 48, 4625–4635. [Google Scholar] [CrossRef] [PubMed]
- Freitas, D.F.; Colón, D.F.; Silva, R.L.; Santos, E.M.; Guimarães, V.H.D.; Ribeiro, G.H.M.; de Paula, A.M.B.; Guimarães, A.L.S.; Dos Reis, S.T.; Cunha, F.Q.; et al. Neutrophil extracellular traps (NETs) modulate inflammatory profile in obese humans and mice: Adipose tissue role on NETs levels. Mol. Biol. Rep. 2022, 49, 3225–3236. [Google Scholar] [CrossRef]
- Cichon, I.; Ortmann, W.; Santocki, M.; Opydo-Chanek, M.; Kolaczkowska, E. Scrutinizing mechanisms of the ‘obesity paradox in sepsis’: Obesity is accompanied by diminished formation of neutrophil extracellular traps (NETs) due to restricted neutrophil-platelet interactions. Cells 2021, 10, 384. [Google Scholar] [CrossRef]
- Carestia, A.; Frechtel, G.; Cerrone, G.; Linari, M.A.; Gonzalez, C.D.; Casais, P.; Schattner, M. NETosis before and after hyperglycemic control in type 2 diabetes mellitus patients. PLoS ONE 2016, 11, e0168647. [Google Scholar] [CrossRef]
- Park, J.H.; Kim, J.E.; Gu, J.Y.; Yoo, H.J.; Park, S.H.; Kim, Y.I.; Nam-Goong, I.S.; Kim, E.S.; Kim, H.K. Evaluation of circulating markers of neutrophil extracellular trap (NET) formation as risk factors for diabetic retinopathy in a case-control association study. Exp. Clin. Endocrinol. Diabetes 2016, 124, 557–561. [Google Scholar] [CrossRef]
- Wang, L.; Zhou, X.; Yin, Y.; Mai, Y.; Wang, D.; Zhang, X. Hyperglycemia induces neutrophil extracellular traps formation through an NADPH oxidase-dependent pathway in diabetic retinopathy. Front. Immunol. 2019, 9, 3076. [Google Scholar] [CrossRef]
- Menegazzo, L.; Ciciliot, S.; Poncina, N.; Mazzucato, M.; Persano, M.; Bonora, B.; Albiero, M.; Vigili de Kreutzenberg, S.; Avogaro, A.; Fadini, G.P. NETosis is induced by high glucose and associated with type 2 diabetes. Acta Diabetol. 2015, 52, 497–503. [Google Scholar] [CrossRef]
- Jafar, N.; Edriss, H.; Nugent, K. The effect of short-term hyperglycemia on the innate immune system. Am. J. Med. Sci. 2016, 351, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Joshi, M.B.; Lad, A.; Bharath Prasad, A.S.; Balakrishnan, A.; Ramachandra, L.; Satyamoorthy, K. High glucose modulates IL-6 mediated immune homeostasis through impeding neutrophil extracellular trap formation. FEBS Lett. 2013, 587, 2241–2246. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.L.; Demers, M.; Martinod, K.; Gallant, M.; Wang, Y.; Goldfine, A.B.; Kahn, C.R.; Wagner, D.D. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat. Med. 2015, 21, 815–819. [Google Scholar] [CrossRef]
- Fadini, G.P.; Menegazzo, L.; Rigato, M.; Scattolini, V.; Poncina, N.; Bruttocao, A.; Ciciliot, S.; Mammano, F.; Ciubotaru, C.D.; Brocco, E.; et al. NETosis delays diabetic wound healing in mice and humans. Diabetes 2016, 65, 1061–1071. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.T.; Chen, L.; Chen, W.L.; Li, N.; Chen, M.J.; Li, X.; Zheng, X.; Zhao, Y.Z.; Wu, Y.X.; Xian, M.; et al. Hydrogen sulfide primes diabetic wound to close through inhibition of NETosis. Mol. Cell. Endocrinol. 2019, 480, 74–82. [Google Scholar] [CrossRef]
- Lavery, L.A.; Oz, O.K.; Bhavan, K.; Wukich, D.K. Diabetic foot syndrome in the twenty-first century. Clin. Podiatr. Med. Surg. 2019, 36, 355–359. [Google Scholar] [CrossRef]
- Lee, M.K.S.; Sreejit, G.; Nagareddy, P.R.; Murphy, A.J. Attack of the NETs! NETosis primes IL-1β-mediated inflammation in diabetic foot ulcers. Clin. Sci. 2020, 134, 1399–1401. [Google Scholar] [CrossRef]
- Liu, C.; Whitener, R.L.; Lin, A.; Xu, Y.; Chen, J.; Savinov, A.; Leiding, J.W.; Wallet, M.A.; Mathews, C.E. Neutrophil cytosolic factor 1 in dendritic cells promotes autoreactive CD8+ T cell activation via cross-presentation in type 1 diabetes. Front. Immunol. 2019, 10, 952. [Google Scholar] [CrossRef]
- Rodríguez-Espinosa, O.; Rojas-Espinosa, O.; Moreno-Altamirano, M.M.; López-Villegas, E.O.; Sánchez-García, F.J. Metabolic requirements for neutrophil extracellular traps formation. Immunology 2015, 145, 213–224. [Google Scholar] [CrossRef]
- Cichon, I.; Ortmann, W.; Kolaczkowska, E. Metabolic pathways involved in formation of spontaneous and lipopolysaccharide-induced neutrophil extracellular traps (NETs) differ in obesity and systemic Inflammation. Int. J. Mol. Sci. 2021, 22, 7718. [Google Scholar] [CrossRef]
- Petersmann, A.; Müller-Wieland, D.; Müller, U.A.; Landgraf, R.; Nauck, M.; Freckmann, G.; Heinemann, L.; Schleicher, E. Definition, classification and diagnosis of diabetes mellitus. Exp. Clin. Endocrinol. Diabetes 2019, 127, S1–S7. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Diabetes (Fact Sheet N°312). Available online: https://web.archive.org/web/20130826174444/http://www.who.int/mediacentre/factsheets/fs312/en/ (accessed on 25 January 2022).
- Demir, S.; Nawroth, P.P.; Herzig, S.; Ekim Üstünel, B. Emerging targets in type 2 diabetes and diabetic complications. Adv. Sci. 2021, 8, e2100275. [Google Scholar] [CrossRef] [PubMed]
- Lotfy, M.; Adeghate, J.; Kalasz, H.; Singh, J.; Adeghate, E. Chronic complications of diabetes mellitus: A mini review. Curr. Diabetes Rev. 2017, 13, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Rohm, T.V.; Meier, D.T.; Olefsky, J.M.; Donath, M.Y. Inflammation in obesity, diabetes, and related disorders. Immunity 2022, 55, 31–55. [Google Scholar] [CrossRef] [PubMed]
- Bollyky, J.B.; Xu, P.; Butte, A.J.; Wilson, D.M.; Beam, C.A.; Greenbaum, C.J.; Type 1 Diabetes TrialNet Study Group. Heterogeneity in recent-onset type 1 diabetes—A clinical trial perspective. Diabetes/Metab. Res. Rev. 2015, 31, 588–594. [Google Scholar] [CrossRef] [PubMed]
- Pearson, J.A.; Wong, F.S.; Wen, L. The importance of the non obese diabetic (NOD) mouse model in autoimmune diabetes. J. Autoimmun. 2016, 66, 76–88. [Google Scholar] [CrossRef]
- Chen, Y.G.; Mathews, C.E.; Driver, J.P. The role of NOD mice in type 1 diabetes research: Lessons from the past and recommendations for the future. Front. Endocrinol. 2018, 9, 51. [Google Scholar] [CrossRef]
- Miao, D.; Yu, L.; Eisenbarth, G.S. Role of autoantibodies in type 1 diabetes. Front. Biosci. 2007, 12, 1889–1898. [Google Scholar] [CrossRef]
- Kawasaki, E. Type 1 diabetes and autoimmunity. Clin. Pediatr. Endocrinol. 2014, 23, 99–105. [Google Scholar] [CrossRef]
- Diana, J.; Simoni, Y.; Furio, L.; Beaudoin, L.; Agerberth, B.; Barrat, F.; Lehuen, A. Crosstalk between neutrophils, B-1a cells and plasmacytoid dendritic cells initiates autoimmune diabetes. Nat. Med. 2013, 19, 65–73. [Google Scholar] [CrossRef]
- Valle, A.; Giamporcaro, G.M.; Scavini, M.; Stabilini, A.; Grogan, P.; Bianconi, E.; Sebastiani, G.; Masini, M.; Maugeri, N.; Porretti, L.; et al. Reduction of circulating neutrophils precedes and accompanies type 1 diabetes. Diabetes 2013, 62, 2072–2077. [Google Scholar] [CrossRef] [PubMed]
- Harsunen, M.H.; Puff, R.; D’Orlando, O.; Giannopoulou, E.; Lachmann, L.; Beyerlein, A.; von Meyer, A.; Ziegler, A.G. Reduced blood leukocyte and neutrophil numbers in the pathogenesis of type 1 diabetes. Horm. Metab. Res. 2013, 45, 467–470. [Google Scholar] [CrossRef] [PubMed]
- Salami, F.; Lee, H.S.; Freyhult, E.; Elding Larsson, H.; Lernmark, Å.; Törn, C.; TEDDY Study Group. Reduction in white blood cell, neutrophil, and red blood cell counts related to sex, HLA, and islet autoantibodies in Swedish TEDDY children at increased risk for type 1 diabetes. Diabetes 2018, 67, 2329–2336. [Google Scholar] [CrossRef] [PubMed]
- Vecchio, F.; Lo Buono, N.; Stabilini, A.; Nigi, L.; Dufort, M.J.; Geyer, S.; Rancoita, P.M.; Cugnata, F.; Mandelli, A.; Valle, A.; et al. Abnormal neutrophil signature in the blood and pancreas of presymptomatic and symptomatic type 1 diabetes. JCI Insight 2018, 3, e122146. [Google Scholar] [CrossRef]
- Huang, J.; Xiao, Y.; Zheng, P.; Zhou, W.; Wang, Y.; Huang, G.; Xu, A.; Zhou, Z. Distinct neutrophil counts and functions in newly diagnosed type 1 diabetes, latent autoimmune diabetes in adults, and type 2 diabetes. Diabetes/Metab. Res. Rev. 2019, 35, e3064. [Google Scholar] [CrossRef]
- Klocperk, A.; Petruzelkova, L.; Pavlikova, M.; Rataj, M.; Kayserova, J.; Pruhova, S.; Kolouskova, S.; Sklenarova, J.; Parackova, Z.; Sediva, A.; et al. Changes in innate and adaptive immunity over the first year after the onset of type 1 diabetes. Acta Diabetol. 2020, 57, 297–307. [Google Scholar] [CrossRef]
- Qin, J.; Fu, S.; Speake, C.; Greenbaum, C.J.; Odegard, J.M. NETosis-associated serum biomarkers are reduced in type 1 diabetes in association with neutrophil count. Clin. Exp. Immunol. 2016, 184, 318–322. [Google Scholar] [CrossRef]
- Dufort, M.J.; Greenbaum, C.J.; Speake, C.; Linsley, P.S. Cell type-specific immune phenotypes predict loss of insulin secretion in new-onset type 1 diabetes. JCI Insight 2019, 4, e125556. [Google Scholar] [CrossRef]
- Shu, L.; Zhong, L.; Xiao, Y.; Wu, X.; Liu, Y.; Jiang, X.; Tang, T.; Hoo, R.; Zhou, Z.; Xu, A. Neutrophil elastase triggers the development of autoimmune diabetes by exacerbating innate immune responses in pancreatic islets of non-obese diabetic mice. Clin. Sci. 2020, 134, 1679–1696. [Google Scholar] [CrossRef]
- Diana, J.; Lehuen, A. Macrophages and β-cells are responsible for CXCR2-mediated neutrophil infiltration of the pancreas during autoimmune diabetes. EMBO Mol. Med. 2014, 6, 1090–1104. [Google Scholar] [CrossRef]
- Garciafigueroa, Y.; Phillips, B.E.; Engman, C.; Trucco, M.; Giannoukakis, N. Neutrophil-associated inflammatory changes in the pre-diabetic pancreas of early-age NOD mice. Front. Endocrinol. 2021, 12, 565981. [Google Scholar] [CrossRef] [PubMed]
- Németh, T.; Mócsai, A. The role of neutrophils in autoimmune diseases. Immunol. Lett. 2012, 143, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xiao, Y.; Zhong, L.; Ye, D.; Zhang, J.; Tu, Y.; Bornstein, S.R.; Zhou, Z.; Lam, K.S.; Xu, A. Increased neutrophil elastase and proteinase 3 and augmented NETosis are closely associated with β-cell autoimmunity in patients with type 1 diabetes. Diabetes 2014, 63, 4239–4248. [Google Scholar] [CrossRef] [PubMed]
- Njeim, R.; Azar, W.S.; Fares, A.H.; Azar, S.T.; Kfoury Kassouf, H.; Eid, A.A. NETosis contributes to the pathogenesis of diabetes and its complications. J. Mol. Endocrinol. 2020, 65, R65–R76. [Google Scholar] [CrossRef] [PubMed]
- Sodré, F.M.C.; Bissenova, S.; Bruggeman, Y.; Tilvawala, R.; Cook, D.P.; Berthault, C.; Mondal, S.; Callebaut, A.; You, S.; Scharfmann, R.; et al. Peptidylarginine deiminase inhibition prevents diabetes development in NOD mice. Diabetes 2021, 70, 516–528. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Wang, X.; He, D.; You, Q.; Zhang, T.; Dong, W.; Fei, J.; Xing, Y.; Wu, J. Ameliorating gut microenvironment through staphylococcal nuclease-mediated intestinal NETs degradation for prevention of type 1 diabetes in NOD mice. Life Sci. 2019, 221, 301–310. [Google Scholar] [CrossRef]
- Parackova, Z.; Zentsova, I.; Vrabcova, P.; Klocperk, A.; Sumnik, Z.; Pruhova, S.; Petruzelkova, L.; Hasler, R.; Sediva, A. Neutrophil extracellular trap-induced dendritic cell activation leads to Th1 polarization in type 1 diabetes. Front. Immunol. 2020, 11, 661. [Google Scholar] [CrossRef]
- Smulski, C.R.; Eibel, H. BAFF and BAFF-receptor in B cell selection and survival. Front. Immunol. 2018, 9, 2285. [Google Scholar] [CrossRef]
- Costa, S.; Bevilacqua, D.; Cassatella, M.A.; Scapini, P. Recent advances on the crosstalk between neutrophils and B or T lymphocytes. Immunology 2019, 156, 23–32. [Google Scholar] [CrossRef]
- Cutler, C.W.; Eke, P.; Arnold, R.R.; Van Dyke, T.E. Defective neutrophil function in an insulin-dependent diabetes mellitus patients. A case report. J. Periodontol. 1991, 62, 394–401. [Google Scholar] [CrossRef]
- Marhoffer, W.; Stein, M.; Schleinkofer, L.; Federlin, K. Evidence of ex vivo and in vitro impaired neutrophil oxidative burst and phagocytic capacity in type 1 diabetes mellitus. Diabetes Res. Clin. Pract. 1993, 19, 183–188. [Google Scholar] [CrossRef]
- Kjersem, H.; Hilsted, J.; Madsbad, S.; Wandall, J.H.; Johansen, K.S.; Borregaard, N. Polymorphonuclear leucocyte dysfunction during short term metabolic changes from normo- to hyperglycemia in type 1 (insulin dependent) diabetic patients. Infection 1988, 16, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Nielson, C.P.; Hindson, D.A. Inhibition of polymorphonuclear leukocyte respiratory burst by elevated glucose concentrations in vitro. Diabetes 1989, 38, 1031–1035. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.B.; Hashim, M.J.; King, J.K.; Govender, R.D.; Mustafa, H.; Al Kaabi, J. Epidemiology of type 2 diabetes—Global burden of disease and forecasted trends. J. Epidemiol. Glob. Health 2020, 10, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Hatanaka, E.; Monteagudo, P.T.; Marrocos, M.S.; Campa, A. Neutrophils and monocytes as potentially important sources of proinflammatory cytokines in diabetes. Clin. Exp. Immunol. 2006, 146, 443–447. [Google Scholar] [CrossRef]
- Good, M.; Newell, F.M.; Haupt, L.M.; Whitehead, J.P.; Hutley, L.J.; Prins, J.B. TNF and TNF receptor expression and insulin sensitivity in human omental and subcutaneous adipose tissue–influence of BMI and adipose distribution. Diabetes Vasc. Dis. Res. 2006, 3, 26–33. [Google Scholar] [CrossRef]
- Bilgic Gazioglu, S.; Akan, G.; Atalar, F.; Erten, G. PAI-1 and TNF-α profiles of adipose tissue in obese cardiovascular disease patients. Int. J. Clin. Exp. Pathol. 2015, 8, 15919–15925. [Google Scholar]
- Meigs, J.B.; Hu, F.B.; Rifai, N.; Manson, J.E. Biomarkers of endothelial dysfunction and risk of type 2 diabetes mellitus. JAMA 2004, 291, 1978–1986. [Google Scholar] [CrossRef]
- Lee, J.; Lee, S.; Zhang, H.; Hill, M.A.; Zhang, C.; Park, Y. Interaction of IL-6 and TNF-α contributes to endothelial dysfunction in type 2 diabetic mouse hearts. PLoS ONE 2017, 12, e0187189. [Google Scholar] [CrossRef]
- Tabit, C.E.; Chung, W.B.; Hamburg, N.M.; Vita, J.A. Endothelial dysfunction in diabetes mellitus: Molecular mechanisms and clinical implications. Rev. Endocr. Metab. Disord. 2010, 11, 61–74. [Google Scholar] [CrossRef]
- Ntaios, G.; Gatselis, N.K.; Makaritsis, K.; Dalekos, G.N. Adipokines as mediators of endothelial function and atherosclerosis. Atherosclerosis 2013, 227, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yang, X.; Bian, F.; Wu, P.; Xing, S.; Xu, G.; Li, W.; Chi, J.; Ouyang, C.; Zheng, T.; et al. TNF-α promotes early atherosclerosis by increasing transcytosis of LDL across endothelial cells: Crosstalk between NF-κB and PPAR-γ. J. Mol. Cell. Cardiol. 2014, 72, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Gandhi, J.K.; Zhong, X.; Wei, Y.; Gong, J.; Duh, E.J.; Vinores, S.A. TNF alpha is required for late BRB breakdown in diabetic retinopathy, and its inhibition prevents leukostasis and protects vessels and neurons from apoptosis. Investig. Ophthalmol. Vis. Sci. 2011, 52, 1336–1344. [Google Scholar] [CrossRef]
- Nordmann, T.M.; Dror, E.; Schulze, F.; Traub, S.; Berishvili, E.; Barbieux, C.; Böni-Schnetzler, M.; Donath, M.Y. The role of inflammation in β-cell dedifferentiation. Sci. Rep. 2017, 7, 6285. [Google Scholar] [CrossRef]
- Talchai, C.; Xuan, S.; Lin, H.V.; Sussel, L.; Accili, D. Pancreatic β cell dedifferentiation as a mechanism of diabetic β cell failure. Cell 2012, 150, 1223–1234. [Google Scholar] [CrossRef] [PubMed]
- Fainsod-Levi, T.; Gershkovitz, M.; Völs, S.; Kumar, S.; Khawaled, S.; Sagiv, J.Y.; Sionov, R.V.; Grunewald, M.; Keshet, E.; Granot, Z. Hyperglycemia impairs neutrophil mobilization leading to enhanced metastatic seeding. Cell Rep. 2017, 21, 2384–2392. [Google Scholar] [CrossRef]
- Gomez, I.; Ward, B.; Souilhol, C.; Recarti, C.; Ariaans, M.; Johnston, J.; Burnett, A.; Mahmoud, M.; Luong, L.A.; West, L.; et al. Neutrophil microvesicles drive atherosclerosis by delivering miR-155 to atheroprone endothelium. Nat. Commun. 2020, 11, 214. [Google Scholar] [CrossRef]
- Rosales, C. Neutrophil: A cell with many roles in inflammation or several cell types? Front. Physiol.-Integr. Physiol. 2018, 9, 113. [Google Scholar] [CrossRef]
- Chen, P.Y.; Cripps, A.W.; West, N.P.; Cox, A.J.; Zhang, P. A correlation-based network for biomarker discovery in obesity with metabolic syndrome. BMC Bioinform. 2019, 20, 477. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uribe-Querol, E.; Rosales, C. Neutrophils Actively Contribute to Obesity-Associated Inflammation and Pathological Complications. Cells 2022, 11, 1883. https://doi.org/10.3390/cells11121883
Uribe-Querol E, Rosales C. Neutrophils Actively Contribute to Obesity-Associated Inflammation and Pathological Complications. Cells. 2022; 11(12):1883. https://doi.org/10.3390/cells11121883
Chicago/Turabian StyleUribe-Querol, Eileen, and Carlos Rosales. 2022. "Neutrophils Actively Contribute to Obesity-Associated Inflammation and Pathological Complications" Cells 11, no. 12: 1883. https://doi.org/10.3390/cells11121883
APA StyleUribe-Querol, E., & Rosales, C. (2022). Neutrophils Actively Contribute to Obesity-Associated Inflammation and Pathological Complications. Cells, 11(12), 1883. https://doi.org/10.3390/cells11121883