
Recent Advances in Microglia Modelling to Address Translational Outcomes in Neurodegenerative Diseases



Abstract
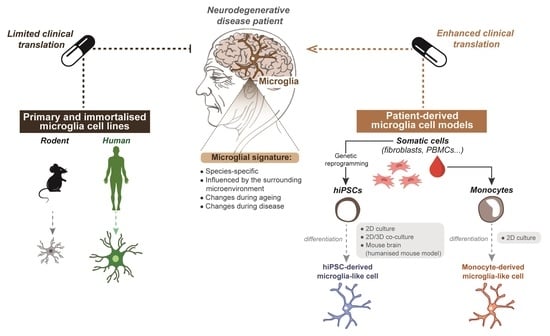
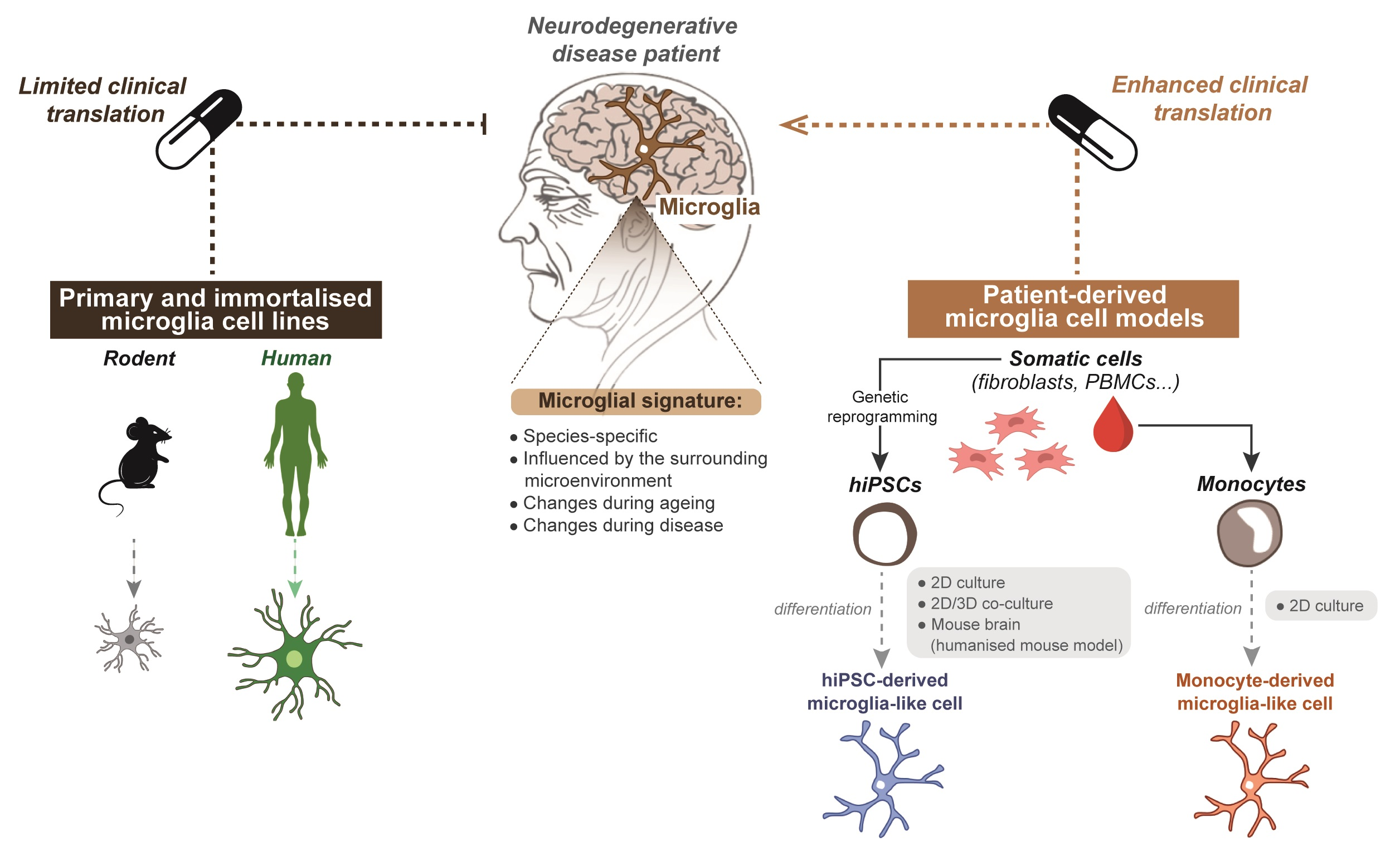
:
1. Introduction
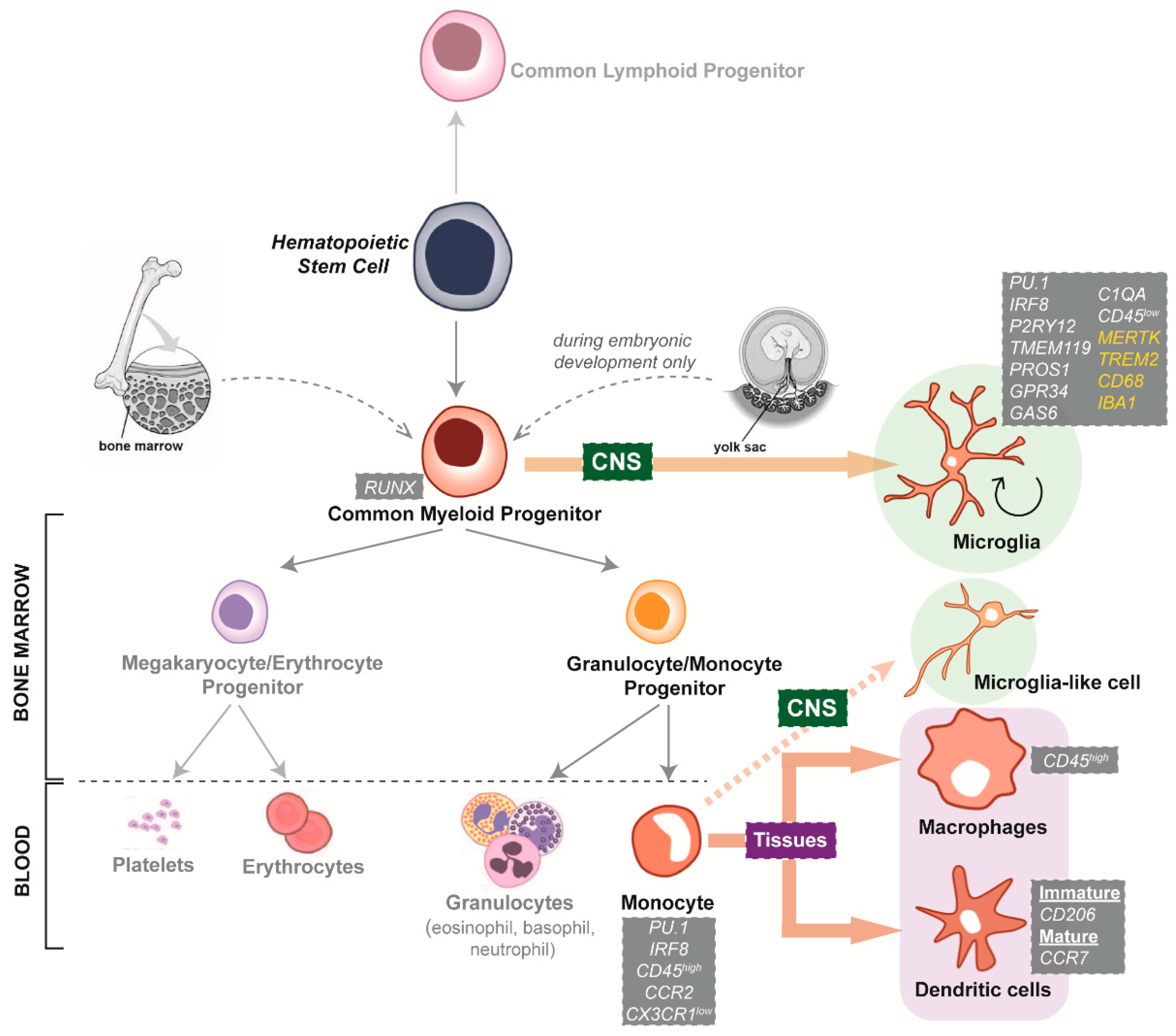
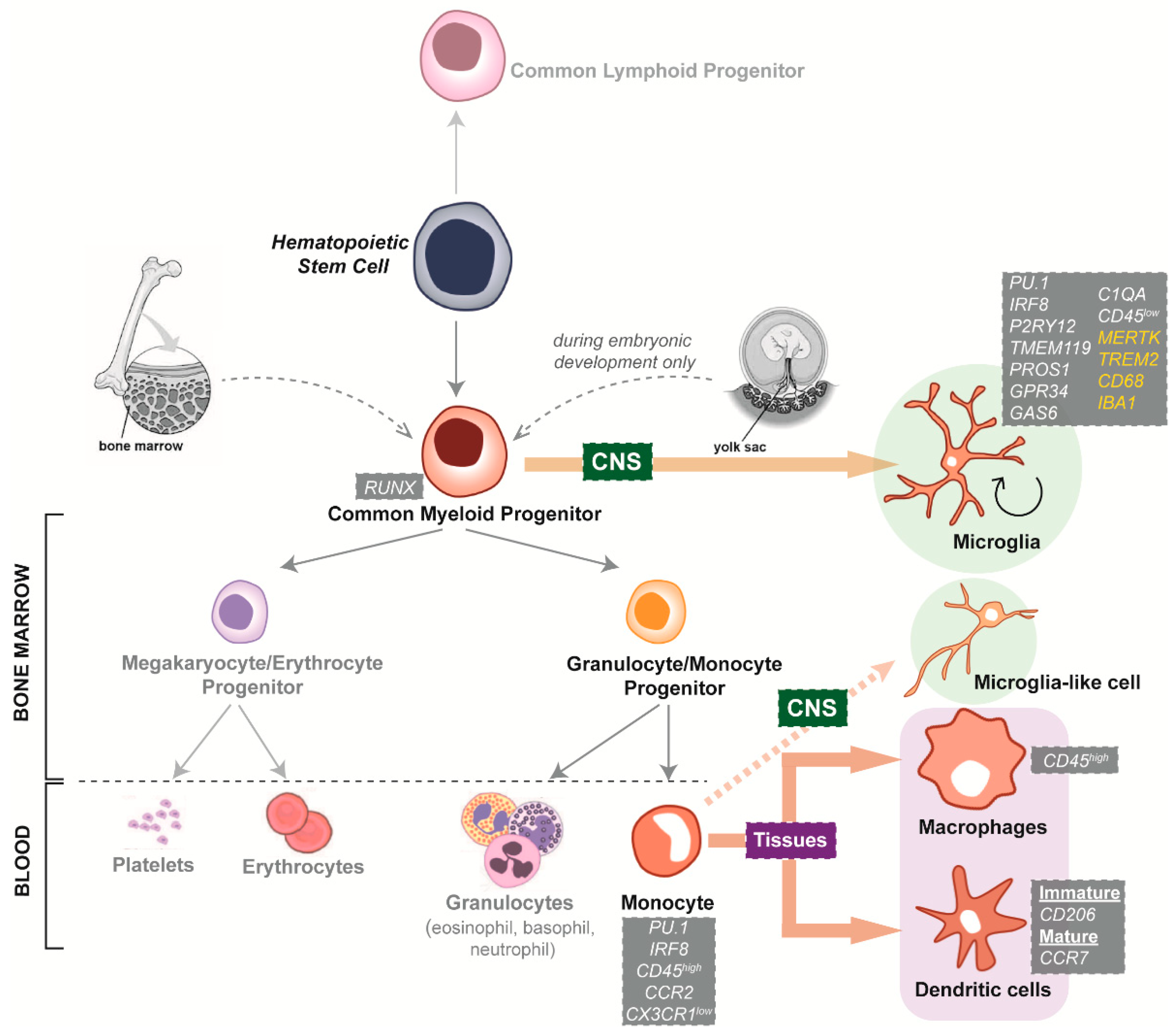
2. The Role of Microglia in Neurodegenerative Diseases
3. Microglia as a Therapeutic Target for Neurodegenerative Diseases
4. Obstacles to Modelling Microglia for Pre-Clinical Studies
4.1. Species-Specific Microglia Signature
4.2. Microglial Dependence on the Surrounding CNS Microenvironment
4.3. Microglial States Associated with Ageing
4.4. Microglial States Associated with Sex
4.5. Microglial States Associated with Neurological Disease
5. Limitations of Primary and Immortalised Microglia In Vitro Models to Study Neurodegenerative Disease

{kind=link}
{kind=link}
| Donor Characteristics | Source | Culture Conditions | Phenotypic Characteristics | Advantages/Disadvantages | Applications | Studies | ||
|---|---|---|---|---|---|---|---|---|
| Primary microglia | ||||||||
| Human |
| Tissue source: autopsy, biopsy Autopsy tissue conditions:
Yield: 200,000–500,000 cells/gram of tissue | 10% FBS in DMEM/F12 | Freshly isolated cells:
Cultured cells:
| Advantages:
Disadvantages:
| Freshly isolated cells:
Culturedcells: Study of microglia physiology in vitro (e.g., surveillance, phagocytosis, immune activation) | [83,94,95,96] | |
| Rodent |
| Tissue conditions:
Yield: 500,000-700,000 cells/gram of tissue | 10% FBS in DMEM/F12 | Advantages:
Disadvantages:
| [79,97] | |||
| Immortalised microglia cell lines | ||||||||
| Human | HMO6 | Embryonic Transformed, v-myc oncogene | 10% FBS in DMEM/F12 | Attenuated or lack of response to inflammatory stimuli (e.g., neither release of IL-1β nor nitric oxide) | Advantages:
Disadvantages:
|
| [98] | |
| HµGlia | Adult Transformed, SV40 large T antigen and hTERT | 10% FBS in DMEM/F12 | Lack expression of microglia-enriched genes | [99] | ||||
| CHME-5 | Embryonic Transformed, SV40 large T antigen | 10% FBS in DMEM/F12 | Uncertain origin (rat origin suggested) | [100] | ||||
| HMC3 | Derived from CHME-5 line | 10% FBS in EMEM | Lack expression of microglia-enriched genes | [101] | ||||
| C13NJ | 10% FBS in DMEM/F12 | [102] | ||||||
| SV40 (IM-HM) | Embryonic Transformed, SV40 large T antigen | 20% FBS | Low expression of microglia-enriched genes | [103] | ||||
| Mouse | BV2 | Neonatal Transformed, v-raf/v-myc oncogene | 10% FBS in DMEM | Attenuated response to inflammatory stimuli (e.g., no release of IL-1β) | [104] | |||
| N9, N11 | Embryonic Transformed, v-myc oncogene | 10% FBS in DMEM | Express a limited number of inflammatory mediators | [105] | ||||
| EOC (subtypes 2, 13.31, 20) | Neonatal Spontaneously immortalised | 10% FBS in DMEM with M-CSF supplement | Some subtypes do not express MHCII | [106] | ||||
| IMG | Adult Transformed, v-raf/v-myc oncogene | 10% FBS in DMEM | Amoeboid morphology | [107] | ||||
| Rat | HAPI | Neonatal Spontaneously immortalised | 10% FBS in DMEM | Attenuated response to inflammatory stimuli | [108] | |||
| Stem cell-derived microglia | ||||||||
| Human |
| hiPSCs (derived from genetically reprogrammed somatic cells, such as skin fibroblasts) | Differentiation towards microglial lineage has been achieved in:
Culture medium is commonly supplemented with M-CSF, IL-34, SCF, VEGF, BMP4, ActivinA and TPO | Best resemble foetal or early postnatal microglia when differentiated under 2D mono-culture conditions (i.e., low expression of TREM2, TMEM119 and P2RY12 compared to adult microglia) | Advantages:
Disadvantages:
|
| [109,110,111,112,113] | |
| Monocytes (isolated from peripheral blood) | RPMI with GM-CSF and IL-34 supplements (Elaborated in Table 2) | [114,115,116,117,118] | ||||||
5.1. Interspecies Differences of Microglia Neurodegenerative Disease Phenotypes
5.2. Limited Availability and Quality of Primary and Immortalised Human Microglia
6. Improving Current Microglia Cell Models
6.1. Generating Microglia from Patient-Derived hiPSCs
6.2. Generating Microglia from Patient-Derived Monocytes
6.3. How Do the Microglia-like Cell Characteristics of hiPSC- and Monocyte-Derived Microglia Models Compare to Each Other?
7. Applications of Patient-Derived Microglia In Vitro Models to Study Neurodegenerative Diseases
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Deuschl, G.; Beghi, E.F.; Fazekas, T.; Varga, K.A.; Christoforidi, E.; Sipido, C.L.; Bassetti, T.V.; Feigin, V.L. The burden of neurological diseases in Europe: An analysis for the Global Burden of Disease Study 2017. Lancet Public Health 2020, 5, e551–e567. [Google Scholar] [CrossRef]
- Soto, C.; Pritzkow, S. Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1332–1340. [Google Scholar] [CrossRef]
- Oxford, A.E.; Stewart, E.S.; Rohn, T.T. Clinical trials in Alzheimer’s disease: A hurdle in the path of remedy. Int. J. Alzheimer’s Dis. 2020, 2020, 5380346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berk, C.; Sabbagh, M.N. Successes and failures for drugs in late-stage development for Alzheimer’s disease. Drugs Aging 2013, 30, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Block, M.L.; Zecca, L.; Hong, J.-S. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Hickman, S.; Izzy, S.; Sen, P.; Morsett, L.; El Khoury, J. Microglia in neurodegeneration. Nat. Neurosci. 2018, 21, 1359–1369. [Google Scholar] [CrossRef] [PubMed]
- Delage, C.I.; Šimončičová, E.; Tremblay, M.-È. Microglial heterogeneity in aging and Alzheimer’s disease: Is sex relevant? J. Pharmacol. Sci. 2021, 146, 169–181. [Google Scholar] [CrossRef]
- Zeiss, C.J. From reproducibility to translation in neurodegenerative disease. ILAR J. 2017, 58, 106–114. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Barres, B.A. Microglia and macrophages in brain homeostasis and disease. Nat. Rev. Immunol. 2018, 18, 225–242. [Google Scholar] [CrossRef]
- Salter, M.W.; Stevens, B. Microglia emerge as central players in brain disease. Nat. Med. 2017, 23, 1018–1027. [Google Scholar] [CrossRef]
- Wolf, S.A.; Boddeke, H.; Kettenmann, H. Microglia in physiology and disease. Annu. Rev. Physiol. 2017, 79, 619–643. [Google Scholar] [CrossRef] [PubMed]
- Hodges, A.K.; TMPiers DCollier Cousins, O.; Pocock, J.M. Pathways linking Alzheimer’s disease risk genes expressed highly in microglia. Neuroimmunol. Neuroinflammation 2021, 8, 245–268. [Google Scholar] [CrossRef]
- Van Cauwenberghe, C.; Van Broeckhoven, C.; Sleegers, K. The genetic landscape of Alzheimer disease: Clinical implications and perspectives. Genet. Med. 2016, 18, 421–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.; Ho, D.-H.; Suk, J.-E.; You, S.; Michael, S.; Kang, J.; Lee, S.J.; Masliah, E.; Hwang, D.; Lee, H.-J. Neuron-released oligomeric α-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat. Commun. 2013, 4, 1562. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; Beers, D.R.; Henkel, J.S.; Zhang, W.; Urushitani, M.; Julien, J.P.; Appel, S.H. Extracellular mutant SOD1 induces microglial-mediated motoneuron injury. Glia 2010, 58, 231–243. [Google Scholar] [CrossRef] [Green Version]
- Liao, B.; Zhao, W.; Beers, D.R.; Henkel, J.S.; Appel, S.H. Transformation from a neuroprotective to a neurotoxic microglial phenotype in a mouse model of ALS. Exp. Neurol. 2012, 237, 147–152. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Guajardo, V.; Febbraro, F.; Kirik, D.; Romero-Ramos, M. Microglia acquire distinct activation profiles depending on the degree of α-synuclein neuropathology in a rAAV based model of Parkinson’s disease. PLoS ONE 2010, 5, e8784. [Google Scholar] [CrossRef]
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L. Inflammation and Alzheimer’s disease. Neurobiol. Aging 2000, 21, 383–421. [Google Scholar] [CrossRef]
- Frank-Cannon, T.C.; Alto, L.T.; McAlpine, F.E.; Tansey, M.G. Does neuroinflammation fan the flame in neurodegenerative diseases? Mol. Neurodegener. 2009, 4, 47. [Google Scholar] [CrossRef] [Green Version]
- Lai, K.S.P.; Liu, C.S.; Rau, A.; Lanctôt, K.L.; Köhler, C.A.; Pakosh, M.; Carvalho, A.F.; Herrmann, N. Peripheral inflammatory markers in Alzheimer’s disease: A systematic review and meta-analysis of 175 studies. J. Neurol. Neurosurg. Psychiatry 2017, 88, 876–882. [Google Scholar] [CrossRef]
- Cummings, J.; Lee, G.; Ritter, A.; Sabbagh, M.; Zhong, K. Alzheimer’s disease drug development pipeline: 2020. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2020, 6, e12050. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. Microglia-targeted candidates push the Alzheimer drug envelope. Nat. Rev. Drug Discov. 2018, 17, 303–306. [Google Scholar] [CrossRef] [PubMed]
- Mora, J.S.; Genge, A.; Chio, A.; Estol, C.J.; Chaverri, D.; Hernández, M.; Marín, S.; Mascias, J.; Rodriguez, G.E.; Povedano, M. Masitinib as an add-on therapy to riluzole in patients with amyotrophic lateral sclerosis: A randomized clinical trial. Amyotroph. Lateral Scler. Front. Degener. 2020, 21, 5–14. [Google Scholar] [CrossRef] [Green Version]
- Therapeutics, D. Denali Therapeutics Announces Decision to Advance DNL151 into Late Stage Clinical Studies in Parkinson’s Patients. 2020. Available online: https://www.denalitherapeutics.com/investors/press-release?id=7661&type=api (accessed on 6 August 2020).
- Takata, K.; Ginhoux, F.; Shimohama, S. Roles of microglia in Alzheimer’s disease and impact of new findings on microglial heterogeneity as a target for therapeutic intervention. Biochem. Pharmacol. 2021, 192, 114754. [Google Scholar] [CrossRef] [PubMed]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masuda, T.; Sankowski, R.; Staszewski, O.; Prinz, M. Microglia Heterogeneity in the Single-Cell Era; Elsevier B.V.: Amsterdam, The Netherlands, 2020; pp. 1271–1281. [Google Scholar]
- Stratoulias, V.; Venero, J.L.; Tremblay, M.È.; Joseph, B. Microglial subtypes: Diversity within the microglial community. EMBO J. 2019, 38, e101997. [Google Scholar] [CrossRef] [PubMed]
- Butovsky, O.; Jedrychowski, M.P.; Moore, C.S.; Cialic, R.; Lanser, A.J.; Gabriely, G.; Koeglsperger, T.; Dake, B.; Wu, P.M.; Doykan, C.E. Identification of a unique TGF-β–dependent molecular and functional signature in microglia. Nat. Neurosci. 2014, 17, 131–143. [Google Scholar] [CrossRef] [Green Version]
- Gosselin, D.; Skola, D.; Coufal, N.G.; Holtman, I.R.; Schlachetzki, J.C.; Sajti, E.; Jaeger, B.N.; O’Connor, C.; Fitzpatrick, C.; Pasillas, M.P. An environment-dependent transcriptional network specifies human microglia identity. Science 2017, 356, eaal3222. [Google Scholar] [CrossRef] [Green Version]
- Galatro, T.F.; Vainchtein, I.D.; Brouwer, N.; Boddeke, E.W.; Eggen, B.J. Isolation of microglia and immune infiltrates from mouse and primate central nervous system. In Inflammation; Springer: Berlin/Heidelberg, Germany, 2017; pp. 333–342. [Google Scholar]
- Geirsdottir, L.; David, E.; Keren-Shaul, H.; Weiner, A.; Bohlen, S.C.; Neuber, J.; Balic, A.; Giladi, A.; Sheban, F.; Dutertre, C.-A. Cross-species single-cell analysis reveals divergence of the primate microglia program. Cell 2019, 179, 1609–1622.e1616. [Google Scholar] [CrossRef] [Green Version]
- Sheridan, G.K.; Murphy, K.J. Neuron–glia crosstalk in health and disease: Fractalkine and CX3CR1 take centre stage. Open Biol. 2013, 3, 130181. [Google Scholar] [CrossRef] [Green Version]
- York, E.M.; Bernier, L.P.; MacVicar, B.A. Microglial modulation of neuronal activity in the healthy brain. Dev. Neurobiol. 2018, 78, 593–603. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Song, W.M.; Andhey, P.S.; Swain, A.; Levy, T.; Miller, K.R.; Poliani, P.L.; Cominelli, M.; Grover, S.; Gilfillan, S. Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat. Med. 2020, 26, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Mendes, M.S.; Majewska, A.K. An overview of microglia ontogeny and maturation in the homeostatic and pathological brain. Eur. J. Neurosci. 2021, 53, 3525–3547. [Google Scholar] [CrossRef]
- Mildner, A.; Huang, H.; Radke, J.; Stenzel, W.; Priller, J. P2Y12 receptor is expressed on human microglia under physiological conditions throughout development and is sensitive to neuroinflammatory diseases. Glia 2017, 65, 375–387. [Google Scholar] [CrossRef] [PubMed]
- Goldmann, T.; Zeller, N.; Raasch, J.; Kierdorf, K.; Frenzel, K.; Ketscher, L.; Basters, A.; Staszewski, O.; Brendecke, S.M.; Spiess, A. USP 18 lack in microglia causes destructive interferonopathy of the mouse brain. EMBO J. 2015, 34, 1612–1629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tay, T.L.; Mai, D.; Dautzenberg, J.; Fernandez-Klett, F.; Lin, G.; Datta, M.; Drougard, A.; Stempfl, T.; Ardura-Fabregat, A.; Staszewski, O. A new fate mapping system reveals context-dependent random or clonal expansion of microglia. Nat. Neurosci. 2017, 20, 793–803. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Nicola, D.; Perry, V.H. Microglial dynamics and role in the healthy and diseased brain: A paradigm of functional plasticity. Neurosci. 2015, 21, 169–184. [Google Scholar] [CrossRef] [Green Version]
- De Biase, L.M.; Schuebel, K.E.; Fusfeld, Z.H.; Jair, K.; Hawes, I.A.; Cimbro, R.; Zhang, H.-Y.; Liu, Q.-R.; Shen, H.; Xi, Z.-X. Local cues establish and maintain region-specific phenotypes of basal ganglia microglia. Neuron 2017, 95, 341–356.e346. [Google Scholar] [CrossRef] [Green Version]
- Wierzba-Bobrowicz, T.; Kosno-Kruszewska, E.; Gwiazda, E.; Lechowicz, W. The comparison of microglia maturation in different structures of the human nervous system. Folia Neuropathol. 1998, 36, 152–160. [Google Scholar]
- Mittelbronn, M.; Dietz, K.; Schluesener, H.; Meyermann, R. Local distribution of microglia in the normal adult human central nervous system differs by up to one order of magnitude. Acta Neuropathol. 2001, 101, 249–255. [Google Scholar] [CrossRef]
- O’Reilly, D.; Addley, M.; Quinn, C.; MacFarlane, A.J.; Gordon, S.; McKnight, A.J.; Greaves, D.R. Functional analysis of the murine Emr1 promoter identifies a novel purine-rich regulatory motif required for high-level gene expression in macrophages. Genomics 2004, 84, 1030–1040. [Google Scholar] [CrossRef] [PubMed]
- Stoessel, M.B.; Majewska, A.K. Little cells of the little brain: Microglia in cerebellar development and function. Trends Neurosci. 2021, 44, 564–578. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.-L.; Yuan, Y.; Tian, L. Microglial regional heterogeneity and its role in the brain. Mol. Psychiatry 2020, 25, 351–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greter, M.; Lelios, I.; Pelczar, P.; Hoeffel, G.; Price, J.; Leboeuf, M.; Kündig, T.M.; Frei, K.; Ginhoux, F.; Merad, M. Stroma-derived interleukin-34 controls the development and maintenance of langerhans cells and the maintenance of microglia. Immunity 2012, 37, 1050–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Szretter, K.J.; Vermi, W.; Gilfillan, S.; Rossini, C.; Cella, M.; Barrow, A.D.; Diamond, M.S.; Colonna, M. IL-34 is a tissue-restricted ligand of CSF1R required for the development of Langerhans cells and microglia. Nat. Immunol. 2012, 13, 753–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, D.G.; Tang, T.M.; Lue, L.F. Studies on Colony Stimulating Factor Receptor-1 and Ligands Colony Stimulating Factor-1 and Interleukin-34 in Alzheimer’s Disease Brains and Human Microglia. Front. Aging Neurosci. 2017, 9, 244. [Google Scholar] [CrossRef] [PubMed]
- Baghdadi, M.; Umeyama, Y.; Hama, N.; Kobayashi, T.; Han, N.; Wada, H.; Seino, K.-I. Interleukin-34, a comprehensive review. J. Leukoc. Biol. 2018, 104, 931–951. [Google Scholar] [CrossRef]
- Vainchtein, I.D.; Chin, G.; Cho, F.S.; Kelley, K.W.; Miller, J.G.; Chien, E.C.; Liddelow, S.A.; Nguyen, P.T.; Nakao-Inoue, H.; Dorman, L.C. Astrocyte-derived interleukin-33 promotes microglial synapse engulfment and neural circuit development. Science 2018, 359, 1269–1273. [Google Scholar] [CrossRef] [Green Version]
- Xuan, F.-L.; Chithanathan, K.; Lilleväli, K.; Yuan, X.; Tian, L. Differences of microglia in the brain and the spinal cord. Front. Cell. Neurosci. 2019, 13, 504. [Google Scholar] [CrossRef]
- Ayata, P.; Badimon, A.; Strasburger, H.J.; Duff, M.K.; Montgomery, S.E.; Loh, Y.-H.E.; Ebert, A.; Pimenova, A.A.; Ramirez, B.R.; Chan, A.T. Epigenetic regulation of brain region-specific microglia clearance activity. Nat. Neurosci. 2018, 21, 1049–1060. [Google Scholar] [CrossRef]
- Long-Smith, C.M.; Sullivan, A.M.; Nolan, Y.M. The influence of microglia on the pathogenesis of Parkinson’s disease. Prog. Neurobiol. 2009, 89, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Matcovitch-Natan, O.; Winter, D.R.; Giladi, A.; Aguilar, S.V.; Spinrad, A.; Sarrazin, S.; Ben-Yehuda, H.; David, E.; González, F.Z.; Perrin, P. Microglia development follows a stepwise program to regulate brain homeostasis. Science 2016, 353, aad8670. [Google Scholar] [CrossRef] [PubMed]
- Hammond, T.R.; Dufort, C.; Dissing-Olesen, L.; Giera, S.; Young, A.; Wysoker, A.; Walker, A.J.; Gergits, F.; Segel, M.; Nemesh, J. Single-cell RNA sequencing of microglia throughout the mouse lifespan and in the injured brain reveals complex cell-state changes. Immunity 2019, 50, 253–271.e256. [Google Scholar] [CrossRef] [Green Version]
- Niraula, A.; Sheridan, J.F.; Godbout, J.P. Microglia Priming with Aging and Stress. Neuropsychopharmacology 2017, 42, 318–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spittau, B. Aging microglia—phenotypes, functions and implications for age-related neurodegenerative diseases. Front. Aging Neurosci. 2017, 9, 194. [Google Scholar] [CrossRef]
- Deczkowska, A.; Amit, I.; Schwartz, M. Microglial immune checkpoint mechanisms. Nat. Neurosci. 2018, 21, 779–786. [Google Scholar] [CrossRef]
- Tichauer, J.E.; Flores, B.; Soler, B.; Bernhardi, L.E.; Ramírez, G.; von Bernhardi, R. Age-dependent changes on TGFβ1 Smad3 pathway modify the pattern of microglial cell activation. Brain Behav. Immun. 2014, 37, 187–196. [Google Scholar] [CrossRef] [Green Version]
- Olah, M.; Patrick, E.; Villani, A.-C.; Xu, J.; White, C.C.; Ryan, K.J.; Piehowski, P.; Kapasi, A.; Nejad, P.; Cimpean, M. A transcriptomic atlas of aged human microglia. Nat. Commun. 2018, 9, 539. [Google Scholar] [CrossRef] [Green Version]
- Streit, W.J.; Sammons, N.W.; Kuhns, A.J.; Sparks, D.L. Dystrophic microglia in the aging human brain. Glia 2004, 45, 208–212. [Google Scholar] [CrossRef]
- Davies, D.S.; Ma, J.; Jegathees, T.; Goldsbury, C. Microglia show altered morphology and reduced arborization in human brain during aging and A lzheimer’s disease. Brain Pathol. 2017, 27, 795–808. [Google Scholar] [CrossRef]
- Damani, M.R.; Zhao, L.; Fontainhas, A.M.; Amaral, J.; Fariss, R.N.; Wong, W.T. Age-related alterations in the dynamic behavior of microglia. Aging Cell 2011, 10, 263–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Safaiyan, S.; Kannaiyan, N.; Snaidero, N.; Brioschi, S.; Biber, K.; Yona, S.; Edinger, A.L.; Jung, S.; Rossner, M.J.; Simons, M. Age-related myelin degradation burdens the clearance function of microglia during aging. Nat. Neurosci. 2016, 19, 995–998. [Google Scholar] [CrossRef] [PubMed]
- Marschallinger, J.; Iram, T.; Zardeneta, M.; Lee, S.E.; Lehallier, B.; Haney, M.S.; Pluvinage, J.V.; Mathur, V.; Hahn, O.; Morgens, D.W. Lipid-droplet-accumulating microglia represent a dysfunctional and proinflammatory state in the aging brain. Nat. Neurosci. 2020, 23, 194–208. [Google Scholar] [CrossRef]
- Frigerio, C.S.; Wolfs, L.; Fattorelli, N.; Thrupp, N.; Voytyuk, I.; Schmidt, I.; Mancuso, R.; Chen, W.-T.; Woodbury, M.E.; Srivastava, G. The major risk factors for Alzheimer’s disease: Age, sex, and genes modulate the microglia response to Aβ plaques. Cell Rep. 2019, 27, 1293–1306.e1296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Réu, P.; Khosravi, A.; Bernard, S.; Mold, J.E.; Salehpour, M.; Alkass, K.; Perl, S.; Tisdale, J.; Possnert, G.; Druid, H. The lifespan and turnover of microglia in the human brain. Cell Rep. 2017, 20, 779–784. [Google Scholar] [CrossRef] [Green Version]
- Montagne, A.; Barnes, S.R.; Sweeney, M.D.; Halliday, M.R.; Sagare, A.P.; Zhao, Z.; Toga, A.W.; Jacobs, R.E.; Liu, C.Y.; Amezcua, L. Blood-brain barrier breakdown in the aging human hippocampus. Neuron 2015, 85, 296–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamatovic, S.M.; Martinez-Revollar, G.; Hu, A.; Choi, J.; Keep, R.F.; Andjelkovic, A.V. Decline in Sirtuin-1 expression and activity plays a critical role in blood-brain barrier permeability in aging. Neurobiol. Dis. 2019, 126, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.D.; Winkler, E.A.; Sagare, A.P.; Singh, I.; LaRue, B.; Deane, R.; Zlokovic, B.V. Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron 2010, 68, 409–427. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, J.M.; Sholar, P.W.; Bilbo, S.D. Sex differences in microglial colonization of the developing rat brain. J. Neurochem. 2012, 120, 948–963. [Google Scholar] [CrossRef]
- Villa, A.; Gelosa, P.; Castiglioni, L.; Cimino, M.; Rizzi, N.; Pepe, G.; Lolli, F.; Marcello, E.; Sironi, L.; Vegeto, E. Sex-specific features of microglia from adult mice. Cell Rep. 2018, 23, 3501–3511. [Google Scholar] [CrossRef]
- Lenz, K.M.; Nugent, B.M.; Haliyur, R.; McCarthy, M.M. Microglia are essential to masculinization of brain and behavior. J. Neurosci. 2013, 33, 2761–2772. [Google Scholar] [CrossRef] [Green Version]
- Hanamsagar, R.; Alter, M.D.; Block, C.S.; Sullivan, H.; Bolton, J.L.; Bilbo, S.D. Generation of a microglial developmental index in mice and in humans reveals a sex difference in maturation and immune reactivity. Glia 2017, 65, 1504–1520. [Google Scholar] [CrossRef]
- Guneykaya, D.; Ivanov, A.; Hernandez, D.P.; Haage, V.; Wojtas, B.; Meyer, N.; Maricos, M.; Jordan, P.; Buonfiglioli, A.; Gielniewski, B. Transcriptional and translational differences of microglia from male and female brains. Cell Rep. 2018, 24, 2773–2783.e2776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thion, M.S.; Low, D.; Silvin, A.; Chen, J.; Grisel, P.; Schulte-Schrepping, J.; Blecher, R.; Ulas, T.; Squarzoni, P.; Hoeffel, G. Microbiome influences prenatal and adult microglia in a sex-specific manner. Cell 2018, 172, 500–516.e516. [Google Scholar] [CrossRef] [Green Version]
- Guillot-Sestier, M.-V.; Araiz, A.R.; Mela, V.; Gaban, A.S.; O’Neill, E.; Joshi, L.; Chouchani, E.T.; Mills, E.L.; Lynch, M.A. Microglial metabolism is a pivotal factor in sexual dimorphism in Alzheimer’s disease. Commun. Biol. 2021, 4, 711. [Google Scholar] [CrossRef]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 2017, 169, 1276–1290.e1217. [Google Scholar] [CrossRef]
- Tu, P.; Gurney, M.E.; Julien, J.-P.; Lee, V.M.; Trojanowski, J.Q. Oxidative stress, mutant SOD1, and neurofilament pathology in transgenic mouse models of human motor neuron disease. Lab. Investig. A J. Tech. Methods Pathol. 1997, 76, 441–456. [Google Scholar]
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J.Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 2019, 570, 332–337. [Google Scholar] [CrossRef]
- Del-Aguila, J.L.; Benitez, B.A.; Li, Z.; Dube, U.; Mihindukulasuriya, K.A.; Budde, J.P.; Farias, F.H.; Fernández, M.V.; Ibanez, L.; Jiang, S. TREM2 brain transcript-specific studies in AD and TREM2 mutation carriers. Mol. Neurodegener. 2019, 14, 18. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, K.; Friedman, B.A.; Etxeberria, A.; Huntley, M.A.; van der Brug, M.P.; Foreman, O.; Paw, J.S.; Modrusan, Z.; Beach, T.G.; Serrano, G.E. Alzheimer’s patient microglia exhibit enhanced aging and unique transcriptional activation. Cell Rep. 2020, 31, 107843. [Google Scholar] [CrossRef]
- Mastroeni, D.; Nolz, J.; Sekar, S.; Delvaux, E.; Serrano, G.; Cuyugan, L.; Liang, W.S.; Beach, T.G.; Rogers, J.; Coleman, P.D. Laser-captured microglia in the Alzheimer’s and Parkinson’s brain reveal unique regional expression profiles and suggest a potential role for hepatitis B in the Alzheimer’s brain. Neurobiol. Aging 2018, 63, 12–21. [Google Scholar] [CrossRef]
- Hirano, K.; Ohgomori, T.; Kobayashi, K.; Tanaka, F.; Matsumoto, T.; Natori, T.; Matsuyama, Y.; Uchimura, K.; Sakamoto, K.; Takeuchi, H. Ablation of keratan sulfate accelerates early phase pathogenesis of ALS. PLoS ONE 2013, 8, e66969. [Google Scholar] [CrossRef] [PubMed]
- Foyez, T.; Takeda-Uchimura, Y.; Ishigaki, S.; Zhang, Z.; Sobue, G.; Kadomatsu, K.; Uchimura, K. Microglial keratan sulfate epitope elicits in central nervous tissues of transgenic model mice and patients with amyotrophic lateral sclerosis. Am. J. Pathol. 2015, 185, 3053–3065. [Google Scholar] [CrossRef]
- Bertolotto, A.; Caterson, B.; Canavese, G.; Migheli, A.; Schiffer, D. Monoclonal antibodies to keratan sulfate immunolocalize ramified microglia in paraffin and cryostat sections of rat brain. J. Histochem. Cytochem. 1993, 41, 481–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertolotto, A.; Agresti, C.; Castello, A.; Manzardo, E.; Riccio, A. 5D4 keratan sulfate epitope identifies a subset of ramified microglia in normal central nervous system parenchyma. J. Neuroimmunol. 1998, 85, 69–77. [Google Scholar] [CrossRef]
- Sousa, C.; Golebiewska, A.; Poovathingal, S.K.; Kaoma, T.; Pires-Afonso, Y.; Martina, S.; Coowar, D.; Azuaje, F.; Skupin, A.; Balling, R. Single-cell transcriptomics reveals distinct inflammation-induced microglia signatures. EMBO Rep. 2018, 19, e46171. [Google Scholar] [CrossRef]
- Jordão, M.J.C.; Sankowski, R.; Brendecke, S.M.; Sagar; Locatelli, G.; Tai, Y.-H.; Tay, T.L.; Schramm, E.; Armbruster, S.; Hagemeyer, N. Single-cell profiling identifies myeloid cell subsets with distinct fates during neuroinflammation. Science 2019, 363, eaat7554. [Google Scholar] [CrossRef]
- Sieger, D.; Peri, F. Animal models for studying microglia: The first, the popular, and the new. Glia 2013, 61, 3–9. [Google Scholar] [CrossRef]
- Sabogal-Guáqueta, A.M.; Marmolejo-Garza, A.; de Pádua, V.P.; Eggen, B.; Boddeke, E.; Dolga, A.M. Microglia alterations in neurodegenerative diseases and their modeling with human induced pluripotent stem cell and other platforms. Prog. Neurobiol. 2020, 190, 101805. [Google Scholar] [CrossRef]
- Doody, R. Developing Disease-Modifying Treatments in Alzheimer’s Disease-A Perspective from Roche and Genentech. J. Prev. Alzheimer’s Dis. 2017, 4, 264–272. [Google Scholar]
- Olah, M.; Raj, D.; Brouwer, N.; de Haas, A.H.; Eggen, B.J.; den Dunnen, W.F.; Biber, K.P.; Boddeke, H.W. An optimized protocol for the acute isolation of human microglia from autopsy brain samples. Glia 2012, 60, 96–111. [Google Scholar] [CrossRef]
- Rustenhoven, J.; Park, T.I.; Schweder, P.; Scotter, J.; Correia, J.; Smith, A.M.; Gibbons, H.M.; Oldfield, R.L.; Bergin, P.S.; Mee, E.W. Isolation of highly enriched primary human microglia for functional studies. Sci. Rep. 2016, 6, 19371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizee, M.R.; Miedema, S.S.; van der Poel, M.; Schuurman, K.G.; van Strien, M.E.; Melief, J.; Smolders, J.; Hendrickx, D.A.; Heutinck, K.M.; Hamann, J. Isolation of primary microglia from the human post-mortem brain: Effects of ante-and post-mortem variables. Acta Neuropathol. Commun. 2017, 5, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rangaraju, S.; Dammer, E.B.; Raza, S.A.; Gao, T.; Xiao, H.; Betarbet, R.; Duong, D.M.; Webster, J.A.; Hales, C.M.; Lah, J.J. Quantitative proteomics of acutely-isolated mouse microglia identifies novel immune Alzheimer’s disease-related proteins. Mol. Neurodegener. 2018, 13, 34. [Google Scholar] [CrossRef]
- Nagai, A.; Nakagawa, E.; Hatori, K.; Choi, H.; McLarnon, J.; Lee, M.; Kim, S. Generation and characterization of immortalized human microglial cell lines: Expression of cytokines and chemokines. Neurobiol. Dis. 2001, 8, 1057–1068. [Google Scholar] [CrossRef]
- Garcia-Mesa, Y.; Jay, T.R.; Checkley, M.A.; Luttge, B.; Dobrowolski, C.; Valadkhan, S.; Landreth, G.E.; Karn, J.; Alvarez-Carbonell, D. Immortalization of primary microglia: A new platform to study HIV regulation in the central nervous system. J. Neurovirology 2017, 23, 47–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janabi, N.; Peudenier, S.; Héron, B.; Ng, K.H.; Tardieu, M. Establishment of human microglial cell lines after transfection of primary cultures of embryonic microglial cells with the SV40 large T antigen. Neurosci. Lett. 1995, 195, 105–108. [Google Scholar] [CrossRef]
- Janabi, N.; di Stefano, M.; Wallon, C.; Hery, C.; Chiodi, F.; Tardieu, M. Induction of human immunodeficiency virus type 1 replication in human glial cells after proinflammatory cytokines stimulation: Effect of IFNγ, IL1β, and TNFα on differentiation and chemokine production in glial cells. Glia 1998, 23, 304–315. [Google Scholar] [CrossRef]
- Bernhart, E.; Kollroser, M.; Rechberger, G.; Reicher, H.; Heinemann, A.; Schratl, P.; Hallström, S.; Wintersperger, A.; Nusshold, C.; DeVaney, T. Lysophosphatidic acid receptor activation affects the C13NJ microglia cell line proteome leading to alterations in glycolysis, motility, and cytoskeletal architecture. Proteomics 2010, 10, 141–158. [Google Scholar] [CrossRef] [Green Version]
- Sellgren, C.M.; Sheridan, S.D.; Gracias, J.; Xuan, D.; Fu, T.; Perlis, R.H. Patient-specific models of microglia-mediated engulfment of synapses and neural progenitors. Mol. Psychiatry 2017, 22, 170–177. [Google Scholar] [CrossRef] [Green Version]
- Horvath, R.J.; Nutile-McMenemy, N.; Alkaitis, M.S.; DeLeo, J.A. Differential migration, LPS-induced cytokine, chemokine, and NO expression in immortalized BV-2 and HAPI cell lines and primary microglial cultures. J. Neurochem. 2008, 107, 557–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hickman, S.E.; Allison, E.K.; el Khoury, J. Microglial dysfunction and defective β-amyloid clearance pathways in aging Alzheimer’s disease mice. J. Neurosci. 2008, 28, 8354–8360. [Google Scholar] [CrossRef] [PubMed]
- Hensley, K.; Fedynyshyn, J.; Ferrell, S.; Floyd, R.A.; Gordon, B.; Grammas, P.; Hamdheydari, L.; Mhatre, M.; Mou, S.; Pye, Q.N. Message and protein-level elevation of tumor necrosis factor α (TNFα) and TNFα-modulating cytokines in spinal cords of the G93A-SOD1 mouse model for amyotrophic lateral sclerosis. Neurobiol. Dis. 2003, 14, 74–80. [Google Scholar] [CrossRef]
- McCarthy, R.C.; Lu, D.-Y.; Alkhateeb, A.; Gardeck, A.M.; Lee, C.-H.; Wessling-Resnick, M. Characterization of a novel adult murine immortalized microglial cell line and its activation by amyloid-beta. J. Neuroinflammation 2016, 13, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Liu, L.; Barger, S.W.; Griffin, W.S.T. Interleukin-1 mediates pathological effects of microglia on tau phosphorylation and on synaptophysin synthesis in cortical neurons through a p38-MAPK pathway. J. Neurosci. 2003, 23, 1605–1611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, M.P.; Hou, Z.; Propson, N.E.; Zhang, J.; Engstrom, C.J.; Costa, V.S.; Jiang, P.; Nguyen, B.K.; Bolin, J.M.; Daly, W.; et al. Human pluripotent stem cell-derived neural constructs for predicting neural toxicity. Proc. Natl. Acad. Sci. USA 2015, 112, 12516–12521. [Google Scholar] [CrossRef] [Green Version]
- Haenseler, W.; Sansom, S.N.; Buchrieser, J.; Newey, S.E.; Moore, C.S.; Nicholls, F.J.; Chintawar, S.; Schnell, C.; Antel, J.P.; Allen, N.D.; et al. A Highly Efficient Human Pluripotent Stem Cell Microglia Model Displays a Neuronal-Co-culture-Specific Expression Profile and Inflammatory Response. Stem Cell Rep. 2017, 8, 1727–1742. [Google Scholar] [CrossRef] [Green Version]
- Abud, E.M.; Ramirez, R.N.; Martinez, E.S.; Healy, L.M.; Nguyen, C.H.H.; Newman, S.A.; Yeromin, A.V.; Scarfone, V.M.; Marsh, S.E.; Fimbres, C.; et al. iPSC-Derived Human Microglia-like Cells to Study Neurological Diseases. Neuron 2017, 94, 278–293. [Google Scholar] [CrossRef] [Green Version]
- Takata, K.; Kozaki, T.; Lee, C.Z.W.; Thion, M.S.; Otsuka, M.; Lim, S.; Utami, K.H.; Fidan, K.; Park, D.S.; Malleret, B.; et al. Induced-Pluripotent-Stem-Cell-Derived Primitive Macrophages Provide a Platform for Modeling Tissue-Resident Macrophage Differentiation and Function. Immunity 2017, 47, 183–198.e186. [Google Scholar] [CrossRef]
- Xu, R.; Li, X.; Boreland, A.J.; Posyton, A.; Kwan, K.; Hart, R.P.; Jiang, P. Human iPSC-derived mature microglia retain their identity and functionally integrate in the chimeric mouse brain. Nat. Commun. 2020, 11, 1577. [Google Scholar] [CrossRef] [Green Version]
- Ohgidani, M.; Kato, T.A.; Setoyama, D.; Sagata, N.; Hashimoto, R.; Shigenobu, K.; Yoshida, T.; Hayakawa, K.; Shimokawa, N.; Miura, D.; et al. Direct induction of ramified microglia-like cells from human monocytes: Dynamic microglial dysfunction in Nasu-Hakola disease. Sci. Rep. 2014, 4, 4957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, K.J.; White, C.C.; Patel, K.; Xu, J.; Olah, M.; Replogle, J.M.; Frangieh, M.; Cimpean, M.; Winn, P.; McHenry, A.; et al. A human microglia-like cellular model for assessing the effects of neurodegenerative disease gene variants. Sci. Transl. Med. 2017, 9, eaai7635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leone, C.; le Pavec, G.; Même, W.; Porcheray, F.; Samah, B.; Dormont, D.; Gras, G. Characterization of human monocyte-derived microglia-like cells. Glia 2006, 54, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Melief, J.; Sneeboer, M.A.; Litjens, M.; Ormel, P.R.; Palmen, S.J.; Huitinga, I.; Kahn, R.S.; Hol, E.M.; de Witte, L.D. Characterizing primary human microglia: A comparative study with myeloid subsets and culture models. Glia 2016, 64, 1857–1868. [Google Scholar] [CrossRef]
- Etemad, S.; Zamin, R.M.; Ruitenberg, M.J.; Filgueira, L. A novel in vitro human microglia model: Characterization of human monocyte-derived microglia. J. Neurosci. Methods 2012, 209, 79–89. [Google Scholar] [CrossRef]
- Ohgidani, M.; Kato, T.A.; Hosoi, M.; Tsuda, M.; Hayakawa, K.; Hayaki, C.; Iwaki, R.; Sagata, N.; Hashimoto, R.; Inoue, K. Fibromyalgia and microglial TNF-α: Translational research using human blood induced microglia-like cells. Sci. Rep. 2017, 7, 11882. [Google Scholar] [CrossRef] [Green Version]
- Rawat, P.; Spector, S.A. Development and characterization of a human microglia cell model of HIV-1 infection. J. Neurovirology 2017, 23, 33–46. [Google Scholar] [CrossRef] [Green Version]
- Sellgren, C.M.; Gracias, J.; Watmuff, B.; Biag, J.D.; Thanos, J.M.; Whittredge, P.B.; Fu, T.; Worringer, K.; Brown, H.E.; Wang, J.; et al. Increased synapse elimination by microglia in schizophrenia patient-derived models of synaptic pruning. Nat. Neurosci. 2019, 22, 374–385. [Google Scholar] [CrossRef]
- Ormel, P.R.; Böttcher, C.; Gigase, F.A.; Missall, R.D.; van Zuiden, W.; Zapata, M.C.F.; Ilhan, D.; de Goeij, M.; Udine, E.; Sommer, I.E. A characterization of the molecular phenotype and inflammatory response of schizophrenia patient-derived microglia-like cells. Brain Behav. Immun. 2020, 90, 196–207. [Google Scholar] [CrossRef]
- Banerjee, A.; Lu, Y.; Do, K.; Mize, T.; Wu, X.; Chen, X.; Chen, J. Validation of Induced Microglia-Like Cells (iMG Cells) for Future Studies of Brain Diseases. Front. Cell. Neurosci. 2021, 15, 85. [Google Scholar] [CrossRef]
- Smit, T.; Ormel, P.R.; Sluijs, J.A.; Hulshof, L.A.; Middeldorp, J.; de Witte, L.D.; Hol, E.M.; Donega, V. Transcriptomic and functional analysis of Aβ1-42 oligomer-stimulated human monocyte-derived microglia-like cells. Brain Behav. Immun. 2022, 100, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Quek, H.; Cuní-López, C.; Stewart, R.; Colletti, T.; Notaro, A.; Nguyen, T.H.; Sun, Y.; Guo, C.C.; Lupton, M.K.; Roberts, T.L.; et al. ALS monocyte-derived microglia-like cells reveal cytoplasmic TDP-43 accumulation, DNA damage, and cell-specific impairment of phagocytosis associated with disease progression. J. Neuroinflammation 2022, 19, 58. [Google Scholar] [CrossRef]
- Friedman, B.A.; Srinivasan, K.; Ayalon, G.; Meilandt, W.J.; Lin, H.; Huntley, M.A.; Cao, Y.; Lee, S.-H.; Haddick, P.C.; Ngu, H. Diverse brain myeloid expression profiles reveal distinct microglial activation states and aspects of Alzheimer’s disease not evident in mouse models. Cell Rep. 2018, 22, 832–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwabe, T.; Srinivasan, K.; Rhinn, H. Shifting paradigms: The central role of microglia in Alzheimer’s disease. Neurobiol. Dis. 2020, 143, 104962. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.M.; Dragunow, M. The human side of microglia. Trends Neurosci. 2014, 37, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Timmerman, R.; Burm, S.M.; Bajramovic, J.J. An overview of in vitro methods to study microglia. Front. Cell. Neurosci. 2018, 12, 242. [Google Scholar] [CrossRef] [PubMed]
- Muffat, J.; Li, Y.; Yuan, B.; Mitalipova, M.; Omer, A.; Corcoran, S.; Bakiasi, G.; Tsai, L.-H.; Aubourg, P.; Ransohoff, R.M. Efficient derivation of microglia-like cells from human pluripotent stem cells. Nat. Med. 2016, 22, 1358–1367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandya, H.; Shen, M.J.; Ichikawa, D.M.; Sedlock, A.B.; Choi, Y.; Johnson, K.R.; Kim, G.; Brown, M.A.; Elkahloun, A.G.; Maric, D. Differentiation of human and murine induced pluripotent stem cells to microglia-like cells. Nat. Neurosci. 2017, 20, 753–759. [Google Scholar] [CrossRef] [PubMed]
- Douvaras, P.; Sun, B.; Wang, M.; Kruglikov, I.; Lallos, G.; Zimmer, M.; Terrenoire, C.; Zhang, B.; Gandy, S.; Schadt, E. Directed differentiation of human pluripotent stem cells to microglia. Stem Cell Rep. 2017, 8, 1516–1524. [Google Scholar] [CrossRef] [Green Version]
- Penney, J.; Ralvenius, W.T.; Tsai, L.-H. Modeling Alzheimer’s disease with iPSC-derived brain cells. Mol. Psychiatry 2020, 25, 148–167. [Google Scholar] [CrossRef] [Green Version]
- Speicher, A.M.; Wiendl, H.; Meuth, S.G.; Pawlowski, M. Generating microglia from human pluripotent stem cells: Novel in vitro models for the study of neurodegeneration. Mol. Neurodegener. 2019, 14, 46. [Google Scholar] [CrossRef] [PubMed]
- Konttinen, H.; Ohtonen, S.; Wojciechowski, S.; Shakirzyanova, A.; Caligola, S.; Giugno, R.; Ishchenko, Y.; Hernández, D.; Fazaludeen, M.F.; Eamen, S. PSEN1ΔE9, APPswe, and APOE4 confer disparate phenotypes in human iPSC-derived microglia. Stem Cell Rep. 2019, 13, 669–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doss, M.X.; Sachinidis, A. Current challenges of iPSC-based disease modeling and therapeutic implications. Cells 2019, 8, 403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravaioli, F.; Bacalini, M.G.; Franceschi, C.; Garagnani, P. Age-related epigenetic derangement upon reprogramming and differentiation of cells from the elderly. Genes 2018, 9, 39. [Google Scholar] [CrossRef]
- Ormel, P.R.; de Sá, R.V.; van Bodegraven, E.J.; Karst, H.; Harschnitz, O.; Sneeboer, M.A.M.; Johansen, L.E.; van Dijk, R.E.; Scheefhals, N.; van Berlekom, A.B.; et al. Microglia innately develop within cerebral organoids. Nat. Commun. 2018, 9, 4167. [Google Scholar] [CrossRef] [PubMed]
- Ginhoux, F.; Lim, S.; Hoeffel, G.; Low, D.; Huber, T. Origin and differentiation of microglia. Front. Cell. Neurosci. 2013, 7, 45. [Google Scholar] [CrossRef] [Green Version]
- Simard, A.R.; Soulet, D.; Gowing, G.; Julien, J.-P.; Rivest, S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer’s disease. Neuron 2006, 49, 489–502. [Google Scholar] [CrossRef] [Green Version]
- Mildner, A.; Schlevogt, B.; Kierdorf, K.; Böttcher, C.; Erny, D.; Kummer, M.P.; Quinn, M.; Brück, W.; Bechmann, I.; Heneka, M.T. Distinct and non-redundant roles of microglia and myeloid subsets in mouse models of Alzheimer’s disease. J. Neurosci. 2011, 31, 11159–11171. [Google Scholar] [CrossRef] [Green Version]
- Cronk, J.C.; Filiano, A.J.; Louveau, A.; Marin, I.; Marsh, R.; Ji, E.; Goldman, D.H.; Smirnov, I.; Geraci, N.; Acton, S. Peripherally derived macrophages can engraft the brain independent of irradiation and maintain an identity distinct from microglia. J. Exp. Med. 2018, 215, 1627–1647. [Google Scholar] [CrossRef] [Green Version]
- Grubman, A.; Vandekolk, T.H.; Schröder, J.; Sun, G.; Hatwell-Humble, J.; Chan, J.; Oksanen, M.; Lehtonen, S.; Hunt, C.; Koistinaho, J.E. A CX3CR1 reporter hESC line facilitates integrative analysis of in-vitro-derived microglia and improved microglia identity upon neuron-glia co-culture. Stem Cell Rep. 2020, 14, 1018–1032. [Google Scholar] [CrossRef]
- Rai, M.A.; Hammonds, J.; Pujato, M.; Mayhew, C.; Roskin, K.; Spearman, P. Comparative analysis of human microglial models for studies of HIV replication and pathogenesis. Retrovirology 2020, 17, 35. [Google Scholar] [CrossRef] [PubMed]
- Pluvinage, J.V.; Sun, J.; Claes, C.; Flynn, R.A.; Haney, M.S.; Iram, T.; Meng, X.; Lindemann, R.; Riley, N.M.; Danhash, E. The CD22-IGF2R interaction is a therapeutic target for microglial lysosome dysfunction in Niemann-Pick type C. Sci. Transl. Med. 2021, 13, eabg2919. [Google Scholar] [CrossRef]
- Piers, T.M.; Cosker, K.; Mallach, A.; Johnson, G.T.; Guerreiro, R.; Hardy, J.; Pocock, J.M. A locked immunometabolic switch underlies TREM2 R47H loss of function in human iPSC-derived microglia. FASEB J. 2020, 34, 2436–2450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rocha, N.P.; Charron, O.; Latham, L.B.; Colpo, G.D.; Zanotti-Fregonara, P.; Yu, M.; Freeman, L.; Stimming, E.F.; Teixeira, A.L. Microglia Activation in Basal Ganglia Is a Late Event in Huntington Disease Pathophysiology. Neurol.-Neuroimmunol. Neuroinflammation 2021, 8, e984. [Google Scholar] [CrossRef]
- Almeida, S.; Zhang, Z.; Coppola, G.; Mao, W.; Futai, K.; Karydas, A.; Geschwind, M.D.; Tartaglia, M.C.; Gao, F.; Gianni, D. Induced pluripotent stem cell models of progranulin-deficient frontotemporal dementia uncover specific reversible neuronal defects. Cell Rep. 2012, 2, 789–798. [Google Scholar] [CrossRef] [Green Version]
- Brownjohn, P.W.; Smith, J.; Solanki, R.; Lohmann, E.; Houlden, H.; Hardy, J.; Dietmann, S.; Livesey, F.J. Functional studies of missense TREM2 mutations in human stem cell-derived microglia. Stem Cell Rep. 2018, 10, 1294–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Reitboeck, P.; Phillips, A.; Piers, T.M.; Villegas-Llerena, C.; Butler, M.; Mallach, A.; Rodrigues, C.; Arber, C.E.; Heslegrave, A.; Zetterberg, H. Human induced pluripotent stem cell-derived microglia-like cells harboring TREM2 missense mutations show specific deficits in phagocytosis. Cell Rep. 2018, 24, 2300–2311. [Google Scholar] [CrossRef] [Green Version]
- Cosker, K.; Mallach, A.; Limaye, J.; Piers, T.M.; Staddon, J.; Neame, S.J.; Hardy, J.; Pocock, J.M. Microglial signalling pathway deficits associated with the patient derived R47H TREM2 variants linked to AD indicate inability to activate inflammasome. Sci. Rep. 2021, 11, 13316. [Google Scholar] [CrossRef]
- Sheridan, S.D.; Thanos, J.M.; de Guzman, R.M.; McCrea, L.T.; Horng, J.E.; Fu, T.; Sellgren, C.M.; Perlis, R.H.; Edlow, A.G. Umbilical cord blood-derived microglia-like cells to model COVID-19 exposure. Transl. Psychiatry 2021, 11, 179. [Google Scholar] [CrossRef]
- Ohgidani, M.; Kato, T.A.; Kanba, S. Introducing directly induced microglia-like (iMG) cells from fresh human monocytes: A novel translational research tool for psychiatric disorders. Front. Cell. Neurosci. 2015, 9, 184. [Google Scholar] [CrossRef] [Green Version]
- Centeno, E.G.Z.; Cimarosti, H.; Bithell, A. 2D versus 3D human induced pluripotent stem cell-derived cultures for neurodegenerative disease modelling. Mol. Neurodegener. 2018, 13, 27. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Wetzel, I.; Marriott, I.; Dréau, D.; D’Avanzo, C.; Kim, D.Y.; Tanzi, R.E.; Cho, H. A 3D human triculture system modeling neurodegeneration and neuroinflammation in Alzheimer’s disease. Nat. Neurosci. 2018, 21, 941–951. [Google Scholar] [CrossRef] [PubMed]
| Leone 2006 [116] | Etemad 2012 [118] | Ohgidani 2014, 2017 [114,119] | Melief 2016 [117] | Ryan 2017 [115] | Sellgren 2017 [103] | Rawat 2017 [120] | Sellgren 2019 [121] | Ormel 2020, [122] | Banerjee 2021 [123] | Smit 2022 [124] | Quek 2022 [125] | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Supplements |
|
|
|
|
|
|
|
|
|
|
|
|
| Days | 12 | 14 | 14 | 14 | 15 | 11 | 10 | 11 | 10 | 10–14 | 10 | 14 |
| Seeding density | T75 flask | 1 × 105 cells/mL | 4 × 105 cells/mL | 4 × 105 cells/mL | 3 × 105 cells/well (24-well plate) | 500,000 cells/well (24-well plate) | 50,000 cells/well (96-well plate) | 1 × 106 cells/well (24-well plate) | 1 × 106 cells/well (48-well plate) | 1 × 106/mL | 600,000 cells/well (48-well plate) | 500,000 cells/ well (48-well plate) |
| Coating | N/A | N/A | N/A | N/A | N/A | Geltrex | N/A | Geltrex | Poly-L-lysine | Geltrex | Poly-L-lysine | Matrigel |
| Monocyteisolation | Counterflow centrifugal elutriation | Adherence to plastic | Adherence to plastic | Anti-CD14+ microbeads | Anti-CD14+ microbeads | Adherence to plastic | Anti-CD14+ microbeads | Adherence to plastic | Anti-CD14+ microbeads | Adherence to plastic | Anti-CD14+ microbeads | Adherence to plastic |
| Transcriptomicprofiling | No | No | No | No | RNAseq | Nanostring | No | Global gene expression by microarray | RNAseq | RNAseq | RNAseq | No |
| Diseasemodelled | N/A | N/A | Nasu–Hakola disease (2014) Fibromyalgia (2017) | N/A | N/A | Schizophrenia | HIV infection | Schizophrenia | Schizophrenia | N/A | N/A | ALS |
| Disease | Microglia Model System | Number of Patients | Disease-Specific Characteristics Compared to Controls | Reference |
|---|---|---|---|---|
| FTD | hiPSC-derivedmicroglia |
|
| [148] |
| FTD-like syndrome Nasu–Hakola disease |
|
| [149] | |
| Nasu–Hakola disease |
|
| [150] | |
| AD (sporadic) |
|
| [146] | |
| [151] | |||
| Familial Mediterranean fever |
|
| [112] | |
| AD (familial and sporadic) |
|
| [135] | |
| Nasu–Hakola disease | Monocyte-derivedmicroglia |
|
| [114] |
| Schizophrenia |
|
| [121] | |
| Fibromyalgia |
|
| [119] | |
| Schizophrenia |
|
| [122] | |
| Huntington’s disease |
|
| [147] | |
| SARS-CoV-2 infection |
|
| [152] | |
| ALS (sporadic) |
|
| [125] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cuní-López, C.; Stewart, R.; Quek, H.; White, A.R. Recent Advances in Microglia Modelling to Address Translational Outcomes in Neurodegenerative Diseases. Cells 2022, 11, 1662. https://doi.org/10.3390/cells11101662
Cuní-López C, Stewart R, Quek H, White AR. Recent Advances in Microglia Modelling to Address Translational Outcomes in Neurodegenerative Diseases. Cells. 2022; 11(10):1662. https://doi.org/10.3390/cells11101662
Chicago/Turabian StyleCuní-López, Carla, Romal Stewart, Hazel Quek, and Anthony R. White. 2022. "Recent Advances in Microglia Modelling to Address Translational Outcomes in Neurodegenerative Diseases" Cells 11, no. 10: 1662. https://doi.org/10.3390/cells11101662
APA StyleCuní-López, C., Stewart, R., Quek, H., & White, A. R. (2022). Recent Advances in Microglia Modelling to Address Translational Outcomes in Neurodegenerative Diseases. Cells, 11(10), 1662. https://doi.org/10.3390/cells11101662