
ATF6 Activation Reduces Amyloidogenic Transthyretin Secretion through Increased Interactions with Endoplasmic Reticulum Proteostasis Factors
 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plasmids, Antibodies, and Reagents
2.2. Cell Culture, Plasmids, Transfections, and Lysates
2.3. [35S] Metabolic Labeling
2.4. Media Conditioning, SDS-PAGE, CN-PAGE, and Immunoblotting
2.5. Quantification of FTTTR Tetramers in Conditioned Media
2.6. Co-Immunoprecipitation of FTTTRA25T
2.7. SILAC-MuDPIT Proteomics
3. Results
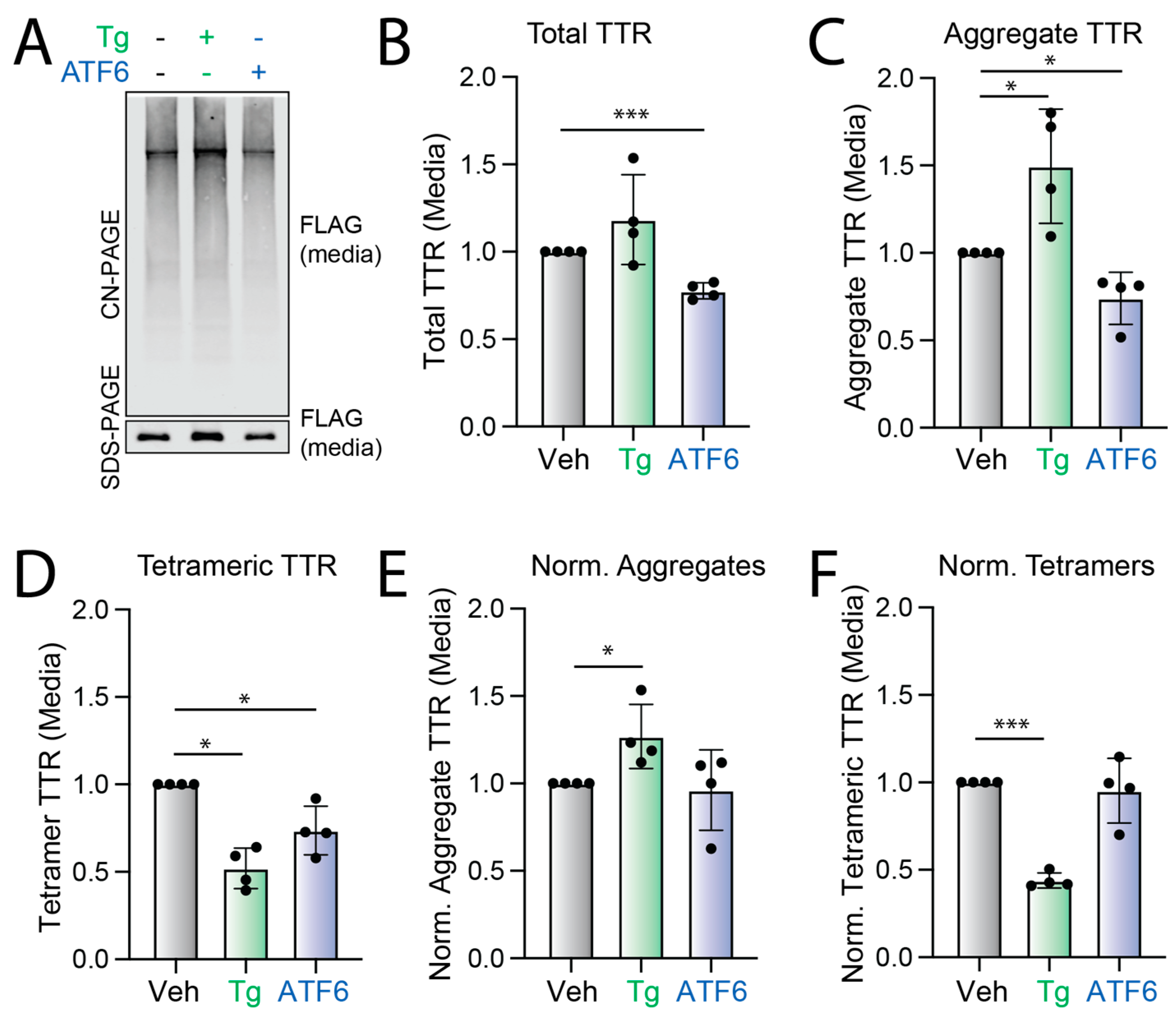
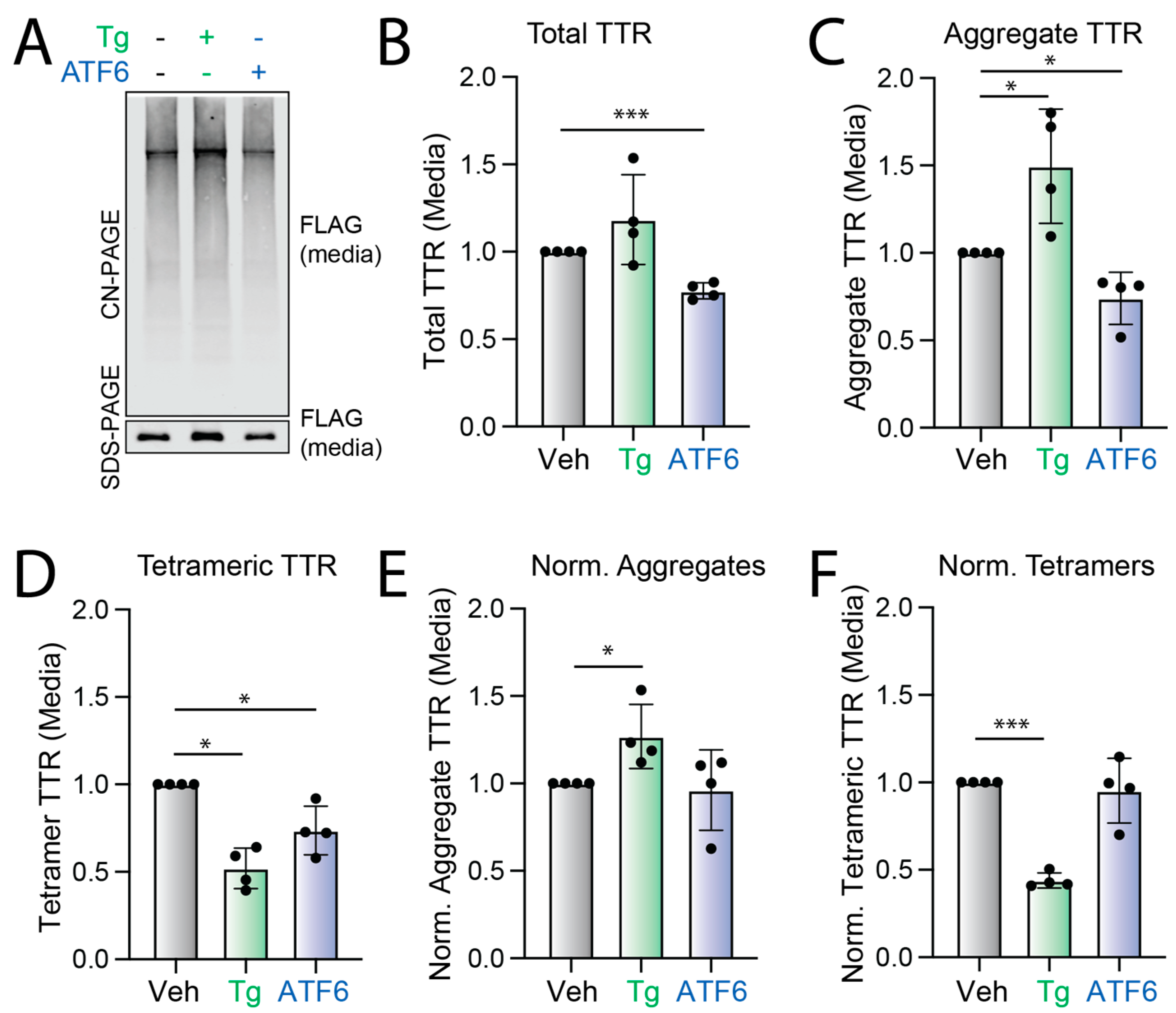
3.1. Stress-Independent ATF6 Activation Does Not Influence the Conformation of Secreted FTTTRA25T
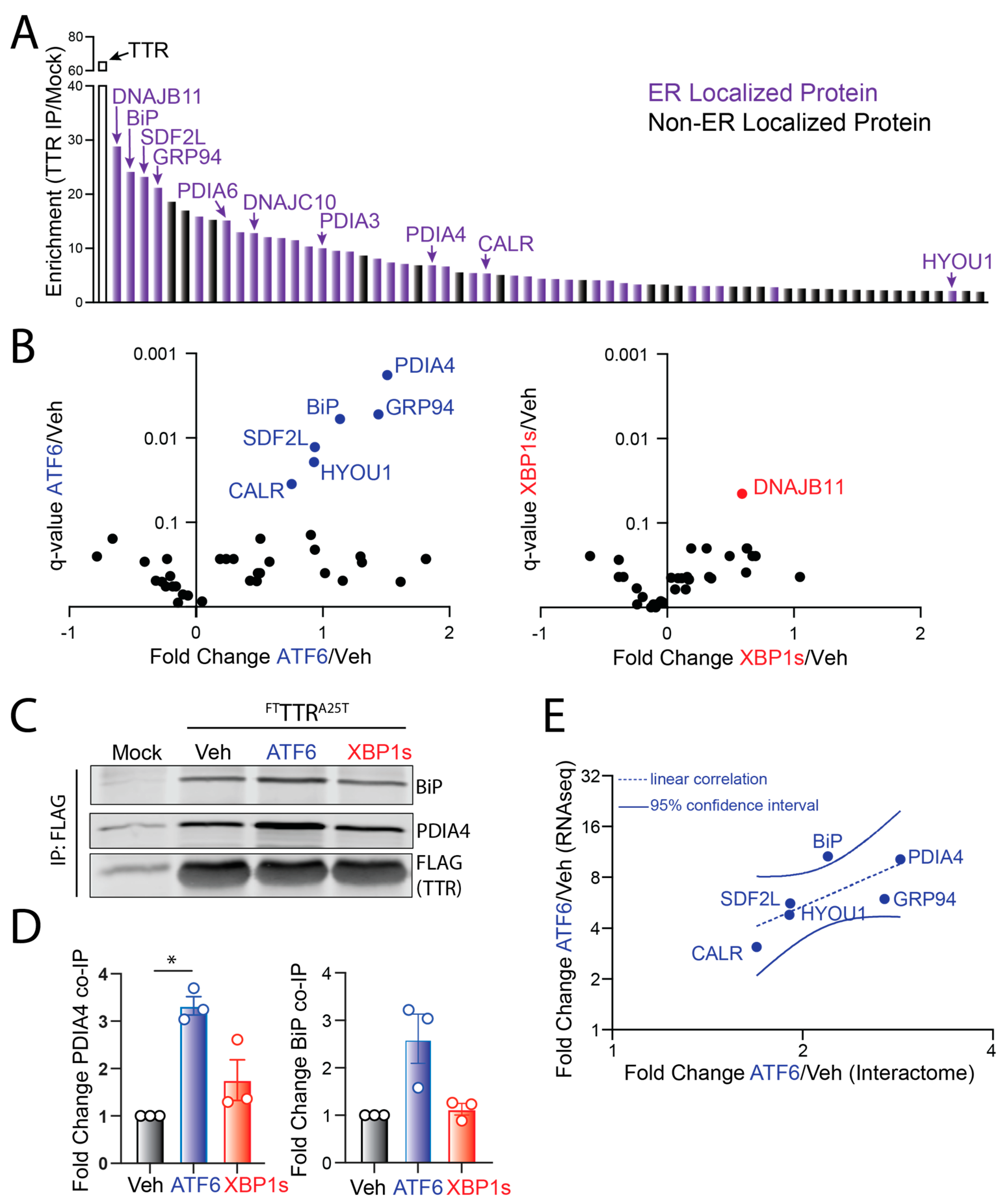
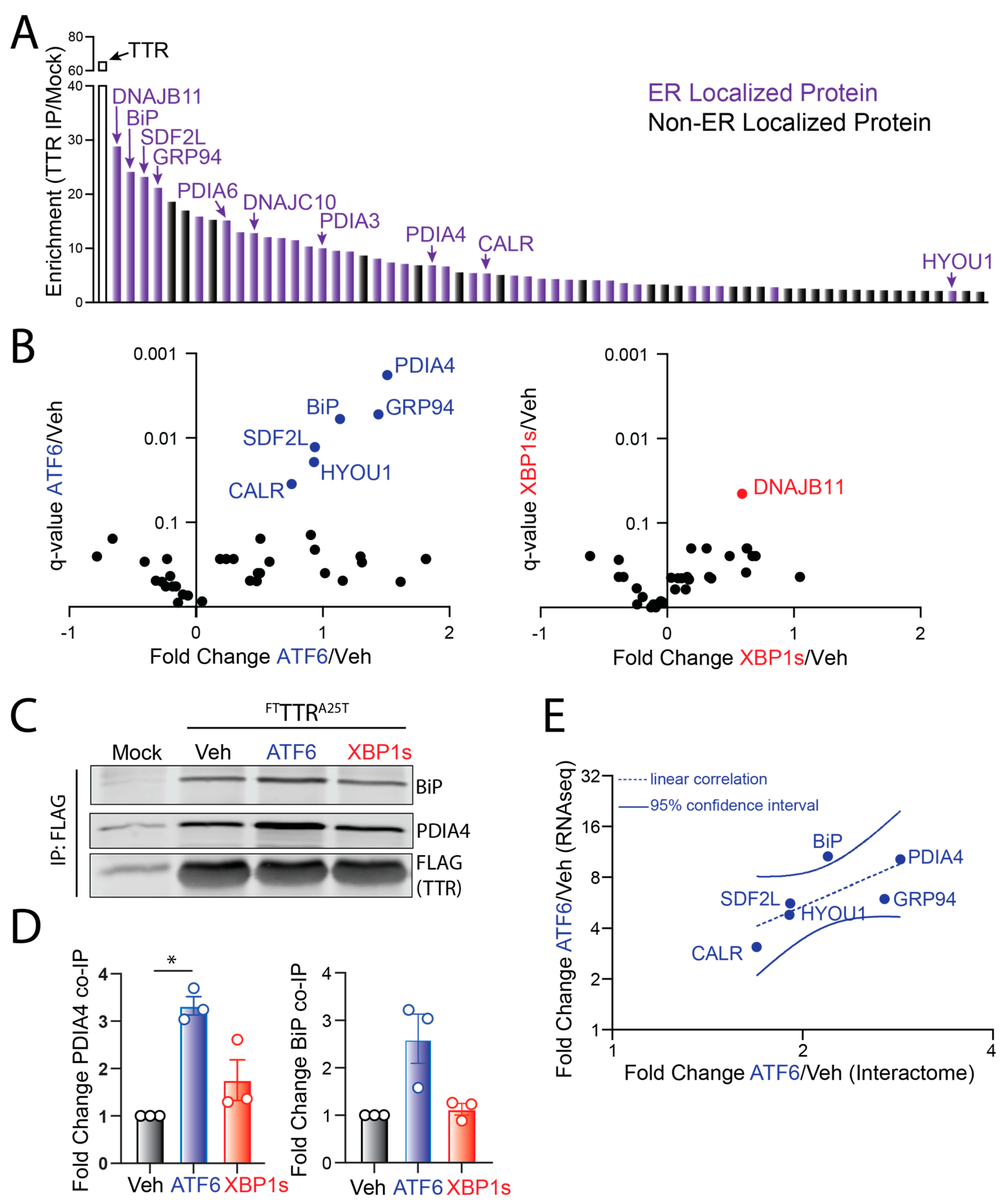
3.2. ATF6 Activation Increases Interaction between FTTTRA25T and ER-Localized Proteostasis Factors
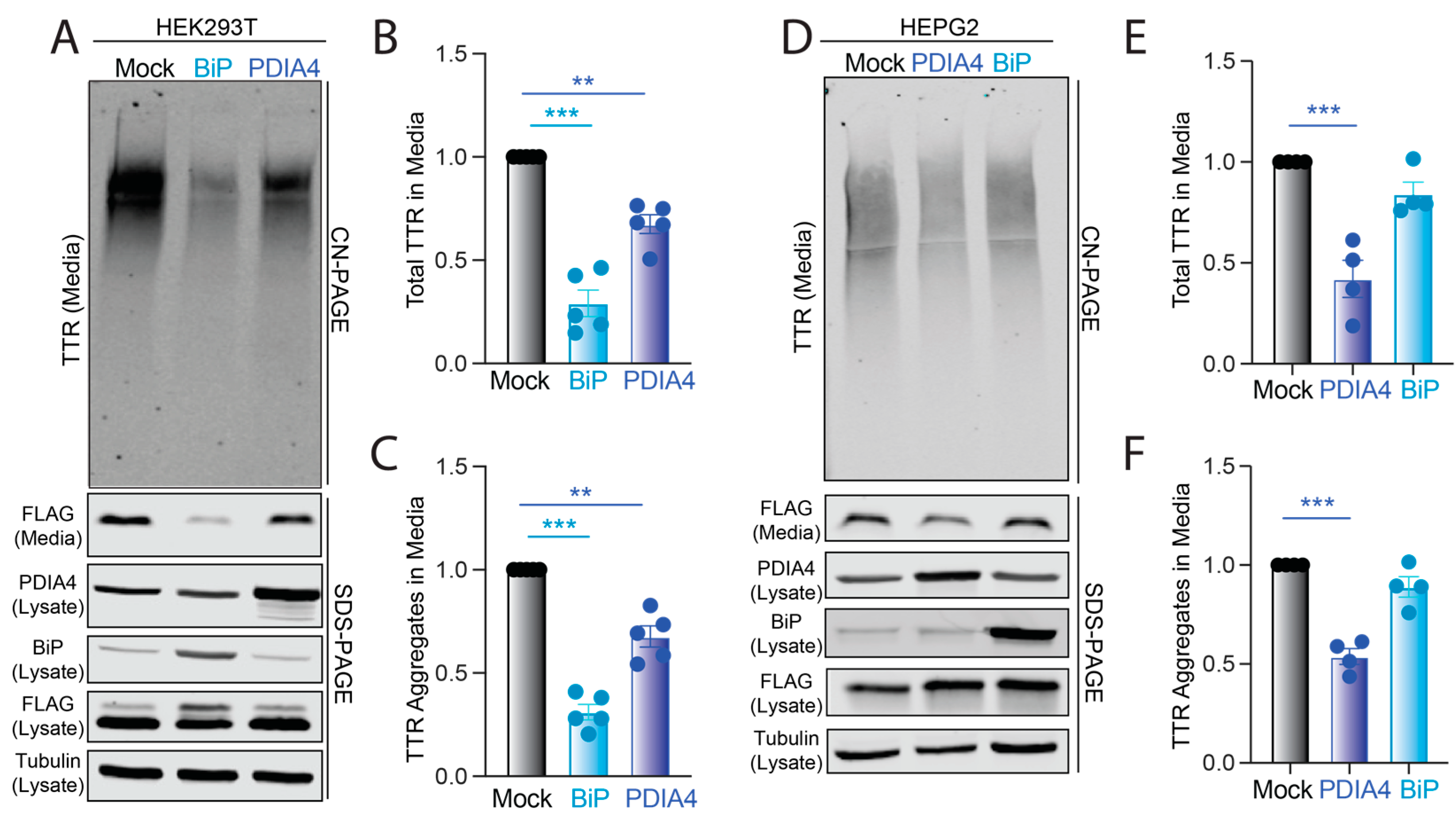
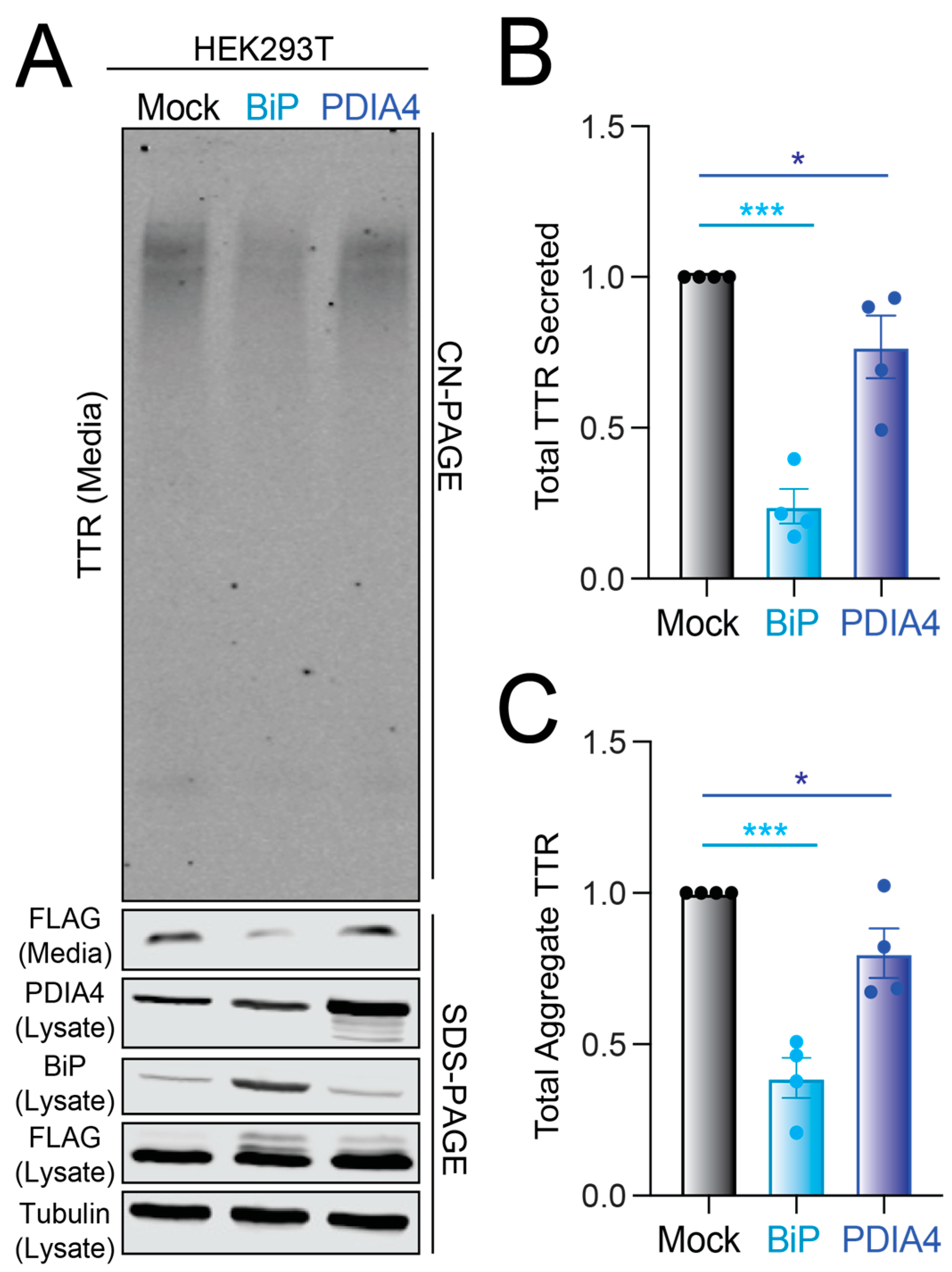
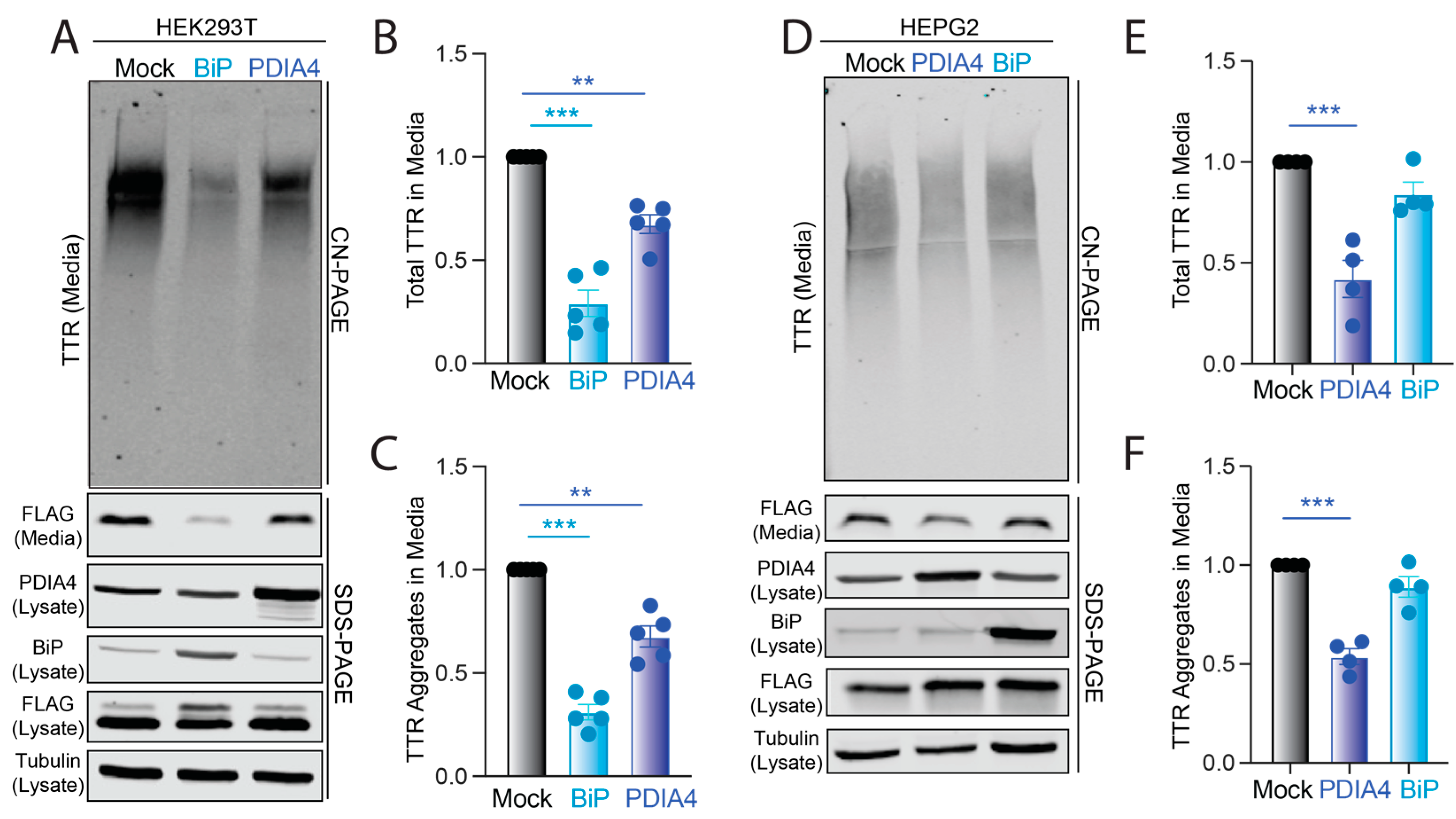
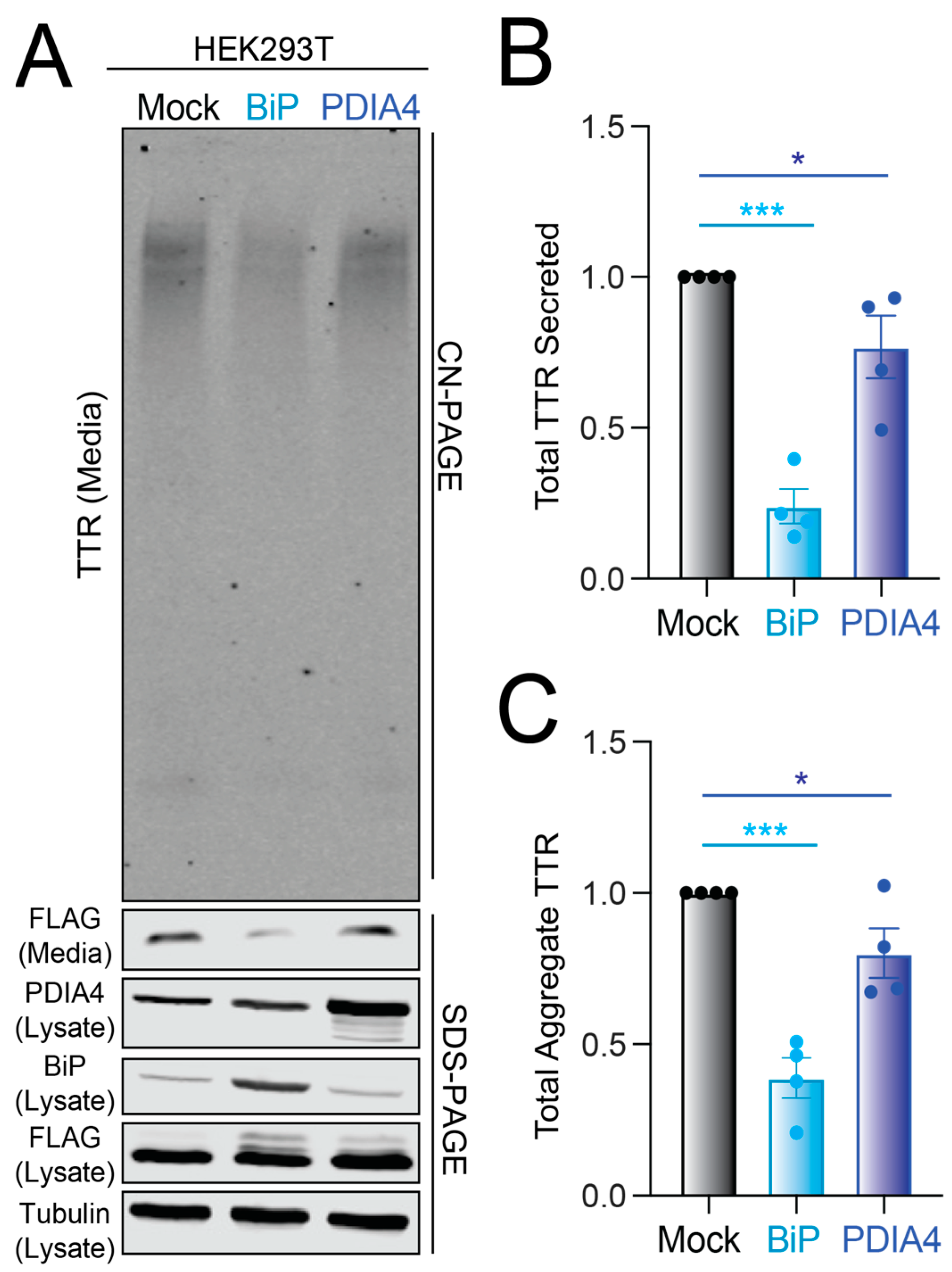
3.3. Overexpression of BiP or PDIA4 Reduces Secretion and Extracellular Aggregation of Destabilized FTTTRA25T
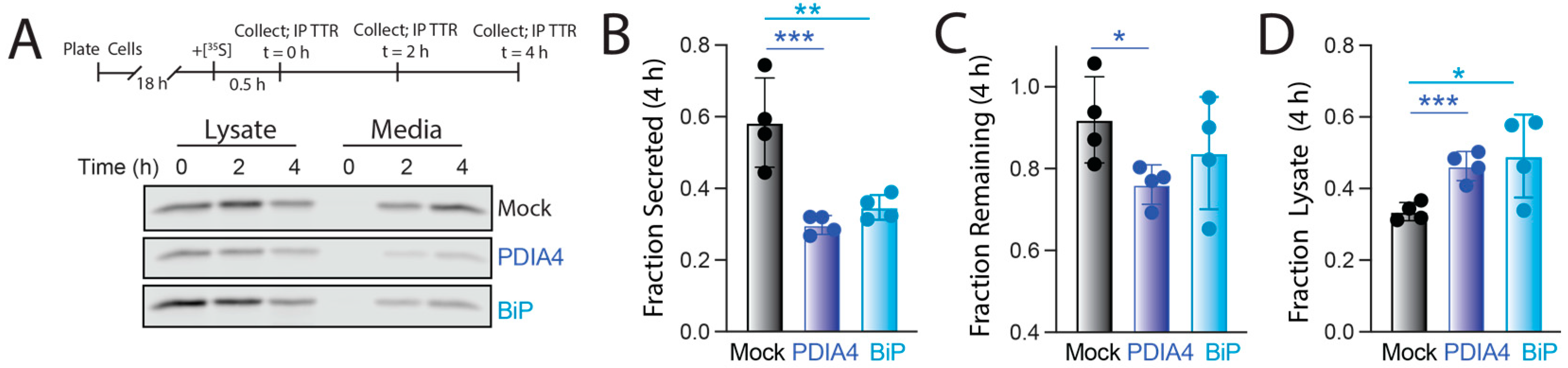
3.4. PDIA4 Overexpression Increases ER Retention of FTTTRA25T
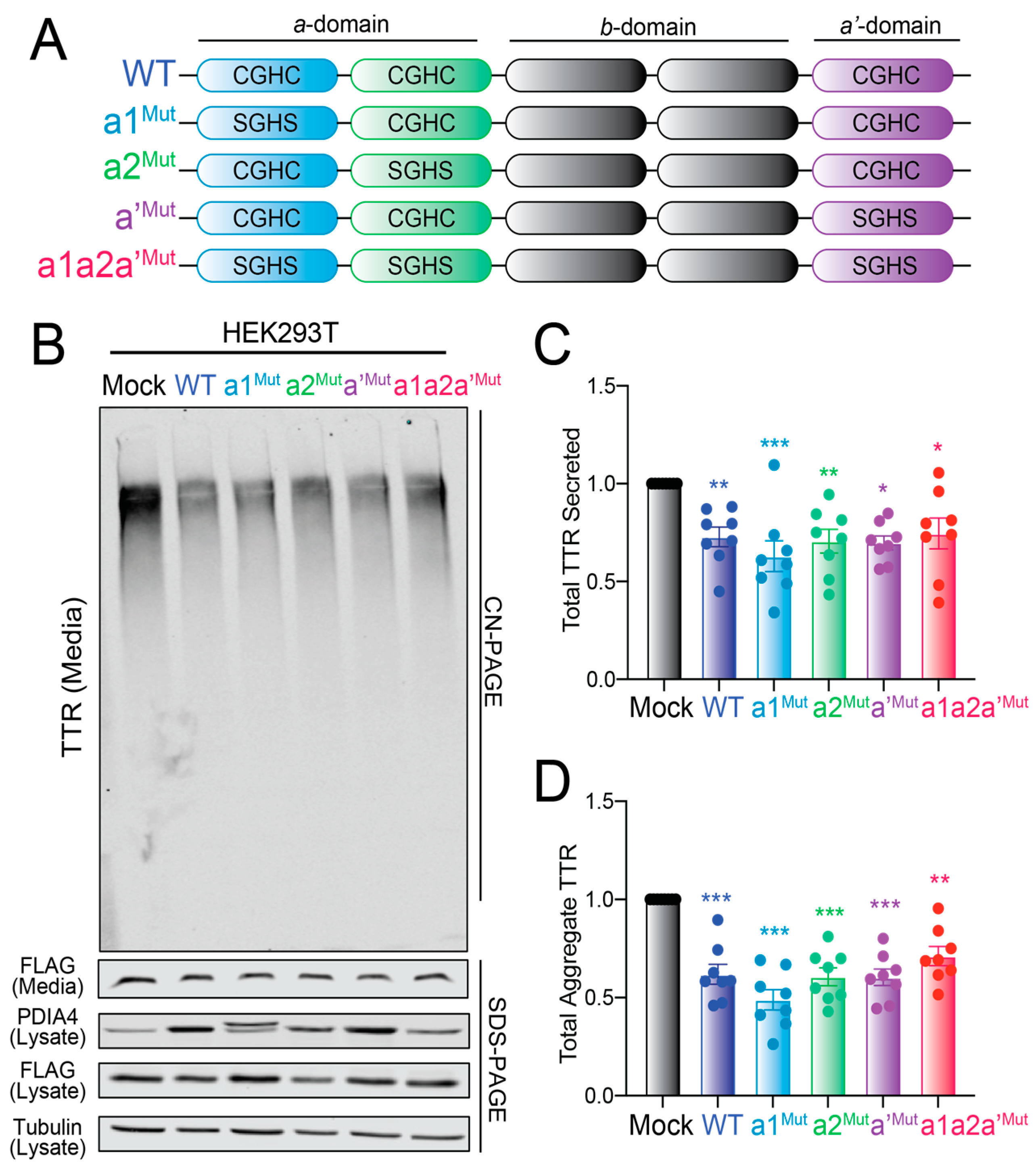
3.5. PDIA4-Dependent Reductions in FTTTRA25T Secretion Is Independent of Cys10
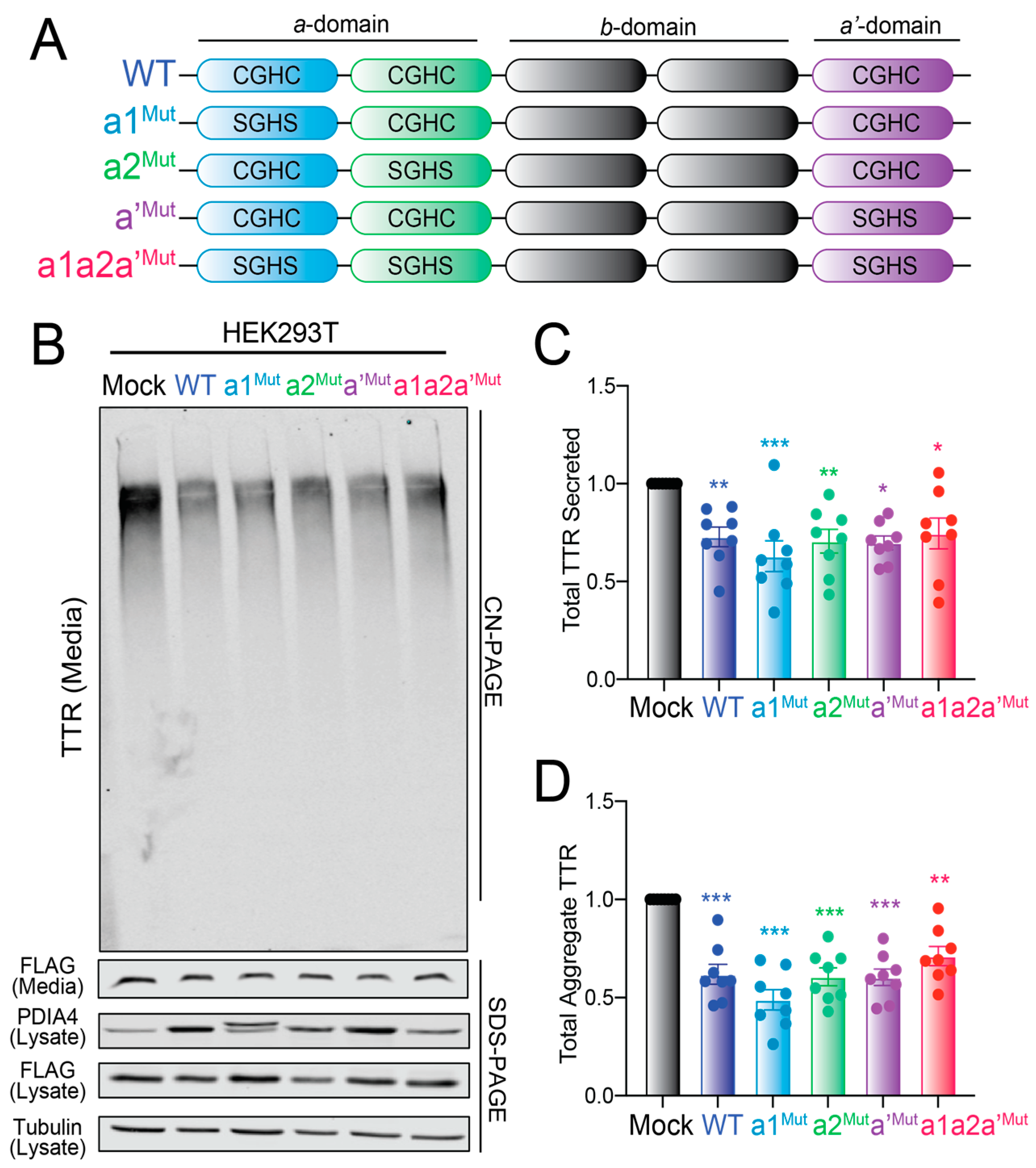
3.6. Overexpression of Redox-Deficient PDIA4 Variants Decrease FTTTRA25T Secretion and Aggregation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Magalhaes, J.; Eira, J.; Liz, M.A. The role of transthyretin in cell biology: Impact on human pathophysiology. Cell Mol. Life Sci. 2021, 78, 6105–6117. [Google Scholar] [CrossRef] [PubMed]
- Buxbaum, J.N.; Reixach, N. Transthyretin: The servant of many masters. Cell Mol. Life Sci. 2009, 66, 3095–3101. [Google Scholar] [CrossRef] [Green Version]
- Ruberg, F.L.; Grogan, M.; Hanna, M.; Kelly, J.W.; Maurer, M.S. Transthyretin Amyloid Cardiomyopathy: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2019, 73, 2872–2891. [Google Scholar] [CrossRef] [PubMed]
- Sekijima, Y. Transthyretin (ATTR) amyloidosis: Clinical spectrum, molecular pathogenesis and disease-modifying treatments. J. Neurol. Neurosurg. Psychiatry 2015, 86, 1036–1043. [Google Scholar] [CrossRef] [PubMed]
- Connors, L.H.; Lim, A.; Prokaeva, T.; Roskens, V.A.; Costello, C.E. Tabulation of human transthyretin (TTR) variants, 2003. Amyloid 2003, 10, 160–184. [Google Scholar] [CrossRef]
- Chiti, F.; Kelly, J.W. Small molecule protein binding to correct cellular folding or stabilize the native state against misfolding and aggregation. Curr. Opin. Struct. Biol. 2022, 72, 267–278. [Google Scholar] [CrossRef]
- Burton, A.; Castano, A.; Bruno, M.; Riley, S.; Schumacher, J.; Sultan, M.B.; See Tai, S.; Judge, D.P.; Patel, J.K.; Kelly, J.W. Drug Discovery and Development in Rare Diseases: Taking a Closer Look at the Tafamidis Story. Drug Des. Devel. Ther. 2021, 15, 1225–1243. [Google Scholar] [CrossRef]
- Coelho, T.; Merlini, G.; Bulawa, C.E.; Fleming, J.A.; Judge, D.P.; Kelly, J.W.; Maurer, M.S.; Plante-Bordeneuve, V.; Labaudiniere, R.; Mundayat, R.; et al. Mechanism of Action and Clinical Application of Tafamidis in Hereditary Transthyretin Amyloidosis. Neurol. Ther. 2016, 5, 1–25. [Google Scholar] [CrossRef] [Green Version]
- Adams, D.; Slama, M. Hereditary transthyretin amyloidosis: Current treatment. Curr. Opin. Neurol. 2020, 33, 553–561. [Google Scholar] [CrossRef]
- Titze-de-Almeida, S.S.; Brandao, P.R.P.; Faber, I.; Titze-de-Almeida, R. Leading RNA Interference Therapeutics Part 1: Silencing Hereditary Transthyretin Amyloidosis, with a Focus on Patisiran. Mol. Diagn. Ther. 2020, 24, 49–59. [Google Scholar] [CrossRef]
- Adams, D.; Cauquil, C.; Labeyrie, C.; Beaudonnet, G.; Algalarrondo, V.; Theaudin, M. TTR kinetic stabilizers and TTR gene silencing: A new era in therapy for familial amyloidotic polyneuropathies. Expert Opin. Pharmacother. 2016, 17, 791–802. [Google Scholar] [CrossRef] [PubMed]
- Benson, M.D.; Dasgupta, N.R.; Monia, B.P. Inotersen (transthyretin-specific antisense oligonucleotide) for treatment of transthyretin amyloidosis. Neurodegener. Dis. Manag. 2019, 9, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Plate, L.; Wiseman, R.L. Regulating Secretory Proteostasis through the Unfolded Protein Response: From Function to Therapy. Trends Cell Biol. 2017, 27, 722–737. [Google Scholar] [CrossRef]
- Eisele, Y.S.; Monteiro, C.; Fearns, C.; Encalada, S.E.; Wiseman, R.L.; Powers, E.T.; Kelly, J.W. Targeting protein aggregation for the treatment of degenerative diseases. Nat. Rev. Drug Discov. 2015, 14, 759–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mesgarzadeh, J.S.; Buxbaum, J.N.; Wiseman, R.L. Stress-responsive regulation of extracellular proteostasis. J. Cell Biol. 2022, 221. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Brodsky, J.L. Protein quality control in the secretory pathway. J. Cell Biol. 2019, 218, 3171–3187. [Google Scholar] [CrossRef] [Green Version]
- Braakman, I.; Bulleid, N.J. Protein folding and modification in the mammalian endoplasmic reticulum. Annu. Rev. Biochem. 2011, 80, 71–99. [Google Scholar] [CrossRef]
- Wiseman, R.L.; Mesgarzadeh, J.S.; Hendershot, L.M. Reshaping endoplasmic reticulum quality control through the unfolded protein response. Mol. Cell 2022, 82, 1477–1491. [Google Scholar] [CrossRef]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef]
- Preissler, S.; Ron, D. Early Events in the Endoplasmic Reticulum Unfolded Protein Response. Cold Spring Harb. Perspect. Biol. 2019, 11, a033894. [Google Scholar] [CrossRef] [Green Version]
- Romine, I.C.; Wiseman, R.L. PERK Signaling Regulates Extracellular Proteostasis of an Amyloidogenic Protein During Endoplasmic Reticulum Stress. Sci. Rep. 2019, 9, 410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.J.; Genereux, J.C.; Suh, E.H.; Vartabedian, V.F.; Rius, B.; Qu, S.; Dendle, M.T.A.; Kelly, J.W.; Wiseman, R.L. Endoplasmic Reticulum Proteostasis Influences the Oligomeric State of an Amyloidogenic Protein Secreted from Mammalian Cells. Cell Chem. Biol. 2016, 23, 1282–1293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buxbaum, J.N.; Tagoe, C.; Gallo, G.; Walker, J.R.; Kurian, S.; Salomon, D.R. Why are some amyloidoses systemic? Does hepatic "chaperoning at a distance" prevent cardiac deposition in a transgenic model of human senile systemic (transthyretin) amyloidosis? FASEB J. 2012, 26, 2283–2293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giadone, R.M.; Liberti, D.C.; Matte, T.M.; Rosarda, J.D.; Torres-Arancivia, C.; Ghosh, S.; Diedrich, J.K.; Pankow, S.; Skvir, N.; Jean, J.C.; et al. Expression of Amyloidogenic Transthyretin Drives Hepatic Proteostasis Remodeling in an Induced Pluripotent Stem Cell Model of Systemic Amyloid Disease. Stem Cell Rep. 2020, 15, 515–528. [Google Scholar] [CrossRef]
- Chen, J.J.; Genereux, J.C.; Qu, S.; Hulleman, J.D.; Shoulders, M.D.; Wiseman, R.L. ATF6 activation reduces the secretion and extracellular aggregation of destabilized variants of an amyloidogenic protein. Chem. Biol. 2014, 21, 1564–1574. [Google Scholar] [CrossRef] [Green Version]
- Shoulders, M.D.; Ryno, L.M.; Genereux, J.C.; Moresco, J.J.; Tu, P.G.; Wu, C.; Yates, J.R., 3rd; Su, A.I.; Kelly, J.W.; Wiseman, R.L. Stress-independent activation of XBP1s and/or ATF6 reveals three functionally diverse ER proteostasis environments. Cell Rep. 2013, 3, 1279–1292. [Google Scholar] [CrossRef] [Green Version]
- Sekijima, Y.; Wiseman, R.L.; Matteson, J.; Hammarstrom, P.; Miller, S.R.; Sawkar, A.R.; Balch, W.E.; Kelly, J.W. The biological and chemical basis for tissue-selective amyloid disease. Cell 2005, 121, 73–85. [Google Scholar] [CrossRef] [Green Version]
- Sekijima, Y.; Hammarstrom, P.; Matsumura, M.; Shimizu, Y.; Iwata, M.; Tokuda, T.; Ikeda, S.; Kelly, J.W. Energetic characteristics of the new transthyretin variant A25T may explain its atypical central nervous system pathology. Lab. Investig. 2003, 83, 409–417. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.; Ong, D.S.; Kelly, J.W. A stilbene that binds selectively to transthyretin in cells and remains dark until it undergoes a chemoselective reaction to create a bright blue fluorescent conjugate. J. Am. Chem. Soc. 2010, 132, 16043–16051. [Google Scholar] [CrossRef] [Green Version]
- Paxman, R.; Plate, L.; Blackwood, E.A.; Glembotski, C.; Powers, E.T.; Wiseman, R.L.; Kelly, J.W. Pharmacologic ATF6 activating compounds are metabolically activated to selectively modify endoplasmic reticulum proteins. eLife 2018, 7, e37168. [Google Scholar] [CrossRef]
- Li, J.; Ni, M.; Lee, B.; Barron, E.; Hinton, D.R.; Lee, A.S.S. The unfolded protein response regulator GRP78/BiP is required for endoplasmic reticulum integrity and stress-induced autophagy in mammalian cells. Cell Death Differ. 2008, 15, 1460–1471. [Google Scholar] [CrossRef] [PubMed]
- Susuki, S.; Sato, T.; Miyata, M.; Momohara, M.; Suico, M.A.; Shuto, T.; Ando, Y.; Kai, H. The Endoplasmic Reticulum-associated Degradation of Transthyretin Variants Is Negatively Regulated by BiP in Mammalian Cells. J. Biol. Chem. 2009, 284, 8312–8321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Zhang, H.; Cheng, Q. PDIA4: The basic characteristics, functions and its potential connection with cancer. Biomed. Pharmacother. 2020, 122, 109688. [Google Scholar] [CrossRef] [PubMed]
- Meunier, L.; Usherwood, Y.K.; Chung, K.T.; Hendershot, L.M. A subset of chaperones and folding enzymes form multiprotein complexes in endoplasmic reticulum to bind nascent proteins. Mol. Biol. Cell 2002, 13, 4456–4469. [Google Scholar] [CrossRef]
- Cai, H.; Wang, C.C.; Tsou, C.L. Chaperone-like activity of protein disulfide isomerase in the refolding of a protein with no disulfide bonds. J. Biol. Chem. 1994, 269, 24550–24552. [Google Scholar] [CrossRef]
- Song, J.L.; Wang, C.C. Chaperone-like activity of protein disulfide-isomerase in the refolding of rhodanese. Eur. J. Biochem. 1995, 231, 312–316. [Google Scholar] [CrossRef]
- Plate, L.; Rius, B.; Nguyen, B.; Genereux, J.C.; Kelly, J.W.; Wiseman, R.L. Quantitative Interactome Proteomics Reveals a Molecular Basis for ATF6-Dependent Regulation of a Destabilized Amyloidogenic Protein. Cell Chem. Biol. 2019, 26, 913–925 e914. [Google Scholar] [CrossRef]
- Sorgjerd, K.; Ghafouri, B.; Jonsson, B.H.; Kelly, J.W.; Blond, S.Y.; Hammarstrom, P. Retention of misfolded mutant transthyretin by the chaperone BiP/GRP78 mitigates amyloidogenesis. J. Mol. Biol. 2006, 356, 469–482. [Google Scholar] [CrossRef]
- Sato, T.; Susuki, S.; Suico, M.A.; Miyata, M.; Ando, Y.; Mizuguchi, M.; Takeuchi, M.; Dobashi, M.; Shuto, T.; Kai, H. Endoplasmic reticulum quality control regulates the fate of transthyretin variants in the cell. EMBO J. 2007, 26, 2501–2512. [Google Scholar] [CrossRef] [Green Version]
- Rius, B.; Mesgarzadeh, J.S.; Romine, I.C.; Paxman, R.J.; Kelly, J.W.; Wiseman, R.L. Pharmacologic targeting of plasma cell endoplasmic reticulum proteostasis to reduce amyloidogenic light chain secretion. Blood Adv. 2021, 5, 1037–1049. [Google Scholar] [CrossRef]
- Grandjean, J.M.D.; Wiseman, R.L. Small molecule strategies to harness the unfolded protein response: Where do we go from here? J. Biol. Chem. 2020, 295, 15692–15711. [Google Scholar] [CrossRef] [PubMed]
- Plate, L.; Cooley, C.B.; Chen, J.J.; Paxman, R.J.; Gallagher, C.M.; Madoux, F.; Genereux, J.C.; Dobbs, W.; Garza, D.; Spicer, T.P.; et al. Small molecule proteostasis regulators that reprogram the ER to reduce extracellular protein aggregation. eLife 2016, 5, e15550. [Google Scholar] [CrossRef] [PubMed]
- Kudo, T.; Kanemoto, S.; Hara, H.; Morimoto, N.; Morihara, T.; Kimura, R.; Tabira, T.; Imaizumi, K.; Takeda, M. A molecular chaperone inducer protects neurons from ER stress. Cell Death Differ. 2008, 15, 364–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mesgarzadeh, J.S.; Romine, I.C.; Smith-Cohen, E.M.; Grandjean, J.M.D.; Kelly, J.W.; Genereux, J.C.; Wiseman, R.L. ATF6 Activation Reduces Amyloidogenic Transthyretin Secretion through Increased Interactions with Endoplasmic Reticulum Proteostasis Factors. Cells 2022, 11, 1661. https://doi.org/10.3390/cells11101661
Mesgarzadeh JS, Romine IC, Smith-Cohen EM, Grandjean JMD, Kelly JW, Genereux JC, Wiseman RL. ATF6 Activation Reduces Amyloidogenic Transthyretin Secretion through Increased Interactions with Endoplasmic Reticulum Proteostasis Factors. Cells. 2022; 11(10):1661. https://doi.org/10.3390/cells11101661
Chicago/Turabian StyleMesgarzadeh, Jaleh S., Isabelle C. Romine, Ethan M. Smith-Cohen, Julia M. D. Grandjean, Jeffery W. Kelly, Joseph C. Genereux, and R. Luke Wiseman. 2022. "ATF6 Activation Reduces Amyloidogenic Transthyretin Secretion through Increased Interactions with Endoplasmic Reticulum Proteostasis Factors" Cells 11, no. 10: 1661. https://doi.org/10.3390/cells11101661
APA StyleMesgarzadeh, J. S., Romine, I. C., Smith-Cohen, E. M., Grandjean, J. M. D., Kelly, J. W., Genereux, J. C., & Wiseman, R. L. (2022). ATF6 Activation Reduces Amyloidogenic Transthyretin Secretion through Increased Interactions with Endoplasmic Reticulum Proteostasis Factors. Cells, 11(10), 1661. https://doi.org/10.3390/cells11101661