
Neuroprotective Effects and Therapeutic Potential of Transcorneal Electrical Stimulation for Depression

 , ,
, ,  and
and
Abstract
:1. Introduction
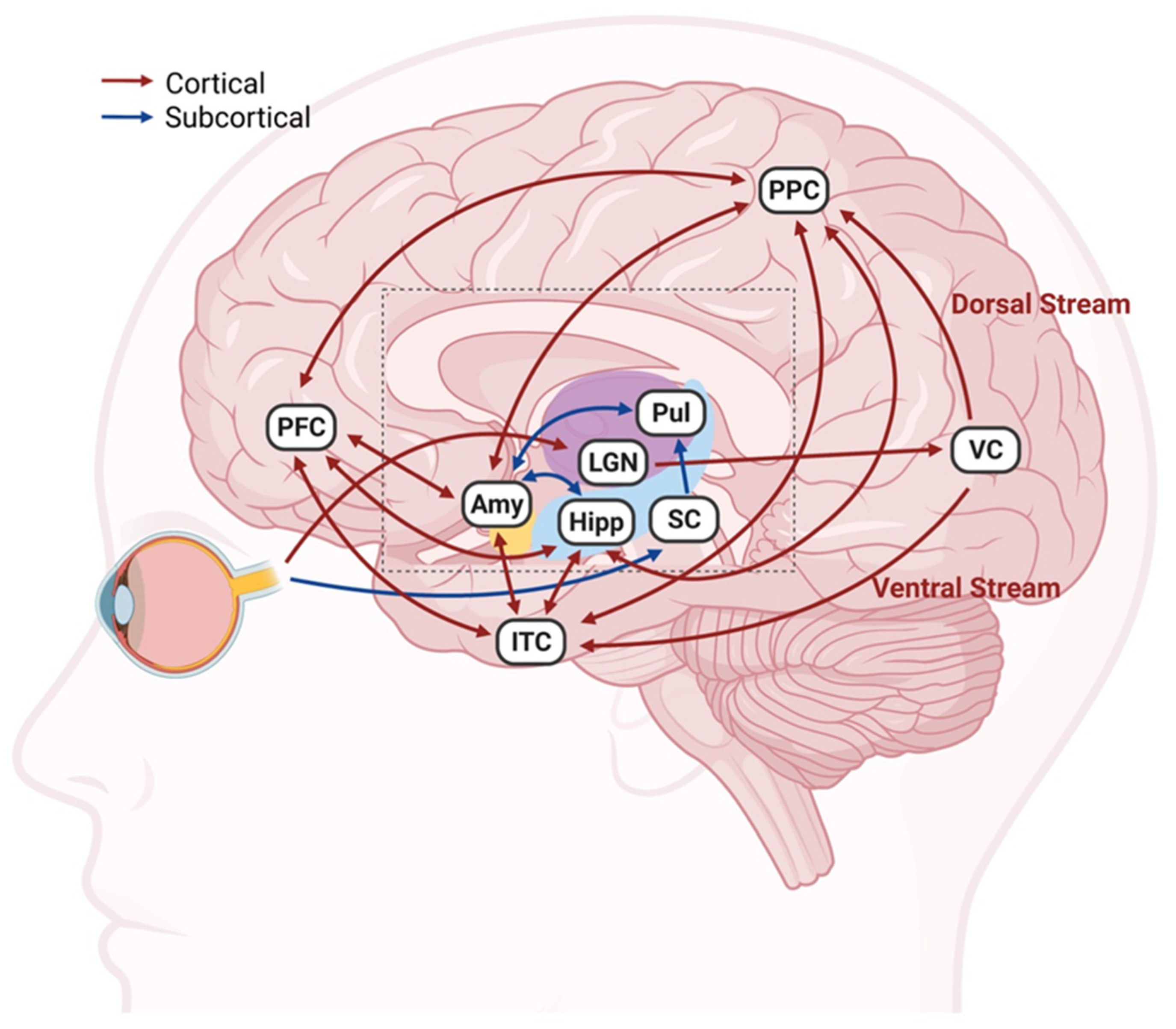
2. Connections between Visual and Emotional Systems
3. Application of TES in Ophthalmology
3.1. Preclinical Studies
3.2. Clinical Studies
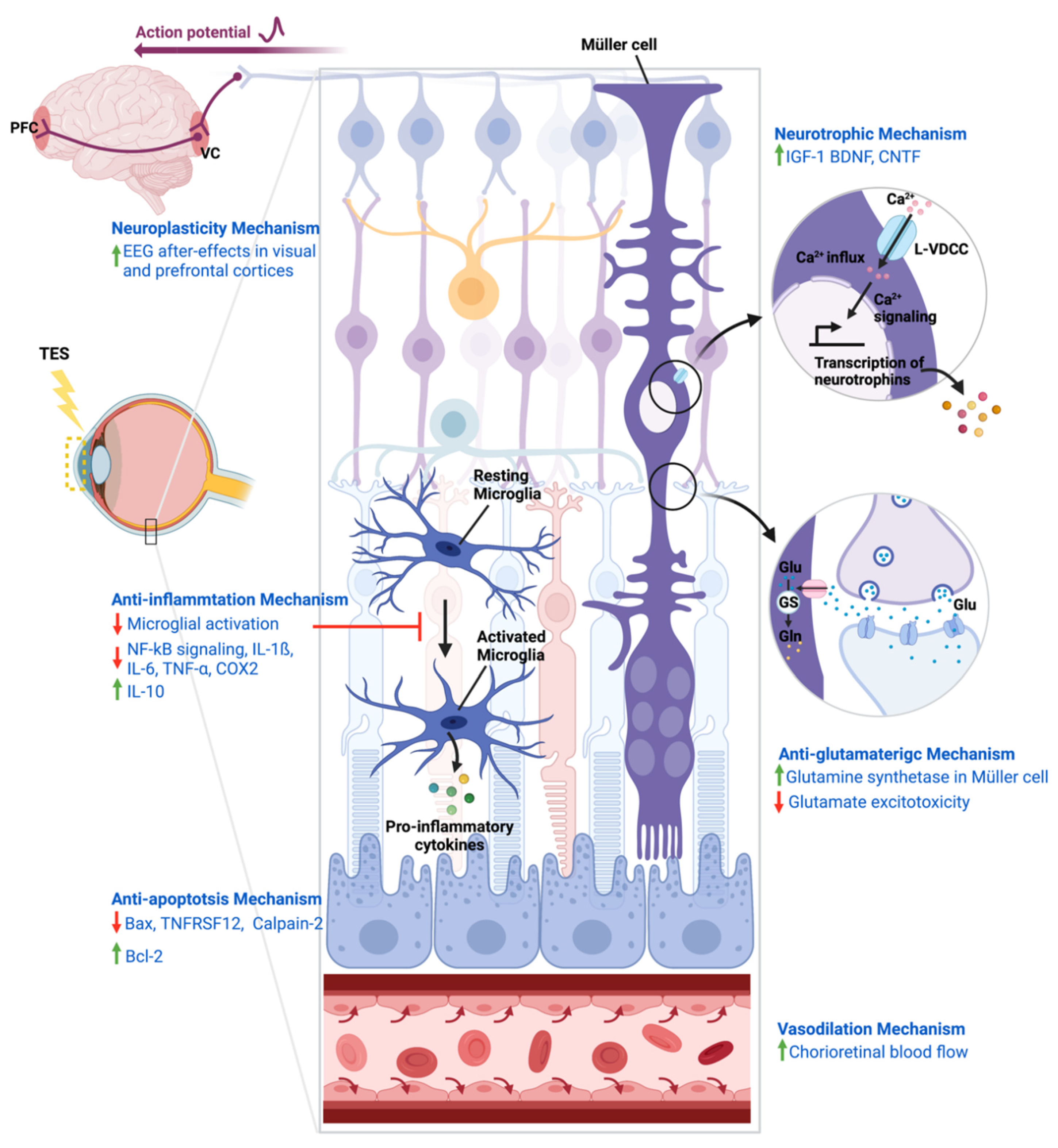
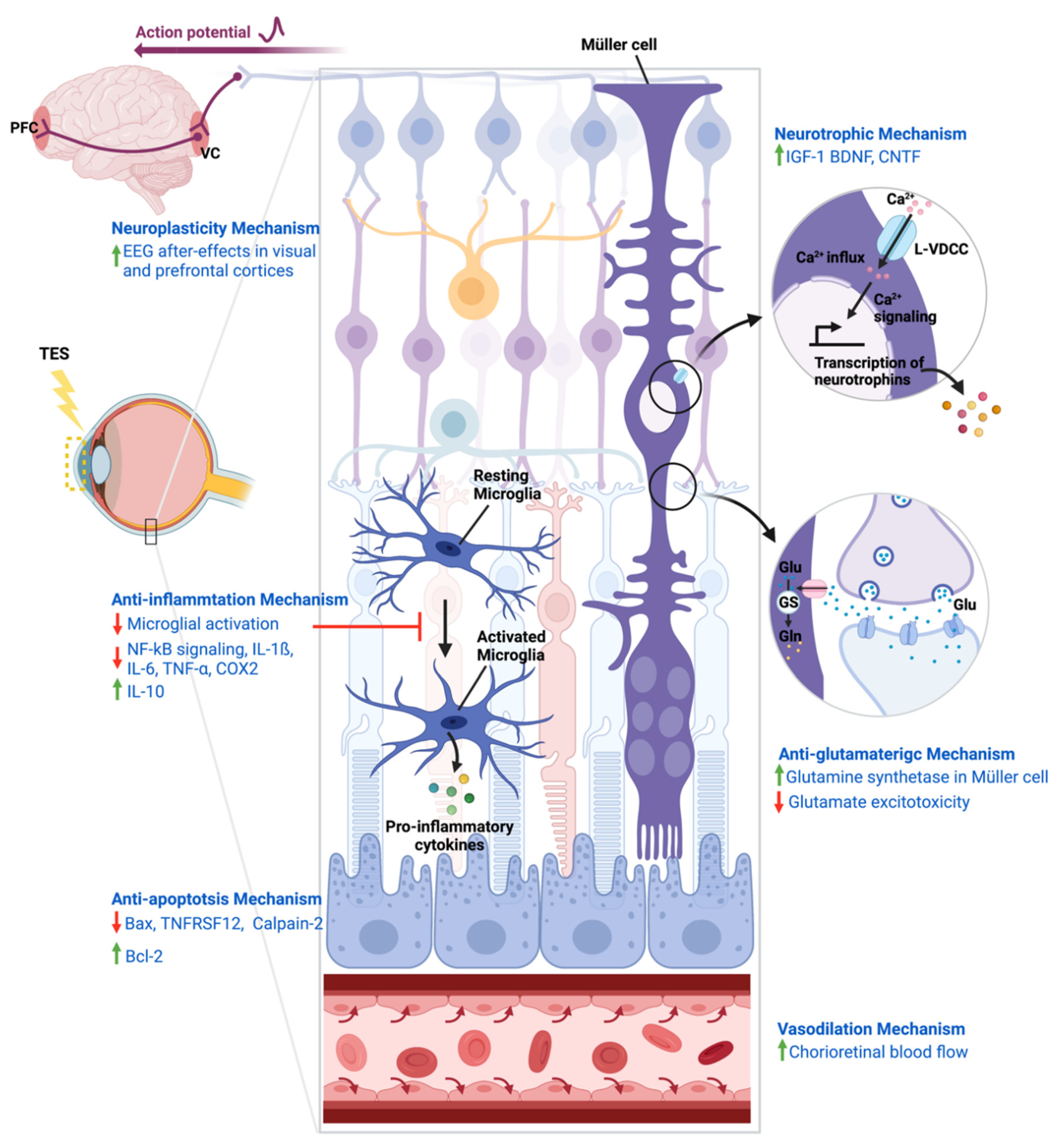
4. Mechanisms of Action of TES
4.1. Neurotrophic Mechanism
4.2. Neuroplasticity Mechanism
4.3. Anti-Inflammatory Mechanism
4.4. Anti-Apoptosis Mechanism
4.5. Anti-Glutamatergic Mechanism
4.6. Vasodilation Mechanism
5. Potential Antidepressant-like Activities of TES
5.1. Brain Regions Stimulated by TES
5.2. TES-Induced Behavioural Changes in Corneally Kindled Models
6. Comparison of FDA-Approved Treatments for Major Depression
7. Benefits and Risks of TES As a Depression Treatment
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- WHO. WHO Depression Fact Sheet; World Health Organization (WHO): Geneva, Switzerland, 2020. [Google Scholar]
- WHO. Depression and Other Common Mental Disorders: Global Health Estimates; World Health Organization (WHO): Geneva, Switzerland, 2017. [Google Scholar]
- Greenberg, P.E.; Fournier, A.A.; Sisitsky, T.; Simes, M.; Berman, R.; Koenigsberg, S.H.; Kessler, R.C. The Economic Burden of Adults with Major Depressive Disorder in the United States (2010 and 2018). Pharmacoeconomics 2021, 39, 653–665. [Google Scholar] [CrossRef] [PubMed]
- Association, A.P. Diagnostic and Statistical Manual of Mental Disorders (DSM-5®); American Psychiatric Pub: Arlington, VA, USA, 2013. [Google Scholar]
- Kang, H.-J.; Kim, S.-Y.; Bae, K.-Y.; Kim, S.-W.; Shin, I.-S.; Yoon, J.-S.; Kim, J.-M. Comorbidity of depression with physical disorders: Research and clinical implications. Chonnam. Med. J. 2015, 51, 8–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroenke, K.; Wu, J.; Bair, M.J.; Krebs, E.E.; Damush, T.M.; Tu, W. Reciprocal relationship between pain and depression: A 12-month longitudinal analysis in primary care. J. Pain 2011, 12, 964–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stubbs, B.; Vancampfort, D.; Veronese, N.; Thompson, T.; Fornaro, M.; Schofield, P.; Solmi, M.; Mugisha, J.; Carvalho, A.F.; Koyanagi, A. Depression and pain: Primary data and meta-analysis among 237,952 people across 47 low-and middle-income countries. Psychol. Med. 2017, 47, 2906–2917. [Google Scholar] [CrossRef] [PubMed]
- Byers, A.L.; Yaffe, K. Depression and risk of developing dementia. Nat. Rev. Neurol. 2011, 7, 323–331. [Google Scholar] [CrossRef] [Green Version]
- Knol, M.; Twisk, J.W.; Beekman, A.T.; Heine, R.; Snoek, F.J.; Pouwer, F. Depression as a risk factor for the onset of type 2 diabetes mellitus. A meta-analysis. Diabetologia 2006, 49, 837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bădescu, S.V.; Tătaru, C.; Kobylinska, L.; Georgescu, E.L.; Zahiu, D.M.; Zăgrean, A.M.; Zăgrean, L. The association between Diabetes mellitus and Depression. J. Med. Life 2016, 9, 120–125. [Google Scholar]
- Van der Kooy, K.; van Hout, H.; Marwijk, H.; Marten, H.; Stehouwer, C.; Beekman, A. Depression and the risk for cardiovascular diseases: Systematic review and meta analysis. Int J. Geriatr. Psychiatry 2007, 22, 613–626. [Google Scholar] [CrossRef]
- Khawaja, I.S.; Westermeyer, J.J.; Gajwani, P.; Feinstein, R.E. Depression and coronary artery disease: The association, mechanisms, and therapeutic implications. Psychiatry 2009, 6, 38–51. [Google Scholar]
- Gump, B.B.; Matthews, K.A.; Eberly, L.E.; Chang, Y.F.; Group, M.R. Depressive symptoms and mortality in men: Results from the Multiple Risk Factor Intervention Trial. Stroke 2005, 36, 98–102. [Google Scholar] [CrossRef] [Green Version]
- Pan, A.; Sun, Q.; Okereke, O.I.; Rexrode, K.M.; Hu, F.B. Depression and risk of stroke morbidity and mortality: A meta-analysis and systematic review. JAMA 2011, 306, 1241–1249. [Google Scholar] [CrossRef] [Green Version]
- Spiegel, D.; Giese-Davis, J. Depression and cancer: Mechanisms and disease progression. Biol. Psychiatry 2003, 54, 269–282. [Google Scholar] [CrossRef]
- Hillhouse, T.M.; Porter, J.H. A brief history of the development of antidepressant drugs: From monoamines to glutamate. Exp. Clin. Psychopharmacol. 2015, 23, 1–21. [Google Scholar] [CrossRef]
- Al-Harbi, K.S. Treatment-resistant depression: Therapeutic trends, challenges, and future directions. Patient Prefer. Adherence 2012, 6, 369. [Google Scholar] [CrossRef] [Green Version]
- Mrazek, D.A.; Hornberger, J.C.; Altar, C.A.; Degtiar, I. A review of the clinical, economic, and societal burden of treatment-resistant depression: 1996–2013. Psychiatr. Serv. 2014, 65, 977–987. [Google Scholar] [CrossRef] [PubMed]
- Morishita, T.; Fayad, S.M.; Higuchi, M.-A.; Nestor, K.A.; Foote, K.D. Deep brain stimulation for treatment-resistant depression: Systematic review of clinical outcomes. Neurotherapeutics 2014, 11, 475–484. [Google Scholar] [CrossRef] [Green Version]
- Temel, Y.; Hescham, S.A.; Jahanshahi, A.; Janssen, M.L.; Tan, S.K.; van Overbeeke, J.J.; Ackermans, L.; Oosterloo, M.; Duits, A.; Leentjens, A.F.; et al. Neuromodulation in psychiatric disorders. Int. Rev. Neurobiol. 2012, 107, 283–314. [Google Scholar] [CrossRef]
- Kolshus, E.; Jelovac, A.; McLoughlin, D. Bitemporal v. high-dose right unilateral electroconvulsive therapy for depression: A systematic review and meta-analysis of randomized controlled trials. Psychol. Med. 2017, 47, 518–530. [Google Scholar] [CrossRef]
- Brus, O.; Cao, Y.; Gustafsson, E.; Hultén, M.; Landen, M.; Lundberg, J.; Nordanskog, P.; Nordenskjöld, A. Self-assessed remission rates after electroconvulsive therapy of depressive disorders. Eur. Psychiatry 2017, 45, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Bahji, A.; Hawken, E.; Sepehry, A.; Cabrera, C.; Vazquez, G. ECT beyond unipolar major depression: Systematic review and meta—Analysis of electroconvulsive therapy in bipolar depression. Acta Psychiatr. Scand. 2019, 139, 214–226. [Google Scholar] [CrossRef]
- Tokutsu, Y.; Umene-Nakano, W.; Shinkai, T.; Yoshimura, R.; Okamoto, T.; Katsuki, A.; Hori, H.; Ikenouchi-Sugita, A.; Hayashi, K.; Atake, K. Follow-up study on electroconvulsive therapy in treatment-resistant depressed patients after remission: A chart review. Clin. Psychopharmacol. Neurosci. 2013, 11, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jelovac, A.; Kolshus, E.; McLoughlin, D.M. Relapse following successful electroconvulsive therapy for major depression: A meta-analysis. Neuropsychopharmacology 2013, 38, 2467–2474. [Google Scholar] [CrossRef] [Green Version]
- Krames, E.S.; Peckham, P.H.; Rezai, A.; Aboelsaad, F. What is neuromodulation? In Neuromodulation; Elsevier: Amsterdam, The Netherlands, 2009; pp. 3–8. [Google Scholar]
- Tao, Y.; Chen, T.; Liu, B.; Wang, L.-Q.; Peng, G.-H.; Qin, L.-M.; Yan, Z.-J.; Huang, Y.-F. The transcorneal electrical stimulation as a novel therapeutic strategy against retinal and optic neuropathy: A review of experimental and clinical trials. Int. J. Ophthalmol. 2016, 9, 914. [Google Scholar] [PubMed]
- Wlaź, P.; Poleszak, E.; Serefko, A.; Wlaź, A.; Rundfeldt, C. Anxiogenic-and antidepressant-like behavior in corneally kindled rats. Pharmacol. Rep. 2015, 67, 349–352. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Wang, G.-J.; Yow, L.; Cela, C.J.; Humayun, M.S.; Weiland, J.D.; Lazzi, G.; Jadvar, H. Modeling and percept of transcorneal electrical stimulation in humans. IEEE Trans. Biomed. Eng. 2011, 58, 1932–1939. [Google Scholar] [CrossRef] [PubMed]
- Agadagba, S.K.; Li, X.; Chan, L.L. Electroencephalogram power alterations in retinal degeneration mice after prolonged transcorneal electrical stimulation. In Proceedings of the 2019 9th International IEEE/EMBS Conference on Neural Engineering (NER), Manhattan, NY, USA, 2019; pp. 219–222. [Google Scholar]
- Agadagba, S.K.; Li, X.; Chan, L.L.H. Excitation of the Pre-frontal and Primary Visual Cortex in Response to Transcorneal Electrical Stimulation in Retinal Degeneration Mice. Front. Neurosci. 2020, 14, 572299:1–572299:13. [Google Scholar] [CrossRef]
- Habel, U.; Klein, M.; Kellermann, T.; Shah, N.J.; Schneider, F. Same or different? Neural correlates of happy and sad mood in healthy males. Neuroimage 2005, 26, 206–214. [Google Scholar] [CrossRef]
- Sergeeva, E.G.; Fedorov, A.B.; Henrich-Noack, P.; Sabel, B.A. Transcorneal alternating current stimulation induces EEG “aftereffects” only in rats with an intact visual system but not after severe optic nerve damage. J. Neurophysiol. 2012, 108, 2494–2500. [Google Scholar] [CrossRef]
- Morimoto, T.; Miyoshi, T.; Matsuda, S.; Tano, Y.; Fujikado, T.; Fukuda, Y. Transcorneal electrical stimulation rescues axotomized retinal ganglion cells by activating endogenous retinal IGF-1 system. Investig. Ophthalmol. Vis. Sci. 2005, 46, 2147–2155. [Google Scholar] [CrossRef] [Green Version]
- Tagami, Y.; Kurimoto, T.; Miyoshi, T.; Morimoto, T.; Sawai, H.; Mimura, O. Axonal regeneration induced by repetitive electrical stimulation of crushed optic nerve in adult rats. Jpn. J. Ophthalmol. 2009, 53, 257–266. [Google Scholar] [CrossRef]
- Ni, Y.-Q.; Gan, D.-K.; Xu, H.-D.; Xu, G.-Z. Neuroprotective effect of transcorneal electrical stimulation on light-induced photoreceptor degeneration. Exp. Neurol. 2009, 219, 439–452. [Google Scholar] [CrossRef]
- Sato, T.; Fujikado, T.; Morimoto, T.; Matsushita, K.; Harada, T.; Tano, Y. Effect of electrical stimulation on IGF-1 transcription by L-type calcium channels in cultured retinal Müller cells. Jpn. J. Ophthalmol. 2008, 52, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Fujikado, T.; Lee, T.-S.; Tano, Y. Direct effect of electrical stimulation on induction of brain-derived neurotrophic factor from cultured retinal Muller cells. Investig. Ophthalmol. Vis. Sci. 2008, 49, 4641–4646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, W.-T.; Ni, Y.-Q.; Jin, Z.-B.; Zhang, M.; Wu, J.-H.; Zhu, Y.; Xu, G.-Z.; Gan, D.-K. Electrical stimulation ameliorates light-induced photoreceptor degeneration in vitro via suppressing the proinflammatory effect of microglia and enhancing the neurotrophic potential of Müller cells. Exp. Neurol. 2012, 238, 192–208. [Google Scholar] [CrossRef] [PubMed]
- Enayati, S.; Chang, K.; Achour, H.; Cho, K.-S.; Xu, F.; Guo, S.; Enayati, K.Z.; Xie, J.; Zhao, E.; Turunen, T. Electrical stimulation induces retinal müller cell proliferation and their progenitor cell potential. Cells 2020, 9, 781. [Google Scholar] [CrossRef] [Green Version]
- Langmann, T. Microglia activation in retinal degeneration. J. Leukoc. Biol. 2007, 81, 1345–1351. [Google Scholar] [CrossRef]
- Fu, L.; Fung, F.K.; Lo, A.C.-Y.; Chan, Y.-K.; So, K.-F.; Wong, I.Y.-H.; Shih, K.C.; Lai, J.S.-M. Transcorneal electrical stimulation inhibits retinal microglial activation and enhances retinal ganglion cell survival after acute ocular hypertensive injury. Transl. Vis. Sci. Technol. 2018, 7, 7. [Google Scholar] [CrossRef]
- Yin, H.; Yin, H.; Zhang, W.; Miao, Q.; Qin, Z.; Guo, S.; Fu, Q.; Ma, J.; Wu, F.; Yin, J. Transcorneal electrical stimulation promotes survival of retinal ganglion cells after optic nerve transection in rats accompanied by reduced microglial activation and TNF-α expression. Brain Res. 2016, 1650, 10–20. [Google Scholar] [CrossRef]
- Jassim, A.H.; Cavanaugh, M.; Shah, J.S.; Willits, R.; Inman, D.M. Transcorneal Electrical Stimulation Reduces Neurodegenerative Process in a Mouse Model of Glaucoma. Ann. Biomed. Eng. 2021, 49, 858–870. [Google Scholar] [CrossRef]
- Willmann, G.; Schäferhoff, K.; Fischer, M.D.; Arango-Gonzalez, B.; Bolz, S.; Naycheva, L.; Röck, T.; Bonin, M.; Bartz-Schmidt, K.U.; Zrenner, E. Gene expression profiling of the retina after transcorneal electrical stimulation in wild-type Brown Norway rats. Investig. Ophthalmol. Vis. Sci. 2011, 52, 7529–7537. [Google Scholar] [CrossRef]
- Tao, Y.; Chen, T.; Liu, Z.-Y.; Wang, L.-Q.; Xu, W.-W.; Qin, L.-M.; Peng, G.-H.; Yi-Fei, H. Topographic Quantification of the Transcorneal Electrical Stimulation (TES)–Induced Protective Effects on N-Methyl-N-Nitrosourea–Treated Retinas. Investig. Ophthalmol. Vis. Sci. 2016, 57, 4614–4624. [Google Scholar] [CrossRef] [Green Version]
- Momeni, H.R. Role of calpain in apoptosis. Cell J. 2011, 13, 65. [Google Scholar]
- Wang, X.; Mo, X.; Li, D.; Wang, Y.; Fang, Y.; Rong, X.; Miao, H.; Shou, T. Neuroprotective effect of transcorneal electrical stimulation on ischemic damage in the rat retina. Exp. Eye Res. 2011, 93, 753–760. [Google Scholar] [CrossRef]
- Kurimoto, T.; Oono, S.; Oku, H.; Tagami, Y.; Kashimoto, R.; Takata, M.; Okamoto, N.; Ikeda, T.; Mimura, O. Transcorneal electrical stimulation increases chorioretinal blood flow in normal human subjects. Clin. Ophthalmol. 2010, 4, 1441. [Google Scholar] [CrossRef] [Green Version]
- Bittner, A.K.; Seger, K.; Salveson, R.; Kayser, S.; Morrison, N.; Vargas, P.; Mendelsohn, D.; Han, J.; Bi, H.; Dagnelie, G. Randomized controlled trial of electro—Stimulation therapies to modulate retinal blood flow and visual function in retinitis pigmentosa. Acta Ophthalmol. 2018, 96, e366–e376. [Google Scholar] [CrossRef] [PubMed]
- Silverstein, D.N.; Ingvar, M. A multi-pathway hypothesis for human visual fear signaling. Front. Syst. Neurosci. 2015, 9, 101. [Google Scholar] [CrossRef] [Green Version]
- McFadyen, J.; Mattingley, J.B.; Garrido, M.I. An afferent white matter pathway from the pulvinar to the amygdala facilitates fear recognition. Elife 2019, 8, e40766. [Google Scholar] [CrossRef] [PubMed]
- Diederich, N.J.; Stebbins, G.; Schiltz, C.; Goetz, C.G. Are patients with Parkinson’s disease blind to blindsight? Brain 2014, 137, 1838–1849. [Google Scholar] [CrossRef] [Green Version]
- Pessoa, L.; Adolphs, R. Emotion processing and the amygdala: From a’low road’to’many roads’ of evaluating biological significance. Nat. Rev. Neurosci. 2010, 11, 773–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsanov, M.; Manahan-Vaughan, D. Synaptic plasticity from visual cortex to hippocampus: Systems integration in spatial information processing. Neuroscientist 2008, 14, 584–597. [Google Scholar] [CrossRef] [PubMed]
- Bird, C.M.; Burgess, N. The hippocampus and memory: Insights from spatial processing. Nat. Rev. Neurosci. 2008, 9, 182–194. [Google Scholar] [CrossRef] [PubMed]
- Veneri, G.; Federico, A.; Rufa, A. Evaluating the influence of motor control on selective attention through a stochastic model: The paradigm of motor control dysfunction in cerebellar patient. BioMed Res. Int. 2014, 2014, 162423:1–162423:13. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Maren, S. Prefrontal-hippocampal interactions in memory and emotion. Front. Syst. Neurosci. 2015, 9, 170. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Wu, X.; Lin, X.; Lin, H. The prevalence of depression and depressive symptoms among eye disease patients: A systematic review and meta-analysis. Sci. Rep. 2017, 7, 46453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, H.G.; Lee, M.J.; Lee, S.M. Visual impairment and risk of depression: A longitudinal follow-up study using a national sample cohort. Sci. Rep. 2018, 8, 2083. [Google Scholar] [CrossRef]
- Hahm, B.-J.; Shin, Y.-W.; Shim, E.-J.; Jeon, H.J.; Seo, J.-M.; Chung, H.; Yu, H.G. Depression and the vision-related quality of life in patients with retinitis pigmentosa. Br. J. Ophthalmol. 2008, 92, 650–654. [Google Scholar] [CrossRef]
- Frank, C.R.; Xiang, X.; Stagg, B.C.; Ehrlich, J.R. Longitudinal associations of self-reported vision impairment with symptoms of anxiety and depression among older adults in the United States. JAMA Ophthalmol. 2019, 137, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, T.; Miyoshi, T.; Sawai, H.; Fujikado, T. Optimal parameters of transcorneal electrical stimulation (TES) to be neuroprotective of axotomized RGCs in adult rats. Exp. Eye Res. 2010, 90, 285–291. [Google Scholar] [CrossRef]
- Henrich-Noack, P.; Lazik, S.; Sergeeva, E.; Wagner, S.; Voigt, N.; Prilloff, S.; Fedorov, A.; Sabel, B.A. Transcorneal alternating current stimulation after severe axon damage in rats results in “long-term silent survivor” neurons. Brain Res. Bull. 2013, 95, 7–14. [Google Scholar] [CrossRef]
- Henrich-Noack, P.; Voigt, N.; Prilloff, S.; Fedorov, A.; Sabel, B.A. Transcorneal electrical stimulation alters morphology and survival of retinal ganglion cells after optic nerve damage. Neurosci. Lett. 2013, 543, 1–6. [Google Scholar] [CrossRef]
- Miyake, K.-I.; Yoshida, M.; Inoue, Y.; Hata, Y. Neuroprotective effect of transcorneal electrical stimulation on the acute phase of optic nerve injury. Investig. Ophthalmol. Vis. Sci. 2007, 48, 2356–2361. [Google Scholar] [CrossRef] [Green Version]
- Osako, T.; Chuman, H.; Maekubo, T.; Ishiai, M.; Kawano, N.; Nao-i, N. Effects of steroid administration and transcorneal electrical stimulation on the anatomic and electrophysiologic deterioration of nonarteritic ischemic optic neuropathy in a rodent model. Jpn. J. Ophthalmol. 2013, 57, 410–415. [Google Scholar] [CrossRef]
- Morimoto, T.; Fujikado, T.; Choi, J.-S.; Kanda, H.; Miyoshi, T.; Fukuda, Y.; Tano, Y. Transcorneal electrical stimulation promotes the survival of photoreceptors and preserves retinal function in royal college of surgeons rats. Investig. Ophthalmol. Vis. Sci. 2007, 48, 4725–4732. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, T.; Kanda, H.; Kondo, M.; Terasaki, H.; Nishida, K.; Fujikado, T. Transcorneal electrical stimulation promotes survival of photoreceptors and improves retinal function in rhodopsin P347L transgenic rabbits. Investig. Ophthalmol. Vis. Sci. 2012, 53, 4254–4261. [Google Scholar] [CrossRef] [Green Version]
- Rahmani, S.; Bogdanowicz, L.; Thomas, J.; Hetling, J.R. Chronic delivery of low-level exogenous current preserves retinal function in pigmented P23H rat. Vis. Res. 2013, 76, 105–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schatz, A.; Röck, T.; Naycheva, L.; Willmann, G.; Wilhelm, B.; Peters, T.; Bartz-Schmidt, K.U.; Zrenner, E.; Messias, A.; Gekeler, F. Transcorneal electrical stimulation for patients with retinitis pigmentosa: A prospective, randomized, sham-controlled exploratory study. Investig. Ophthalmol. Vis. Sci. 2011, 52, 4485–4496. [Google Scholar] [CrossRef]
- Schatz, A.; Pach, J.; Gosheva, M.; Naycheva, L.; Willmann, G.; Wilhelm, B.; Peters, T.; Bartz-Schmidt, K.U.; Zrenner, E.; Messias, A. Transcorneal electrical stimulation for patients with retinitis pigmentosa: A prospective, randomized, sham-controlled follow-up study over 1 year. Investig. Ophthalmol. Vis. Sci. 2017, 58, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Naycheva, L.; Schatz, A.; Willmann, G.; Bartz-Schmidt, K.U.; Zrenner, E.; Röck, T.; Gekeler, F. Transcorneal electrical stimulation in patients with retinal artery occlusion: A prospective, randomized, sham-controlled pilot study. Ophthalmol. Ther. 2013, 2, 25–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inomata, K.; Shinoda, K.; Ohde, H.; Tsunoda, K.; Hanazono, G.; Kimura, I.; Yuzawa, M.; Tsubota, K.; Miyake, Y. Transcorneal electrical stimulation of retina to treat longstanding retinal artery occlusion. Graefe’s Arch. Clin. Exp. Ophthalmol. 2007, 245, 1773–1780. [Google Scholar] [CrossRef] [PubMed]
- Oono, S.; Kurimoto, T.; Kashimoto, R.; Tagami, Y.; Okamoto, N.; Mimura, O. Transcorneal electrical stimulation improves visual function in eyes with branch retinal artery occlusion. Clin. Ophthalmol. 2011, 5, 397. [Google Scholar]
- Fujikado, T.; Morimoto, T.; Matsushita, K.; Shimojo, H.; Okawa, Y.; Tano, Y. Effect of transcorneal electrical stimulation in patients with nonarteritic ischemic optic neuropathy or traumatic optic neuropathy. Jpn. J. Ophthalmol. 2006, 50, 266–273. [Google Scholar] [CrossRef]
- Huang, E.J.; Reichardt, L.F. Neurotrophins: Roles in neuronal development and function. Annu. Rev. Neurosci. 2001, 24, 677–736. [Google Scholar] [CrossRef] [Green Version]
- Sabel, B.A.; Fedorov, A.B.; Naue, N.; Borrmann, A.; Herrmann, C.; Gall, C. Non-invasive alternating current stimulation improves vision in optic neuropathy. Restor. Neurol. Neurosci. 2011, 29, 493–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foik, A.T.; Kublik, E.; Sergeeva, E.G.; Tatlisumak, T.; Rossini, P.M.; Sabel, B.A.; Waleszczyk, W.J. Retinal origin of electrically evoked potentials in response to transcorneal alternating current stimulation in the rat. Investig. Ophthalmol. Vis. Sci. 2015, 56, 1711–1718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osborne, N.N.; Casson, R.J.; Wood, J.P.; Chidlow, G.; Graham, M.; Melena, J. Retinal ischemia: Mechanisms of damage and potential therapeutic strategies. Prog. Retin. Eye Res. 2004, 23, 91–147. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, M. Abnormalities in glutamate metabolism and excitotoxicity in the retinal diseases. Scientifica 2013, 2013, 528940:1–528940:13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muniyappa, R.; Walsh, M.; Rangi, J.; Zayas, R.; Standley, P.R.; Ram, J.; Sowers, J. Insulin like growth factor 1 increases vascular smooth muscle nitric oxide production. Life Sci. 1997, 61, 925–931. [Google Scholar] [CrossRef]
- Haylor, J.; Singh, I.; El Nahas, A.M. Nitric oxide synthesis inhibitor prevents vasodilation by insulin-like growth factor I. Kidney Int. 1991, 39, 333–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atalay, B.; Bolay, H.; Dalkara, T.; Soylemezoglu, F.; Oge, K.; Ozcan, O.E. Transcorneal stimulation of trigeminal nerve afferents to increase cerebral blood flow in rats with cerebral vasospasm: A noninvasive method to activate the trigeminovascular reflex. J. Neurosurg. 2002, 97, 1179–1183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayberg, H.S.; Brannan, S.K.; Tekell, J.L.; Silva, J.A.; Mahurin, R.K.; McGinnis, S.; Jerabek, P.A. Regional metabolic effects of fluoxetine in major depression: Serial changes and relationship to clinical response. Biol. Psychiatry 2000, 48, 830–843. [Google Scholar] [CrossRef]
- Brody, A.L.; Saxena, S.; Mandelkern, M.A.; Fairbanks, L.A.; Ho, M.L.; Baxter, L.R. Brain metabolic changes associated with symptom factor improvement in major depressive disorder. Biol. Psychiatry 2001, 50, 171–178. [Google Scholar] [CrossRef]
- Lévesque, J.; Eugène, F.; Joanette, Y.; Paquette, V.; Mensour, B.; Beaudoin, G.; Leroux, J.-M.; Bourgouin, P.; Beauregard, M. Neural circuitry underlying voluntary suppression of sadness. Biol. Psychiatry 2003, 53, 502–510. [Google Scholar] [CrossRef]
- Phan, K.L.; Fitzgerald, D.A.; Nathan, P.J.; Moore, G.J.; Uhde, T.W.; Tancer, M.E. Neural substrates for voluntary suppression of negative affect: A functional magnetic resonance imaging study. Biol. Psychiatry 2005, 57, 210–219. [Google Scholar] [CrossRef]
- Zeng, L.-L.; Shen, H.; Liu, L.; Wang, L.; Li, B.; Fang, P.; Zhou, Z.; Li, Y.; Hu, D. Identifying major depression using whole-brain functional connectivity: A multivariate pattern analysis. Brain 2012, 135, 1498–1507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albertini, G.; Walrave, L.; Demuyser, T.; Massie, A.; De Bundel, D.; Smolders, I. 6 Hz corneal kindling in mice triggers neurobehavioral comorbidities accompanied by relevant changes in c—Fos immunoreactivity throughout the brain. Epilepsia 2018, 59, 67–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koshal, P.; Kumar, P. Effect of liraglutide on corneal kindling epilepsy induced depression and cognitive impairment in mice. Neurochem. Res. 2016, 41, 1741–1750. [Google Scholar] [CrossRef]
- Dwyer, J.B.; Bloch, M.H. Antidepressants for pediatric patients. Curr. Psychiatr. 2019, 18, 26. [Google Scholar] [PubMed]
- Segi-Nishida, E. The effect of serotonin-targeting antidepressants on neurogenesis and neuronal maturation of the hippocampus mediated via 5-HT1A and 5-HT4 receptors. Front. Cell. Neurosci. 2017, 11, 142. [Google Scholar] [CrossRef]
- Hardy, S.; Bastick, L.; O’Neill-Kerr, A.; Sabesan, P.; Lankappa, S.; Palaniyappan, L. Transcranial magnetic stimulation in clinical practice. BJPsych Adv. 2016, 22, 373–379. [Google Scholar] [CrossRef] [Green Version]
- Turi, Z.; Normann, C.; Domschke, K.; Vlachos, A. Transcranial magnetic stimulation in psychiatry: Is there a need for electric field standardization? Front. Hum. Neurosci. 2021, 15, 639640:1–639640:7. [Google Scholar] [CrossRef]
- Salik, I.; Marwaha, R. Electroconvulsive Therapy. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Cattaneo, A.; Bocchio-Chiavetto, L.; Zanardini, R.; Milanesi, E.; Placentino, A.; Gennarelli, M. Reduced peripheral brain-derived neurotrophic factor mRNA levels are normalized by antidepressant treatment. Int. J. Neuropsychopharmacol. 2010, 13, 103–108. [Google Scholar] [CrossRef]
- Martocchia, A.; Curto, M.; Scaccianoce, S.; Comite, F.; Xenos, D.; Nasca, C.; Falaschi, G.M.; Ferracuti, S.; Girardi, P.; Nicoletti, F. Effects of escitalopram on serum BDNF levels in elderly patients with depression: A preliminary report. Aging Clin. Exp. Res. 2014, 26, 461–464. [Google Scholar] [CrossRef]
- Zanardini, R.; Gazzoli, A.; Ventriglia, M.; Perez, J.; Bignotti, S.; Rossini, P.M.; Gennarelli, M.; Bocchio-Chiavetto, L. Effect of repetitive transcranial magnetic stimulation on serum brain derived neurotrophic factor in drug resistant depressed patients. J. Affect. Disord. 2006, 91, 83–86. [Google Scholar] [CrossRef]
- Yukimasa, T.; Yoshimura, R.; Tamagawa, A.; Uozumi, T.; Shinkai, K.; Ueda, N.; Tsuji, S.; Nakamura, J. High-frequency repetitive transcranial magnetic stimulation improves refractory depression by influencing catecholamine and brain-derived neurotrophic factors. Pharmacopsychiatry 2006, 39, 52–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, S.-F.; Shi, T.-Y.; Wang, W.-N.; Chen, Y.-C.; Tan, Q.-R. Long-lasting effects of chronic rTMS to treat chronic rodent model of depression. Behav. Brain Res. 2012, 232, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-N.; Wang, L.; Zhang, R.-G.; Chen, Y.-C.; Liu, L.; Gao, F.; Nie, H.; Hou, W.-G.; Peng, Z.-W.; Tan, Q. Anti-depressive mechanism of repetitive transcranial magnetic stimulation in rat: The role of the endocannabinoid system. J. Psychiatr. Res. 2014, 51, 79–87. [Google Scholar] [CrossRef]
- Zhao, X.; Li, Y.; Tian, Q.; Zhu, B.; Zhao, Z. Repetitive transcranial magnetic stimulation increases serum brain-derived neurotrophic factor and decreases interleukin-1β and tumor necrosis factor-α in elderly patients with refractory depression. J. Int. Med Res. 2019, 47, 1848–1855. [Google Scholar] [CrossRef] [PubMed]
- Taliaz, D.; Nagaraj, V.; Haramati, S.; Chen, A.; Zangen, A. Altered brain-derived neurotrophic factor expression in the ventral tegmental area, but not in the hippocampus, is essential for antidepressant-like effects of electroconvulsive therapy. Biol. Psychiatry 2013, 74, 305–312. [Google Scholar] [CrossRef]
- Brunoni, A.R.; Baeken, C.; Machado-Vieira, R.; Gattaz, W.F.; Vanderhasselt, M.-A. BDNF blood levels after electroconvulsive therapy in patients with mood disorders: A systematic review and meta-analysis. World J. Biol. Psychiatry 2014, 15, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Vanicek, T.; Kranz, G.S.; Vyssoki, B.; Fugger, G.; Komorowski, A.; Höflich, A.; Saumer, G.; Milovic, S.; Lanzenberger, R.; Eckert, A. Acute and subsequent continuation electroconvulsive therapy elevates serum BDNF levels in patients with major depression. Brain Stimul. 2019, 12, 1041–1050. [Google Scholar] [CrossRef]
- Kosten, T.A.; Galloway, M.P.; Duman, R.S.; Russell, D.S.; D’sa, C. Repeated unpredictable stress and antidepressants differentially regulate expression of the bcl-2 family of apoptotic genes in rat cortical, hippocampal, and limbic brain structures. Neuropsychopharmacology 2008, 33, 1545–1558. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Hu, Z.; Du, X.; Davies, H.; Huo, X.; Fang, M. miR-16 and fluoxetine both reverse autophagic and apoptotic change in chronic unpredictable mild stress model rats. Front. Neurosci. 2017, 11, 428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.; Ren, H.; Gu, S.; Li, X.; Jiang, C.; Li, J.; Zhang, M.; Mu, J.; Li, W.; Wang, W. rTMS ameliorated depressive-like behaviors by restoring HPA axis balance and prohibiting hippocampal neuron apoptosis in a rat model of depression. Psychiatry Res. 2018, 269, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Chavda, N.; Kantharia, N.; Charan, J. Effects of fluoxetine and escitalopram on C-reactive protein in patients of depression. J. Pharmacol. Pharmacother. 2011, 2, 11–16. [Google Scholar] [CrossRef] [Green Version]
- Hannestad, J.; DellaGioia, N.; Bloch, M. The effect of antidepressant medication treatment on serum levels of inflammatory cytokines: A meta-analysis. Neuropsychopharmacology 2011, 36, 2452–2459. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Sun, S.-S.; Cui, L.-B.; Wang, S.-Q.; Peng, Z.-W.; Tan, Q.-R.; Hou, W.-G.; Cai, M. Repetitive transcranial magnetic stimulation elicits antidepressant-and anxiolytic-like effect via nuclear factor-E2-related factor 2-mediated anti-inflammation mechanism in rats. Neuroscience 2020, 429, 119–133. [Google Scholar] [CrossRef]
- Hestad, K.A.; Tønseth, S.; Støen, C.D.; Ueland, T.; Aukrust, P. Raised plasma levels of tumor necrosis factor α in patients with depression: Normalization during electroconvulsive therapy. J. ECT 2003, 19, 183–188. [Google Scholar] [CrossRef]
- Rotter, A.; Biermann, T.; Stark, C.; Decker, A.; Demling, J.; Zimmermann, R.; Sperling, W.; Kornhuber, J.; Henkel, A. Changes of cytokine profiles during electroconvulsive therapy in patients with major depression. J. ECT 2013, 29, 162–169. [Google Scholar] [CrossRef]
- Zincir, S.; Öztürk, P.; Bilgen, A.E.; Izci, F.; Yükselir, C. Levels of serum immunomodulators and alterations with electroconvulsive therapy in treatment-resistant major depression. Neuropsychiatr. Dis. Treat. 2016, 12, 1389. [Google Scholar] [CrossRef] [Green Version]
- Sorri, A.; Järventausta, K.; Kampman, O.; Lehtimäki, K.; Björkqvist, M.; Tuohimaa, K.; Hämäläinen, M.; Moilanen, E.; Leinonen, E. Low tumor necrosis factor-α levels predict symptom reduction during electroconvulsive therapy in major depressive disorder. Brain Behav. 2018, 8, e00933. [Google Scholar] [CrossRef]
- Belge, J.-B.; Van Diermen, L.; Sabbe, B.; Parizel, P.; Morrens, M.; Coppens, V.; Constant, E.; de Timary, P.; Sienaert, P.; Schrijvers, D. Inflammation, Hippocampal Volume, and Therapeutic Outcome following Electroconvulsive Therapy in Depressive Patients: A Pilot Study. Neuropsychobiology 2020, 79, 222–232. [Google Scholar] [CrossRef]
- Yang, T.; Nie, Z.; Shu, H.; Kuang, Y.; Chen, X.; Cheng, J.; Yu, S.; Liu, H. The role of BDNF on neural plasticity in depression. Front. Cell. Neurosci. 2020, 14, 82. [Google Scholar] [CrossRef]
- Hennigan, A.; O’callaghan, R.; Kelly, A. Neurotrophins and their receptors: Roles in plasticity, neurodegeneration and neuroprotection. Biochem. Soc. Trans. 2007, 35, 424–427. [Google Scholar] [CrossRef]
- Bathina, S.; Das, U.N. Brain-derived neurotrophic factor and its clinical implications. Arch. Med Sci. 2015, 11, 1164. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-K.; Lee, H.-P.; Won, S.-D.; Park, E.-Y.; Lee, H.-Y.; Lee, B.-H.; Lee, S.-W.; Yoon, D.; Han, C.; Kim, D.-J. Low plasma BDNF is associated with suicidal behavior in major depression. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2007, 31, 78–85. [Google Scholar] [CrossRef]
- McKernan, D.P.; Dinan, T.G.; Cryan, J.F. “Killing the Blues”: A role for cellular suicide (apoptosis) in depression and the antidepressant response? Prog. Neurobiol. 2009, 88, 246–263. [Google Scholar] [CrossRef] [PubMed]
- Duman, R.S. Neuronal damage and protection in the pathophysiology and treatment of psychiatric illness: Stress and depression. Dialogues Clin. Neurosci. 2009, 11, 239. [Google Scholar] [PubMed]
- Kale, J.; Osterlund, E.J.; Andrews, D.W. BCL-2 family proteins: Changing partners in the dance towards death. Cell Death Differ. 2018, 25, 65–80. [Google Scholar] [CrossRef] [Green Version]
- Pandya, M.; Altinay, M.; Malone, D.A.; Anand, A. Where in the brain is depression? Curr. psychiatry Rep. 2012, 14, 634–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, A. The role of inflammation in depression. Psychiatr. Danub. 2013, 25, S216–S223. [Google Scholar]
- Gałecki, P.; Talarowska, M. Inflammatory theory of depression. Psychiatr. Pol. 2018, 52, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Roque, S.; Correia-Neves, M.; Mesquita, A.R.; Palha, J.A.; Sousa, N. Interleukin-10: A key cytokine in depression? Cardiovasc. Psychiatry Neurol. 2009, 2009, 187894:1–187894:5. [Google Scholar] [CrossRef] [Green Version]
- Lozano, A.M.; Lipsman, N.; Bergman, H.; Brown, P.; Chabardes, S.; Chang, J.W.; Matthews, K.; McIntyre, C.C.; Schlaepfer, T.E.; Schulder, M.; et al. Deep brain stimulation: Current challenges and future directions. Nat. Rev. Neurol. 2019, 15, 148–160. [Google Scholar] [CrossRef] [PubMed]
- Fenoy, A.J.; Simpson, R.K., Jr. Risks of common complications in deep brain stimulation surgery: Management and avoidance. J. Neurosurg. 2014, 120, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Hamurcu, M.S.; Aydogmuş, S.A.; Saricaoğlu, M.S. Evaluation of the efficacy of transcorneal electric stimulation therapy in retinitis pigmentosa patients with electrophysiological and structural tests. Int. J. Clin. Exp. Ophthalmol. 2020, 4, 31–37. [Google Scholar] [CrossRef]
- Bae, E.H.; Schrader, L.M.; Machii, K.; Alonso-Alonso, M.; Riviello, J.J., Jr.; Pascual-Leone, A.; Rotenberg, A. Safety and tolerability of repetitive transcranial magnetic stimulation in patients with epilepsy: A review of the literature. Epilepsy Behav. 2007, 10, 521–528. [Google Scholar] [CrossRef]
- Brakemeier, E.-L.; Berman, R.; Prudic, J.; Zwillenberg, K.; Sackeim, H.A. Self-evaluation of the cognitive effects of electroconvulsive therapy. J. ECT 2011, 27, 59–66. [Google Scholar] [CrossRef]
- Porter, R.J.; Baune, B.T.; Morris, G.; Hamilton, A.; Bassett, D.; Boyce, P.; Hopwood, M.J.; Mulder, R.; Parker, G.; Singh, A.B. Cognitive side-effects of electroconvulsive therapy: What are they, how to monitor them and what to tell patients. BJPsych Open 2020, 6, e40:1–e40:7. [Google Scholar] [CrossRef]
- Holtzheimer, P.E.; Mayberg, H.S. Neuromodulation for treatment-resistant depression. F1000 Med. Rep. 2012, 4. [Google Scholar] [CrossRef]
- Uher, R.; Farmer, A.; Henigsberg, N.; Rietschel, M.; Mors, O.; Maier, W.; Kozel, D.; Hauser, J.; Souery, D.; Placentino, A. Adverse reactions to antidepressants. Br. J. Psychiatry 2009, 195, 202–210. [Google Scholar] [CrossRef]
- Carvalho, A.F.; Sharma, M.S.; Brunoni, A.R.; Vieta, E.; Fava, G.A. The Safety, Tolerability and Risks Associated with the Use of Newer Generation Antidepressant Drugs: A Critical Review of the Literature. Psychother. Psychosom. 2016, 85, 270–288. [Google Scholar] [CrossRef] [PubMed]
- Jolly, J.K.; Wagner, S.K.; Martus, P.; MacLaren, R.E.; Wilhelm, B.; Webster, A.R.; Downes, S.M.; Charbel Issa, P.; Kellner, U.; Jägle, H.; et al. Transcorneal Electrical Stimulation for the Treatment of Retinitis Pigmentosa: A Multicenter Safety Study of the OkuStim® System (TESOLA-Study). Ophthalmic Res. 2020, 63, 234–243. [Google Scholar] [CrossRef]
- Morimoto, T.; Fukui, T.; Matsushita, K.; Okawa, Y.; Shimojyo, H.; Kusaka, S.; Tano, Y.; Fujikado, T. Evaluation of residual retinal function by pupillary constrictions and phosphenes using transcorneal electrical stimulation in patients with retinal degeneration. Graefe’s Arch. Clin. Exp. Ophthalmol. 2006, 244, 1283. [Google Scholar] [CrossRef] [PubMed]
- Wagner, S.K.; Jolly, J.K.; Pefkianaki, M.; Gekeler, F.; Webster, A.R.; Downes, S.M.; Maclaren, R.E. Transcorneal electrical stimulation for the treatment of retinitis pigmentosa: Results from the TESOLAUK trial. BMJ open Ophthalmol. 2017, 2, e000096. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
| Treatment | Study | Subject | Administration Protocol | Results |
|---|---|---|---|---|
| Neurotrophic Mechanism | ||||
| Transcorneal electrical stimulation (TES) | [34] | Male Wistar rats, model of optic nerve transection | 100 μA, 20 Hz, 3 ms/phase, 1 h, 1 session | TES increased survival of axotomized RGCs and expression of IGF-1 in Müller cells in the retina. |
| [37] | Retinal Müller cell culture | 1–10 mA, 20 Hz, 1 ms/phase, 30 min, 1 session | Stimulation increased IGF-1 mRNA and Ca2+ reflux which were suppressed by an L-VDCC blocker. | |
| [38] | Retinal Müller cell culture | 10 mA, 20 Hz, 1 ms/phase, 30 min, 1 session | Stimulation increased BDNF mRNA and intracellular protein levels which were suppressed by L-VDCC blocker. | |
| [36] | Male Sprague Dawley rats, model of light-induced photoreceptor degeneration | Pre-TES: 100–500 μA, 20–100 Hz, 3 ms/phase, 1.5 h, 1 session. Post-TES: 200/300 μA, 20 Hz, 3 ms/phase, 1 session every 3 days for up to 14 days | Post-TES better preserved ONL and retinal functions. TES increased gene and protein expressions of CNTF and BDNF. | |
| [35] | Male Wistar rats; model of optic nerve crush | 100 μA, 20 Hz, 1 ms/phase, 1 h, 1 session on day 0; 2 sessions on day 0 and 7; 4 sessions on day 0, 4, 7, and 10; daily sessions on day 0–12 | TES increased IGF-1 immunoreactivity in the retina, and promoted axonal regeneration and survival of RGCs. The axonal regeneration was inhibited with an IGF-1 receptor antagonist. | |
| [39] | Retinal microglia and Müller cell culture with intense light exposure | 300/500/1000/1600 μA, 20 Hz, 3 ms/phase, 1 h, 1 session | Stimulation increased secretion of BDNF and CNTF by Müller cells. | |
| [46] | C57/BL mice, MNU model of photoreceptor degeneration | 100/200 μA, 20 Hz, 3 ms/phase, 3 sessions on days 1, 3, and 6 after MNU injection | TES ameliorated photoreceptor degeneration and increased mRNA levels of BDNF and CNTF. | |
| [40] | Retinal Müller cell culture | 10–500 μA, 10–100 Hz, 0.5–5 ms/phase | TES induced Müller cell proliferation which was blocked by an L-type calcium channel blocker. TES also increased CNTF mRNA level. | |
| Selective serotonin reuptake inhibitors (SSRIs) | [97] | 21 patients with depression 23 healthy controls | Escitalopram, 12 weeks | Escitalopram in depressed patients increased leukocyte BDNF mRNA and serum BDNF to similar levels as in controls. Changes in BDNF levels were correlated with symptoms improvement. |
| [98] | 5 patients with depression 10 healthy controls | Escitalopram, 10 mg/day, 8 weeks | Escitalopram in depressed patients improved GDS scores and increased serum BDNF beyond the levels in controls. The increase in BDNF was correlated to the improved GDS scores. | |
| Repeat transcranial magnetic stimulation (rTMS) | [99] | 16 patients with treatment-resistant depression | 1 Hz/17 Hz, 5 consecutive sessions with 24 h interval | rTMS improved HDRS scores and increased serum BDNF. |
| [100] | 26 patients with treatment-resistant depression | Stimulated in the left PFC, intensity 80% MT, 20 Hz, 800 pulses/day, 10 days | rTMS improved HDRS scores and increased plasma BDNF by 23% in responders. A trend of an association between changes in HDRS scores and BDNF levels was found with rTMS. | |
| [101] | Male Sprague Dawley rats; CUMS model of depression | 15 Hz, intensity 100% of device’s maximum power, 60 pulses/train, 15 s train duration, 15 s intertrain interval, 17 trains/day, 1000 pulses/day, 21 consecutive days | rTMS improved depressive-like behaviour and increased BDNF protein levels in the hippocampus. | |
| [102] | Male Sprague Dawley rats; CUMS model of depression | 15 Hz, intensity 100% of device’s maximum power, 15 trains of 60 pulses with 15 s inter-train interval, 900 pulses/day, 7 days | rTMS improved depressive-like behaviour and increased BDNF protein levels in the hippocampus. | |
| [103] | 58 patients with treatment-resistant depression: rTMS (n = 19); non-rTMS controls (n = 19) 30 healthy individuals: rTMS | Stimulated in left dlPFC, intensity 80% MT, 10 Hz, 1200 pulses of 1 s with 11 s interval, 20 min/session, 5 sessions/week, 4 weeks. | rTMS in depressed patients improved HDRS scores and increased serum BDNF, which was negatively correlated with HDRS scores. | |
| Electroconvulsive therapy (ECT) | [104] | Male Sprague Dawley rats | Stimulated with ear clip electrodes, 100 V, 50 Hz, 1.5 s, duration, 1 session/day, 10 days | ECT increased BDNF protein level in the hippocampus. ECT also decreased BDNF protein level in the VTA, which was necessary for the antidepressant-like effects of ECT. |
| [105] | Meta-analysis of 11 studies enrolled patients with unipolar, bipolar and psychotic depression | Stimulated unilaterally or bilaterally on frontal/temporal/frontotemporal positions, 2–3 sessions/week, 6–12 sessions | ECT increased blood BDNF levels. | |
| [106] | 24 patients with depression | Stimulated unilaterally in right frontotemporal and parietal position, maximum charge 1000 mC, 3 sessions/week, 6–13 sessions | ECT improved HDRS scores in responders and remitters, and increased serum BDNF up to 1 month after the last treatment. | |
| Anti-apoptosis Mechanism | ||||
| Transcorneal electrical stimulation (TES) | [36] | Male Sprague Dawley rats with light-induced photoreceptor degeneration | Pre-TES: 100–500 μA, 20–100 Hz, 3 ms/phase, 1.5 h, 1 session. Post-TES: 200/300 μA, 20 Hz, 3 ms/phase, 1 session every 3 days for up to 14 days | Post-TES better preserved ONL and retinal functions. TES increased gene and protein expression of Bcl-2 and decreased expression of Bax. |
| [45] | Male brown Norway rats | 200 μA, 20 Hz, 1 ms/phase, 1 h, one session | TES differentially regulated 490 genes, including downregulation of Bax and Tnfrs12a. | |
| [46] | C57/BL mice; MNU model of photoreceptor degeneration | 100/200 μA, 20 Hz, 3 ms/phase, 3 sessions on days 1, 3, and 6 after MNU injection | TES ameliorated photoreceptor degeneration, increased mRNA Bcl-2 mRNA, and decreased Bax and Calpain-2 mRNA levels. | |
| Selective serotonin reuptake inhibitors (SSRIs) | [107] | Male Sprague Dawley rats; CUMS model of depression | Fluoxetine, 5 mg/kg/day, i.p., 21 days | Fluoxetine decreased Bax mRNA in the hippocampus and increased Bcl-2 mRNA in CeA, Cg, and Fr. |
| [108] | Male Sprague Dawley rats; CUMS model of depression | Fluoxetine, 3 weeks | Fluoxetine improved depressive-like behaviour, increased Bcl-2 and decreased caspase-3 protein levels in the hippocampus. | |
| Repeat transcranial magnetic stimulation (rTMS) | [102] | Male Sprague Dawley rats; CUMS model of depression | 15 Hz, intensity 100% of device’s maximum power, 15 trains of 60 pulses with 15 s inter-train interval, 900 pulses/day, 7 days | rTMS improved depressive-like behaviour, increased Bcl-2, and decreased Bax protein levels in the hippocampus. |
| [109] | Male Sprague Dawley rats; CUMS model of depression | 10 Hz, intensity 50% of MT, 1 s stimulation duration, 10 s inter-pulse interval, 500 pulses/day, 10 min/day, 5 daily sessions with 2-day intervals for 3 weeks | rTMS improved depressive-like behaviour, decreased Bax mRNA and protein levels in the hippocampus. | |
| Electroconvulsive therapy (ECT) | [107] | Male Sprague Dawley rats; CUMS model of depression | Stimulated with ear clip electrodes, 60 mA, 0.3 s duration, 1 session daily for 21 days | ECT increased Bcl-2 mRNA in Cg, lateral Fr, CeA, and Bcl-xl mRNA in the hippocampus. |
| Anti-inflammation Mechanism | ||||
| Transcorneal electrical stimulation (TES) | [39] | Retinal microglia and Müller cells culture with intense light exposure | 300/500/1000/1600 μA, 20 Hz, 3 ms/phase, 1 h, 1 session | Stimulation inhibited microglial activation and microglial secretion of IL-1β and TNF-α. |
| [43] | Male Sprague Dawley rats; model of optic nerve transection | 200 μA, 20 Hz, 2 ms/phase, 1 h, 2 sessions on day 0 and day 4; or 4 sessions on day 0, 4, 7 and 10 after optic nerve transection | TES increased survival of RGCs, suppressed microglial activation, and reduced TNF-α expression in microglia. | |
| [42] | Mongolian gerbils; model of acute ocular hypertensive injury | 100 μA, 20 Hz, 1 ms/phase, 1 h, 2 sessions on day 1 and 4 for the 1-week experiment; or 2 sessions on day 1 and 4 each week for the 1-month experiment | TES improved retinal function and survival of RGCs, decreased pNF-κB, IL-6, Cox2 and TNF-α expressions, increased IL-10 levels, and suppressed microglial activation. | |
| [44] | DBA/2 J mice, model of glaucoma | 100 μA, 20 Hz, 1 ms/phase, 10 min, every 3 days for 8 weeks | TES improved RGC axonal survival and reduced the number of microglia. | |
| Selective serotonin reuptake inhibitors (SSRIs) | [110] | 98 patients with depression | Fluoxetine, 20 mg/day, 8 weeks Or escitalopram, 20 mg/day, 8 weeks | Fluoxetine and escitalopram reduced inflammatory markers, including C-reactive protein concentration, erythrocyte sedimentation rate, and white blood cell count. |
| [111] | Meta-analysis of 22 studies on enrolled patients with depression who were taking FDA-approved pharmacological treatments | Different classes of antidepressants were included, primarily SSRIs | SSRIs in depressed patients reduced serum IL-6, IL-1ß, and TNF-α. | |
| Repeat transcranial magnetic stimulation (rTMS) | [112] | Male Sprague Dawley rats; CUMS model of depression | 15 Hz, intensity 30% of device’s maximum power, 20 s train duration, 15 min intertrain interval, 900 pulses/day, seven consecutive days | rTMS improved depressive-like behaviour, increased Nrf2 proteins and decreased TNF-a, iNOS, IL-1ß, and IL-6 expression in the hippocampus. These effects were reversed by Nrf2 silencing. |
| [103] | 58 patients with treatment-resistant depression: rTMS (n = 19); non-rTMS controls (n = 19) 30 healthy individuals: rTMS | Stimulated in left dlPFC, intensity 80% MT, 10 Hz, 1200 pulses of 1 s with 11 s interval, 20 min/session, 5 sessions/week, 4 weeks | rTMS in depressed patients improved HDRS scores and decreased serum IL-1ß and TNF-a to levels similar to healthy individuals. IL-1ß and TNF-a levels were positively correlated with HAMD scores. | |
| Electroconvulsive therapy (ECT) | [113] | 23 patients with depression: ECT (n = 15); non-ECT control (n = 8) 15 healthy controls | Maximum charge 504 mC, intensity 0.9 A, max duration 7.9 s, 3 sessions/week, 4–18 sessions | ECT in depressed patients decreased plasma levels of TNF-α to levels comparable with healthy controls. |
| [114] | 15 patients with depression | Stimulated unilaterally on the right d’Elia position, intensity 0.9 A, 10–70 Hz, 0.5–1.0 ms/phase, 6–8 s stimulus duration, max charge 964 mC, 2–3 sessions/week, 12 sessions | ECT decreased serum levels of eotaxin-3 and IL-5 24 h after the last ECT. | |
| [115] | 50 patients with treatment-resistant depression | Stimulated bilaterally in temporal position, square wave pulse, intensity 550–800 mA, 40–90 Hz, 1–2 ms/pulse, 0.5–4 s stimulation duration, maximum charge 1172 mC, 3 sessions/week, 5–12 sessions | ECT increased serum levels of IL-1 and IL-10, and decreased levels of IL-4 and IFN-γ. | |
| [116] | 30 patients with depression | Intensity at 1.5 times of seizure threshold, 3 sessions/week, 5–17 sessions | ECT decreased plasma TNF-α level 2 and 4 h after treatment in the 1st, 5th, and last ECT sessions. | |
| [117] | 62 patients with depression | Stimulated unilaterally on the right, 2 sessions/week until patients were asymptomatic/did not improve further for 3 sessions/intolerable side effects occurred | ECT decreased plasma IL-6 level. TNFα level remained unchanged after ECT. | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, W.-S.; Kwon, S.-H.; Agadagba, S.K.; Chan, L.-L.-H.; Wong, K.-H.; Lim, L.-W. Neuroprotective Effects and Therapeutic Potential of Transcorneal Electrical Stimulation for Depression. Cells 2021, 10, 2492. https://doi.org/10.3390/cells10092492
Yu W-S, Kwon S-H, Agadagba SK, Chan L-L-H, Wong K-H, Lim L-W. Neuroprotective Effects and Therapeutic Potential of Transcorneal Electrical Stimulation for Depression. Cells. 2021; 10(9):2492. https://doi.org/10.3390/cells10092492
Chicago/Turabian StyleYu, Wing-Shan, So-Hyun Kwon, Stephen Kugbere Agadagba, Leanne-Lai-Hang Chan, Kah-Hui Wong, and Lee-Wei Lim. 2021. "Neuroprotective Effects and Therapeutic Potential of Transcorneal Electrical Stimulation for Depression" Cells 10, no. 9: 2492. https://doi.org/10.3390/cells10092492
APA StyleYu, W.-S., Kwon, S.-H., Agadagba, S. K., Chan, L.-L.-H., Wong, K.-H., & Lim, L.-W. (2021). Neuroprotective Effects and Therapeutic Potential of Transcorneal Electrical Stimulation for Depression. Cells, 10(9), 2492. https://doi.org/10.3390/cells10092492