
The Dually Localized EF-Hand Domain-Containing Protein TgEFP1 Regulates the Lytic Cycle of Toxoplasma gondii



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Host Cell and Parasite Maintenance
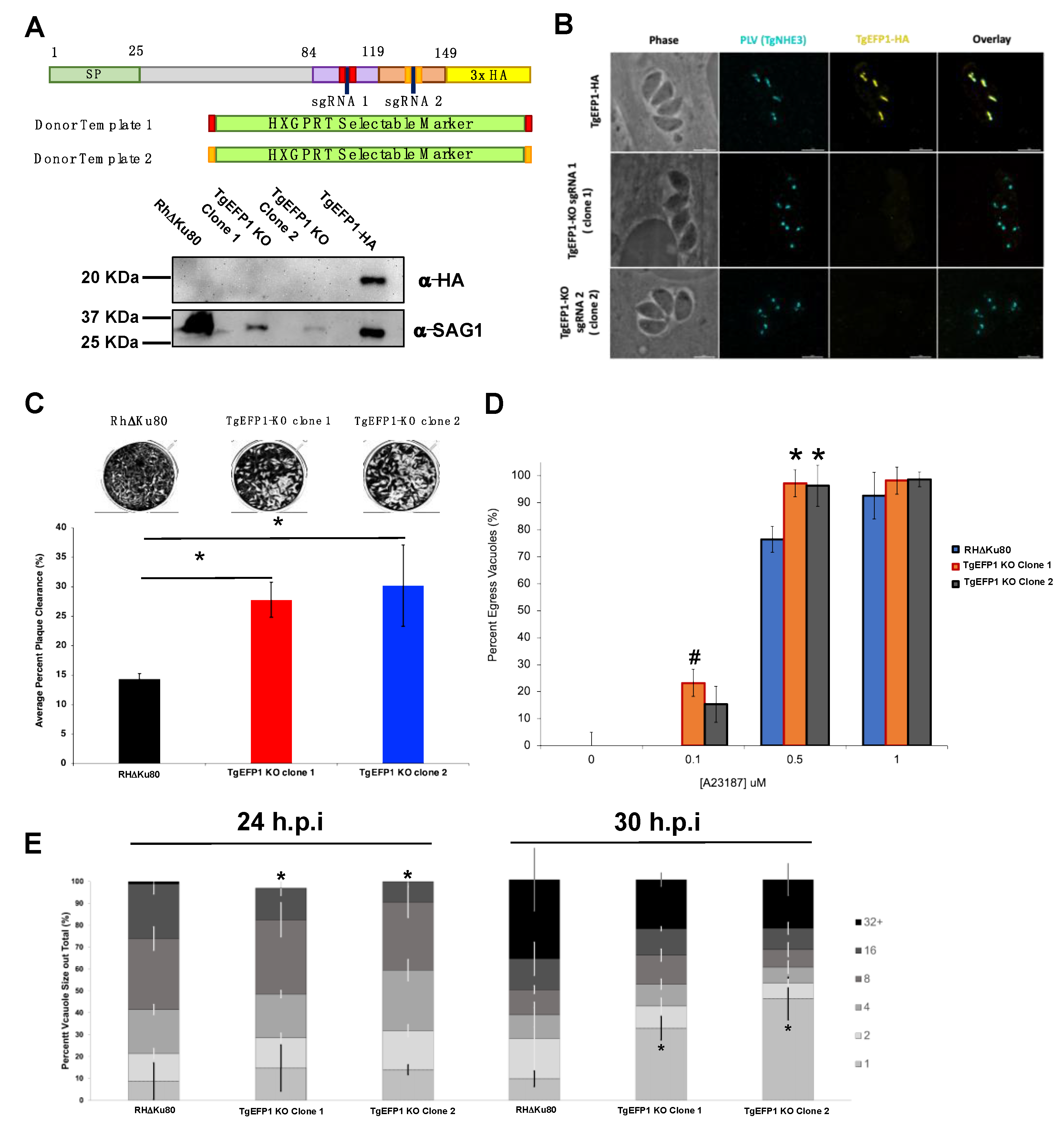
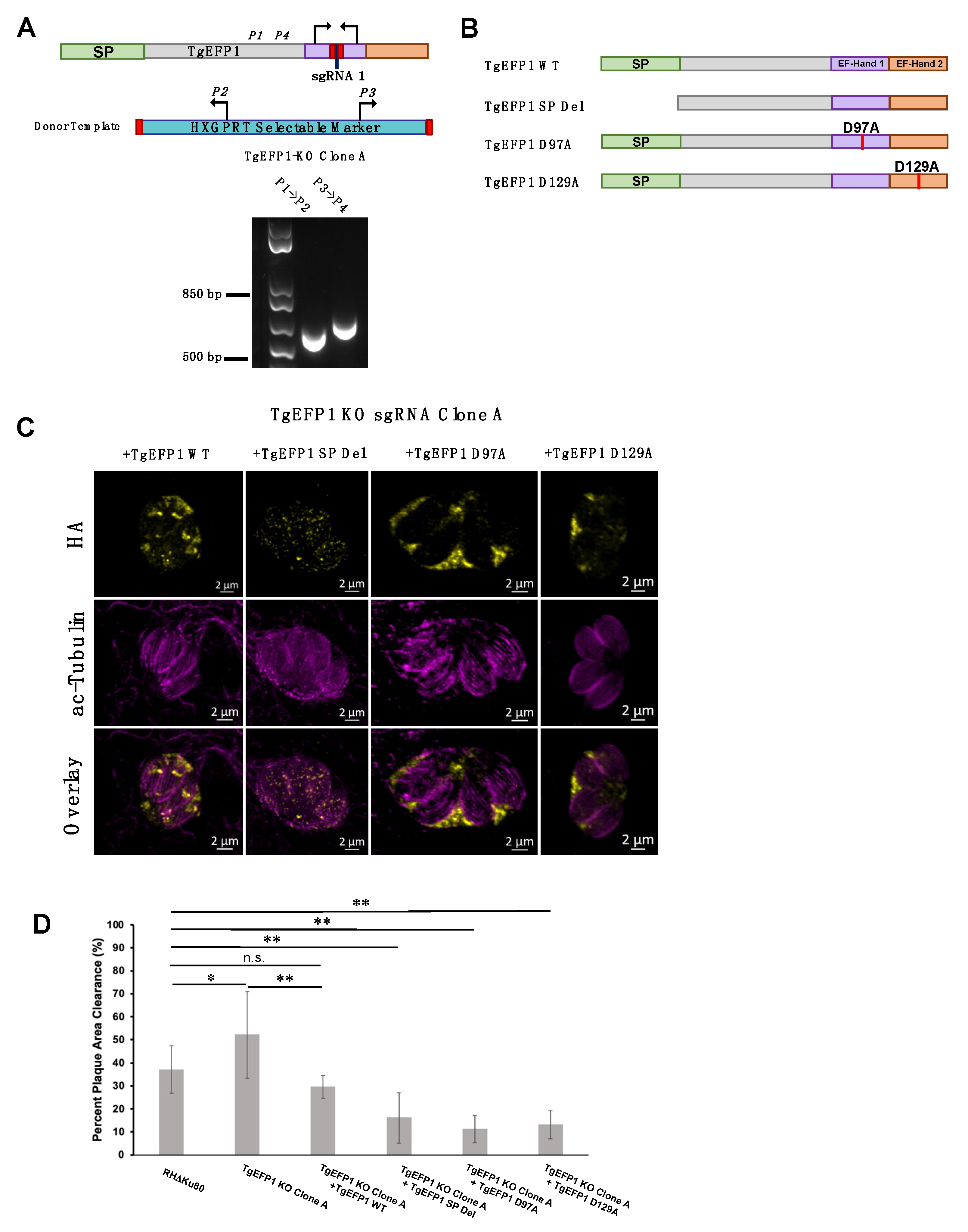
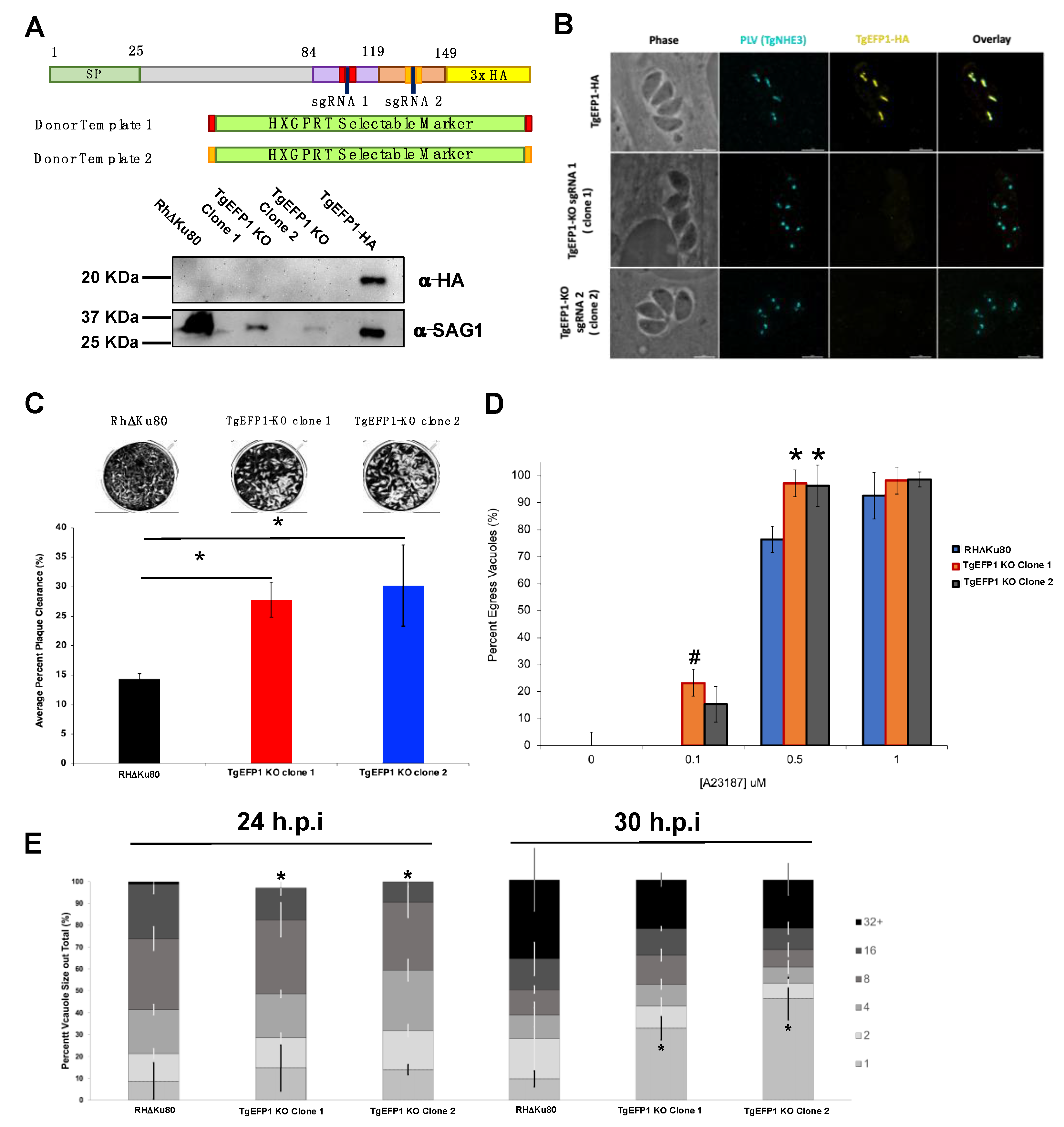
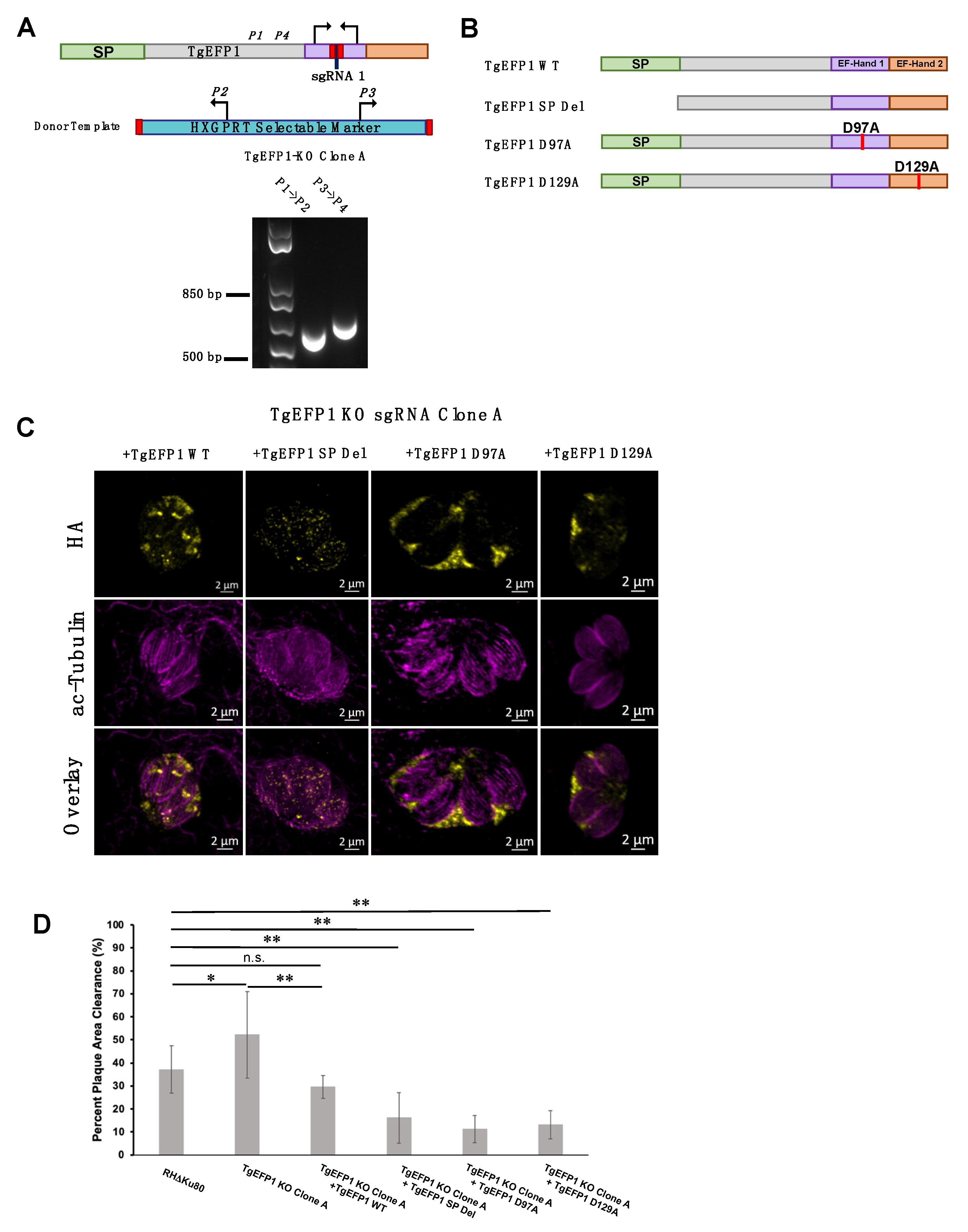
2.2. Generation of Transgenic Parasite Lines
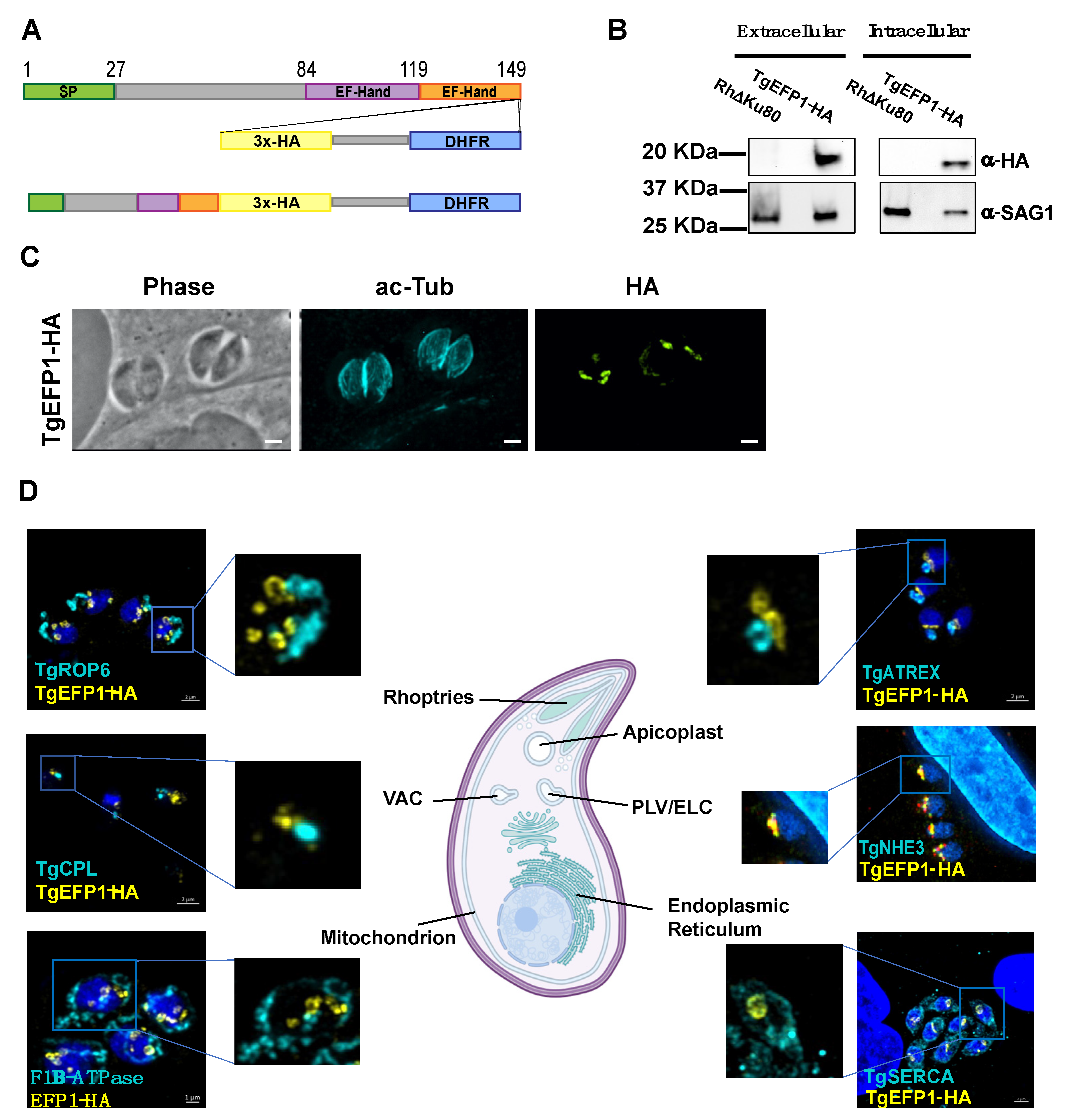
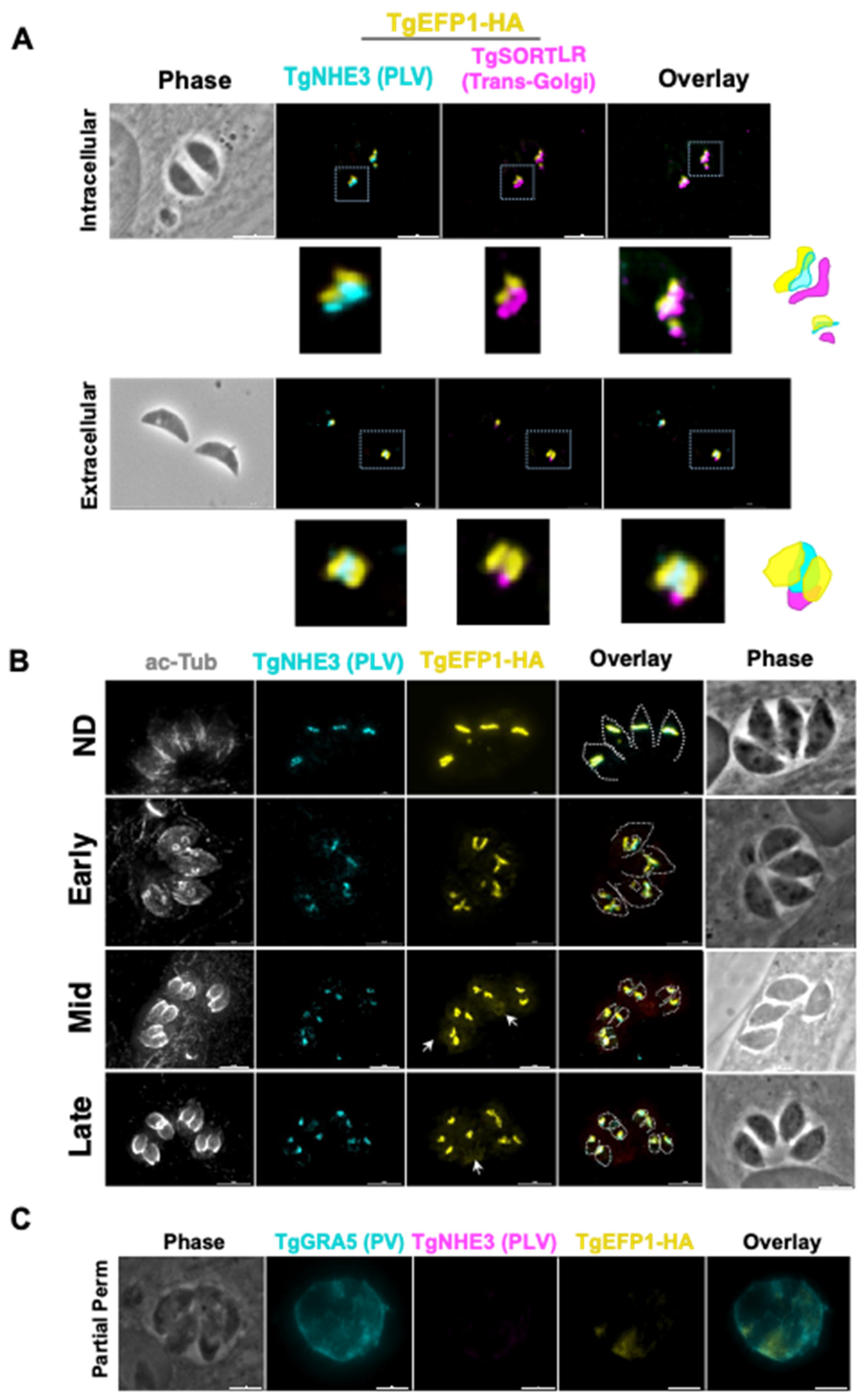
2.3. Immunofluorescence Assays and Western Blot
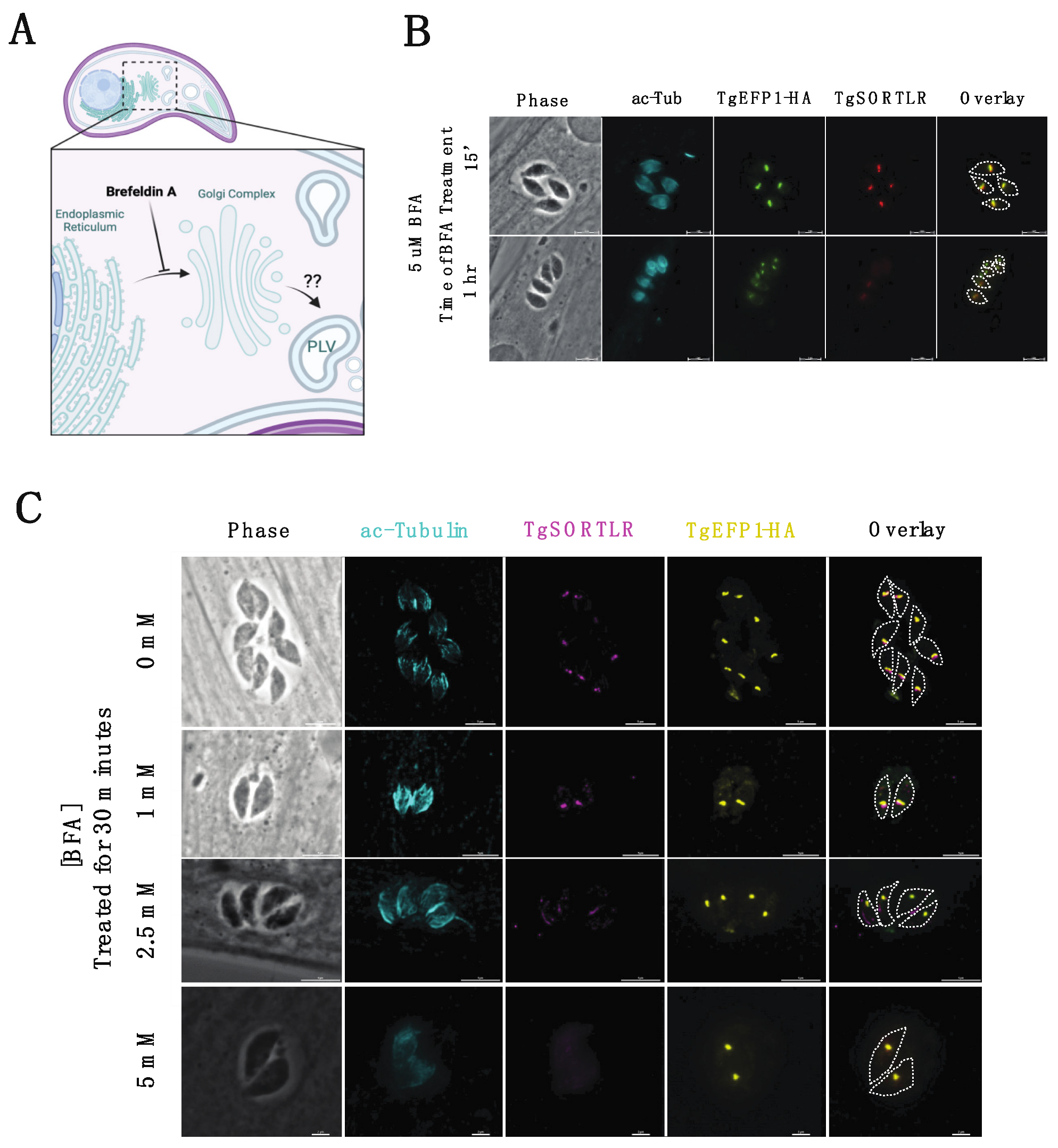
2.4. Brefeldin A (BFA) Experiments
2.5. Ultrastructure Expansion Microscopy
2.6. Phenotypic Assays
2.7. Statistical Analysis
3. Results
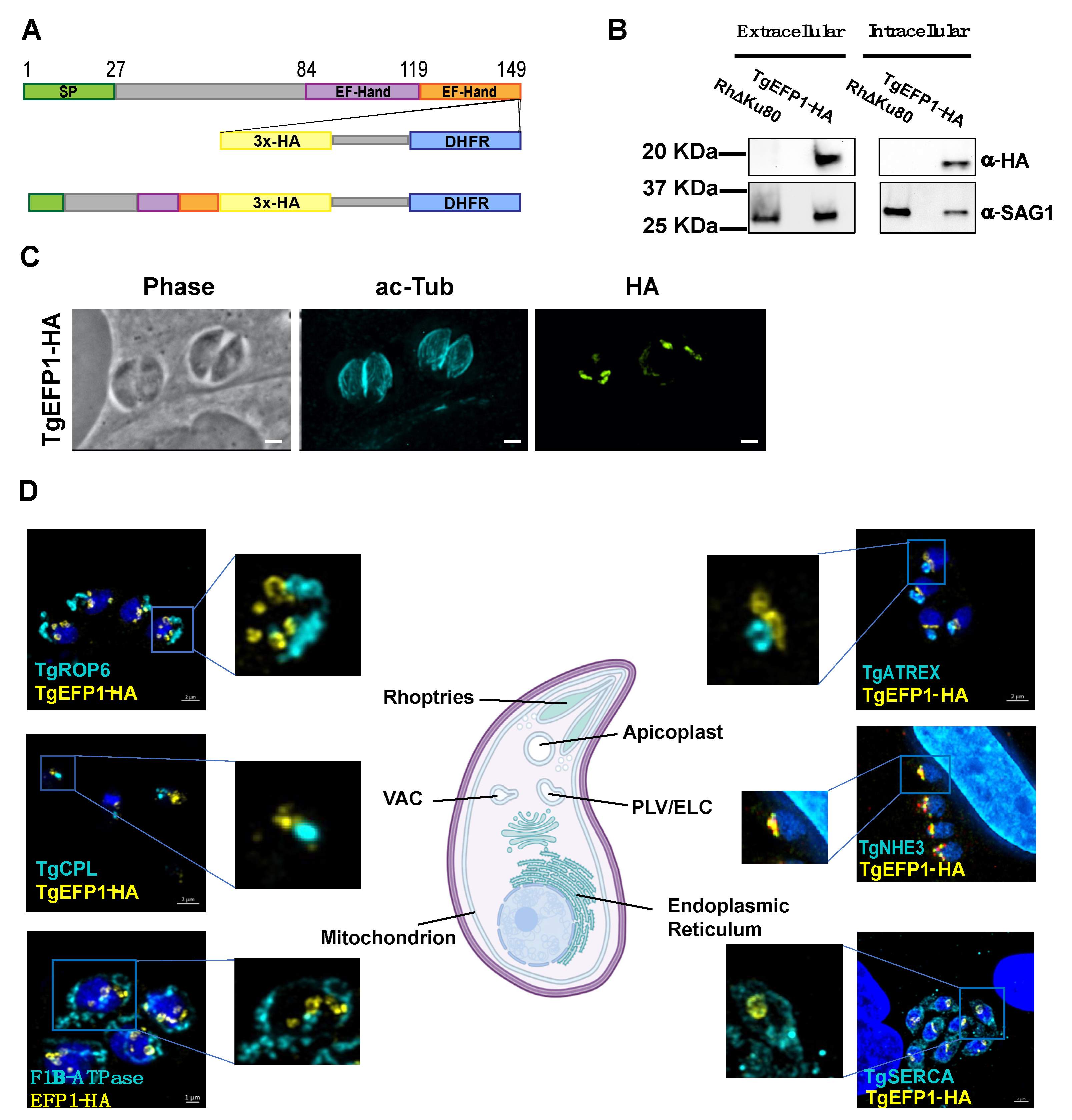
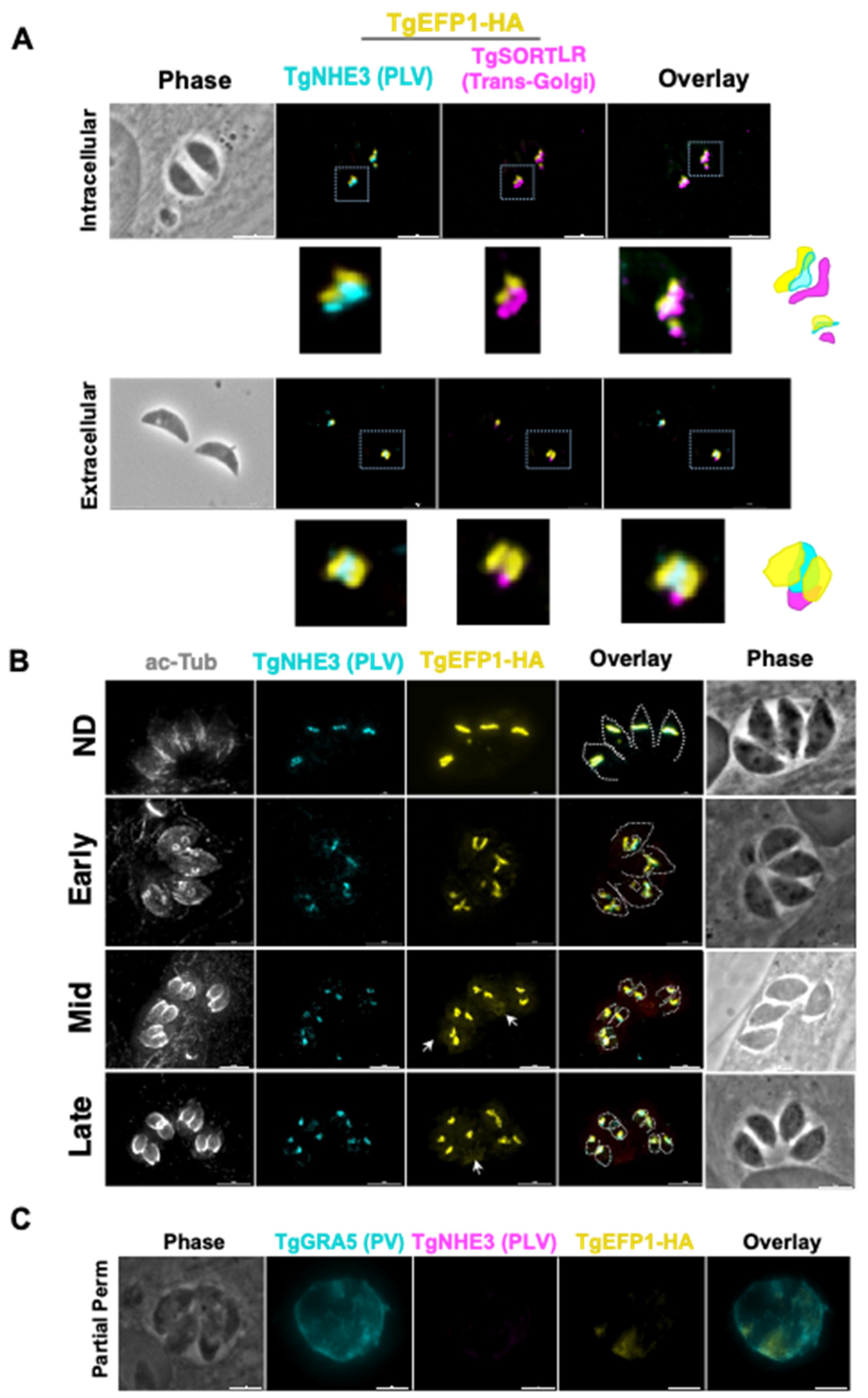
3.1. TgEFP1-HA Is Localized to the PLV/ELC and the Parasitophorous Vacuole
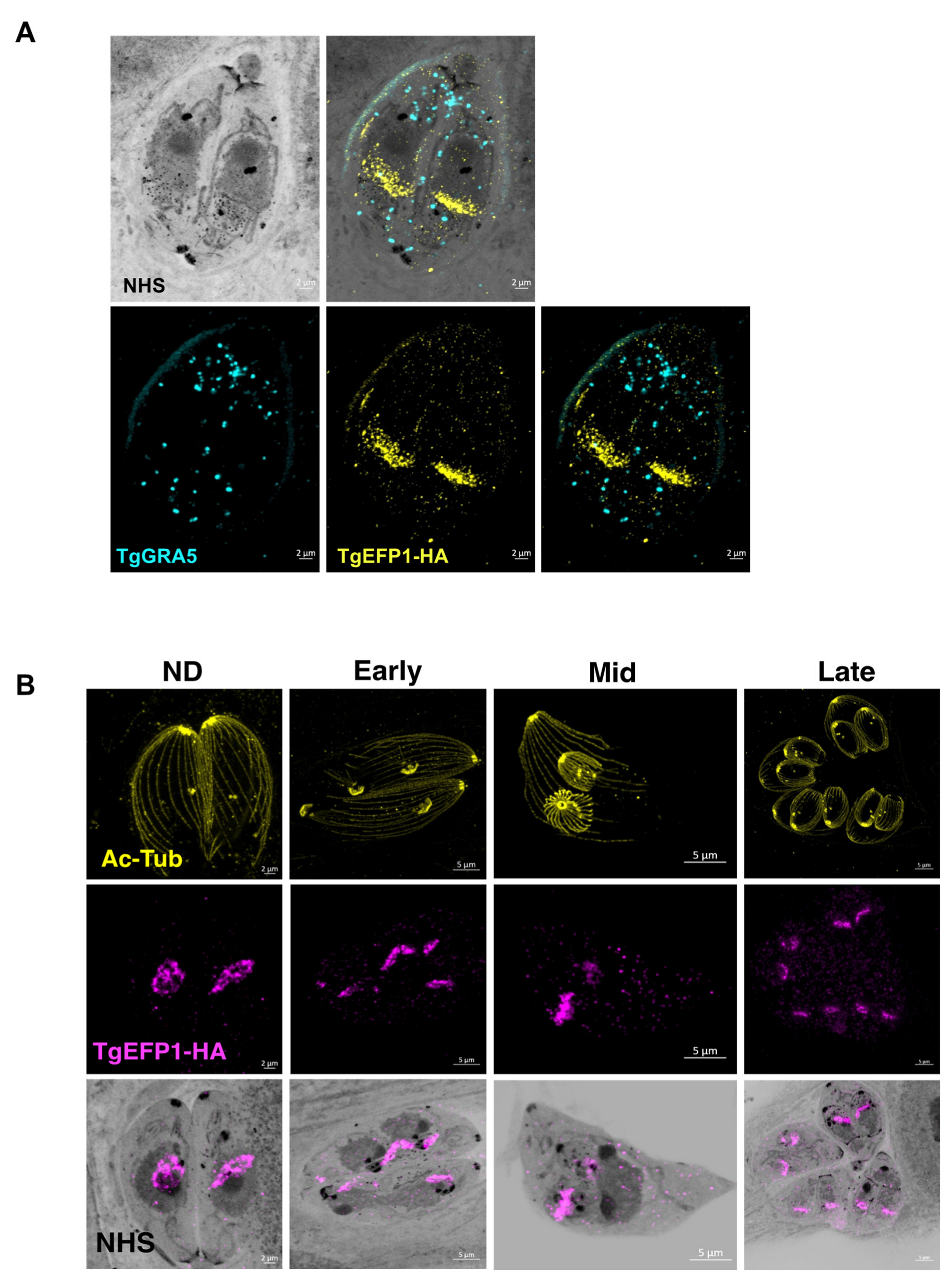
3.2. Monitoring of TgEFP1 Localization by Ultrastructure Expansion Microscopy
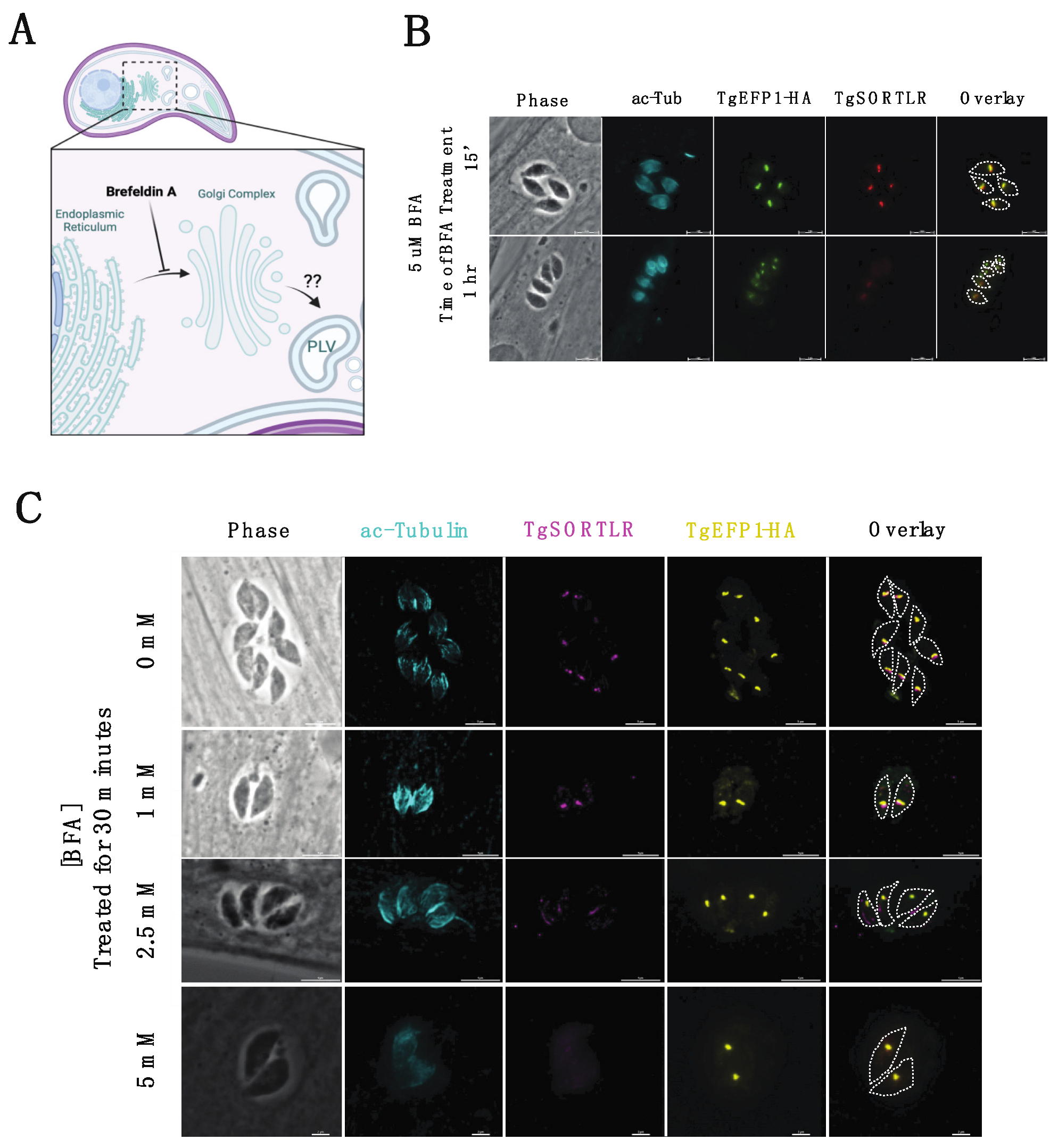
3.3. Brefeldin A (BFA) Treatment on Intracellular Parasites Depletes PV Localization but Does Not Affect PLV/ELC Localization of TgEFP1-HA
3.4. TgEFP1 Knockout (TgEFP1-KO) Parasites Have a Faster Lytic Cycle
3.5. Parasites Lacking TgEFP1 Are More Sensitive to Ionophore Induced Egress
3.6. Mutating the Signal Peptide or EF-Hands Disrupts the Localization Pattern of TgEFP1
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bagur, R.; Hajnóczky, G. Intracellular Ca2+ Sensing: Its Role in Calcium Homeostasis and Signaling. Mol. Cell 2017, 66, 780–788. [Google Scholar] [CrossRef] [Green Version]
- Prins, D.; Michalak, M. Organellar calcium buffers. Cold Spring Harb. Perspect. Biol. 2011, 3, a004069. [Google Scholar] [CrossRef] [Green Version]
- Hortua Triana, M.A.; Márquez-Nogueras, K.M.; Vella, S.A.; Moreno, S.N.J. Calcium signaling and the lytic cycle of the Apicomplexan parasite Toxoplasma gondii. Biochim. Biophys. Acta—Mol. Cell Res. 2018, 1865, 1846–1856. [Google Scholar] [CrossRef]
- Robert-Gangneux, F.; Dardé, M.L. Epidemiology of and diagnostic strategies for toxoplasmosis. Clin. Microbiol. Rev. 2012, 25, 264–296. [Google Scholar] [CrossRef] [Green Version]
- Montoya, J.G.; Rosso, F. Diagnosis and management of toxoplasmosis. Clin. Perinatol. 2005, 32, 705–726. [Google Scholar] [CrossRef]
- Borges-Pereira, L.; Budu, A.; McKnight, C.A.; Moore, C.A.; Vella, S.A.; Hortua Triana, M.A.; Liu, J.; Garcia, C.R.; Pace, D.A.; Moreno, S.N. Calcium Signaling throughout the Toxoplasma gondii Lytic Cycle: A study using genetically encoded calcium indicators. J. Biol. Chem. 2015, 290, 26914–26926. [Google Scholar] [CrossRef] [Green Version]
- Pace, D.A.; McKnight, C.A.; Liu, J.; Jimenez, V.; Moreno, S.N. Calcium entry in Toxoplasma gondii and its enhancing effect of invasion-linked traits. J. Biol. Chem. 2014, 289, 19637–19647. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.E. A ubiquitous intracellular parasite: The cellular biology of Toxoplasma gondii. Int. J. Parasitol. 1995, 25, 1301–1309. [Google Scholar] [CrossRef]
- Rohloff, P.; Miranda, K.; Rodrigues, J.C.; Fang, J.; Galizzi, M.; Plattner, H.; Hentschel, J.; Moreno, S.N. Calcium uptake and proton transport by acidocalcisomes of Toxoplasma gondii. PLoS ONE 2011, 6, e18390. [Google Scholar] [CrossRef]
- Liu, J.; Pace, D.; Dou, Z.; King, T.P.; Guidot, D.; Li, Z.H.; Carruthers, V.B.; Moreno, S.N. A vacuolar-H(+) -pyrophosphatase (TgVP1) is required for microneme secretion, host cell invasion, and extracellular survival of Toxoplasma gondii. Mol. Microbiol. 2014, 93, 698–712. [Google Scholar] [CrossRef] [Green Version]
- Luo, S.; Ruiz, F.A.; Moreno, S.N. The acidocalcisome Ca2+-ATPase (TgA1) of Toxoplasma gondii is required for polyphosphate storage, intracellular calcium homeostasis and virulence. Mol. Microbiol. 2005, 55, 1034–1045. [Google Scholar] [CrossRef] [PubMed]
- Francia, M.E.; Wicher, S.; Pace, D.A.; Sullivan, J.; Moreno, S.N.; Arrizabalaga, G. A Toxoplasma gondii protein with homology to intracellular type Na⁺/H⁺ exchangers is important for osmoregulation and invasion. Exp. Cell Res. 2011, 317, 1382–1396. [Google Scholar] [CrossRef] [Green Version]
- Miranda, K.; Pace, D.A.; Cintron, R.; Rodrigues, J.C.; Fang, J.; Smith, A.; Rohloff, P.; Coelho, E.; de Haas, F.; de Souza, W.; et al. Characterization of a novel organelle in Toxoplasma gondii with similar composition and function to the plant vacuole. Mol. Microbiol. 2010, 76, 1358–1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaFavers, K.A.; Márquez-Nogueras, K.M.; Coppens, I.; Moreno, S.N.J.; Arrizabalaga, G. A novel dense granule protein, GRA41, regulates timing of egress and calcium sensitivity in Toxoplasma gondii. Cell. Microbiol. 2017, 19, e12749. [Google Scholar] [CrossRef] [Green Version]
- Shen, B.; Brown, K.; Long, S.; Sibley, L.D. Development of CRISPR/Cas9 for Efficient Genome Editing in Toxoplasma gondii. Methods Mol. Biol. 2017, 1498, 79–103. [Google Scholar] [CrossRef]
- Long, S.; Brown, K.M.; Sibley, L.D. CRISPR-mediated Tagging with BirA Allows Proximity Labeling in. Bio-Protoc. 2018, 8, e2768. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Broncel, M.; Dominicus, C.; Sampson, E.; Blakely, W.J.; Treeck, M.; Arrizabalaga, G. A plasma membrane localized protein phosphatase in Toxoplasma gondii, PPM5C, regulates attachment to host cells. Sci. Rep. 2019, 9, 5924. [Google Scholar] [CrossRef] [Green Version]
- Blakely, W.J.; Holmes, M.J.; Arrizabalaga, G. The Secreted Acid Phosphatase Domain-Containing GRA44 from Toxoplasma gondii Is Required for c-Myc Induction in Infected Cells. mSphere 2020, 5, e00877-19. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, K.; Charvat, R.; Arrizabalaga, G. Identification of Fis1 Interactors in Toxoplasma gondii Reveals a Novel Protein Required for Peripheral Distribution of the Mitochondrion. mBio 2020, 11, e02732-19. [Google Scholar] [CrossRef] [Green Version]
- Liffner, B.; Absalon, S. Expansion Microscopy Reveals. Microorganisms 2021, 9, 2306. [Google Scholar] [CrossRef]
- Heredero-Bermejo, I.; Varberg, J.M.; Charvat, R.; Jacobs, K.; Garbuz, T.; Sullivan, W.J.; Arrizabalaga, G. TgDrpC, an atypical dynamin-related protein in Toxoplasma gondii, is associated with vesicular transport factors and parasite division. Mol. Microbiol. 2019, 111, 46–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzman, C.; Bagga, M.; Kaur, A.; Westermarck, J.; Abankwa, D. ColonyArea: An ImageJ plugin to automatically quantify colony formation in clonogenic assays. PLoS ONE 2014, 9, e92444. [Google Scholar] [CrossRef] [PubMed]
- Vakili, O.; Khatami, S.H.; Maleksabet, A.; Movahedpour, A.; Fana, S.E.; Sadegh, R.; Salmanzadeh, A.H.; Razeghifam, H.; Nourdideh, S.; Tehrani, S.S.; et al. Finding Appropriate Signal Peptides for Secretory Production of Recombinant Glucarpidase: An In Silico Method. Recent Pat. Biotechnol. 2021, 15, 302–315. [Google Scholar] [CrossRef]
- De Castro, E.; Sigrist, C.J.; Gattiker, A.; Bulliard, V.; Langendijk-Genevaux, P.S.; Gasteiger, E.; Bairoch, A.; Hulo, N. ScanProsite: Detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucleic Acids Res. 2006, 34, W362–W365. [Google Scholar] [CrossRef]
- Sigrist, C.J.; Cerutti, L.; Hulo, N.; Gattiker, A.; Falquet, L.; Pagni, M.; Bairoch, A.; Bucher, P. PROSITE: A documented database using patterns and profiles as motif descriptors. Brief. Bioinform. 2002, 3, 265–274. [Google Scholar] [CrossRef]
- Huang, R.; Que, X.; Hirata, K.; Brinen, L.S.; Lee, J.H.; Hansell, E.; Engel, J.; Sajid, M.; Reed, S. The cathepsin L of Toxoplasma gondii (TgCPL) and its endogenous macromolecular inhibitor, toxostatin. Mol. Biochem. Parasitol. 2009, 164, 86–94. [Google Scholar] [CrossRef] [Green Version]
- Nishi, M.; Hu, K.; Murray, J.M.; Roos, D.S. Organellar dynamics during the cell cycle of Toxoplasma gondii. J. Cell Sci. 2008, 121, 1559–1568. [Google Scholar] [CrossRef] [Green Version]
- Carmeille, R.; Schiano Lomoriello, P.; Devarakonda, P.M.; Kellermeier, J.A.; Heaslip, A.T. Actin and an unconventional myosin motor, TgMyoF, control the organization and dynamics of the endomembrane network in Toxoplasma gondii. PLoS Pathog. 2021, 17, e1008787. [Google Scholar] [CrossRef]
- Halling, D.B.; Liebeskind, B.J.; Hall, A.W.; Aldrich, R.W. Conserved properties of individual Ca2+-binding sites in calmodulin. Proc. Natl. Acad. Sci. USA 2016, 113, E1216–E1225. [Google Scholar] [CrossRef] [Green Version]
- Strynadka, N.C.; James, M.N. Crystal structures of the helix-loop-helix calcium-binding proteins. Annu. Rev. Biochem. 1989, 58, 951–998. [Google Scholar] [CrossRef]
- Schwaller, B. Cytosolic Ca2+ buffers. Cold Spring Harb. Perspect. Biol. 2010, 2, a004051. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Dykes, E.J.; Li, J.; Moreno, S.N.J.; Hortua Triana, M.A. Characterization of Two EF-hand Domain-containing Proteins from Toxoplasma gondii. J. Eukaryot. Microbiol. 2019, 66, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Asai, S.; Ichikawa, T.; Nomura, H.; Kobayashi, M.; Kamiyoshihara, Y.; Mori, H.; Kadota, Y.; Zipfel, C.; Jones, J.D.G.; Yoshioka, H. The variable domain of a plant calcium-dependent protein kinase (CDPK) confers subcellular localization and substrate recognition for NADPH oxidase. J. Biol. Chem. 2013, 288, 14332–14340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardew, E.M.; Verlinde, C.L.M.J.; Pohl, E. The calcium-dependent protein kinase 1 from Toxoplasma gondii as target for structure-based drug design. Parasitology 2018, 145, 210–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaji, R.Y.; Johnson, D.E.; Treeck, M.; Wang, M.; Hudmon, A.; Arrizabalaga, G. Phosphorylation of a Myosin Motor by TgCDPK3 Facilitates Rapid Initiation of Motility during Toxoplasma gondii egress. PLoS Pathog. 2015, 11, e1005268. [Google Scholar] [CrossRef] [Green Version]
- McCoy, J.M.; Whitehead, L.; van Dooren, G.G.; Tonkin, C.J. TgCDPK3 regulates calcium-dependent egress of Toxoplasma gondii from host cells. PLoS Pathog. 2012, 8, e1003066. [Google Scholar] [CrossRef] [Green Version]
- McGovern, O.L.; Rivera-Cuevas, Y.; Kannan, G.; Narwold, A.J.; Carruthers, V.B. Intersection of endocytic and exocytic systems in Toxoplasma gondii. Traffic 2018, 19, 336–353. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Zhao, Z.; Gu, M.; Feng, X.; Xu, H. Release and uptake mechanisms of vesicular Ca2+ stores. Protein Cell 2019, 10, 8–19. [Google Scholar] [CrossRef] [Green Version]
- Mohanta, T.K.; Yadav, D.; Khan, A.L.; Hashem, A.; Abd Allah, E.F.; Al-Harrasi, A. Molecular Players of EF-hand Containing Calcium Signaling Event in Plants. Int. J. Mol. Sci. 2019, 20, 1476. [Google Scholar] [CrossRef] [Green Version]
- Day, I.S.; Reddy, V.S.; Shad Ali, G.; Reddy, A.S. Analysis of EF-hand-containing proteins in Arabidopsis. Genome Biol. 2002, 3, research0056.1. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dave, N.; LaFavers, K.; Arrizabalaga, G. The Dually Localized EF-Hand Domain-Containing Protein TgEFP1 Regulates the Lytic Cycle of Toxoplasma gondii. Cells 2022, 11, 1709. https://doi.org/10.3390/cells11101709
Dave N, LaFavers K, Arrizabalaga G. The Dually Localized EF-Hand Domain-Containing Protein TgEFP1 Regulates the Lytic Cycle of Toxoplasma gondii. Cells. 2022; 11(10):1709. https://doi.org/10.3390/cells11101709
Chicago/Turabian StyleDave, Noopur, Kaice LaFavers, and Gustavo Arrizabalaga. 2022. "The Dually Localized EF-Hand Domain-Containing Protein TgEFP1 Regulates the Lytic Cycle of Toxoplasma gondii" Cells 11, no. 10: 1709. https://doi.org/10.3390/cells11101709
APA StyleDave, N., LaFavers, K., & Arrizabalaga, G. (2022). The Dually Localized EF-Hand Domain-Containing Protein TgEFP1 Regulates the Lytic Cycle of Toxoplasma gondii. Cells, 11(10), 1709. https://doi.org/10.3390/cells11101709