
LFA1 Activation: Insights from a Single-Molecule Approach


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
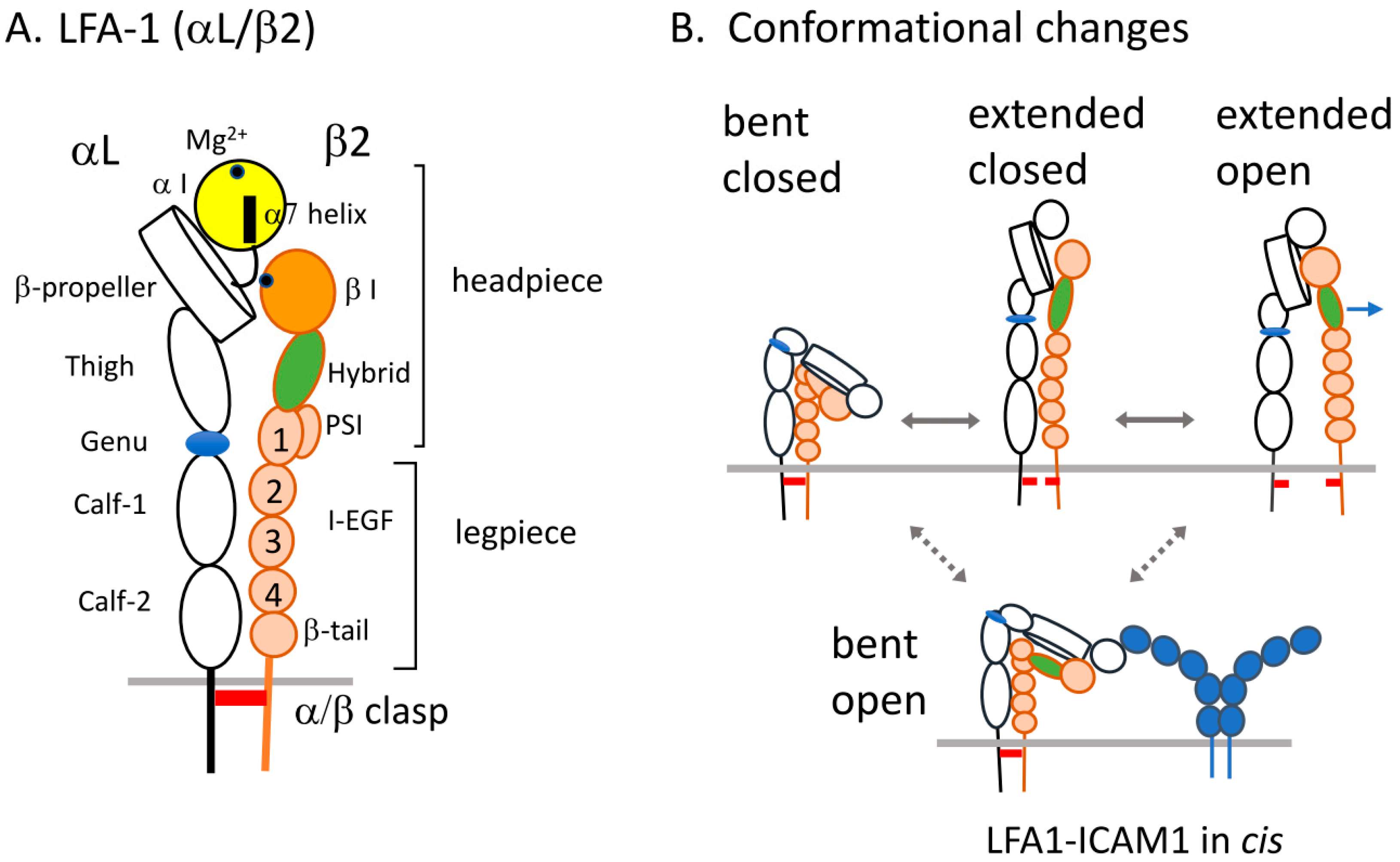
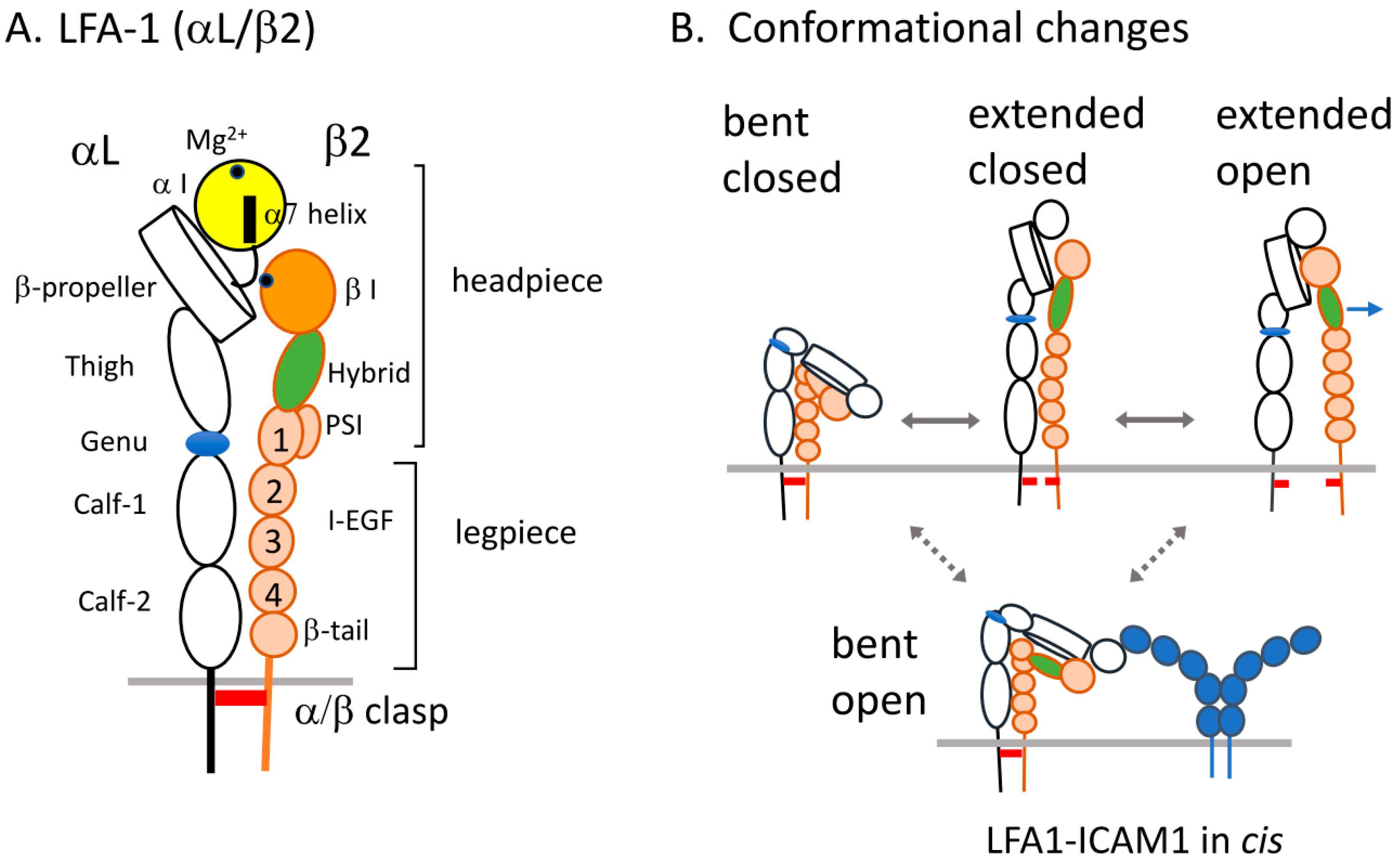
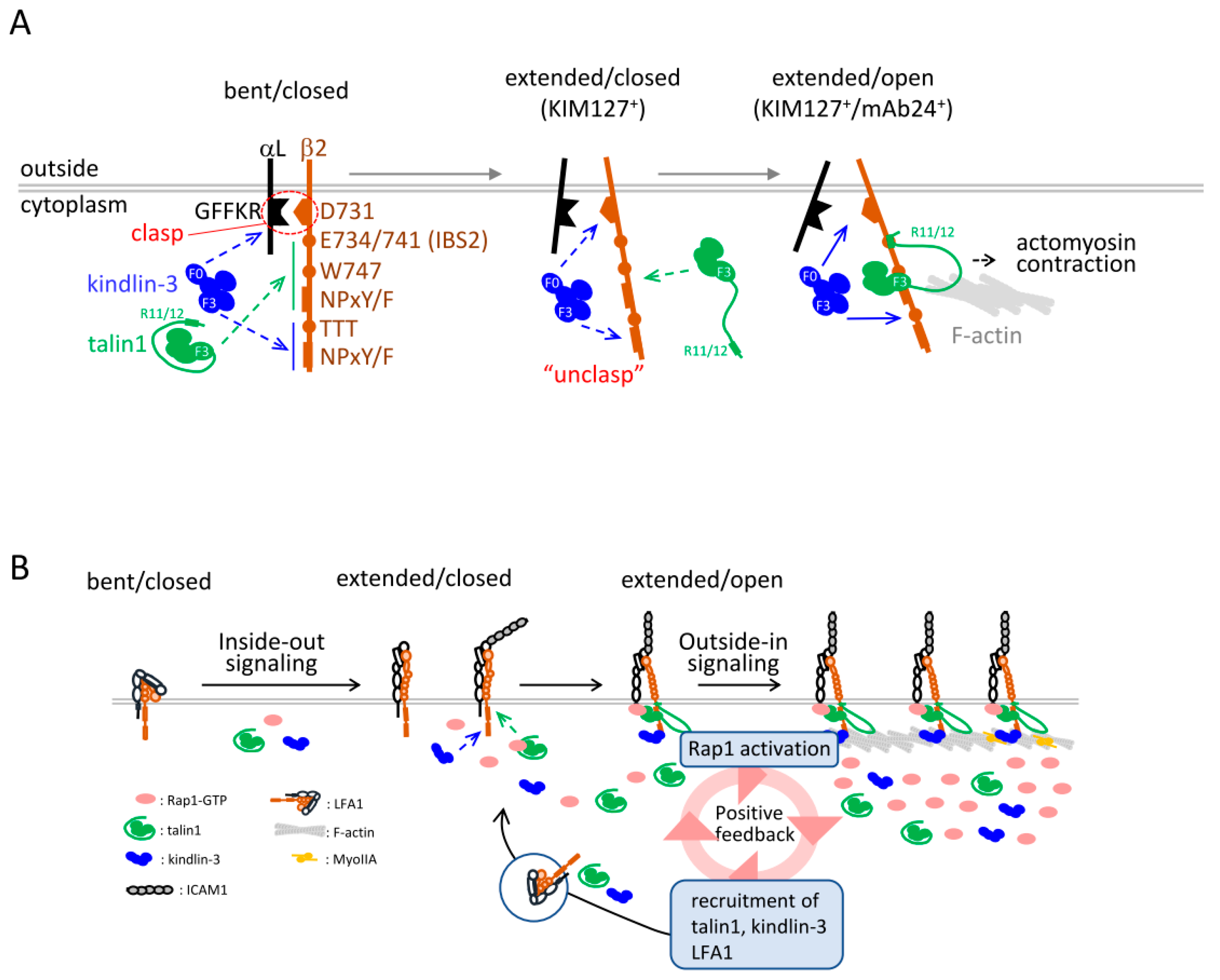
2. Structure of LFA1 and Conformational Changes
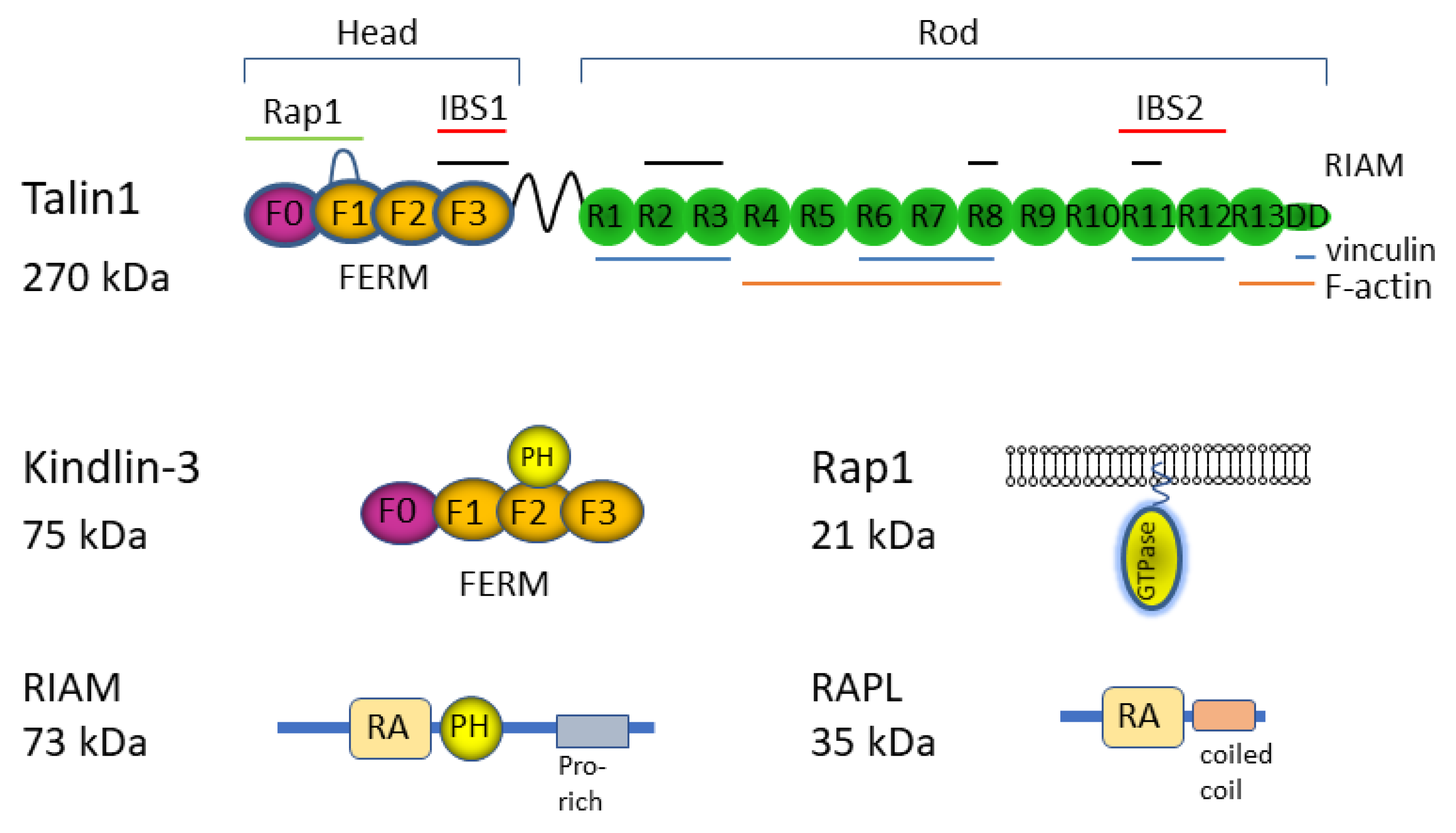
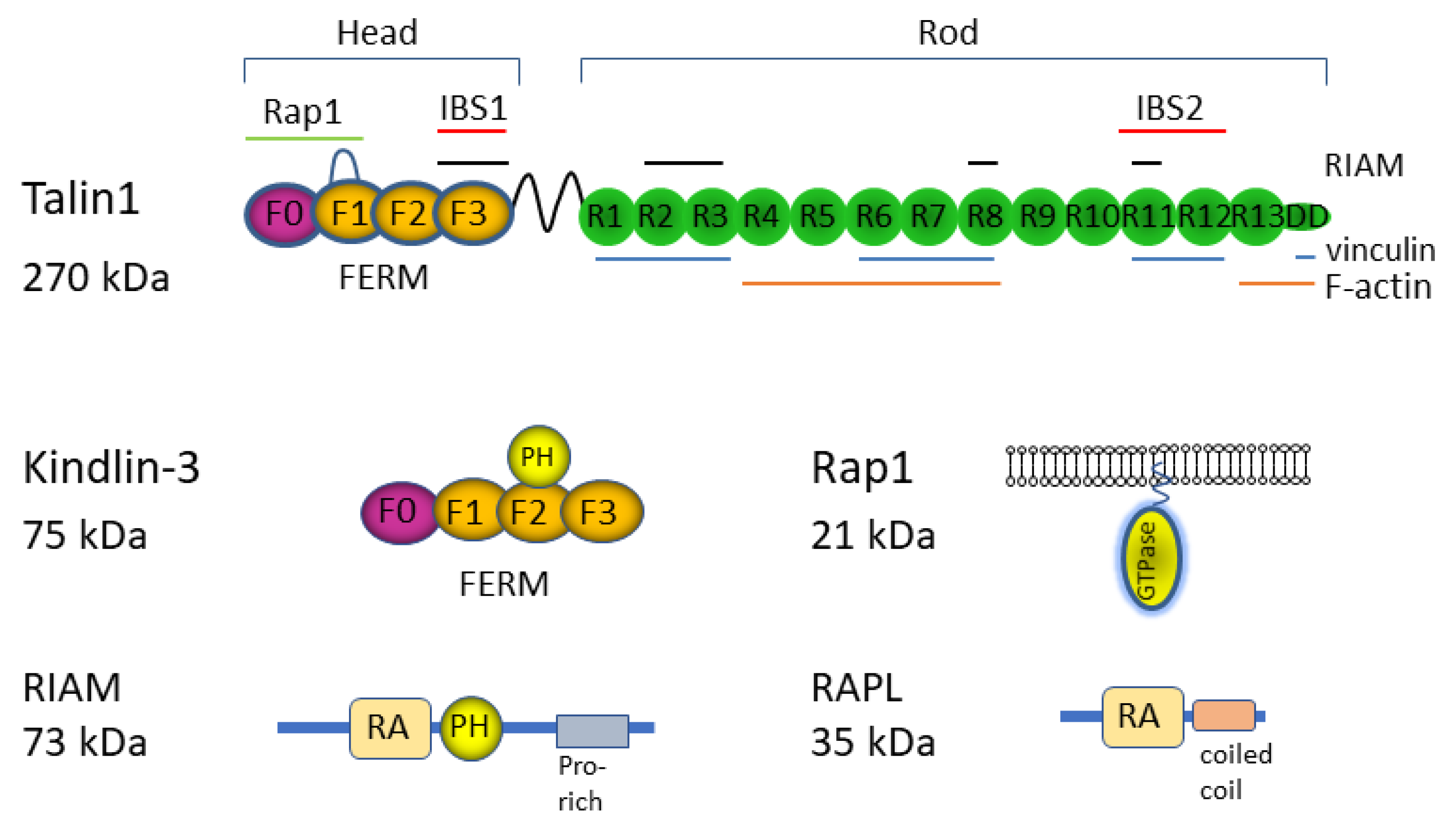
3. Intracellular Regulators of LFA1 Activation
3.1. Rap1
3.2. Talin1
3.3. Kindlin-3
3.4. RIAM
3.5. RAPL
4. Affinity Measurements of LFA1 and ICAM1 Interactions
5. Single-Molecule Measurements of Ligand Binding and Integrin Adaptor Proteins
5.1. LFA1 and ICAM1 Interactions
5.2. Single-Molecule Measurements of Integrin Adaptors to LFA1
5.3. Translocation and Binding Kinetics of Integrin Adaptors
5.4. Coupling of Affinity and Avidity Modulation of LFA1
6. Summary
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hanna, S.; Etzioni, A. Leukocyte adhesion deficiencies. Ann. N. Y. Acad. Sci. 2012, 1250, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Hogg, N.; Patzak, I.; Willenbrock, F. The insider’s guide to leukocyte integrin signalling and function. Nat. Rev. Immunol. 2011, 11, 416–426. [Google Scholar] [CrossRef] [PubMed]
- Dustin, M.; Springer, T.A. T-cell receptor cross-linking transiently stimulates adhesiveness through LFA-1. Nature 1989, 341, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Kinashi, T. Intracellular signalling controlling integrin activation in lymphocytes. Nat. Rev. Immunol. 2005, 5, 546–559. [Google Scholar] [CrossRef]
- Moser, M.; Legate, K.R.; Zent, R.; Fassler, R. The tail of integrins, talin, and kindlins. Science 2009, 324, 895–899. [Google Scholar] [CrossRef]
- Calderwood, D.A.; Campbell, I.D.; Critchley, D.R. Talins and kindlins: Partners in integrin-mediated adhesion. Nat. Rev. Mol. Cell Biol. 2013, 14, 503–517. [Google Scholar] [CrossRef] [Green Version]
- Rognoni, E.; Ruppert, R.; Fässler, R. The kindlin family: Functions, signaling properties and implications for human disease. J. Cell Sci. 2016, 129, 17–27. [Google Scholar] [CrossRef] [Green Version]
- Takagi, J.; Springer, T.A. Integrin activation and structural rearrangement. Immunol. Rev. 2002, 186, 141–163. [Google Scholar] [CrossRef]
- Luo, B.-H.; Carman, C.V.; Springer, T.A. Structural Basis of Integrin Regulation and Signaling. Annu. Rev. Immunol. 2007, 25, 619–647. [Google Scholar] [CrossRef] [Green Version]
- Arnaout, M.A. Biology and structure of leukocyte β2 integrins and their role in inflammation. F1000Research 2016, 5, 2433. [Google Scholar] [CrossRef]
- Shimaoka, M.; Xiao, T.; Liu, J.-H.; Yang, Y.; Dong, Y.; Jun, C.-D.; McCormack, A.; Zhang, R.; Joachimiak, A.; Takagi, J.; et al. Structures of the αL I Domain and Its Complex with ICAM-1 Reveal a Shape-Shifting Pathway for Integrin Regulation. Cell 2003, 112, 99–111. [Google Scholar] [CrossRef] [Green Version]
- Springer, T.A. Traffic signals on endothelium for lymphocyte recirculation and leukocyte emigration. Annu. Rev. Physiol. 1995, 57, 827–872. [Google Scholar] [CrossRef] [PubMed]
- Lebedeva, T.; Dustin, M.; Sykulev, Y. ICAM-1 co-stimulates target cells to facilitate antigen presentation. Curr. Opin. Immunol. 2005, 17, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.P.; Stehle, T.; Zhang, R.; Joachimiak, A.; Frech, M.; Goodman, S.L.; Arnaout, M.A. Crystal structure of the extracellular segment of integrin alpha Vbeta3 in complex with an Arg-Gly-Asp ligand. Science 2002, 296, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Adair, B.D.; Xiong, J.-P.; Maddock, C.; Goodman, S.L.; Arnaout, M.A.; Yeager, M. Three-dimensional EM structure of the ectodomain of integrin αVβ3 in a complex with fibronectin. J. Cell Biol. 2005, 168, 1109–1118. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Zhu, J.; Mi, L.-Z.; Walz, T.; Sun, H.; Chen, J.; Springer, T.A. Structural specializations of α4β7, an integrin that mediates rolling adhesion. J. Cell Biol. 2012, 196, 131–146. [Google Scholar] [CrossRef] [Green Version]
- Mould, A.P.; Symonds, E.J.H.; Buckley, P.A.; Grossmann, J.G.; McEwan, P.A.; Barton, S.J.; Askari, J.A.; Craig, S.E.; Bella, J.; Humphries, M. Structure of an Integrin-Ligand Complex Deduced from Solution X-ray Scattering and Site-directed Mutagenesis. J. Biol. Chem. 2003, 278, 39993–39999. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Salas, A.; A Springer, T. Bistable regulation of integrin adhesiveness by a bipolar metal ion cluster. Nat. Struct. Mol. Biol. 2003, 10, 995–1001. [Google Scholar] [CrossRef]
- Takagi, J.; Petre, B.M.; Walz, T.; Springer, T.A. Global Conformational Rearrangements in Integrin Extracellular Domains in Outside-In and Inside-Out Signaling. Cell 2002, 110, 599–611. [Google Scholar] [CrossRef] [Green Version]
- Nishida, N.; Xie, C.; Shimaoka, M.; Cheng, Y.; Walz, T.; Springer, T.A. Activation of Leukocyte β2 Integrins by Conversion from Bent to Extended Conformations. Immunity 2006, 25, 583–594. [Google Scholar] [CrossRef] [Green Version]
- Sen, M.; Koksal, A.; Yuki, K.; Wang, J.; Springer, T.A. Ligand- and cation-induced structural alterations of the leukocyte integrin LFA-1. J. Biol. Chem. 2018, 293, 6565–6577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Su, Y.; Xia, W.; Qin, Y.; Humphries, M.; Vestweber, D.; Cabañas, C.; Lu, C.; A Springer, T. Conformational equilibria and intrinsic affinities define integrin activation. EMBO J. 2017, 36, 629–645. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Springer, T.A. Energy landscape differences among integrins establish the framework for understanding activation. J. Cell Biol. 2017, 217, 397–412. [Google Scholar] [CrossRef]
- Wegener, K.L.; Campbell, I.D. Transmembrane and cytoplasmic domains in integrin activation and protein-protein interactions (Review). Mol. Membr. Biol. 2008, 25, 376–387. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.F.; A Springer, T. The alpha subunit cytoplasmic domain regulates the assembly and adhesiveness of integrin lymphocyte function-associated antigen-1. J. Immunol. 1997, 159, 268–278. [Google Scholar] [PubMed]
- Kim, M.; Carman, C.V.; Springer, T.A. Bidirectional Transmembrane Signaling by Cytoplasmic Domain Separation in Integrins. Science 2003, 301, 1720–1725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chigaev, A.; Buranda, T.; Dwyer, D.C.; Prossnitz, E.R.; Sklar, L.A. FRET Detection of Cellular α4-Integrin Conformational Activation. Biophys. J. 2003, 85, 3951–3962. [Google Scholar] [CrossRef] [Green Version]
- Ye, F.; Hu, G.; Taylor, D.; Ratnikov, B.; Bobkov, A.A.; McLean, M.A.; Sligar, S.G.; Taylor, K.A.; Ginsberg, M.H. Recreation of the terminal events in physiological integrin activation. J. Cell Biol. 2010, 188, 157–173. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Ferzly, M.; Takagi, J.; Springer, T.A. Epitope mapping of antibodies to the C-terminal region of the integrin beta 2 subunit reveals regions that become exposed upon receptor activation. J. Immunol. 2001, 166, 5629–5637. [Google Scholar] [CrossRef] [Green Version]
- Fan, Z.; McArdle, S.; Mark, G.; Mikulski, Z.; Gutierrez, E.; Engelhardt, B.; Deutsch, U.; Ginsberg, M.; Groisman, A.; Ley, K. Neutrophil recruitment limited by high-affinity bent β2 integrin binding ligand in cis. Nat. Commun. 2016, 7, 12658. [Google Scholar] [CrossRef] [Green Version]
- Fan, Z.; Kiosses, W.B.; Sun, H.; Orecchioni, M.; Ghosheh, Y.; Zajonc, D.M.; Arnaout, M.A.; Gutierrez, E.; Groisman, A.; Ginsberg, M.H.; et al. High-Affinity Bent β2-Integrin Molecules in Arresting Neutrophils Face Each Other through Binding to ICAMs In cis. Cell Rep. 2019, 26, 119–130.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bos, J.L.; De Rooij, J.; Reedquist, K.A. Rap1 signalling: Adhering to new models. Nat. Rev. Mol. Cell Biol. 2001, 2, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Chrzanowska-Wodnicka, M.; Smyth, S.S.; Schoenwaelder, S.M.; Fischer, T.H.; White, G.C. Rap1b is required for normal platelet function and hemostasis in mice. J. Clin. Investig. 2005, 115, 680–687. [Google Scholar] [CrossRef] [Green Version]
- Chu, H.; Awasthi, A.; White, G.C.; Chrzanowska-Wodnicka, M.; Malarkannan, S. Rap1b Regulates B Cell Development, Homing, and T Cell-Dependent Humoral Immunity. J. Immunol. 2008, 181, 3373–3383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondo, N.; Ueda, Y.; Kita, T.; Ozawa, M.; Tomiyama, T.; Yasuda, K.; Lim, D.-S.; Kinashi, T. NDR1-Dependent Regulation of Kindlin-3 Controls High-Affinity LFA-1 Binding and Immune Synapse Organization. Mol. Cell. Biol. 2017, 37, e00424-16. [Google Scholar] [CrossRef] [Green Version]
- Ishihara, S.; Nishikimi, A.; Umemoto, E.; Miyasaka, M.; Saegusa, M.; Katagiri, K. Dual functions of Rap1 are crucial for T-cell homeostasis and prevention of spontaneous colitis. Nat. Commun. 2015, 6, 8982. [Google Scholar] [CrossRef] [PubMed]
- Su, W.; Wynne, J.; Pinheiro, E.M.; Strazza, M.; Mor, A.; Montenont, E.; Berger, J.; Paul, D.S.; Bergmeier, W.; Gertler, F.B.; et al. Rap1 and its effector RIAM are required for lymphocyte trafficking. Blood 2015, 126, 2695–2703. [Google Scholar] [CrossRef] [Green Version]
- Ueda, Y.; Kondo, N.; Ozawa, M.; Yasuda, K.; Tomiyama, T.; Kinashi, T. Sema3e/Plexin D1 Modulates Immunological Synapse and Migration of Thymocytes by Rap1 Inhibition. J. Immunol. 2016, 196, 3019–3031. [Google Scholar] [CrossRef] [Green Version]
- Crittenden, J.R.; Bergmeier, W.; Zhang, Y.; Piffath, C.L.; Liang, Y.; Wagner, D.D.; Housman, D.E.; Graybiel, A.M. CalDAG-GEFI integrates signaling for platelet aggregation and thrombus formation. Nat. Med. 2004, 10, 982–986. [Google Scholar] [CrossRef]
- Reedquist, K.A.; Ross, E.; Koop, E.A.; Wolthuis, R.; Zwartkruis, F.J.; Van Kooyk, Y.; Salmon, M.; Buckley, C.D.; Bos, J.L. The Small Gtpase, Rap1, Mediates Cd31-Induced Integrin Adhesion. J. Cell Biol. 2000, 148, 1151–1158. [Google Scholar] [CrossRef]
- Azoulay-Alfaguter, I.; Strazza, M.; Peled, M.; Novak, H.K.; Muller, J.; Dustin, M.L.; Mor, A. The tyrosine phosphatase SHP-1 promotes T cell adhesion by activating the adaptor protein CrkII in the immunological synapse. Sci. Signal. 2017, 10, eaal2880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stefanini, L.; Bergmeier, W. RAP1-GTPase signaling and platelet function. J. Mol. Med. 2016, 94, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Katagiri, K.; Maeda, A.; Shimonaka, M.; Kinashi, T. RAPL, a Rap1-binding molecule that mediates Rap1-induced adhesion through spatial regulation of LFA-1. Nat. Immunol. 2003, 4, 741–748. [Google Scholar] [CrossRef]
- Lafuente, E.M.; van Puijenbroek, A.A.; Krause, M.; Carman, C.V.; Freeman, G.J.; Berezovskaya, A.; Constantine, E.; Springer, T.A.; Gertler, F.B.; Boussiotis, V.A. RIAM, an Ena/VASP and Profilin Ligand, Interacts with Rap1-GTP and Mediates Rap1-Induced Adhesion. Dev. Cell 2004, 7, 585–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plak, K.; Pots, H.; Van Haastert, P.J.M.; Kortholt, A. Direct Interaction between TalinB and Rap1 is necessary for adhesion of Dictyostelium cells. BMC Cell Biol. 2016, 17, 1. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Yang, J.; Bromberger, T.; Holly, A.; Lu, F.; Liu, H.; Sun, K.; Klapproth, S.; Hirbawi, J.; Byzova, T.; et al. Structure of Rap1b bound to talin reveals a pathway for triggering integrin activation. Nat. Commun. 2017, 8, 1744. [Google Scholar] [CrossRef] [Green Version]
- Bromberger, T.; Zhu, L.; Klapproth, S.; Qin, J.; Moser, M. Rap1 and membrane lipids cooperatively recruit talin to trigger integrin activation. J. Cell Sci. 2019, 132. [Google Scholar] [CrossRef]
- Bromberger, T.; Klapproth, S.; Rohwedder, I.; Zhu, L.; Mittmann, L.; Reichel, C.A.; Sperandio, M.; Qin, J.; Moser, M. Direct Rap1/Talin1 interaction regulates platelet and neutrophil integrin activity in mice. Blood 2018, 132, 2754–2762. [Google Scholar] [CrossRef] [Green Version]
- Lagarrigue, F.; Gingras, A.R.; Paul, D.S.; Valadez, A.J.; Cuevas, M.N.; Sun, H.; Lopez-Ramirez, M.A.; Goult, B.T.; Shattil, S.J.; Bergmeier, W.; et al. Rap1 binding to the talin 1 F0 domain makes a minimal contribution to murine platelet GPIIb-IIIa activation. Blood Adv. 2018, 2, 2358–2368. [Google Scholar] [CrossRef] [Green Version]
- Tadokoro, S.; Shattil, S.J.; Eto, K.; Tai, V.; Liddington, R.C.; de Pereda, J.M.; Ginsberg, M.H.; Calderwood, D.A. Talin binding to integrin beta tails: A final common step in integrin activation. Science 2003, 302, 103–106. [Google Scholar] [CrossRef]
- Shattil, S.J.; Kim, C.; Ginsberg, M.H. The final steps of integrin activation: The end game. Nat. Rev. Mol. Cell Biol. 2010, 11, 288–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieswandt, B.; Moser, M.; Pleines, I.; Varga-Szabo, D.; Monkley, S.; Critchley, D.; Fassler, R. Loss of talin1 in platelets abrogates integrin activation, platelet aggregation, and thrombus formation in vitro and in vivo. J. Exp. Med. 2007, 204, 3113–3118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wernimont, S.A.; Wiemer, A.J.; Bennin, D.A.; Monkley, S.J.; Ludwig, T.; Critchley, D.R.; Huttenlocher, A. Contact-Dependent T Cell Activation and T Cell Stopping Require Talin1. J. Immunol. 2011, 187, 6256–6267. [Google Scholar] [CrossRef]
- Calderwood, D.A.; Zent, R.; Grant, R.; Rees, D.J.; Hynes, R.O.; Ginsberg, M.H. The talin head domain binds to integrin beta subunit cytoplasmic tails and regulates integrin activation. J. Biol. Chem. 1999, 274, 28071–28074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anthis, N.J.; Wegener, K.L.; Ye, F.; Kim, C.; Goult, B.T.; Lowe, E.D.; Vakonakis, I.; Bate, N.; Critchley, D.R.; Ginsberg, M.H.; et al. The structure of an integrin/talin complex reveals the basis of inside-out signal transduction. EMBO J. 2009, 28, 3623–3632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodius, S.; Chaloin, O.; Moes, M.; Schaffner-Reckinger, E.; Landrieu, I.; Lippens, G.; Lin, M.; Zhang, J.; Kieffer, N. The Talin Rod IBS2 α-Helix Interacts with the β3 Integrin Cytoplasmic Tail Membrane-proximal Helix by Establishing Charge Complementary Salt Bridges. J. Biol. Chem. 2008, 283, 24212–24223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goksoy, E.; Ma, Y.-Q.; Wang, X.; Kong, X.; Perera, D.; Plow, E.F.; Qin, J. Structural Basis for the Autoinhibition of Talin in Regulating Integrin Activation. Mol. Cell 2008, 31, 124–133. [Google Scholar] [CrossRef] [Green Version]
- Goult, B.T.; Xu, X.-P.; Gingras, A.R.; Swift, M.; Patel, B.; Bate, N.; Kopp, P.M.; Barsukov, I.L.; Critchley, D.R.; Volkmann, N.; et al. Structural studies on full-length talin1 reveal a compact auto-inhibited dimer: Implications for talin activation. J. Struct. Biol. 2013, 184, 21–32. [Google Scholar] [CrossRef] [Green Version]
- Song, X.; Yang, J.; Hirbawi, J.; Ye, S.; Perera, H.D.; Goksoy, E.; Dwivedi, P.; Plow, E.F.; Zhang, R.; Qin, J. A novel membrane-dependent on/off switch mechanism of talin FERM domain at sites of cell adhesion. Cell Res. 2012, 22, 1533–1545. [Google Scholar] [CrossRef] [Green Version]
- Legate, K.R.; Takahashi, S.; Bonakdar, N.; Fabry, B.; Boettiger, D.; Zent, R.; Fässler, R. Integrin adhesion and force coupling are independently regulated by localized PtdIns(4,5)2synthesis. EMBO J. 2011, 30, 4539–4553. [Google Scholar] [CrossRef] [Green Version]
- Yago, T.; Zhang, N.; Zhao, L.; Abrams, C.S.; McEver, R.P. Selectins and chemokines use shared and distinct signals to activate β2 integrins in neutrophils. Blood Adv. 2018, 2, 731–744. [Google Scholar] [CrossRef] [Green Version]
- Moser, M.; Nieswandt, B.; Ussar, S.; Pozgajova, M.; Fässler, R. Kindlin-3 is essential for integrin activation and platelet aggregation. Nat. Med. 2008, 14, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Malinin, N.L.; Zhang, L.; Choi, J.; Ciocea, A.; Razorenova, O.; Ma, Y.-Q.; A Podrez, E.; Tosi, M.; Lennon, D.P.; I Caplan, A.; et al. A point mutation in KINDLIN3 ablates activation of three integrin subfamilies in humans. Nat. Med. 2009, 15, 313–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moser, M.; Bauer, M.; Schmid, S.; Ruppert, R.; Schmidt, S.; Sixt, M.; Wang, H.V.; Sperandio, M.; Fassler, R. Kindlin-3 is required for beta2 integrin-mediated leukocyte adhesion to endothelial cells. Nat. Med. 2009, 15, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Svensson, L.; Howarth, K.; McDowall, A.; Patzak, I.; Evans, R.; Ussar, S.; Moser, M.; Metin, A.; Fried, M.; Tomlinson, I.; et al. Leukocyte adhesion deficiency-III is caused by mutations in KINDLIN3 affecting integrin activation. Nat. Med. 2009, 15, 306–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefort, C.T.; Ley, K. Neutrophil arrest by LFA-1 activation. Front. Immunol. 2012, 3, 157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bledzka, K.; Liu, J.; Xu, Z.; Perera, H.D.; Yadav, S.P.; Bialkowska, K.; Qin, J.; Ma, Y.-Q.; Plow, E.F. Spatial Coordination of Kindlin-2 with Talin Head Domain in Interaction with Integrin β Cytoplasmic Tails. J. Biol. Chem. 2012, 287, 24585–24594. [Google Scholar] [CrossRef] [Green Version]
- Hart, R.; Stanley, P.; Chakravarty, P.; Hogg, N. The Kindlin 3 Pleckstrin Homology Domain Has an Essential Role in Lymphocyte Function-associated Antigen 1 (LFA-1) Integrin-mediated B Cell Adhesion and Migration. J. Biol. Chem. 2013, 288, 14852–14862. [Google Scholar] [CrossRef] [Green Version]
- Wen, L.; Marki, A.; Roy, P.; McArdle, S.; Sun, H.; Fan, Z.; Gingras, A.R.; Ginsberg, M.H.; Ley, K. Kindlin-3 recruitment to the plasma membrane precedes high-affinity β2-integrin and neutrophil arrest from rolling. Blood 2021, 137, 29–38. [Google Scholar] [CrossRef]
- Theodosiou, M.; Widmaier, M.; Böttcher, R.T.; Rognoni, E.; Veelders, M.; Bharadwaj, M.; Lambacher, A.; Austen, K.; Muller, D.J.; Zent, R.; et al. Kindlin-2 cooperates with talin to activate integrins and induces cell spreading by directly binding paxillin. eLife 2016, 5, e10130. [Google Scholar] [CrossRef]
- Nguyen, H.T.T.; Xu, Z.; Shi, X.; Liu, S.; Schulte, M.L.; White, G.C.; Ma, Y. Paxillin binding to the PH domain of kindlin-3 in platelets is required to support integrin αIIbβ3 outside-in signaling. J. Thromb. Haemost. 2021, 19, 3126–3138. [Google Scholar] [CrossRef] [PubMed]
- Böttcher, R.T.; Böttcher, R.T.; Veelders, M.; Veelders, M.; Rombaut, P.; Rombaut, P.; Faix, J.; Faix, J.; Theodosiou, M.; Theodosiou, M.; et al. Kindlin-2 recruits paxillin and Arp2/3 to promote membrane protrusions during initial cell spreading. J. Cell Biol. 2017, 216, 3785–3798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Huang, M.; Lai, J.; Mao, K.; Sun, P.; Cao, Z.; Hu, Y.; Zhang, Y.; Schulte, M.L.; Jin, C.; et al. Kindlin supports platelet integrin αIIbβ3 activation by interacting with paxillin. J. Cell Sci. 2017, 130, 3764–3775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, L.; Liu, H.; Lu, F.; Yang, J.; Byzova, T.V.; Qin, J. Structural Basis of Paxillin Recruitment by Kindlin-2 in Regulating Cell Adhesion. Structure 2019, 27, 1686–1697.e5. [Google Scholar] [CrossRef]
- Bledzka, K.; Bialkowska, K.; Sossey-Alaoui, K.; Vaynberg, J.; Pluskota, E.; Qin, J.; Plow, E.F. Kindlin-2 directly binds actin and regulates integrin outside-in signaling. J. Cell Biol. 2016, 213, 97–108. [Google Scholar] [CrossRef] [Green Version]
- Ye, F.; Petrich, B.G.; Anekal, P.; Lefort, C.T.; Kasirer-Friede, A.; Shattil, S.J.; Ruppert, R.; Moser, M.; Fässler, R.; Ginsberg, M.H. The Mechanism of Kindlin-Mediated Activation of Integrin alphaIIbbeta3. Curr. Biol. 2013, 23, 2288–2295. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Deng, Y.; Sun, K.; Yang, H.; Liu, J.; Wang, M.; Zhang, Z.; Lin, J.; Wu, C.; Wei, Z.; et al. Structural basis of kindlin-mediated integrin recognition and activation. Proc. Natl. Acad. Sci. USA 2017, 114, 9349–9354. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Xiao, D.; Ni, Y.; Zhang, T.; Cao, Z.; Xu, Z.; Nguyen, H.; Zhang, J.; White, G.C.; Ding, J.; et al. Structure basis of the FERM domain of kindlin-3 in supporting integrin αIIbβ3 activation in platelets. Blood Adv. 2020, 4, 3128–3135. [Google Scholar] [CrossRef]
- Bu, W.; Levitskaya, Z.; Loh, Z.Y.; Jin, S.; Basu, S.; Ero, R.; Yan, X.; Wang, M.; Ngan, S.F.C.; Sze, S.K.; et al. Structural basis of human full-length kindlin-3 homotrimer in an auto-inhibited state. PLoS Biol. 2020, 18, e3000755. [Google Scholar] [CrossRef]
- Wynne, J.P.; Wu, J.; Su, W.; Mor, A.; Patsoukis, N.; Boussiotis, V.A.; Hubbard, S.R.; Philips, M.R. Rap1-interacting adapter molecule (RIAM) associates with the plasma membrane via a proximity detector. J. Cell Biol. 2012, 199, 317–329. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Zhu, L.; Zhang, H.; Hirbawi, J.; Fukuda, K.; Dwivedi, P.; Liu, J.; Byzova, T.; Plow, E.F.; Wu, J.; et al. Conformational activation of talin by RIAM triggers integrin-mediated cell adhesion. Nat. Commun. 2014, 5, 5880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagarrigue, F.; Kim, C.; Ginsberg, M.H. The Rap1-RIAM-talin axis of integrin activation and blood cell function. Blood 2016, 128, 479–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klapproth, S.; Sperandio, M.; Pinheiro, E.M.; Prünster, M.; Soehnlein, O.; Gertler, F.B.; Fässler, R.; Moser, M. Loss of the Rap1 effector RIAM results in leukocyte adhesion deficiency due to impaired β2 integrin function in mice. Blood 2015, 126, 2704–2712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stritt, S.; Wolf, K.; Lorenz, V.; Vögtle, T.; Gupta, S.; Bösl, M.R.; Nieswandt, B. Rap1-GTP–interacting adaptor molecule (RIAM) is dispensable for platelet integrin activation and function in mice. Blood 2015, 125, 219–222. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Lagarrigue, F.; Wang, H.; Fan, Z.; Lopez-Ramirez, M.A.; Chang, J.T.; Ginsberg, M.H. Distinct integrin activation pathways for effector and regulatory T cell trafficking and function. J. Exp. Med. 2020, 218, e20201524. [Google Scholar] [CrossRef]
- Krause, M.; Leslie, J.D.; Stewart, M.; Lafuente, E.M.; Valderrama, F.; Jagannathan, R.; Strasser, G.A.; Rubinson, D.A.; Liu, H.; Way, M.; et al. Lamellipodin, an Ena/VASP Ligand, Is Implicated in the Regulation of Lamellipodial Dynamics. Dev. Cell 2004, 7, 571–583. [Google Scholar] [CrossRef] [Green Version]
- Law, A.-L.; Vehlow, A.; Kotini, M.; Dodgson, L.; Soong, D.; Theveneau, E.; Bodo, C.; Taylor, E.; Navarro, C.; Perera, U.; et al. Lamellipodin and the Scar/WAVE complex cooperate to promote cell migration in vivo. J. Cell Biol. 2013, 203, 673–689. [Google Scholar] [CrossRef] [Green Version]
- Richter, A.M.; Pfeifer, G.P.; Dammann, R.H. The RASSF proteins in cancer; from epigenetic silencing to functional characterization. Biochim. Biophys. Acta 2009, 1796, 114–128. [Google Scholar] [CrossRef]
- Katagiri, K.; Imamura, M.; Kinashi, T. Spatiotemporal regulation of the kinase Mst1 by binding protein RAPL is critical for lymphocyte polarity and adhesion. Nat. Immunol. 2006, 7, 919–928. [Google Scholar] [CrossRef]
- Katagiri, K.; Ohnishi, N.; Kabashima, K.; Iyoda, T.; Takeda, N.; Shinkai, Y.; Inaba, K.; Kinashi, T. Crucial functions of the Rap1 effector molecule RAPL in lymphocyte and dendritic cell trafficking. Nat. Immunol. 2004, 5, 1045–1051. [Google Scholar] [CrossRef]
- Katagiri, K.; Katakai, T.; Ebisuno, Y.; Ueda, Y.; Okada, T.; Kinashi, T. Mst1 controls lymphocyte trafficking and interstitial motility within lymph nodes. EMBO J. 2009, 28, 1319–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mou, F.; Praskova, M.; Xia, F.; Van Buren, D.; Hock, H.; Avruch, J.; Zhou, D. The Mst1 and Mst2 kinases control activation of rho family GTPases and thymic egress of mature thymocytes. J. Exp. Med. 2012, 209, 741–759. [Google Scholar] [CrossRef]
- Tang, F.; Gill, J.; Ficht, X.; Barthlott, T.; Cornils, H.; Schmitz-Rohmer, D.; Hynx, D.; Zhou, D.; Zhang, L.; Xue, G.; et al. The kinases NDR1/2 act downstream of the Hippo homolog MST1 to mediate both egress of thymocytes from the thymus and lymphocyte motility. Sci. Signal. 2015, 8, ra100. [Google Scholar] [CrossRef] [PubMed]
- Nishikimi, A.; Ishihara, S.; Ozawa, M.; Etoh, K.; Fukuda, M.; Kinashi, T.; Katagiri, K. Rab13 acts downstream of the kinase Mst1 to deliver the integrin LFA-1 to the cell surface for lymphocyte trafficking. Sci. Signal. 2014, 7, ra72. [Google Scholar] [CrossRef] [PubMed]
- Capece, T.; Walling, B.L.; Lim, K.; Kim, K.-D.; Bae, S.; Chung, H.-L.; Topham, D.J.; Kim, M. A novel intracellular pool of LFA-1 is critical for asymmetric CD8+ T cell activation and differentiation. J. Cell Biol. 2017, 216, 3817–3829. [Google Scholar] [CrossRef] [PubMed]
- Schürpf, T.; A Springer, T. Regulation of integrin affinity on cell surfaces. EMBO J. 2011, 30, 4712–4727. [Google Scholar] [CrossRef] [Green Version]
- Shen, H.; Tauzin, L.J.; Baiyasi, R.; Wang, W.; Moringo, N.; Shuang, B.; Landes, C.F. Single Particle Tracking: From Theory to Biophysical Applications. Chem. Rev. 2017, 117, 7331–7376. [Google Scholar] [CrossRef]
- Grakoui, A.; Bromley, S.K.; Sumen, C.; Davis, M.M.; Shaw, A.S.; Allen, P.M.; Dustin, M.L. The Immunological Synapse: A Molecular Machine Controlling T Cell Activation. Science 1999, 285, 221–227. [Google Scholar] [CrossRef] [Green Version]
- E Labadia, M.; Jeanfavre, D.D.; O Caviness, G.; Morelock, M.M. Molecular regulation of the interaction between leukocyte function-associated antigen-1 and soluble ICAM-1 by divalent metal cations. J. Immunol. 1998, 161, 836–842. [Google Scholar]
- Comrie, W.A.; Babich, A.; Burkhardt, J.K. F-actin flow drives affinity maturation and spatial organization of LFA-1 at the immunological synapse. J. Cell Biol. 2015, 208, 475–491. [Google Scholar] [CrossRef]
- Cairo, C.W.; Mirchev, R.; Golan, D.E. Cytoskeletal Regulation Couples LFA-1 Conformational Changes to Receptor Lateral Mobility and Clustering. Immunity 2006, 25, 297–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondo, N.; Ueda, Y.; Kinashi, T. Kindlin-3 disrupts an intersubunit association in the integrin LFA1 to trigger positive feedback activation by Rap1 and talin1. Sci. Signal. 2021, 14. [Google Scholar] [CrossRef] [PubMed]
- Anthis, N.J.; Wegener, K.L.; Critchley, D.R.; Campbell, I.D. Structural Diversity in Integrin/Talin Interactions. Structure 2010, 18, 1654–1666. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kondo, N.; Ueda, Y.; Kinashi, T. LFA1 Activation: Insights from a Single-Molecule Approach. Cells 2022, 11, 1751. https://doi.org/10.3390/cells11111751
Kondo N, Ueda Y, Kinashi T. LFA1 Activation: Insights from a Single-Molecule Approach. Cells. 2022; 11(11):1751. https://doi.org/10.3390/cells11111751
Chicago/Turabian StyleKondo, Naoyuki, Yoshihiro Ueda, and Tatsuo Kinashi. 2022. "LFA1 Activation: Insights from a Single-Molecule Approach" Cells 11, no. 11: 1751. https://doi.org/10.3390/cells11111751
APA StyleKondo, N., Ueda, Y., & Kinashi, T. (2022). LFA1 Activation: Insights from a Single-Molecule Approach. Cells, 11(11), 1751. https://doi.org/10.3390/cells11111751