
Induced CAR-Macrophages as a Novel Therapeutic Cell Type for Cancer Immune Cell Therapies


 ,
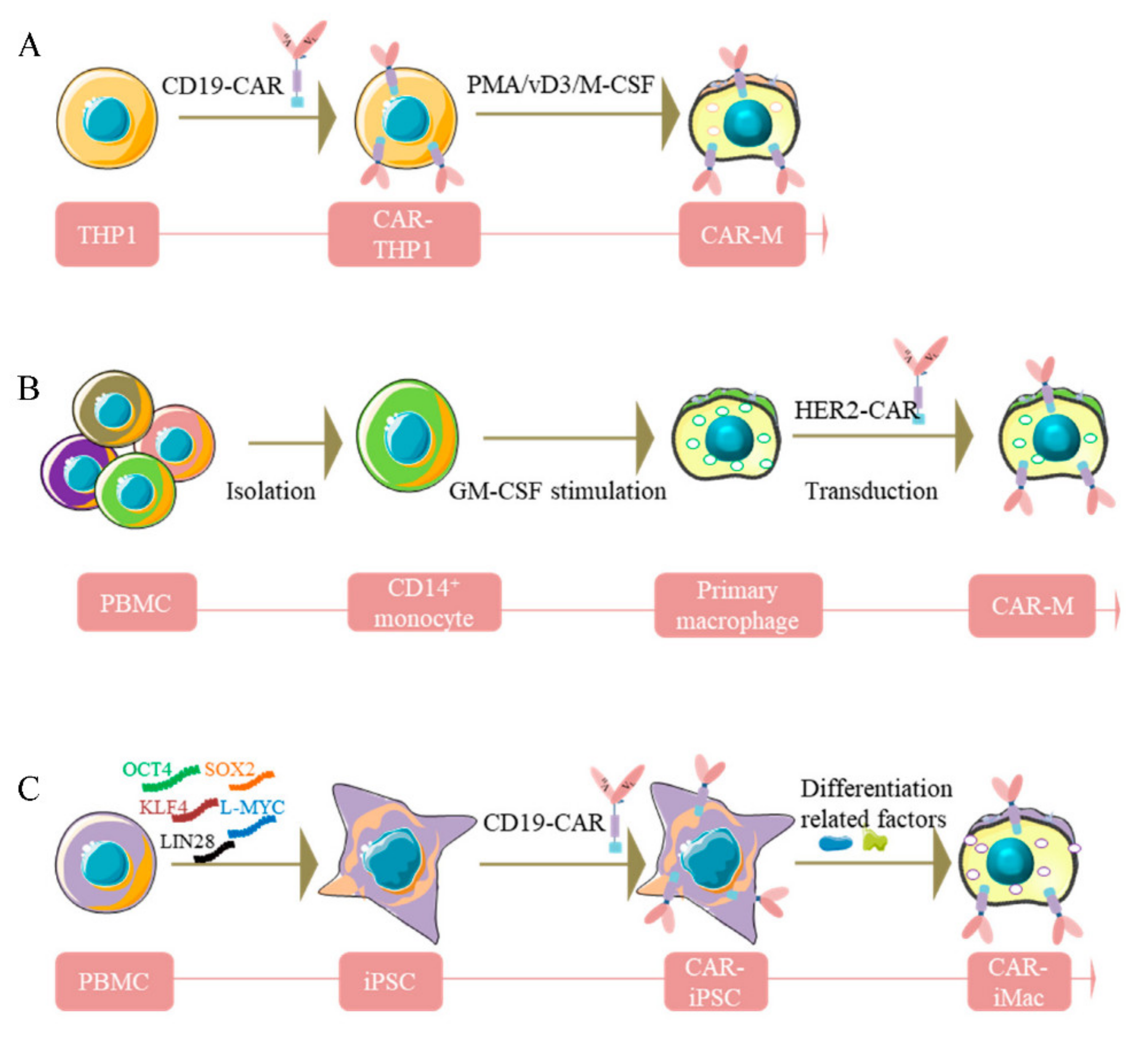
, 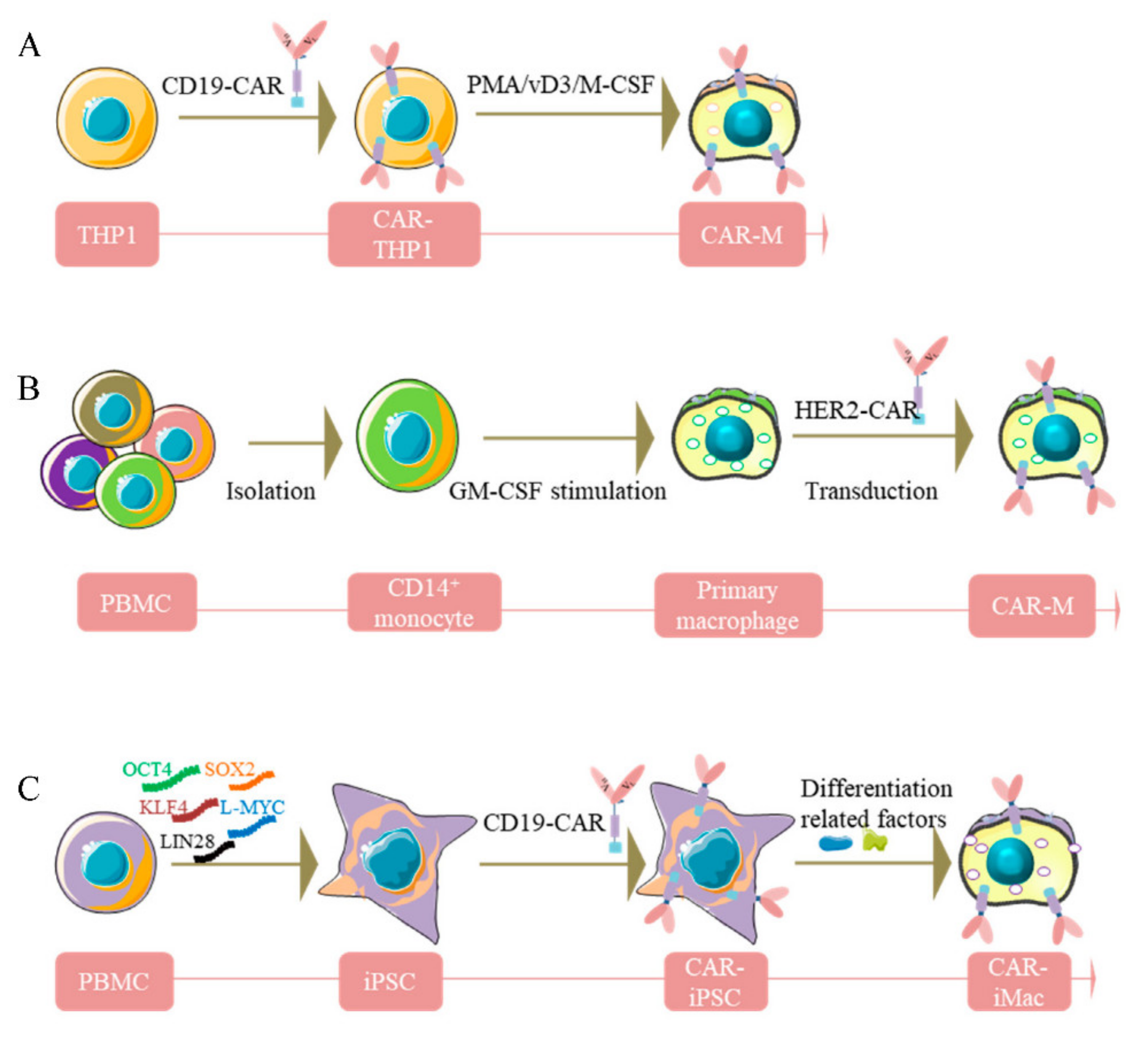{kind=link}
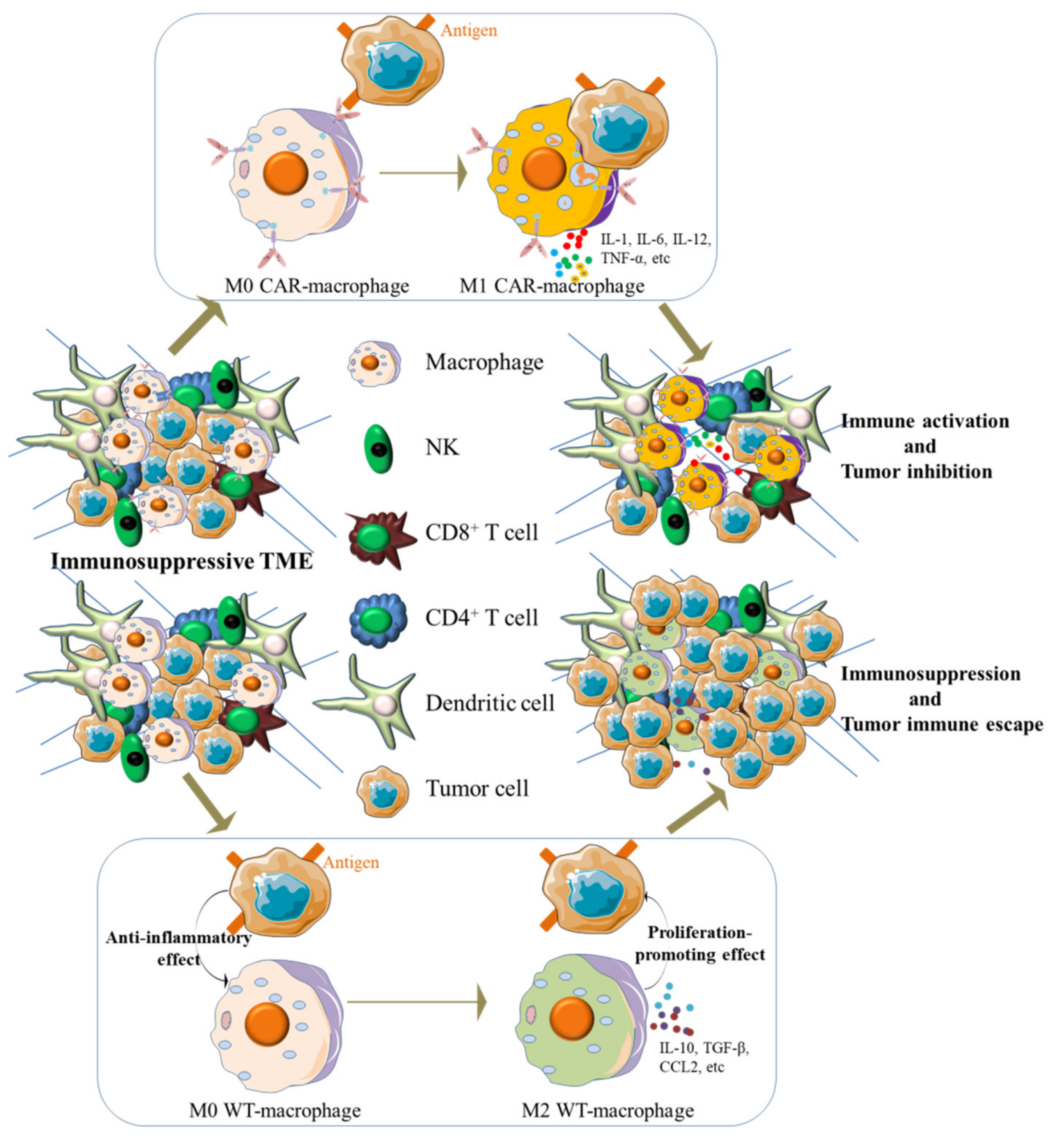
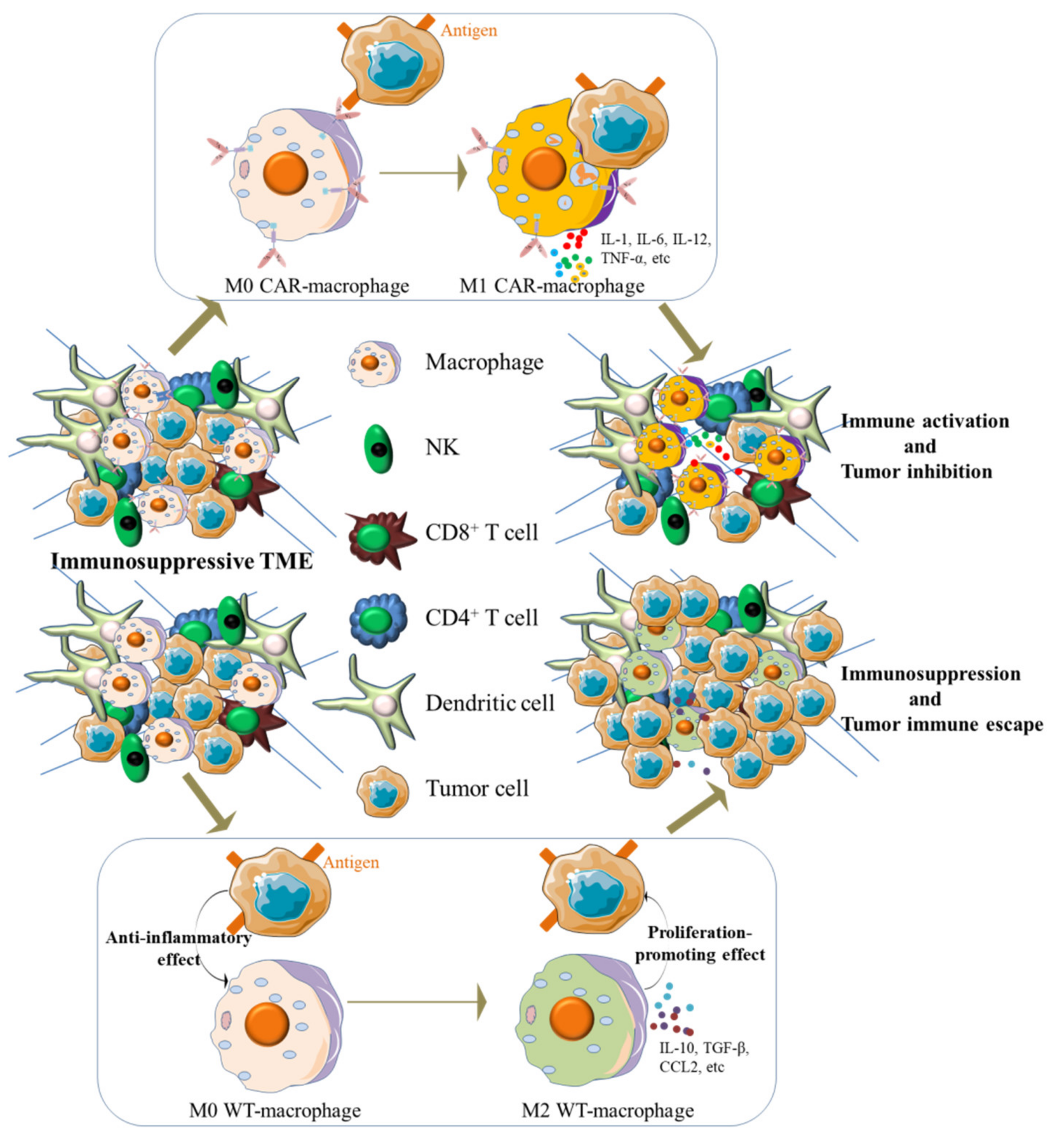{kind=link}
Abstract
:1. Introduction
2. Crosstalk between Macrophages and Tumor Microenvironment
3. Sources of Human CAR-Macrophages Production
3.1. Human THP-1 Macrophage Cell Line
3.2. Human Macrophages from PBMC
3.3. Pluripotent Stem Cell-Derived Car-Macrophage Cells
4. Applications Assessment
4.1. Human Macrophage THP-1 Cells and Human Macrophages from PBMC-Derived CAR-Ms
4.1.1. In Vitro Experiments
4.1.2. In Vivo Experiments
4.2. Pluripotent Stem Cell-Derived CAR-Macrophage Cell: CAR-IMac
4.2.1. In Vitro Experiment
4.2.2. In Vivo Experiment
5. Advantages and Challenges of CAR-Macrophages Immunotherapy
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sadelain, M.; Brentjens, R.; Rivière, I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013, 3, 388–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F.; et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 2013, 368, 1509–1518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daniyan, A.F.; Brentjens, R.J. At the Bench: Chimeric antigen receptor (CAR) T cell therapy for the treatment of B cell malignancies. J. Leukoc. Biol. 2016, 100, 1255–1264. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Han, X.; Bo, J.; Han, W. Target selection for CAR-T therapy. J. Hematol. Oncol. 2019, 12, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Yan, L.; Zhou, M. Target selection of CAR T cell therapy in accordance with the TME for solid tumors. Am. J. Cancer Res. 2019, 9, 228–241. [Google Scholar]
- Majzner, R.G.; Mackall, C.L. Tumor Antigen Escape from CAR T-cell Therapy. Cancer Discov. 2018, 8, 1219–1226. [Google Scholar] [CrossRef] [Green Version]
- Jin, R.; Hao, J.; Yi, Y.; Sauter, E.; Li, B. Regulation of macrophage functions by FABP-mediated inflammatory and metabolic pathways. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2021, 1866, 158964. [Google Scholar] [CrossRef]
- Chen, Y.; Yu, Z.; Tan, X.; Jiang, H.; Xu, Z.; Fang, Y.; Han, D.; Hong, W.; Wei, W.; Tu, J. CAR-macrophage: A new immunotherapy candidate against solid tumors. Biomed. Pharmacother. 2021, 139, 111605. [Google Scholar] [CrossRef]
- Varol, C.; Mildner, A.; Jung, S. Macrophages: Development and tissue specialization. Annu. Rev. Immunol. 2015, 33, 643–675. [Google Scholar] [CrossRef]
- Orecchioni, M.; Ghosheh, Y.; Pramod, A.B.; Ley, K. Macrophage Polarization: Different Gene Signatures in M1(LPS+) vs. Classically and M2(LPS−) vs. alternatively activated macrophages. Front. Immunol. 2019, 10, 1084. [Google Scholar] [CrossRef]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell. Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef] [PubMed]
- Silveira, L.S.; Antunes Bde, M.; Minari, A.L.; Dos Santos, R.V.; Neto, J.C.; Lira, F.S. Macrophage Polarization: Implications on Metabolic Diseases and the Role of Exercise. Crit. Rev. Eukaryot. Gene Expr. 2016, 26, 115–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, N.; Bai, X.; Shu, Y.; Ahmad, O.; Shen, P. Targeting tumor-associated macrophages as an antitumor strategy. Biochem. Pharmacol. 2021, 183, 114354. [Google Scholar] [CrossRef]
- Cassetta, L.; Pollard, J.W. Targeting macrophages: Therapeutic approaches in cancer. Nat. Rev. Drug Discov. 2018, 17, 887–904. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Qi, Q.; Qian, X.; Han, J.; Zhu, X.; Zhang, Q.; Xia, R. The role of PD-1/PD-L1 axis and macrophage in the progression and treatment of cancer. J. Cancer Res. Clin. Oncol. 2019, 145, 1377–1385. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, N.; Li, Q.; Zhang, W.; Ke, F.; Leng, Q.; Wang, H.; Chen, J.; Wang, H. Tumor-associated macrophages recruit CCR6+ regulatory T cells and promote the development of colorectal cancer via enhancing CCL20 production in mice. PLoS ONE 2011, 6, e19495. [Google Scholar] [CrossRef]
- Curiel, T.J.; Coukos, G.; Zou, L.; Alvarez, X.; Cheng, P.; Mottram, P.; Evdemon-Hogan, M.; Conejo-Garcia, J.R.; Zhang, L.; Burow, M.; et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 2004, 10, 942–949. [Google Scholar] [CrossRef]
- De Nardo, D.G.; Ruffell, B. Macrophages as regulators of tumour immunity and immunotherapy. Nat. Rev. Immunol. 2019, 19, 369–382. [Google Scholar] [CrossRef]
- Cao, Q.; Wang, Y.; Zheng, D.; Sun, Y.; Wang, Y.; Lee, V.W.; Zheng, G.; Tan, T.K.; Ince, J.; Alexander, S.I.; et al. IL-10/TGF-beta-modified macrophages induce regulatory T cells and protect against adriamycin nephrosis. J. Am. Soc. Nephrol. 2010, 21, 933–942. [Google Scholar] [CrossRef] [Green Version]
- Tomita, K.; Freeman, B.L.; Bronk, S.F.; LeBrasseur, N.K.; White, T.A.; Hirsova, P.; Ibrahim, S.H. CXCL10-Mediates Macrophage, but not Other Innate Immune Cells-Associated Inflammation in Murine Nonalcoholic Steatohepatitis. Sci. Rep. 2016, 6, 28786. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Li, X.; Zhu, L.; Ming, S.; Wang, H.; Xie, J.; Ren, L.; Huang, J.; Liang, D.; Xiong, L.; et al. CD163+ macrophages suppress T cell response by producing TGF-β in pediatric colorectal polyps. Int. Immunopharmacol. 2021, 96, 107644. [Google Scholar] [CrossRef] [PubMed]
- Kashfi, K.; Kannikal, J.; Nath, N. Macrophage Reprogramming and Cancer Therapeutics: Role of iNOS-Derived NO. Cells 2021, 10, 3194. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Ju, X.; Zhang, H.; Zhou, Z.; Chen, M.; Wang, Q. Tumor-associated macrophages induce PD-L1 expression in gastric cancer cells through IL-6 and TNF-ɑ signaling. Exp. Cell Res. 2020, 396, 112315. [Google Scholar] [CrossRef] [PubMed]
- Xia, Q.; Jia, J.; Hu, C.; Lu, J.; Li, J.; Xu, H.; Fang, J.; Feng, D.; Wang, L.; Chen, Y. Tumor-associated macrophages promote PD-L1 expression in tumor cells by regulating PKM2 nuclear translocation in pancreatic ductal adenocarcinoma. Oncogene 2022, 41, 865–877. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Liang, M.; Xiong, L.; Su, N.; Gao, X.; Jiang, Z. PD-L1 induces macrophage polarization toward the M2 phenotype via Erk/Akt/mTOR. Exp. Cell Res. 2021, 402, 112575. [Google Scholar] [CrossRef]
- Gordon, S.R.; Maute, R.L.; Dulken, B.W.; Hutter, G.; George, B.M.; McCracken, M.N.; Gupta, R.; Tsai, J.M.; Sinha, R.; Corey, D.; et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 2017, 545, 495–499. [Google Scholar] [CrossRef]
- Hofer, F.; Di Sario, G.; Musiu, C.; Sartoris, S.; De Sanctis, F.; Ugel, S. A Complex Metabolic Network Confers Immunosuppressive Functions to Myeloid-Derived Suppressor Cells (MDSCs) within the Tumour Microenvironment. Cells 2021, 10, 2700. [Google Scholar] [CrossRef]
- Baumann, T.; Dunkel, A.; Schmid, C.; Schmitt, S.; Hiltensperger, M.; Lohr, K.; Laketa, V.; Donakonda, S.; Ahting, U.; Lorenz-Depiereux, B.; et al. Regulatory myeloid cells paralyze T cells through cell-cell transfer of the metabolite methylglyoxal. Nat. Immunol. 2020, 21, 555–566. [Google Scholar] [CrossRef]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef]
- Raghavan, S.; Mehta, P.; Xie, Y.; Lei, Y.L.; Mehta, G. Ovarian cancer stem cells and macrophages reciprocally interact through the WNT pathway to promote pro-tumoral and malignant phenotypes in 3D engineered microenvironments. J. Immunother. Cancer 2019, 7, 190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Hodge, J.; Wang, J.; Wang, Y.; Wang, L.; Singh, U.; Li, Y.; Yao, Y.; Wang, D.; Ai, W.; et al. Emodin reduces Breast Cancer Lung Metastasis by suppressing Macrophage-induced Breast Cancer Cell Epithelial-mesenchymal transition and Cancer Stem Cell formation. Theranostics 2020, 10, 8365–8381. [Google Scholar] [CrossRef] [PubMed]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teresa Pinto, A.; Laranjeiro Pinto, M.; Patrícia Cardoso, A.; Monteiro, C.; Teixeira Pinto, M.; Filipe Maia, A.; Castro, P.; Figueira, R.; Monteiro, A.; Marques, M.; et al. Ionizing radiation modulates human macrophages towards a pro-inflammatory phenotype preserving their pro-invasive and pro-angiogenic capacities. Sci. Rep. 2016, 6, 18765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lobera, M.; Madauss, K.P.; Pohlhaus, D.T.; Wright, Q.G.; Trocha, M.; Schmidt, D.R.; Baloglu, E.; Trump, R.P.; Head, M.S.; Hofmann, G.A.; et al. Selective class IIa histone deacetylase inhibition via a nonchelating zinc-binding group. Nat. Chem. Biol. 2013, 9, 319–325. [Google Scholar] [CrossRef]
- Paulus, P.; Stanley, E.R.; Schäfer, R.; Abraham, D.; Aharinejad, S. Colony-stimulating factor-1 antibody reverses chemoresistance in human MCF-7 breast cancer xenografts. Cancer Res. 2006, 66, 4349–4356. [Google Scholar] [CrossRef] [Green Version]
- Hughes, R.; Qian, B.Z.; Rowan, C.; Muthana, M.; Keklikoglou, I.; Olson, O.C.; Tazzyman, S.; Danson, S.; Addison, C.; Clemons, M.; et al. Perivascular M2 Macrophages Stimulate Tumor Relapse after Chemotherapy. Cancer Res. 2015, 75, 3479–3491. [Google Scholar] [CrossRef] [Green Version]
- Vonderheide, R.H.; Flaherty, K.T.; Khalil, M.; Stumacher, M.S.; Bajor, D.L.; Hutnick, N.A.; Sullivan, P.; Mahany, J.J.; Gallagher, M.; Kramer, A.; et al. Clinical activity and immune modulation in cancer patients treated with CP-870,893, a novel CD40 agonist monoclonal antibody. J. Clin. Oncol. 2007, 25, 876–883. [Google Scholar] [CrossRef]
- Willingham, S.B.; Volkmer, J.P.; Gentles, A.J.; Sahoo, D.; Dalerba, P.; Mitra, S.S.; Wang, J.; Contreras-Trujillo, H.; Martin, R.; Cohen, J.D.; et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 6662–6667. [Google Scholar] [CrossRef] [Green Version]
- Edris, B.; Weiskopf, K.; Volkmer, A.K.; Volkmer, J.P.; Willingham, S.B.; Contreras-Trujillo, H.; Liu, J.; Majeti, R.; West, R.B.; Fletcher, J.A.; et al. Antibody therapy targeting the CD47 protein is effective in a model of aggressive metastatic leiomyosarcoma. Proc. Natl. Acad. Sci. USA 2012, 109, 6656–6661. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Z.; Chung, H.; Banan, B.; Manning, P.T.; Ott, K.C.; Lin, S.; Capoccia, B.J.; Subramanian, V.; Hiebsch, R.R.; Upadhya, G.A.; et al. Antibody mediated therapy targeting CD47 inhibits tumor progression of hepatocellular carcinoma. Cancer Lett. 2015, 360, 302–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macciò, A.; Gramignano, G.; Cherchi, M.C.; Tanca, L.; Melis, L.; Madeddu, C. Role of M1-polarized tumor-associated macrophages in the prognosis of advanced ovarian cancer patients. Sci. Rep. 2020, 10, 6096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, X.; Zhang, J.; Li, D.; Mao, Y.; Mo, F.; Du, W.; Ma, X. Prognostic significance of tumor-associated macrophages in ovarian cancer: A meta-analysis. Gynecol. Oncol. 2017, 147, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Sloas, C.; Gill, S.; Klichinsky, M. Engineered CAR-Macrophages as Adoptive Immunotherapies for Solid Tumors. Front. Immunol. 2021, 12, 783305. [Google Scholar] [CrossRef]
- Klichinsky, M.; Ruella, M.; Shestova, O.; Lu, X.M.; Best, A.; Zeeman, M.; Schmierer, M.; Gabrusiewicz, K.; Anderson, N.R.; Petty, N.E.; et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat. Biotechnol. 2020, 38, 947–953. [Google Scholar] [CrossRef] [PubMed]
- Alvey, C.; Discher, D.E. Engineering macrophages to eat cancer: From “marker of self” CD47 and phagocytosis to differentiation. J. Leukoc. Biol. 2017, 102, 31–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franken, L.; Schiwon, M.; Kurts, C. Macrophages: Sentinels and regulators of the immune system. Cell. Microbiol. 2016, 18, 475–487. [Google Scholar] [CrossRef]
- Tsuchiya, S.; Yamabe, M.; Yamaguchi, Y.; Kobayashi, Y.; Konno, T.; Tada, K. Establishment and characterization of a human acute monocytic leukemia cell line (THP-1). Int. J. Cancer 1980, 26, 171–176. [Google Scholar] [CrossRef]
- Sakamoto, H.; Aikawa, M.; Hill, C.C.; Weiss, D.; Taylor, W.R.; Libby, P.; Lee, R.T. Biomechanical strain induces class a scavenger receptor expression in human monocyte/macrophages and THP-1 cells: A potential mechanism of increased atherosclerosis in hypertension. Circulation 2001, 104, 109–114. [Google Scholar] [CrossRef] [Green Version]
- Daigneault, M.; Preston, J.A.; Marriott, H.M.; Whyte, M.K.; Dockrell, D.H. The identification of markers of macrophage differentiation in PMA-stimulated THP-1 cells and monocyte-derived macrophages. PLoS ONE 2010, 5, e8668. [Google Scholar] [CrossRef]
- Biriken, D.; Yazıhan, N.; Yılmaz, Ş. Değişik konsantrasyonlarda ve sürelerde Phorbol-12-Myristate-13-Acetate ile uyarılan insan THP-1 lösemi hücrelerinin sitokin ve midkin yanıtlarının araştırılması [Investigation of cytokine and midkine responses of human THP-1 leukemia cells induced by phorbol-12-Myristate-13-Acetate (PMA) at different concentrations and times]. Mikrobiyol. Bul. 2018, 52, 147–155. [Google Scholar] [PubMed]
- Maeß, M.B.; Wittig, B.; Cignarella, A.; Lorkowski, S. Reduced PMA enhances the responsiveness of transfected THP-1 macrophages to polarizing stimuli. J. Immunol. Methods 2014, 402, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Schwende, H.; Fitzke, E.; Ambs, P.; Dieter, P. Differences in the state of differentiation of THP-1 cells induced by phorbol ester and 1,25-dihydroxyvitamin D3. J. Leukoc. Biol. 1996, 59, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Phillips, R.J.; Lutz, M.; Premack, B. Differential signaling mechanisms regulate expression of CC chemokine receptor-2 during monocyte maturation. J. Inflamm. 2005, 2, 14. [Google Scholar] [CrossRef] [Green Version]
- Chanput, W.; Mes, J.J.; Savelkoul, H.F.; Wichers, H.J. Characterization of polarized THP-1 macrophages and polarizing ability of LPS and food compounds. Food Funct. 2013, 4, 266–276. [Google Scholar] [CrossRef]
- Caras, I.; Tucureanu, C.; Lerescu, L.; Pitica, R.; Melinceanu, L.; Neagu, S.; Salageanu, A. Influence of tumor cell culture supernatants on macrophage functional polarization: In vitro models of macrophage-tumor environment interaction. Tumori J. 2011, 97, 647–654. [Google Scholar] [CrossRef]
- Mangan, D.F.; Wahl, S.M. Differential regulation of human monocyte programmed cell death (apoptosis) by chemotactic factors and pro-inflammatory cytokines. J. Immunol. 1991, 147, 3408–3412. [Google Scholar]
- Mangan, D.F.; Welch, G.R.; Wahl, S.M. Lipopolysaccharide, tumor necrosis factor-alpha, and IL-1 beta prevent programmed cell death (apoptosis) in human peripheral blood monocytes. J. Immunol. 1991, 146, 1541–1546. [Google Scholar]
- Cousins, R.J.; Blanchard, R.K.; Popp, M.P.; Liu, L.; Cao, J.; Moore, J.B.; Green, C.L. A global view of the selectivity of zinc deprivation and excess on genes expressed in human THP-1 mononuclear cells. Proc. Natl. Acad. Sci. USA 2003, 100, 6952–6957. [Google Scholar] [CrossRef] [Green Version]
- Rogers, P.D.; Thornton, J.; Barker, K.S.; McDaniel, D.O.; Sacks, G.S.; Swiatlo, E.; McDaniel, L.S. Pneumolysin-dependent and -independent gene expression identified by cDNA microarray analysis of THP-1 human mononuclear cells stimulated by Streptococcus pneumoniae. Infect. Immun. 2003, 71, 2087–2094. [Google Scholar] [CrossRef] [Green Version]
- Chanput, W.; Mes, J.J.; Wichers, H.J. THP-1 cell line: An in vitro cell model for immune modulation approach. Int. Immunopharmacol. 2014, 23, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Takashiba, S.; Van Dyke, T.E.; Amar, S.; Murayama, Y.; Soskolne, A.W.; Shapira, L. Differentiation of monocytes to macrophages primes cells for lipopolysaccharide stimulation via accumulation of cytoplasmic nuclear factor kappaB. Infect. Immun. 1999, 67, 5573–5578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zarember, K.A.; Godowski, P.J. Tissue expression of human Toll-like receptors and differential regulation of Toll-like receptor mRNAs in leukocytes in response to microbes, their products, and cytokines. J. Immunol. 2002, 168, 554–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novakovic, B.; Habibi, E.; Wang, S.Y.; Arts, R.J.W.; Davar, R.; Megchelenbrink, W.; Kim, B.; Kuznetsova, T.; Kox, M.; Zwaag, J.; et al. β-Glucan Reverses the Epigenetic State of LPS-Induced Immunological Tolerance. Cell 2016, 167, 1354–1368.e14. [Google Scholar] [CrossRef] [Green Version]
- Seeley, J.J.; Ghosh, S. Molecular mechanisms of innate memory and tolerance to LPS. J. Leukoc. Biol. 2017, 101, 107–119. [Google Scholar] [CrossRef]
- Schildberger, A.; Rossmanith, E.; Eichhorn, T.; Strassl, K.; Weber, V. Monocytes, peripheral blood mononuclear cells, and THP-1 cells exhibit different cytokine expression patterns following stimulation with lipopolysaccharide. Mediat. Inflamm. 2013, 2013, 697972. [Google Scholar] [CrossRef] [Green Version]
- Fuss, I.J.; Kanof, M.E.; Smith, P.D.; Zola, H. Isolation of whole mononuclear cells from peripheral blood and cord blood. Curr. Protoc. Immunol. 2009, 85, 1–7. [Google Scholar] [CrossRef]
- Bruckmeier, M.; Kuehnl, A.; Culmes, M.; Pelisek, J.; Eckstein, H.H. Impact of oxLDL and LPS on C-type natriuretic peptide system is different between THP-1 cells and human peripheral blood monocytic cells. Cell. Physiol. Biochem. 2012, 30, 199–209. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, M.; Ljungberg, J.; Richter, J.; Kiefer, T.; Magnusson, M.; Lieber, A.; Widegren, B.; Karlsson, S.; Fan, X. Development of an adenoviral vector system with adenovirus serotype 35 tropism; efficient transient gene transfer into primary malignant hematopoietic cells. J. Gene Med. 2004, 6, 631–641. [Google Scholar] [CrossRef]
- Zhang, L.; Tian, L.; Dai, X.; Yu, H.; Wang, J.; Lei, A.; Zhu, M.; Xu, J.; Zhao, W.; Zhu, Y.; et al. Pluripotent stem cell-derived CAR-macrophage cells with antigen-dependent anti-cancer cell functions. J. Hematol. Oncol. 2020, 13, 153. [Google Scholar] [CrossRef]
- Lam, E.; Stein, S.; Falck-Pedersen, E. Adenovirus detection by the cGAS/STING/TBK1 DNA sensing cascade. J. Virol. 2014, 88, 974–981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muruve, D.A.; Pétrilli, V.; Zaiss, A.K.; White, L.R.; Clark, S.A.; Ross, P.J.; Parks, R.J.; Tschopp, J. The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature 2008, 452, 103–107. [Google Scholar] [CrossRef]
- Marofi, F.; Motavalli, R.; Safonov, V.A.; Thangavelu, L.; Yumashev, A.V.; Alexander, M.; Shomali, N.; Chartrand, M.S.; Pathak, Y.; Jarahian, M.; et al. CAR T cells in solid tumors: Challenges and opportunities. Stem Cell Res. Ther. 2021, 12, 81. [Google Scholar] [CrossRef] [PubMed]
- Brudno, J.N.; Kochenderfer, J.N. Recent advances in CAR T-cell toxicity: Mechanisms, manifestations and management. Blood Rev. 2019, 34, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.N.; Fry, T.J. Mechanisms of resistance to CAR T cell therapy. Nat. Rev. Clin. Oncol. 2019, 16, 372–385. [Google Scholar] [CrossRef]
- Yoon, D.H.; Osborn, M.J.; Tolar, J.; Kim, C.J. Incorporation of Immune Checkpoint Blockade into Chimeric Antigen Receptor T Cells (CAR-Ts): Combination or Built-In CAR-T. Int. J. Mol. Sci. 2018, 19, 340. [Google Scholar] [CrossRef] [Green Version]
- Wu, K.; Lin, K.; Li, X.; Yuan, X.; Xu, P.; Ni, P.; Xu, D. Redefining Tumor-Associated Macrophage Subpopulations and Functions in the Tumor Microenvironment. Front. Immunol. 2020, 11, 1731. [Google Scholar] [CrossRef]
- Van der Heide, D.; Weiskirchen, R.; Bansal, R. Therapeutic Targeting of Hepatic Macrophages for the Treatment of Liver Diseases. Front. Immunol. 2019, 10, 2852. [Google Scholar] [CrossRef] [Green Version]
- Toor, S.M.; Sasidharan Nair, V.; Decock, J.; Elkord, E. Immune checkpoints in the tumor microenvironment. Semin. Cancer Biol. 2020, 65, 1–12. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, S.; Lei, A.; Wang, X.; Lu, H.; Wang, S.; Yang, Y.; Li, N.; Zhang, Y.; Zhang, J. Induced CAR-Macrophages as a Novel Therapeutic Cell Type for Cancer Immune Cell Therapies. Cells 2022, 11, 1652. https://doi.org/10.3390/cells11101652
Su S, Lei A, Wang X, Lu H, Wang S, Yang Y, Li N, Zhang Y, Zhang J. Induced CAR-Macrophages as a Novel Therapeutic Cell Type for Cancer Immune Cell Therapies. Cells. 2022; 11(10):1652. https://doi.org/10.3390/cells11101652
Chicago/Turabian StyleSu, Siyu, Anhua Lei, Xudong Wang, Hengxing Lu, Shuhang Wang, Yuqi Yang, Ning Li, Yi Zhang, and Jin Zhang. 2022. "Induced CAR-Macrophages as a Novel Therapeutic Cell Type for Cancer Immune Cell Therapies" Cells 11, no. 10: 1652. https://doi.org/10.3390/cells11101652
APA StyleSu, S., Lei, A., Wang, X., Lu, H., Wang, S., Yang, Y., Li, N., Zhang, Y., & Zhang, J. (2022). Induced CAR-Macrophages as a Novel Therapeutic Cell Type for Cancer Immune Cell Therapies. Cells, 11(10), 1652. https://doi.org/10.3390/cells11101652