
The Role of Endothelial-to-Mesenchymal Transition in Cardiovascular Disease

 ,
,  ,
, 

Abstract
:1. Introduction
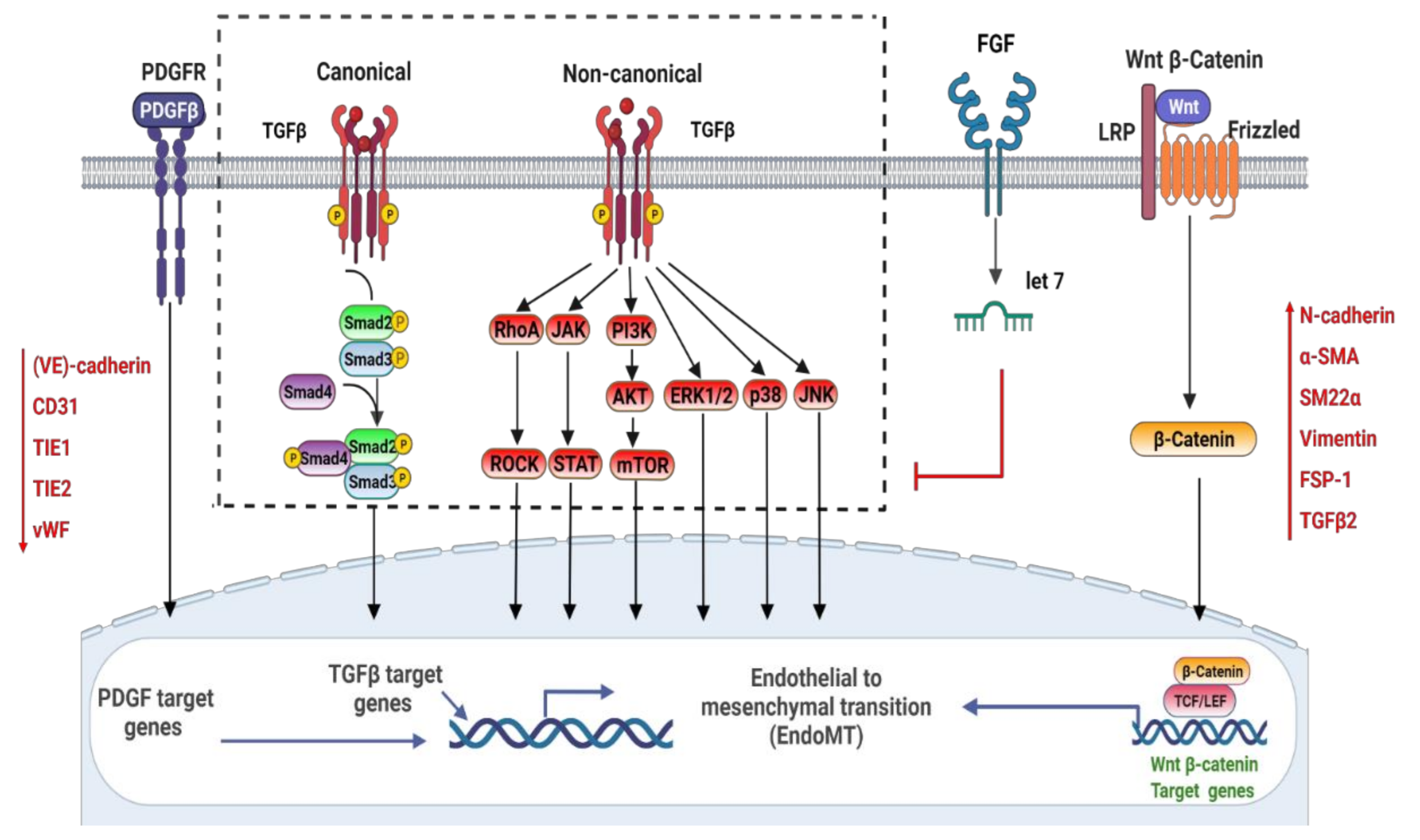
2. Signaling Pathways Involved in the Regulation of EndoMT
2.1. TGFβ (Transforming Growth Factor-β)
2.2. PDGF
2.3. Wnt/β-Catenin
2.4. FGF
3. EndoMT in In Vitro Studies
4. EndoMT in In Vivo Studies
5. Partial and Reversible EndoMT
6. EndoMT in Heart and Valve Development
7. EndoMT in Atherosclerosis
8. EndoMT in Adult Valve Disease
9. EndoMT in Myocardial Fibrosis
10. EndoMT in Pulmonary Arterial Hypertension
11. Perspective
12. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dela Paz, N.G.; D’Amore, P.A. Arterial versus venous endothelial cells. Cell Tissue Res. 2009, 335, 5–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishii, Y.; Langberg, J.; Rosborough, K.; Mikawa, T. Endothelial cell lineages of the heart. Cell Tissue Res. 2009, 335, 67–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyuno, D.; Yamaguchi, H.; Ito, T.; Kono, T.; Kimura, Y.; Imamura, M.; Konno, T.; Hirata, K.; Sawada, N.; Kojima, T. Targeting tight junctions during epithelial to mesenchymal transition in human pancreatic cancer. World J. Gastroenterol. 2014, 20, 10813–10824. [Google Scholar] [CrossRef] [PubMed]
- Deanfield, J.E.; Halcox, J.P.; Rabelink, T.J. Endothelial function and dysfunction: Testing and clinical relevance. Circulation 2007, 115, 1285–1295. [Google Scholar] [CrossRef] [PubMed]
- Peiris, H.; Bonder, C.S.; Coates, P.T.; Keating, D.J.; Jessup, C.F. The β-cell/EC axis: How do islet cells talk to each other? Diabetes 2014, 63, 3–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadry, H.; Noorani, B.; Cucullo, L. A blood-brain barrier overview on structure, function, impairment, and biomarkers of integrity. Fluids Barriers CNS 2020, 17, 69. [Google Scholar] [CrossRef] [PubMed]
- Navarro, P.; Ruco, L.; Dejana, E. Differential Localization of VE- and N-Cadherins in Human Endothelial Cells: VE-Cadherin Competes with N-Cadherin for Junctional Localization. J. Cell Biol. 1998, 140, 1475–1484. [Google Scholar] [CrossRef]
- Loh, C.Y.; Chai, J.Y.; Tang, T.F.; Wong, W.F.; Sethi, G.; Shanmugam, M.K.; Chong, P.P.; Looi, C.Y. The E-Cadherin and N-Cadherin Switch in Epithelial-to-Mesenchymal Transition: Signaling, Therapeutic Implications, and Challenges. Cells 2019, 8, 1118. [Google Scholar] [CrossRef] [Green Version]
- Coultas, L.; Chawengsaksophak, K.; Rossant, J. Endothelial cells and VEGF in vascular development. Nature 2005, 438, 937–945. [Google Scholar] [CrossRef]
- Salva, K.A.; Haemel, A.K.; Pincus, L.B.; Liu, J.; Sundram, U.; Guitart, J.; Longley, B.J.; Wood, G.S. Expression of CD31/PECAM-1 (platelet endothelial cell adhesion molecule 1) by blastic plasmacytoid dendritic cell neoplasms. JAMA Dermatol. 2014, 150, 73–76. [Google Scholar] [CrossRef] [Green Version]
- Baldwin, H.S.; Shen, H.M.; Yan, H.C.; DeLisser, H.M.; Chung, A.; Mickanin, C.; Trask, T.; Kirschbaum, N.E.; Newman, P.J.; Albelda, S.M. Platelet endothelial cell adhesion molecule-1 (PECAM-1/CD31): Alternatively spliced, functionally distinct isoforms expressed during mammalian cardiovascular development. Development 1994, 120, 2539–2553. [Google Scholar] [CrossRef] [PubMed]
- Jakobsen, K.R.; Demuth, C.; Sorensen, B.S.; Nielsen, A.L. The role of epithelial to mesenchymal transition in resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Transl. Lung Cancer Res. 2016, 5, 172–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dmitrieva, N.I.; Burg, M.B. Secretion of von Willebrand factor by endothelial cells links sodium to hypercoagulability and thrombosis. Proc. Natl. Acad. Sci. USA 2014, 111, 6485–6490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saharinen, P.; Jeltsch, M.; Santoyo, M.M.; Leppänen, V.-M.; Alitalo, K. The TIE Receptor Family. In Receptor Tyrosine Kinases: Family and Subfamilies; Springer: Cham, Switzerland, 2015; pp. 743–775. [Google Scholar]
- Garcia, J.; Sandi, M.J.; Cordelier, P.; Binetruy, B.; Pouyssegur, J.; Iovanna, J.L.; Tournaire, R. Tie1 deficiency induces endothelial-mesenchymal transition. EMBO Rep. 2012, 13, 431–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadler, T.; Scarpa, M.; Rieder, F.; West, G.; Stylianou, E. Cytokine-induced chromatin modifications of the type I collagen alpha 2 gene during intestinal endothelial-to-mesenchymal transition. Inflamm. Bowel Dis. 2013, 19, 1354–1364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Somoza, R.A.; Welter, J.F.; Correa, D.; Caplan, A.I. Chondrogenic differentiation of mesenchymal stem cells: Challenges and unfulfilled expectations. Tissue Eng. Part B Rev. 2014, 20, 596–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munir, H.; Ward, L.S.C.; Sheriff, L.; Kemble, S.; Nayar, S.; Barone, F.; Nash, G.B.; McGettrick, H.M. Adipogenic Differentiation of Mesenchymal Stem Cells Alters Their Immunomodulatory Properties in a Tissue-Specific Manner. Stem Cells 2017, 35, 1636–1646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohamed-Ahmed, S.; Fristad, I.; Lie, S.A.; Suliman, S.; Mustafa, K.; Vindenes, H.; Idris, S.B. Adipose-derived and bone marrow mesenchymal stem cells: A donor-matched comparison. Stem Cell Res. Ther. 2018, 9, 168. [Google Scholar] [CrossRef]
- Chen, Q.; Shou, P.; Zheng, C.; Jiang, M.; Cao, G.; Yang, Q.; Cao, J.; Xie, N.; Velletri, T.; Zhang, X.; et al. Fate decision of mesenchymal stem cells: Adipocytes or osteoblasts? Cell Death Differ. 2016, 23, 1128–1139. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Fuhr, J.; Boye, E.; Gyorffy, S.; Soker, S.; Atala, A.; Mulliken, J.B.; Bischoff, J. Mesenchymal stem cells and adipogenesis in hemangioma involution. Stem Cells 2006, 24, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Mahmoud, M.M.; Serbanovic-Canic, J.; Feng, S.; Souilhol, C.; Xing, R.; Hsiao, S.; Mammoto, A.; Chen, J.; Ariaans, M.; Francis, S.E.; et al. Shear stress induces endothelial-to-mesenchymal transition via the transcription factor Snail. Sci. Rep. 2017, 7, 3375. [Google Scholar] [CrossRef] [PubMed]
- Haynes, B.A.; Yang, L.F.; Huyck, R.W.; Lehrer, E.J.; Turner, J.M.; Barabutis, N.; Correll, V.L.; Mathiesen, A.; McPheat, W.; Semmes, O.J.; et al. Endothelial-to-Mesenchymal Transition in Human Adipose Tissue Vasculature Alters the Particulate Secretome and Induces Endothelial Dysfunction. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 2168–2191. [Google Scholar] [CrossRef] [PubMed]
- Talele, N.P.; Fradette, J.; Davies, J.E.; Kapus, A.; Hinz, B. Expression of α-Smooth Muscle Actin Determines the Fate of Mesenchymal Stromal Cells. Stem Cell Rep. 2015, 4, 1016–1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Sun, C.; Liang, Z.; Li, H.; Chen, L.; Luo, H.; Zhang, H.; Ding, P.; Sun, X.; Qin, Z.; et al. FSP1+ fibroblast subpopulation is essential for the maintenance and regeneration of medullary thymic epithelial cells. Sci. Rep. 2015, 5, 14871. [Google Scholar] [CrossRef] [PubMed]
- Mendez, M.G.; Kojima, S.; Goldman, R.D. Vimentin induces changes in cell shape, motility, and adhesion during the epithelial to mesenchymal transition. FASEB J. 2010, 24, 1838–1851. [Google Scholar] [CrossRef] [Green Version]
- Kasten, A.; Naser, T.; Brullhoff, K.; Fiedler, J.; Muller, P.; Moller, M.; Rychly, J.; Groll, J.; Brenner, R.E. Guidance of mesenchymal stem cells on fibronectin structured hydrogel films. PLoS ONE 2014, 9, e109411. [Google Scholar] [CrossRef]
- Friedenstein, A. Stromal-Hematopoietic Interrelationships: Maximov’s Ideas and Modern Models; Springer: Berlin/Heidelberg, Germany, 1989; pp. 159–167. [Google Scholar]
- Fiocchi, C.; Ina, K.; Danese, S.; Leite, A.Z.; Vogel, J.D. Alterations of mesenchymal and endothelial cells in inflammatory bowel diseases. Adv. Exp. Med. Biol. 2006, 579, 168–176. [Google Scholar] [CrossRef]
- Abu El-Asrar, A.M.; De Hertogh, G.; van den Eynde, K.; Alam, K.; Van Raemdonck, K.; Opdenakker, G.; Van Damme, J.; Geboes, K.; Struyf, S. Myofibroblasts in proliferative diabetic retinopathy can originate from infiltrating fibrocytes and through endothelial-to-mesenchymal transition (EndoMT). Exp. Eye Res. 2015, 132, 179–189. [Google Scholar] [CrossRef]
- Zeisberg, E.M.; Tarnavski, O.; Zeisberg, M.; Dorfman, A.L.; McMullen, J.R.; Gustafsson, E.; Chandraker, A.; Yuan, X.; Pu, W.T.; Roberts, A.B.; et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 2007, 13, 952–961. [Google Scholar] [CrossRef]
- Bischoff, J. Endothelial-to-Mesenchymal Transition. Circ. Res. 2019, 124, 1163–1165. [Google Scholar] [CrossRef]
- Baumann, K. Mechanotransduction: Kindlin’ the fate of mesenchymal stem cells. Nat. Rev. Mol. Cell Biol. 2018, 19, 278–279. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ogbu, S.C.; Musich, P.R.; Thewke, D.P.; Yao, Z.; Jiang, Y. The Contribution of Endothelial-Mesenchymal Transition to Atherosclerosis. Int. J. Transl. Med. 2021, 1, 39–54. [Google Scholar] [CrossRef]
- Rosa, I.; Romano, E.; Fioretto, B.S.; Manetti, M. The contribution of mesenchymal transitions to the pathogenesis of systemic sclerosis. Eur. J. Rheumatol. 2020, 7, S157–S164. [Google Scholar] [CrossRef] [PubMed]
- Von Gise, A.; Pu, W.T. Endocardial and epicardial epithelial to mesenchymal transitions in heart development and disease. Circ. Res. 2012, 110, 1628–1645. [Google Scholar] [CrossRef] [PubMed]
- Stenmark, K.R.; Frid, M.; Perros, F. Endothelial-to-Mesenchymal Transition: An Evolving Paradigm and a Promising Therapeutic Target in PAH. Circulation 2016, 133, 1734–1737. [Google Scholar] [CrossRef]
- Cheng, W.; Li, X.; Liu, D.; Cui, C.; Wang, X. Endothelial-to-Mesenchymal Transition: Role in Cardiac Fibrosis. J. Cardiovasc. Pharmacol. Ther. 2021, 26, 3–11. [Google Scholar] [CrossRef]
- Bruijn, L.E.; van den Akker, B.; van Rhijn, C.M.; Hamming, J.F.; Lindeman, J.H.N. Extreme Diversity of the Human Vascular Mesenchymal Cell Landscape. J. Am. Heart Assoc. 2020, 9, e017094. [Google Scholar] [CrossRef]
- Ichim, T.E.; O’Heeron, P.; Kesari, S. Fibroblasts as a practical alternative to mesenchymal stem cells. J. Transl. Med. 2018, 16, 212. [Google Scholar] [CrossRef] [Green Version]
- Islam, S.; Bostrom, K.I.; Di Carlo, D.; Simmons, C.A.; Tintut, Y.; Yao, Y.; Hsu, J.J. The Mechanobiology of Endothelial-to-Mesenchymal Transition in Cardiovascular Disease. Front. Physiol. 2021, 12, 734215. [Google Scholar] [CrossRef]
- Jimenez, S.A.; Piera-Velazquez, S. Endothelial to mesenchymal transition (EndoMT) in the pathogenesis of Systemic Sclerosis-associated pulmonary fibrosis and pulmonary arterial hypertension. Myth or reality? Matrix Biol. 2016, 51, 26–36. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, N.; Phan, S.H.; Imaizumi, K.; Matsuo, M.; Nakashima, H.; Kawabe, T.; Shimokata, K.; Hasegawa, Y. Endothelial-mesenchymal transition in bleomycin-induced pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2010, 43, 161–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, S.; Zhang, M.; Yi, Z.; Zhang, H.; Shen, T.; Yu, X.; Zhang, C.; Zheng, X.; Yu, L.; Ma, C.; et al. The role of PDGF-B/TGF-β1/neprilysin network in regulating endothelial-to-mesenchymal transition in pulmonary artery remodeling. Cell. Signal. 2016, 28, 1489–1501. [Google Scholar] [CrossRef] [PubMed]
- Greaves, D.; Calle, Y. Epithelial Mesenchymal Transition (EMT) and Associated Invasive Adhesions in Solid and Haematological Tumours. Cells 2022, 11, 649. [Google Scholar] [CrossRef] [PubMed]
- Auersperg, N.; Pan, J.; Grove, B.D.; Peterson, T.; Fisher, J.; Maines-Bandiera, S.; Somasiri, A.; Roskelley, C.D. E-cadherin induces mesenchymal-to-epithelial transition in human ovarian surface epithelium. Proc. Natl. Acad. Sci. USA 1999, 96, 6249–6254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, T.; Maruyama, K.; Kawamura, T.; Urade, Y.; Wada, Y. PERK participates in cardiac valve development via fatty acid oxidation and endocardial-mesenchymal transformation. Sci. Rep. 2020, 10, 20094. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.M.; Wumaier, G.; Zhu, N.; Dong, L.; Li, C.W.; Xia, J.W.; Zhang, Y.Z.; Zhang, P.; Zhang, X.J.; Zhang, Y.Y.; et al. Protein tyrosine phosphatase L1 represses endothelial-mesenchymal transition by inhibiting IL-1β/NF-κB/Snail signaling. Acta Pharmacol. Sin. 2020, 41, 1102–1110. [Google Scholar] [CrossRef]
- Lee, J.G.; Kay, E.P. FGF-2-mediated signal transduction during endothelial mesenchymal transformation in corneal endothelial cells. Exp. Eye Res. 2006, 83, 1309–1316. [Google Scholar] [CrossRef]
- Tian, D.; Zeng, X.; Wang, W.; Wang, Z.; Zhang, Y.; Wang, Y. Protective effect of rapamycin on endothelial-to-mesenchymal transition in HUVECs through the Notch signaling pathway. Vascul. Pharmacol. 2019, 113, 20–26. [Google Scholar] [CrossRef]
- Li, C.; Dong, F.; Jia, Y.; Du, H.; Dong, N.; Xu, Y.; Wang, S.; Wu, H.; Liu, Z.; Li, W. Notch signal regulates corneal endothelial-to-mesenchymal transition. Am. J. Pathol. 2013, 183, 786–795. [Google Scholar] [CrossRef]
- Chang, A.C.; Fu, Y.; Garside, V.C.; Niessen, K.; Chang, L.; Fuller, M.; Setiadi, A.; Smrz, J.; Kyle, A.; Minchinton, A.; et al. Notch initiates the endothelial-to-mesenchymal transition in the atrioventricular canal through autocrine activation of soluble guanylyl cyclase. Dev. Cell 2011, 21, 288–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsura, A.; Suzuki, H.I.; Ueno, T.; Mihira, H.; Yamazaki, T.; Yasuda, T.; Watabe, T.; Mano, H.; Yamada, Y.; Miyazono, K. MicroRNA-31 is a positive modulator of endothelial-mesenchymal transition and associated secretory phenotype induced by TGF-β. Genes Cells 2016, 21, 99–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Dong, F.; Jeong, J.; Masuda, T.; Lobe, C.G. Constitutively active Notch1 signaling promotes endothelialmesenchymal transition in a conditional transgenic mouse model. Int. J. Mol. Med. 2014, 34, 669–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossato, F.A.; Su, Y.; Mackey, A.; Ng, Y.S.E. Fibrotic Changes and Endothelial-to-Mesenchymal Transition Promoted by VEGFR2 Antagonism Alter the Therapeutic Effects of VEGFA Pathway Blockage in a Mouse Model of Choroidal Neovascularization. Cells 2020, 9, 2057. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Sanchez-Duffhues, G.; Goumans, M.-J.; Ten Dijke, P. TGF-β-Induced Endothelial to Mesenchymal Transition in Disease and Tissue Engineering. Front. Cell Dev. Biol. 2020, 8, 260. [Google Scholar] [CrossRef] [PubMed]
- Sporn, M.B.; Todaro, G.J. Autocrine Secretion and Malignant Transformation of Cells. N. Engl. J. Med. 1980, 303, 878–880. [Google Scholar] [CrossRef]
- Pinto, M.T.; Ferreira Melo, F.U.; Malta, T.M.; Rodrigues, E.S.; Plaça, J.R.; Silva, W.A., Jr.; Panepucci, R.A.; Covas, D.T.; de Oliveira Rodrigues, C.; Kashima, S. Endothelial cells from different anatomical origin have distinct responses during SNAIL/TGF-β2-mediated endothelial-mesenchymal transition. Am. J. Transl. Res. 2018, 10, 4065–4081. [Google Scholar]
- Diez, M.; Musri, M.M.; Ferrer, E.; Barbera, J.A.; Peinado, V.I. Endothelial progenitor cells undergo an endothelial-to-mesenchymal transition-like process mediated by TGFβRI. Cardiovasc. Res. 2010, 88, 502–511. [Google Scholar] [CrossRef] [Green Version]
- Doerr, M.; Morrison, J.; Bergeron, L.; Coomber, B.L.; Viloria-Petit, A. Differential effect of hypoxia on early endothelial-mesenchymal transition response to transforming growth beta isoforms 1 and 2. Microvasc. Res. 2016, 108, 48–63. [Google Scholar] [CrossRef]
- Shao, E.S.; Lin, L.; Yao, Y.; Bostrom, K.I. Expression of vascular endothelial growth factor is coordinately regulated by the activin-like kinase receptors 1 and 5 in endothelial cells. Blood 2009, 114, 2197–2206. [Google Scholar] [CrossRef] [Green Version]
- Xie, F.; Zhang, Z.; van Dam, H.; Zhang, L.; Zhou, F. Regulation of TGF-β Superfamily Signaling by SMAD Mono-Ubiquitination. Cells 2014, 3, 981–993. [Google Scholar] [CrossRef] [PubMed]
- Gwon, M.G.; An, H.J.; Kim, J.Y.; Kim, W.H.; Gu, H.; Kim, H.J.; Leem, J.; Jung, H.J.; Park, K.K. Anti-fibrotic effects of synthetic TGF-β1 and Smad oligodeoxynucleotide on kidney fibrosis in vivo and in vitro through inhibition of both epithelial dedifferentiation and endothelial-mesenchymal transitions. FASEB J. 2020, 34, 333–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Feng, Y.; Wang, Y.; Xiang, D.; Zhang, X.; Yuan, F. Autophagy regulates Endothelial-Mesenchymal transition by decreasing the phosphorylation level of Smad3. Biochem. Biophys. Res. Commun. 2017, 487, 740–747. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Yu, L.; He, A.; Liu, Q. Klotho Inhibits Unilateral Ureteral Obstruction-Induced Endothelial-to-Mesenchymal Transition via TGF-β1/Smad2/Snail1 Signaling in Mice. Front. Pharmacol. 2019, 10, 348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tecalco-Cruz, A.C.; Rios-Lopez, D.G.; Vazquez-Victorio, G.; Rosales-Alvarez, R.E.; Macias-Silva, M. Transcriptional cofactors Ski and SnoN are major regulators of the TGF-β/Smad signaling pathway in health and disease. Signal. Transduct. Target Ther. 2018, 3, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyazawa, K.; Miyazono, K. Regulation of TGF-β Family Signaling by Inhibitory Smads. Cold Spring Harb. Perspect. Biol. 2017, 9, a022095. [Google Scholar] [CrossRef] [Green Version]
- Walton, K.L.; Johnson, K.E.; Harrison, C.A. Targeting TGF-β Mediated SMAD Signaling for the Prevention of Fibrosis. Front. Pharmacol. 2017, 8, 461. [Google Scholar] [CrossRef] [Green Version]
- Batlle, R.; Andres, E.; Gonzalez, L.; Llonch, E.; Igea, A.; Gutierrez-Prat, N.; Berenguer-Llergo, A.; Nebreda, A.R. Regulation of tumor angiogenesis and mesenchymal-endothelial transition by p38α through TGF-β and JNK signaling. Nat. Commun. 2019, 10, 3071. [Google Scholar] [CrossRef]
- Kumarswamy, R.; Volkmann, I.; Jazbutyte, V.; Dangwal, S.; Park, D.H.; Thum, T. Transforming growth factor-β-induced endothelial-to-mesenchymal transition is partly mediated by microRNA-21. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 361–369. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Ma, W.; Xu, H.; Huang, M.; Zhang, D.; He, Z.; Zhang, L.; Brem, S.; O’Rourke, D.M.; Gong, Y.; et al. PDGF-mediated mesenchymal transformation renders endothelial resistance to anti-VEGF treatment in glioblastoma. Nat. Commun. 2018, 9, 3439. [Google Scholar] [CrossRef]
- Ng, F.; Boucher, S.; Koh, S.; Sastry, K.S.; Chase, L.; Lakshmipathy, U.; Choong, C.; Yang, Z.; Vemuri, M.C.; Rao, M.S.; et al. PDGF, TGF-β, and FGF signaling is important for differentiation and growth of mesenchymal stem cells (MSCs): Transcriptional profiling can identify markers and signaling pathways important in differentiation of MSCs into adipogenic, chondrogenic, and osteogenic lineages. Blood 2008, 112, 295–307. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.H.; Chen, X.; He, X. Platelet-derived growth factors and their receptors: Structural and functional perspectives. Biochim. Biophys. Acta 2013, 1834, 2176–2186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrae, J.; Gallini, R.; Betsholtz, C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 2008, 22, 1276–1312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrison, D.K.; Kaplan, D.R.; Rhee, S.G.; Williams, L.T. Platelet-derived growth factor (PDGF)-dependent association of phospholipase C-gamma with the PDGF receptor signaling complex. Mol. Cell. Biol. 1990, 10, 2359–2366. [Google Scholar] [CrossRef]
- Yokota, J.; Chosa, N.; Sawada, S.; Okubo, N.; Takahashi, N.; Hasegawa, T.; Kondo, H.; Ishisaki, A. PDGF-induced PI3K-mediated signaling enhances the TGF-β-induced osteogenic differentiation of human mesenchymal stem cells in a TGF-β-activated MEK-dependent manner. Int. J. Mol. Med. 2014, 33, 534–542. [Google Scholar] [CrossRef] [Green Version]
- Nakata, S.; Fujita, N.; Kitagawa, Y.; Okamoto, R.; Ogita, H.; Takai, Y. Regulation of platelet-derived growth factor receptor activation by afadin through SHP-2: Implications for cellular morphology. J. Biol. Chem. 2007, 282, 37815–37825. [Google Scholar] [CrossRef] [Green Version]
- Vignais, M.L.; Sadowski, H.B.; Watling, D.; Rogers, N.C.; Gilman, M. Platelet-derived growth factor induces phosphorylation of multiple JAK family kinases and STAT proteins. Mol. Cell. Biol. 1996, 16, 1759–1769. [Google Scholar] [CrossRef] [Green Version]
- Fischer, A.N.; Fuchs, E.; Mikula, M.; Huber, H.; Beug, H.; Mikulits, W. PDGF essentially links TGF-β signaling to nuclear β-catenin accumulation in hepatocellular carcinoma progression. Oncogene 2007, 26, 3395–3405. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Rahman, O. Targeting platelet-derived growth factor (PDGF) signaling in gastrointestinal cancers: Preclinical and clinical considerations. Tumour Biol. 2015, 36, 21–31. [Google Scholar] [CrossRef]
- Shang, S.; Hua, F.; Hu, Z.-W. The regulation of β-catenin activity and function in cancer: Therapeutic opportunities. Oncotarget 2017, 8, 33972–33989. [Google Scholar] [CrossRef] [Green Version]
- Aisagbonhi, O.; Rai, M.; Ryzhov, S.; Atria, N.; Feoktistov, I.; Hatzopoulos, A.K. Experimental myocardial infarction triggers canonical Wnt signaling and endothelial-to-mesenchymal transition. Dis. Models Mech. 2011, 4, 469–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cadigan, K.M.; Waterman, M.L. TCF/LEFs and Wnt signaling in the nucleus. Cold Spring Harb. Perspect. Biol. 2012, 4, a007906. [Google Scholar] [CrossRef] [PubMed]
- Liebner, S.; Cattelino, A.; Gallini, R.; Rudini, N.; Iurlaro, M.; Piccolo, S.; Dejana, E. β-catenin is required for endothelial-mesenchymal transformation during heart cushion development in the mouse. J. Cell Biol. 2004, 166, 359–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.G.; Kay, E.P. Cross-talk among Rho GTPases acting downstream of PI 3-kinase induces mesenchymal transformation of corneal endothelial cells mediated by FGF-2. Invest. Ophthalmol. Vis. Sci. 2006, 47, 2358–2368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, M.K.; Kay, E.P. Regulatory role of FGF-2 on type I collagen expression during endothelial mesenchymal transformation. Invest. Ophthalmol. Vis. Sci. 2005, 46, 4495–4503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Correia, A.C.; Moonen, J.R.; Brinker, M.G.; Krenning, G. FGF2 inhibits endothelial-mesenchymal transition through microRNA-20a-mediated repression of canonical TGF-β signaling. J. Cell Sci. 2016, 129, 569–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, P.Y.; Qin, L.; Barnes, C.; Charisse, K.; Yi, T.; Zhang, X.; Ali, R.; Medina, P.P.; Yu, J.; Slack, F.J.; et al. FGF regulates TGF-β signaling and endothelial-to-mesenchymal transition via control of let-7 miRNA expression. Cell Rep. 2012, 2, 1684–1696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terzuoli, E.; Nannelli, G.; Giachetti, A.; Morbidelli, L.; Ziche, M.; Donnini, S. Targeting endothelial-to-mesenchymal transition: The protective role of hydroxytyrosol sulfate metabolite. Eur. J. Nutr. 2020, 59, 517–527. [Google Scholar] [CrossRef]
- Yoshimatsu, Y.; Watabe, T. Emerging roles of inflammation-mediated endothelial-mesenchymal transition in health and disease. Inflamm. Regen. 2022, 42, 9. [Google Scholar] [CrossRef]
- Krenning, G.; Moonen, J.A.J.; van Luyn, M.J.A.; Harmsen, M.C. Vascular smooth muscle cells for use in vascular tissue engineering obtained by endothelial-to-mesenchymal transdifferentiation (EnMT) on collagen matrices. Biomaterials 2008, 29, 3703–3711. [Google Scholar] [CrossRef]
- Arkonac, B.M.; Foster, L.C.; Sibinga, N.E.; Patterson, C.; Lai, K.; Tsai, J.C.; Lee, M.E.; Perrella, M.A.; Haber, E. Vascular endothelial growth factor induces heparin-binding epidermal growth factor-like growth factor in vascular endothelial cells. J. Biol. Chem. 1998, 273, 4400–4405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noseda, M.; McLean, G.; Niessen, K.; Chang, L.; Pollet, I.; Montpetit, R.; Shahidi, R.; Dorovini-Zis, K.; Li, L.; Beckstead, B.; et al. Notch activation results in phenotypic and functional changes consistent with endothelial-to-mesenchymal transformation. Circ. Res. 2004, 94, 910–917. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Hu, J.; Liu, L. MiR-200a modulates TGF-β1-induced endothelial-to-mesenchymal shift via suppression of GRB2 in HAECs. Biomed. Pharmacother. 2017, 95, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Peng, Z.; Zu, C.; Ma, J.; Lu, S.; Zhong, J.; Zhang, S. Losartan Attenuates Myocardial Endothelial-To-Mesenchymal Transition in Spontaneous Hypertensive Rats via Inhibiting TGF-β/Smad Signaling. PLoS ONE 2016, 11, e0155730. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lui, K.O.; Zhou, B. Reassessing endothelial-to-mesenchymal transition in cardiovascular diseases. Nat. Rev. Cardiol. 2018, 15, 445–456. [Google Scholar] [CrossRef] [PubMed]
- Piera-Velazquez, S.; Li, Z.; Jimenez, S.A. Role of endothelial-mesenchymal transition (EndoMT) in the pathogenesis of fibrotic disorders. Am. J. Pathol. 2011, 179, 1074–1080. [Google Scholar] [CrossRef]
- Helmke, A.; Casper, J.; Nordlohne, J.; David, S.; Haller, H.; Zeisberg, E.M.; von Vietinghoff, S. Endothelial-to-mesenchymal transition shapes the atherosclerotic plaque and modulates macrophage function. FASEB J. 2019, 33, 2278–2289. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Zhang, C.; Chen, J.; Yang, M.; Afzal, T.A.; An, W.; Maguire, E.M.; He, S.; Luo, J.; Wang, X.; et al. miRNA-200c-3p promotes endothelial to mesenchymal transition and neointimal hyperplasia in artery bypass grafts. J. Pathol. 2021, 253, 209–224. [Google Scholar] [CrossRef]
- Zhang, J.; Rojas, S.; Singh, S.; Musich, P.R.; Gutierrez, M.; Yao, Z.; Thewke, D.; Jiang, Y. Wnt2 Contributes to the Development of Atherosclerosis. Front. Cardiovasc. Med. 2021, 8, 751720. [Google Scholar] [CrossRef]
- Yang, J.H.; Wylie-Sears, J.; Bischoff, J. Opposing actions of Notch1 and VEGF in post-natal cardiac valve endothelial cells. Biochem. Biophys. Res. Commun. 2008, 374, 512–516. [Google Scholar] [CrossRef] [Green Version]
- Ranchoux, B.; Antigny, F.; Rucker-Martin, C.; Hautefort, A.; Pechoux, C.; Bogaard, H.J.; Dorfmuller, P.; Remy, S.; Lecerf, F.; Plante, S.; et al. Endothelial-to-mesenchymal transition in pulmonary hypertension. Circulation 2015, 131, 1006–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, M.; Yang, F.; Zhang, D.; Lin, M.; Duan, H.; El-Mayta, R.; Zhang, L.; Qin, L.; Shewale, S.V.; Pei, L.; et al. Endothelial plasticity drives aberrant vascularization and impedes cardiac repair after myocardial infarction. Nat. Cardiovasc. Res. 2022, 1, 372–388. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.Y.; Qin, L.; Baeyens, N.; Li, G.; Afolabi, T.; Budatha, M.; Tellides, G.; Schwartz, M.A.; Simons, M. Endothelial-to-mesenchymal transition drives atherosclerosis progression. J. Clin. Invest. 2015, 125, 4514–4528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Violin, J.D.; DeWire, S.M.; Yamashita, D.; Rominger, D.H.; Nguyen, L.; Schiller, K.; Whalen, E.J.; Gowen, M.; Lark, M.W. Selectively engaging β-arrestins at the angiotensin II type 1 receptor reduces blood pressure and increases cardiac performance. J. Pharmacol. Exp. Ther. 2010, 335, 572–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorelova, A.; Berman, M.; Al Ghouleh, I. Endothelial-to-Mesenchymal Transition in Pulmonary Arterial Hypertension. Antioxid. Redox Signal. 2021, 34, 891–914. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.S.; Hultgren, N.W.; Hughes, C.C.W. Regulation of Partial and Reversible Endothelial-to-Mesenchymal Transition in Angiogenesis. Front. Cell Dev. Biol. 2021, 9, 702021. [Google Scholar] [CrossRef]
- Medici, D.; Hay, E.D.; Olsen, B.R. Snail and Slug Promote Epithelial-Mesenchymal Transition through β-Catenin–T-Cell Factor-4-dependent Expression of Transforming Growth Factor-β3. Mol. Biol. Cell 2008, 19, 4875–4887. [Google Scholar] [CrossRef] [Green Version]
- Viñals, F.; Pouysségur, J. Transforming growth factor β1 (TGF-β1) promotes endothelial cell survival during in vitro angiogenesis via an autocrine mechanism implicating TGF-α signaling. Mol. Cell. Biol. 2001, 21, 7218–7230. [Google Scholar] [CrossRef] [Green Version]
- Quijada, P.; Trembley, M.A.; Small, E.M. The Role of the Epicardium During Heart Development and Repair. Circ. Res. 2020, 126, 377–394. [Google Scholar] [CrossRef]
- Piera-Velazquez, S.; Jimenez, S.A. Endothelial to Mesenchymal Transition: Role in Physiology and in the Pathogenesis of Human Diseases. Physiol. Rev. 2019, 99, 1281–1324. [Google Scholar] [CrossRef]
- Bloomekatz, J.; Singh, R.; Prall, O.W.; Dunn, A.C.; Vaughan, M.; Loo, C.S.; Harvey, R.P.; Yelon, D. Platelet-derived growth factor (PDGF) signaling directs cardiomyocyte movement toward the midline during heart tube assembly. eLife 2017, 6, e21172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tallquist, M.D.; Soriano, P. Cell autonomous requirement for PDGFRα in populations of cranial and cardiac neural crest cells. Development 2003, 130, 507–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Widyantoro, B.; Emoto, N.; Nakayama, K.; Anggrahini, D.W.; Adiarto, S.; Iwasa, N.; Yagi, K.; Miyagawa, K.; Rikitake, Y.; Suzuki, T.; et al. Endothelial cell-derived endothelin-1 promotes cardiac fibrosis in diabetic hearts through stimulation of endothelial-to-mesenchymal transition. Circulation 2010, 121, 2407–2418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, S.; Qian, J.; Wu, G.; Qian, Y.; Wang, Z.; Chen, T.; Wang, J.; Huang, W.; Liang, G. Schizandrin B attenuates angiotensin II induced endothelial to mesenchymal transition in vascular endothelium by suppressing NF-κB activation. Phytomedicine 2019, 62, 152955. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.H.; Suriguga, G.M.; Liu, W.J.; Cui, N.X.; Wang, Y.; Du, X.; Yi, Z.C. High glucose induced endothelial to mesenchymal transition in human umbilical vein endothelial cell. Exp. Mol. Pathol. 2017, 102, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Tsai, P.S.; Chiu, C.Y.; Sheu, M.L.; Yang, C.Y.; Lan, K.C.; Liu, S.H. Advanced glycation end products activated endothelial-to-mesenchymal transition in pancreatic islet endothelial cells and triggered islet fibrosis in diabetic mice. Chem. Biol. Interact. 2021, 345, 109562. [Google Scholar] [CrossRef]
- Zhang, B.; Niu, W.; Dong, H.-Y.; Liu, M.-L.; Luo, Y.; Li, Z.-C. Hypoxia induces endothelial-mesenchymal transition in pulmonary vascular remodeling. Int. J. Mol. Med. 2018, 42, 270–278. [Google Scholar] [CrossRef] [Green Version]
- Yao, J.; Guihard, P.J.; Blazquez-Medela, A.M.; Guo, Y.; Moon, J.H.; Jumabay, M.; Bostrom, K.I.; Yao, Y. Serine Protease Activation Essential for Endothelial-Mesenchymal Transition in Vascular Calcification. Circ. Res. 2015, 117, 758–769. [Google Scholar] [CrossRef] [Green Version]
- Souilhol, C.; Harmsen, M.C.; Evans, P.C.; Krenning, G. Endothelial-mesenchymal transition in atherosclerosis. Cardiovasc. Res. 2018, 114, 565–577. [Google Scholar] [CrossRef]
- Chen, P.Y.; Schwartz, M.A.; Simons, M. Endothelial-to-Mesenchymal Transition, Vascular Inflammation, and Atherosclerosis. Front. Cardiovasc. Med. 2020, 7, 53. [Google Scholar] [CrossRef]
- Mintet, E.; Lavigne, J.; Paget, V.; Tarlet, G.; Buard, V.; Guipaud, O.; Sabourin, J.C.; Iruela-Arispe, M.L.; Milliat, F.; Francois, A. Endothelial Hey2 deletion reduces endothelial-to-mesenchymal transition and mitigates radiation proctitis in mice. Sci. Rep. 2017, 7, 4933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, N.A.; Hong, X.; Doulamis, I.P.; Meibalan, E.; Peiseler, T.; Melero-Martin, J.; Garcia-Cardena, G.; Del Nido, P.J.; Friehs, I. Abnormal Flow Conditions Promote Endocardial Fibroelastosis Via Endothelial-to-Mesenchymal Transition, Which Is Responsive to Losartan Treatment. JACC Basic Transl. Sci. 2021, 6, 984–999. [Google Scholar] [CrossRef] [PubMed]
- Boyer, A.S.; Ayerinskas, I.I.; Vincent, E.B.; McKinney, L.A.; Weeks, D.L.; Runyan, R.B. TGFβ2 and TGFβ3 have separate and sequential activities during epithelial-mesenchymal cell transformation in the embryonic heart. Dev. Biol. 1999, 208, 530–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goumans, M.J.; Ten Dijke, P. TGF-β Signaling in Control of Cardiovascular Function. Cold Spring Harb. Perspect. Biol. 2018, 10, a022210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, C.S.; Chen, L.L.; Hsu, T.A.; Chen, C.C.; Chua, K.V.; Li, C.P.; Huang, T.S. Endothelial-mesenchymal transition harnesses HSP90α-secreting M2-macrophages to exacerbate pancreatic ductal adenocarcinoma. J. Hematol. Oncol. 2019, 12, 138. [Google Scholar] [CrossRef]
- Good, R.B.; Gilbane, A.J.; Trinder, S.L.; Denton, C.P.; Coghlan, G.; Abraham, D.J.; Holmes, A.M. Endothelial to Mesenchymal Transition Contributes to Endothelial Dysfunction in Pulmonary Arterial Hypertension. Am. J. Pathol. 2015, 185, 1850–1858. [Google Scholar] [CrossRef]
- Andueza, A.; Kumar, S.; Kim, J.; Kang, D.W.; Mumme, H.L.; Perez, J.I.; Villa-Roel, N.; Jo, H. Endothelial Reprogramming by Disturbed Flow Revealed by Single-Cell RNA and Chromatin Accessibility Study. Cell Rep. 2020, 33, 108491. [Google Scholar] [CrossRef]
- Li, F.; Yan, K.; Wu, L.; Zheng, Z.; Du, Y.; Liu, Z.; Zhao, L.; Li, W.; Sheng, Y.; Ren, L.; et al. Single-cell RNA-seq reveals cellular heterogeneity of mouse carotid artery under disturbed flow. Cell Death Discov. 2021, 7, 180. [Google Scholar] [CrossRef]
- Paranya, G.; Vineberg, S.; Dvorin, E.; Kaushal, S.; Roth, S.J.; Rabkin, E.; Schoen, F.J.; Bischoff, J. Aortic valve endothelial cells undergo transforming growth factor-β-mediated and non-transforming growth factor-β-mediated transdifferentiation in vitro. Am. J. Pathol. 2001, 159, 1335–1343. [Google Scholar] [CrossRef]
- Zhang, H.; Lui, K.O.; Zhou, B. Endocardial Cell Plasticity in Cardiac Development, Diseases and Regeneration. Circ. Res. 2018, 122, 774–789. [Google Scholar] [CrossRef]
- Nakajima, Y.; Yamagishi, T.; Hokari, S.; Nakamura, H. Mechanisms involved in valvuloseptal endocardial cushion formation in early cardiogenesis: Roles of transforming growth factor (TGF)-β and bone morphogenetic protein (BMP). Anat. Rec. 2000, 258, 119–127. [Google Scholar] [CrossRef]
- Yutzey, K.E.; Demer, L.L.; Body, S.C.; Huggins, G.S.; Towler, D.A.; Giachelli, C.M.; Hofmann-Bowman, M.A.; Mortlock, D.P.; Rogers, M.B.; Sadeghi, M.M.; et al. Calcific aortic valve disease: A consensus summary from the Alliance of Investigators on Calcific Aortic Valve Disease. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2387–2393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wirrig, E.E.; Yutzey, K.E. Conserved transcriptional regulatory mechanisms in aortic valve development and disease. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 737–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bischoff, J.; Casanovas, G.; Wylie-Sears, J.; Kim, D.H.; Bartko, P.E.; Guerrero, J.L.; Dal-Bianco, J.P.; Beaudoin, J.; Garcia, M.L.; Sullivan, S.M.; et al. CD45 Expression in Mitral Valve Endothelial Cells After Myocardial Infarction. Circ. Res. 2016, 119, 1215–1225. [Google Scholar] [CrossRef]
- Pardali, E.; Sanchez-Duffhues, G.; Gomez-Puerto, M.C.; Ten Dijke, P. TGF-β-Induced Endothelial-Mesenchymal Transition in Fibrotic Diseases. Int. J. Mol. Sci. 2017, 18, 2157. [Google Scholar] [CrossRef] [Green Version]
- Turkeli, A.; Yilmaz, O.; Karaman, M.; Kanik, E.T.; Firinci, F.; Inan, S.; Yuksel, H. Anti-VEGF treatment suppresses remodeling factors and restores epithelial barrier function through the E-cadherin/β-catenin signaling axis in experimental asthma models. Exp. Ther. Med. 2021, 22, 689. [Google Scholar] [CrossRef]
- Welch-Reardon, K.M.; Wu, N.; Hughes, C.C. A role for partial endothelial-mesenchymal transitions in angiogenesis? Arterioscler. Thromb. Vasc. Biol. 2015, 35, 303–308. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.; Chen, H.; Ge, D.; Xu, Y.; Xu, H.; Yang, Y.; Gu, M.; Zhou, Y.; Zhu, J.; Ge, T.; et al. Mir-21 Promotes Cardiac Fibrosis after Myocardial Infarction via Targeting Smad7. Cell. Physiol. Biochem. 2017, 42, 2207–2219. [Google Scholar] [CrossRef]
- Wang, J.; Huang, W.; Xu, R.; Nie, Y.; Cao, X.; Meng, J.; Xu, X.; Hu, S.; Zheng, Z. MicroRNA-24 regulates cardiac fibrosis after myocardial infarction. J. Cell. Mol. Med. 2012, 16, 2150–2160. [Google Scholar] [CrossRef]
- Rooij, E.V.; Sutherland, L.B.; Thatcher, J.E.; DiMaio, J.M.; Naseem, R.H.; Marshall, W.S.; Hill, J.A.; Olson, E.N. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13027–13032. [Google Scholar] [CrossRef] [Green Version]
- Xiong, J. To be EndMT or not to be, that is the question in pulmonary hypertension. Protein Cell 2015, 6, 547–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Benedetto, P.; Ruscitti, P.; Berardicurti, O.; Vomero, M.; Navarini, L.; Dolo, V.; Cipriani, P.; Giacomelli, R. Endothelial-to-mesenchymal transition in systemic sclerosis. Clin. Exp. Immunol. 2021, 205, 12–27. [Google Scholar] [CrossRef] [PubMed]
- Woo, K.V.; Shen, I.Y.; Weinheimer, C.J.; Kovacs, A.; Nigro, J.; Lin, C.Y.; Chakinala, M.; Byers, D.E.; Ornitz, D.M. Endothelial FGF signaling is protective in hypoxia-induced pulmonary hypertension. J. Clin. Invest. 2021, 131, e141467. [Google Scholar] [CrossRef] [PubMed]
- Yun, E.; Kook, Y.; Yoo, K.H.; Kim, K.I.; Lee, M.S.; Kim, J.; Lee, A. Endothelial to Mesenchymal Transition in Pulmonary Vascular Diseases. Biomedicines 2020, 8, 639. [Google Scholar] [CrossRef]
- Jin, Y.; Cheng, X.; Lu, J.; Li, X. Exogenous BMP-7 Facilitates the Recovery of Cardiac Function after Acute Myocardial Infarction through Counteracting TGF-β1 Signaling Pathway. Tohoku J. Exp. Med. 2018, 244, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Xia, R.; Li, Z.; Zhang, L.; Xia, C.; Ai, H.; Yang, Z.; Guo, Y. Mesenchymal Stem Cells Combined with Hepatocyte Growth Factor Therapy for Attenuating Ischaemic Myocardial Fibrosis: Assessment using Multimodal Molecular Imaging. Sci. Rep. 2016, 6, 33700. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Chen, X.; Chen, L.; Zhou, X.; Zheng, G.; Zhang, H.; Huang, W.; Cai, J. Anti-fibrosis effect of scutellarin via inhibition of endothelial-mesenchymal transition on isoprenaline-induced myocardial fibrosis in rats. Molecules 2014, 19, 15611–15623. [Google Scholar] [CrossRef]
- Wang, D.; Zhu, H.; Yang, Q.; Sun, Y. Effects of relaxin on cardiac fibrosis, apoptosis, and tachyarrhythmia in rats with myocardial infarction. Biomed. Pharmacother. 2016, 84, 348–355. [Google Scholar] [CrossRef]
- Lai, B.; Li, Z.; He, M.; Wang, Y.; Chen, L.; Zhang, J.; Yang, Y.; Shyy, J.Y. Atheroprone flow enhances the endothelial-to-mesenchymal transition. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1293–H1303. [Google Scholar] [CrossRef] [Green Version]
- Chowkwale, M.; Mahler, G.J.; Huang, P.; Murray, B.T. A multiscale in silico model of endothelial to mesenchymal transformation in a tumor microenvironment. J. Theor. Biol. 2019, 480, 229–240. [Google Scholar] [CrossRef]
- Tripathi, S.; Xing, J.; Levine, H.; Jolly, M.K. Mathematical Modeling of Plasticity and Heterogeneity in EMT. In The Epithelial-to Mesenchymal Transition: Methods and Protocols; Campbell, K., Theveneau, E., Eds.; Springer: New York, NY, USA, 2021; pp. 385–413. [Google Scholar]
- Weinstein, N.; Mendoza, L.; Álvarez-Buylla, E.R. A Computational Model of the Endothelial to Mesenchymal Transition. Front. Genet. 2020, 11, 40. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Molecules Involved | Functional Changes Seen Related to EndoMT | Model System (In Vitro/In Vivo) | Cardiac Disease Studied | References |
|---|---|---|---|---|
| TGFβ | Cells lost endothelial markers; developed spindly shapes; gained the capacity to produce a variety of fibroblast-specific molecules. Regulated cell phenotypes and lipid uptake and cell signaling, acted as a profibrotic switch in cardiac fibrosis diseases, thereby affecting the surface structure and internal atherosclerotic plaque. | In vitro: mitral valve endothelial cells; HUVECs; HCAECs; and HAECs. In vivo: frozen sections of aortic valves from mature sheep; in atherosclerotic plaque in the mouse model. | Atherosclerosis; adult valve disease; cardiac fibrosis; pulmonary arterial hypertension. | [23,31,91,92,93,95,96,105] |
| miR-200a overexpression; miR-200c-3p | miR-200a overexpression blocked EndoMT: inhibited α-SMA, FSP-1, CD31, and VE-cadherin expression. miRNA-200c-3p promoted EndoMT. | In vitro: HAECs; HUVECs. In vivo: In human femoral arteries with atherosclerotic lesions; in the mouse model. | Cardiac fibrosis; atherosclerosis. | [95,100] |
| ERK pathway↓ | Losartan suppressed EndoMT by blocking the TGFβ-induced phosphorylation of the ERK pathway. | In vitro: mitral valve endothelial cells. | Myocardial fibrosis | [96,106] |
| TGFβ1 treatment: PDGF-BB signaling↑; SM22α ↑; α-SMA↑ | Unable to prevent thrombin formation; acquired and enhanced the migratory capacity. | In vitro: HUVECs; HCAECs; HAECs. In vivo: frozen sections of aortic valves from mature sheep. | Adult valve disease | [92,93,95] |
| Wnt2↑ | Expressed significantly high in atherosclerotic lesions. | In vivo: in atherosclerotic lesions in the mouse model. | Atherosclerosis | [101] |
| BMPR2 | BMPR2 mutated gene was related to idiopathic PAH. | In vivo: familial PAH in rats. | Pulmonary arterial hypertension | [37,103,107] |
| PDGFR-β↑ VE-cadherin↓ | The PDGF–NF-κB–HIF1-α–Snail axis promoted VE-cadherin down-expression. | In vivo: in the mouse model. | Myocardial infarction | [104] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peng, Q.; Shan, D.; Cui, K.; Li, K.; Zhu, B.; Wu, H.; Wang, B.; Wong, S.; Norton, V.; Dong, Y.; et al. The Role of Endothelial-to-Mesenchymal Transition in Cardiovascular Disease. Cells 2022, 11, 1834. https://doi.org/10.3390/cells11111834
Peng Q, Shan D, Cui K, Li K, Zhu B, Wu H, Wang B, Wong S, Norton V, Dong Y, et al. The Role of Endothelial-to-Mesenchymal Transition in Cardiovascular Disease. Cells. 2022; 11(11):1834. https://doi.org/10.3390/cells11111834
Chicago/Turabian StylePeng, Qianman, Dan Shan, Kui Cui, Kathryn Li, Bo Zhu, Hao Wu, Beibei Wang, Scott Wong, Vikram Norton, Yunzhou Dong, and et al. 2022. "The Role of Endothelial-to-Mesenchymal Transition in Cardiovascular Disease" Cells 11, no. 11: 1834. https://doi.org/10.3390/cells11111834
APA StylePeng, Q., Shan, D., Cui, K., Li, K., Zhu, B., Wu, H., Wang, B., Wong, S., Norton, V., Dong, Y., Lu, Y. W., Zhou, C., & Chen, H. (2022). The Role of Endothelial-to-Mesenchymal Transition in Cardiovascular Disease. Cells, 11(11), 1834. https://doi.org/10.3390/cells11111834