
Potential Therapeutic Candidates for Age-Related Macular Degeneration (AMD)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Overview of Potential Therapeutic Candidates for AMD
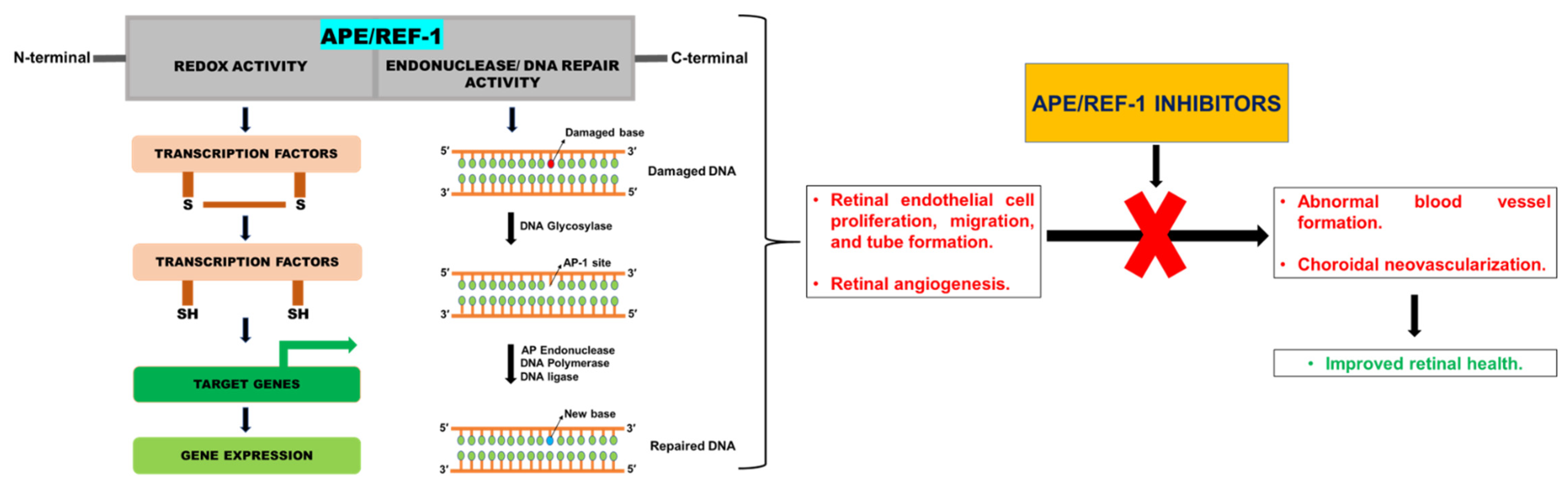
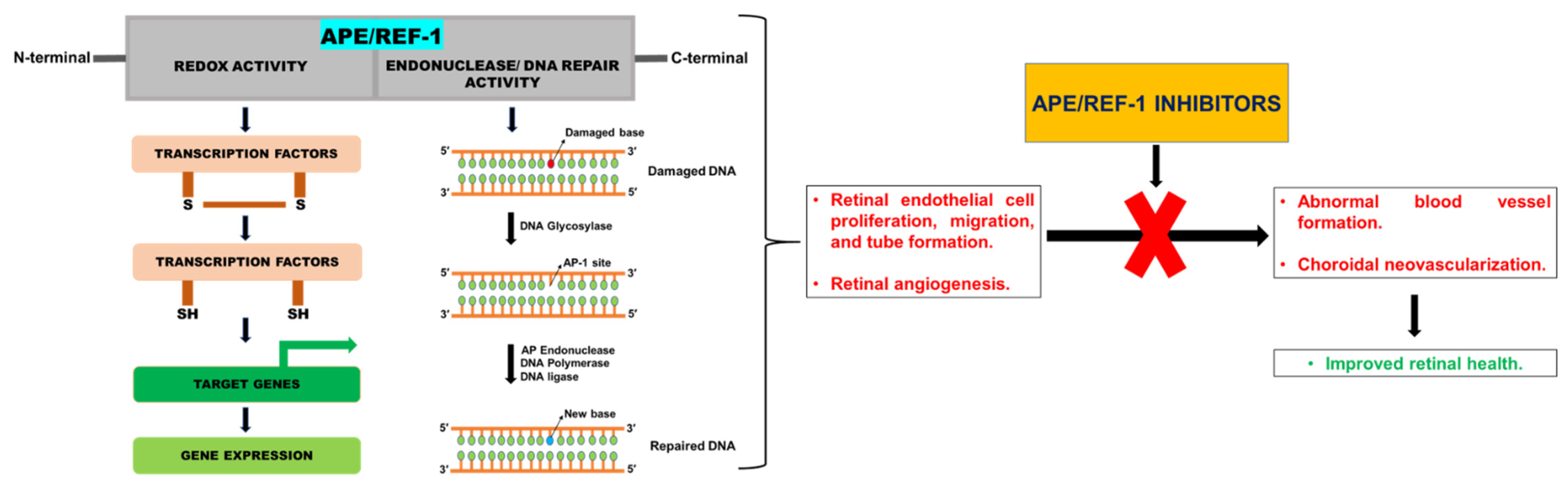
2.1. APE/REF-1
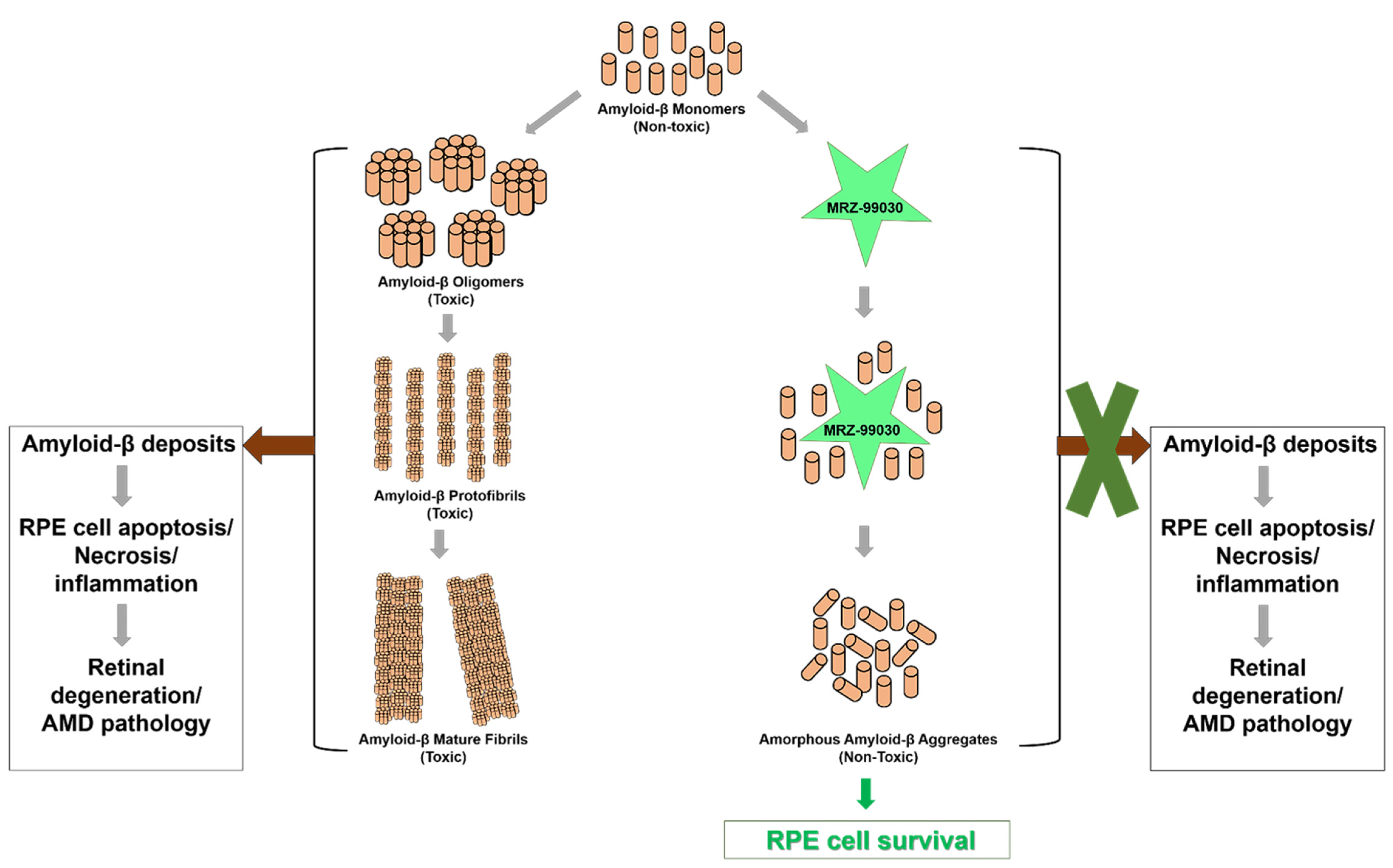
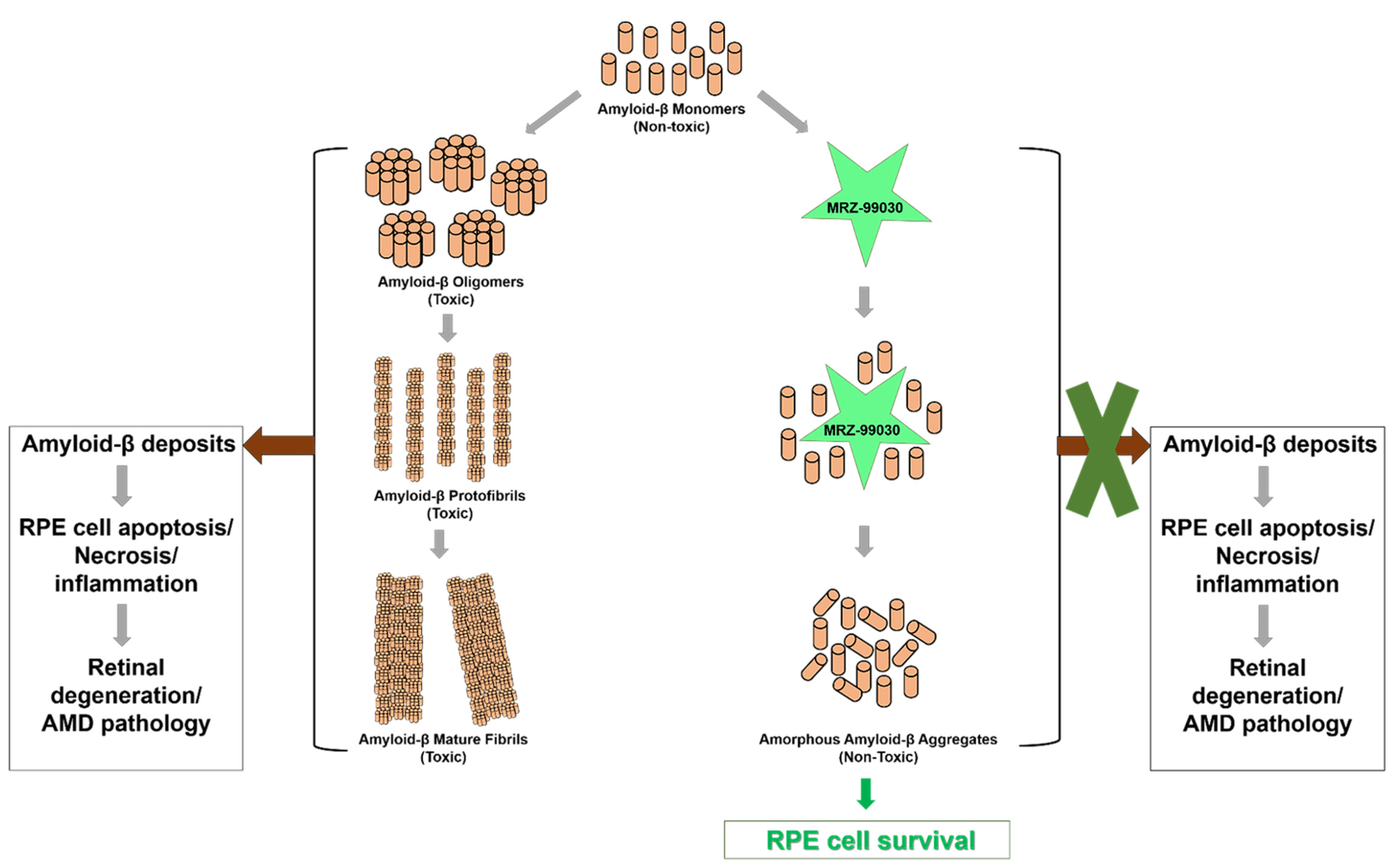
2.2. MRZ-99030
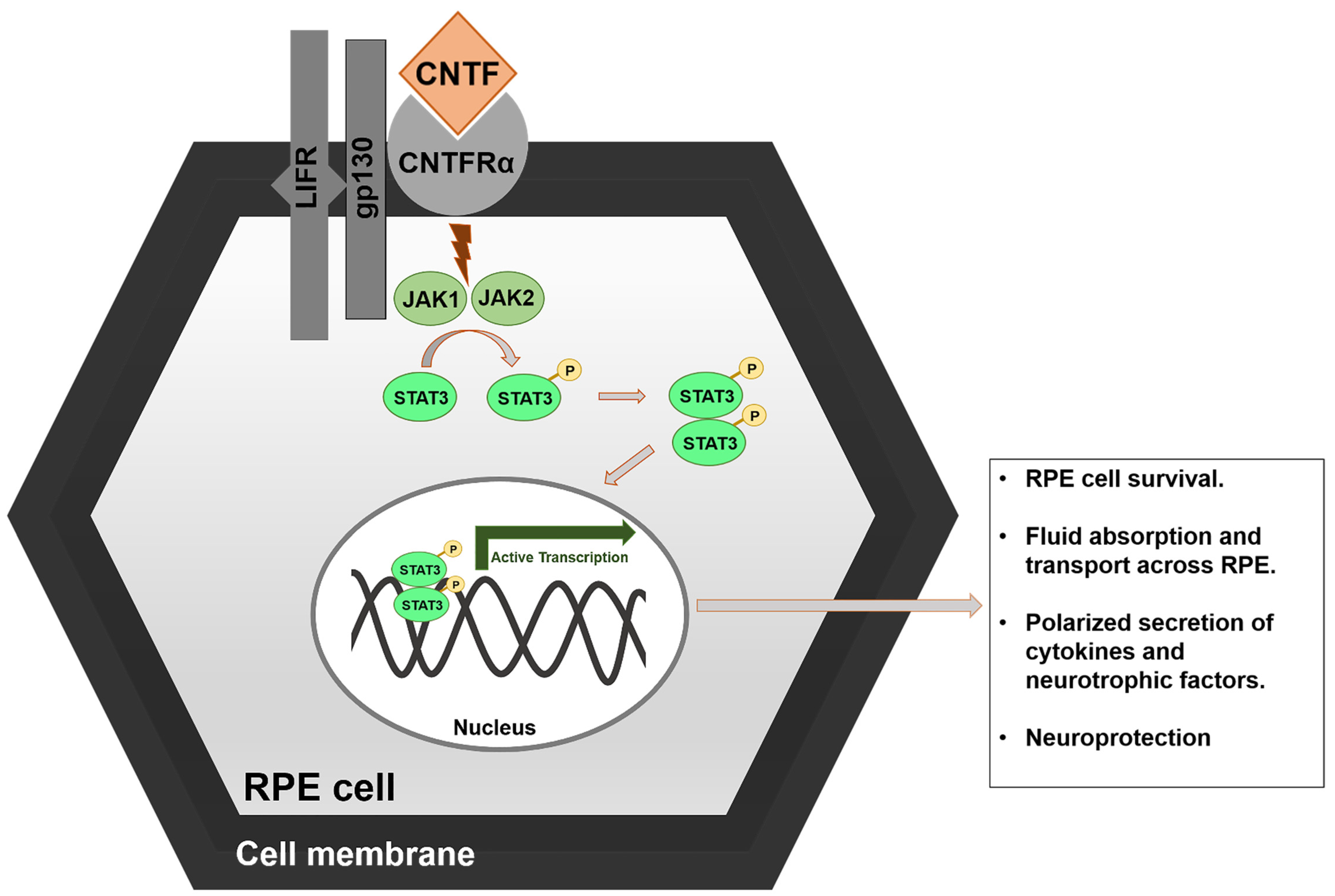
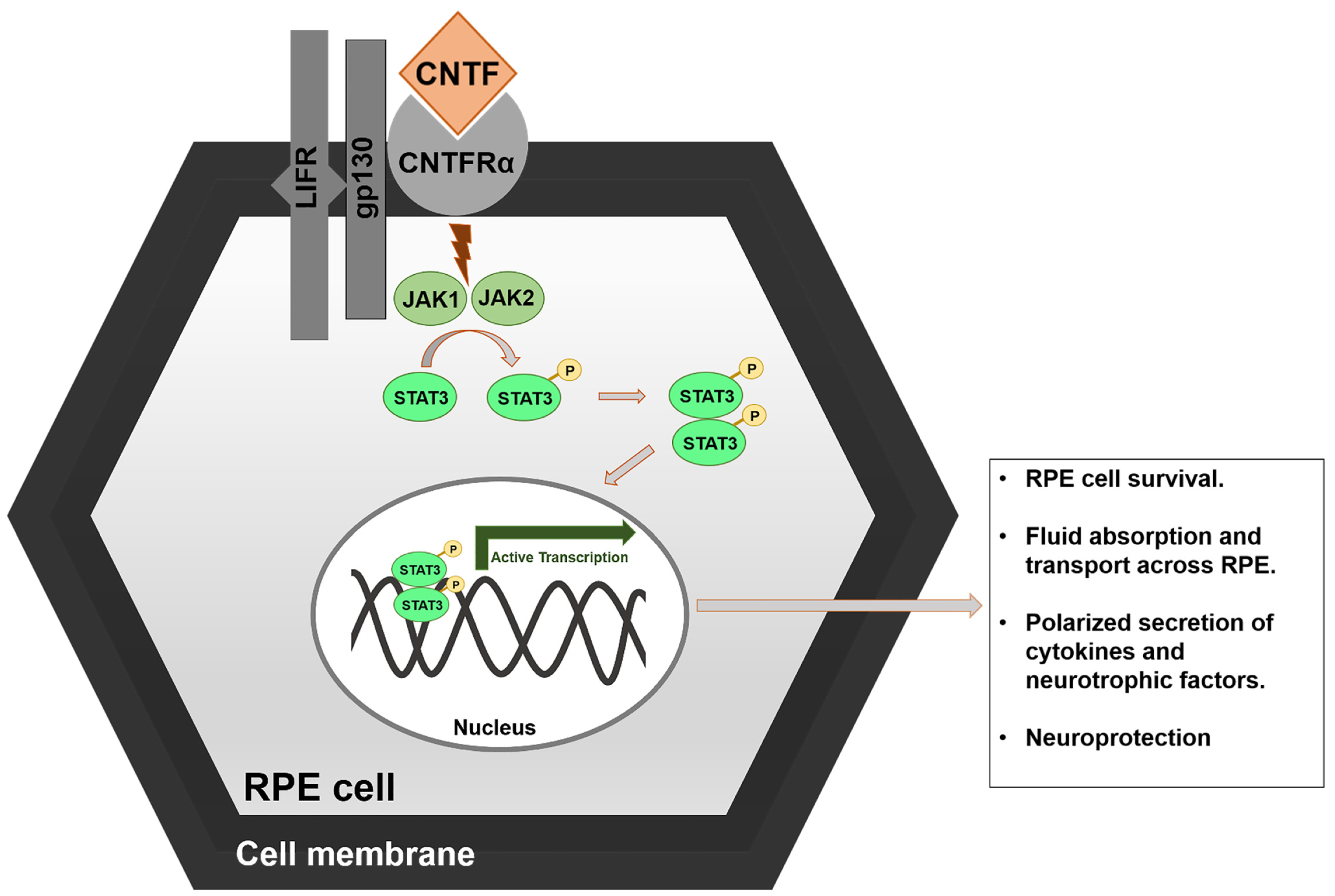
2.3. Ciliary NeuroTrophic Factor (CNTF)
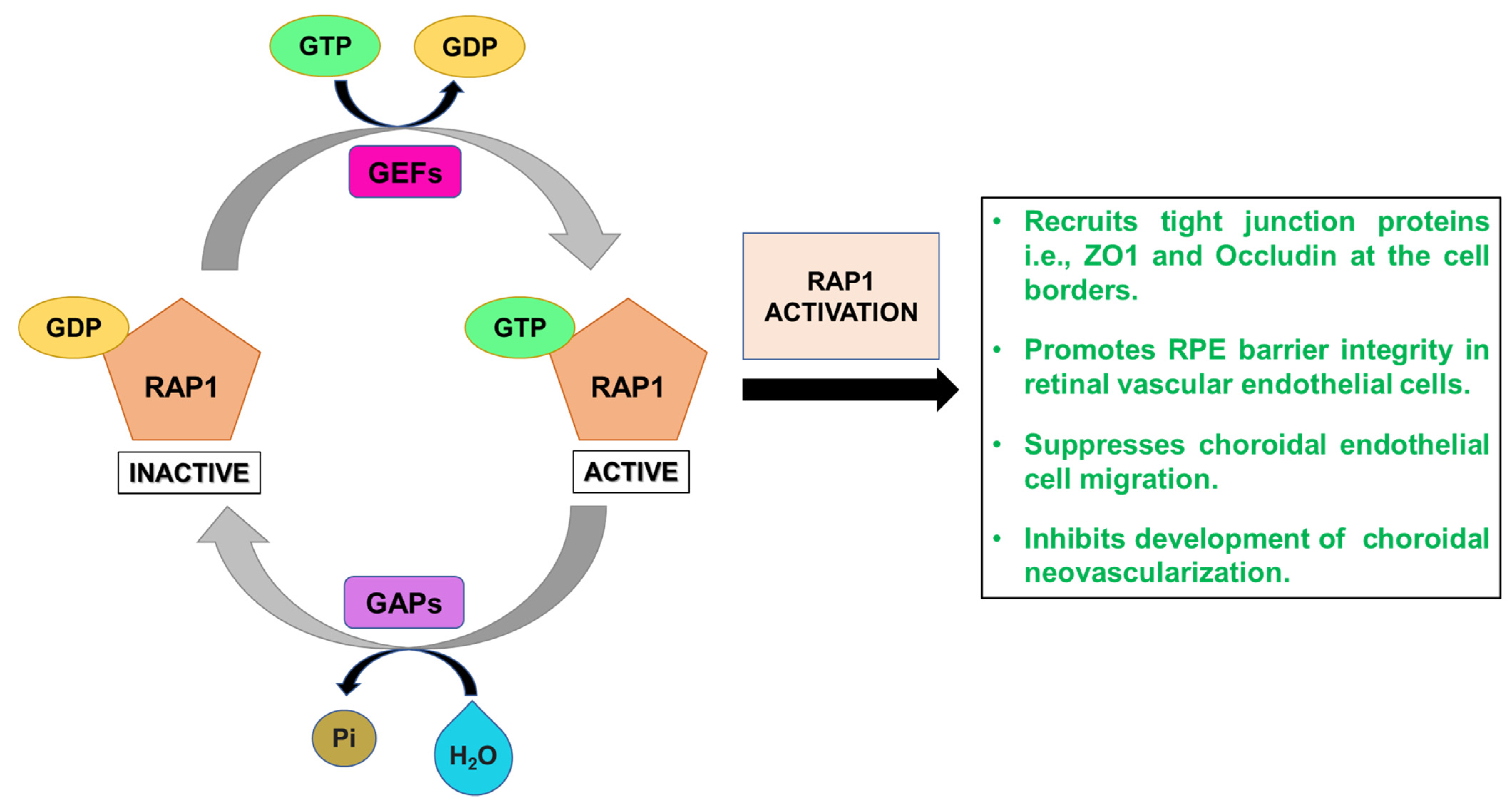
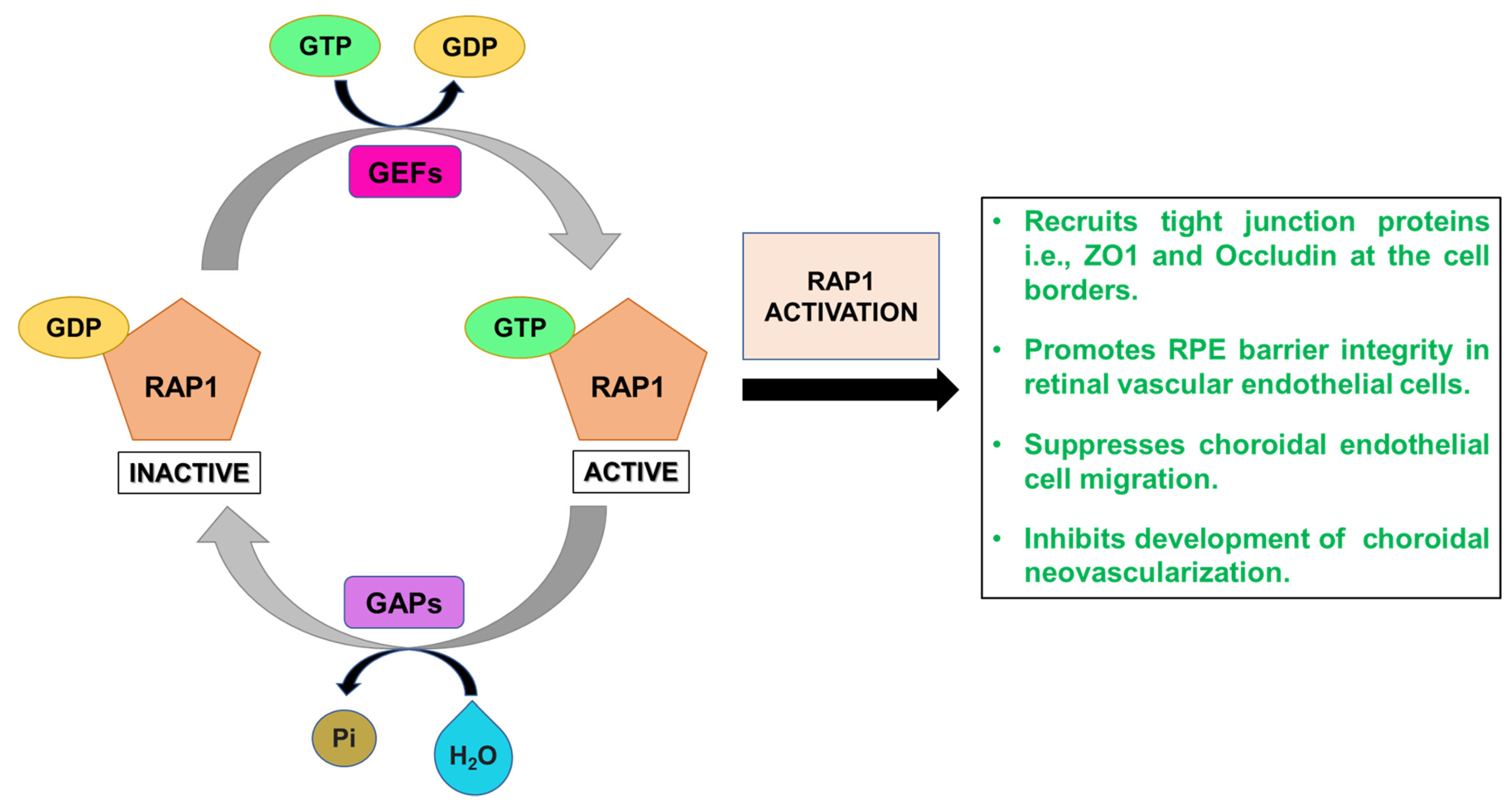
2.4. RAP1
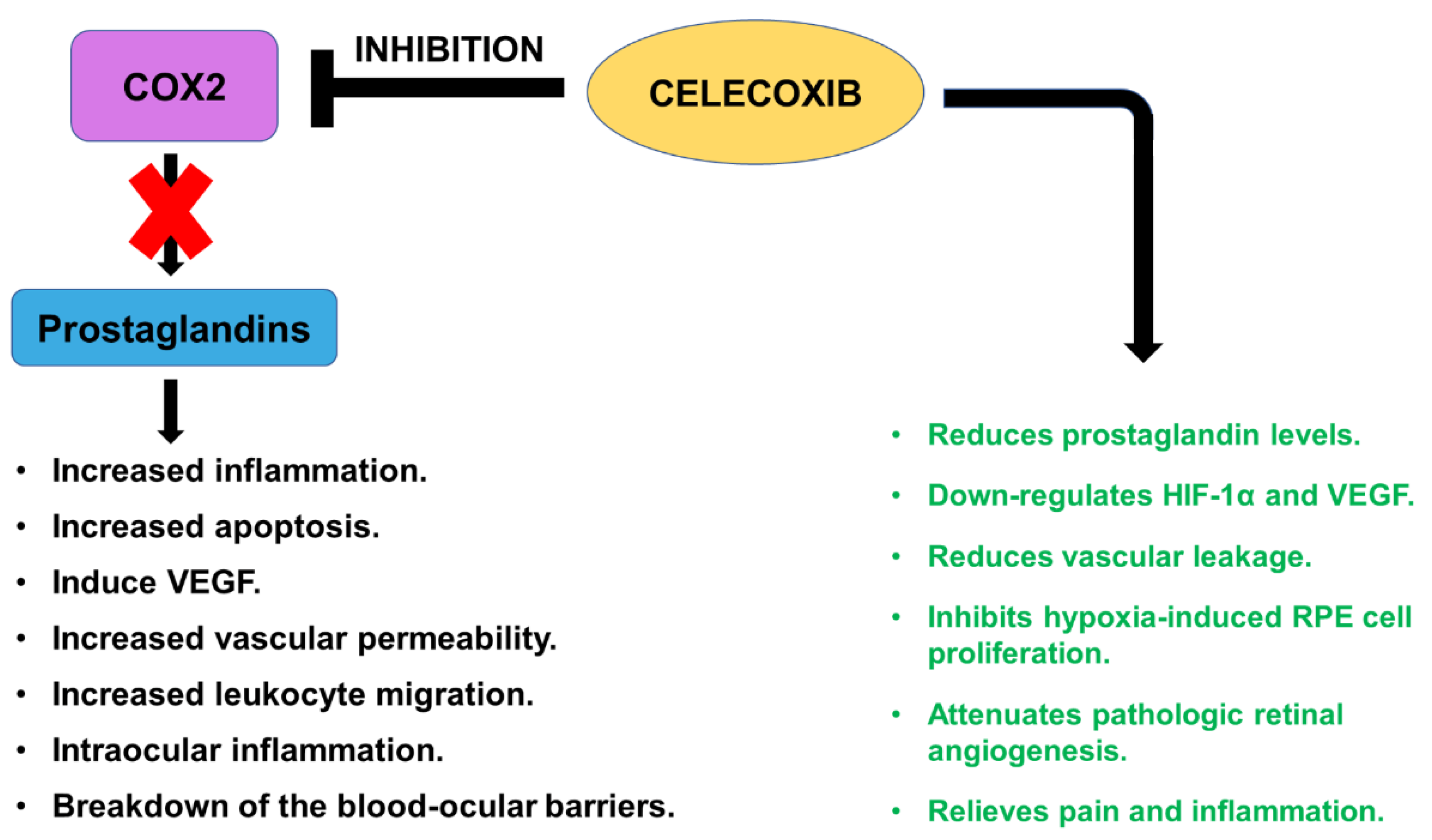
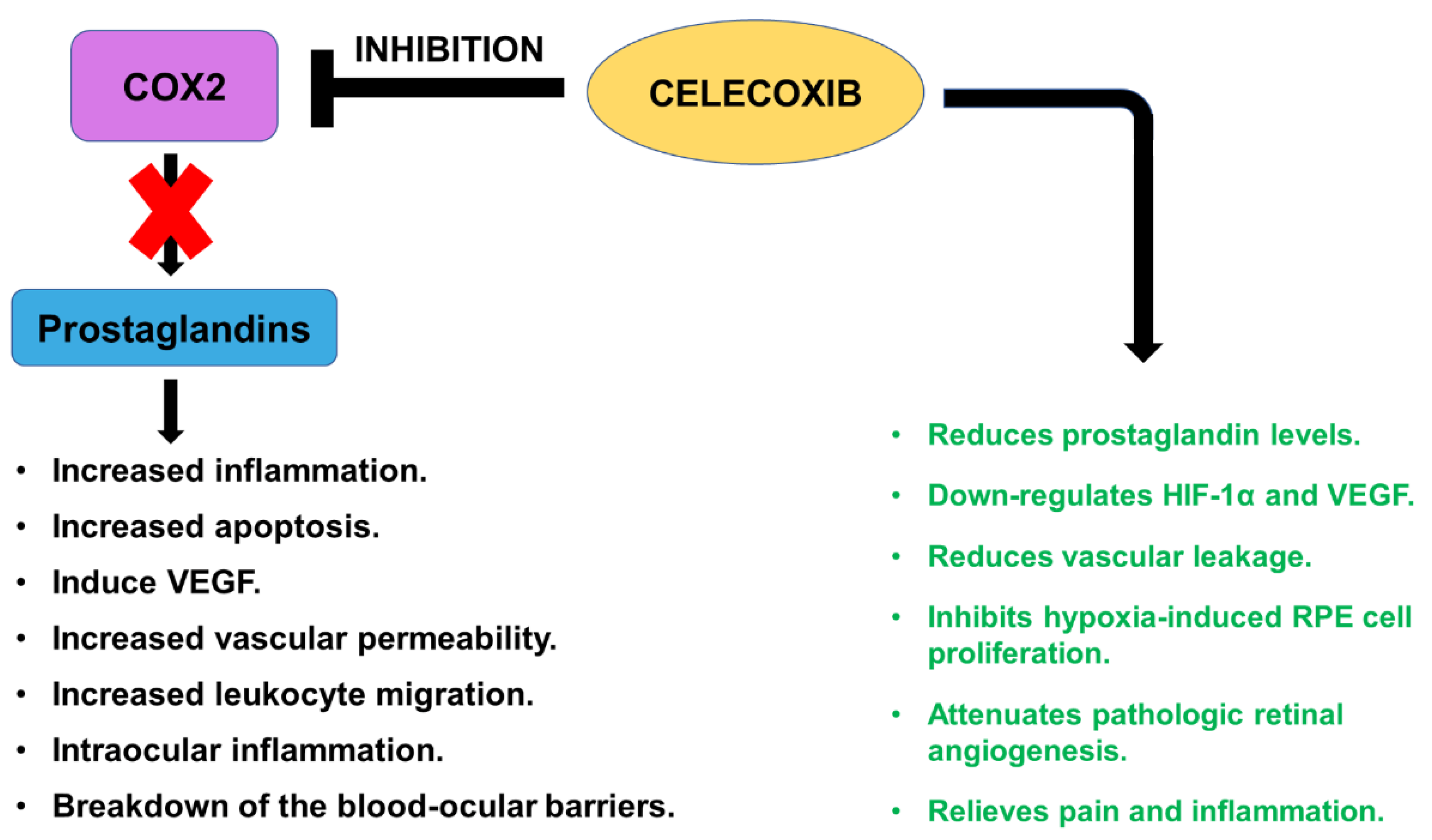
2.5. Celecoxib
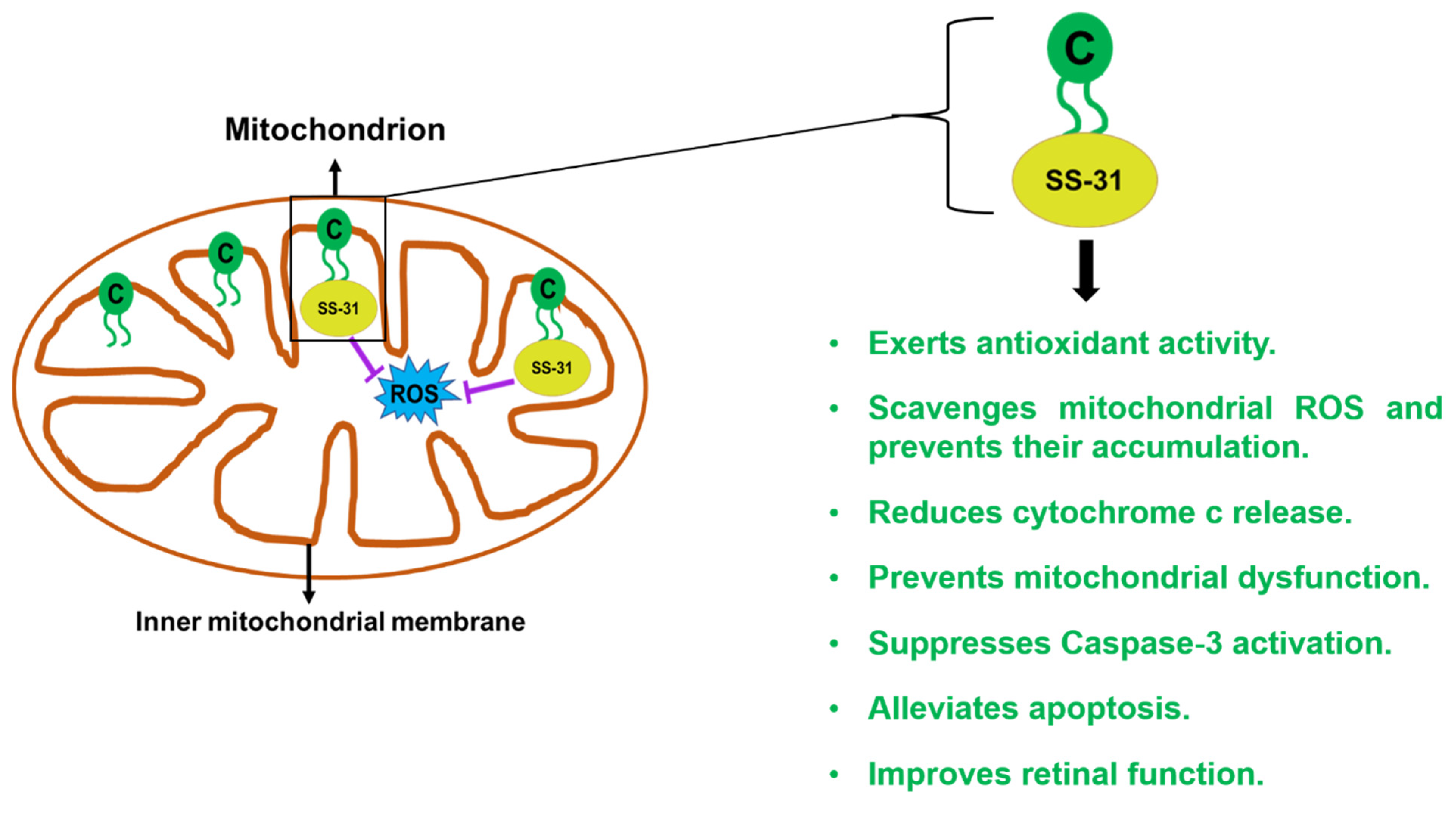
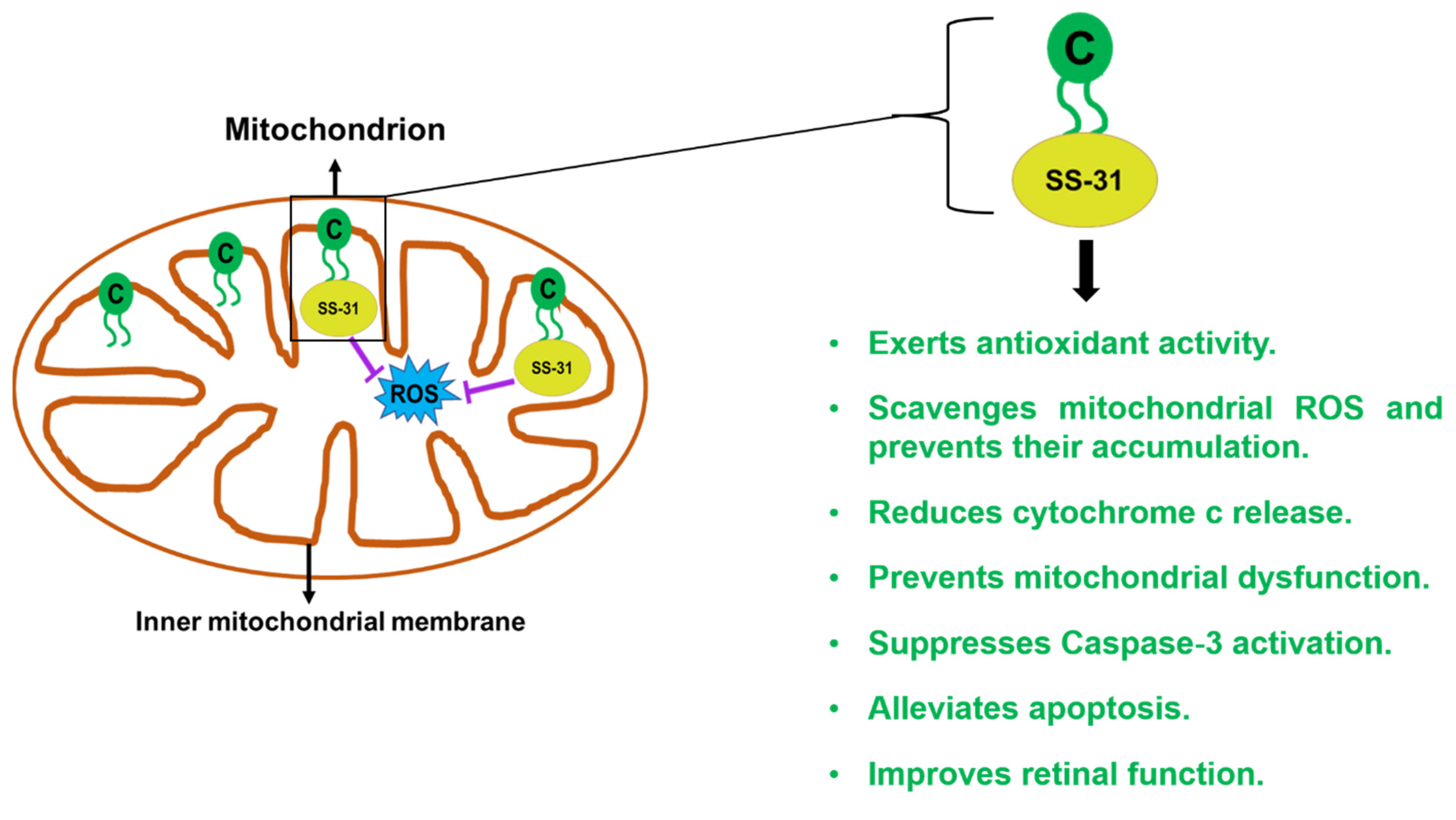
2.6. SS-31/Elamipretide
3. Conclusions and Future Directions
Funding
Conflicts of Interest
References
- Khan, S.S.; Singer, B.D.; Vaughan, D.E. Molecular and physiological manifestations and measurement of aging in humans. Aging Cell. 2017, 16, 624–633. [Google Scholar] [CrossRef]
- Flatt, T.; Partridge, L. Horizons in the evolution of aging. BMC Biol. 2018, 16, 93. [Google Scholar] [CrossRef]
- Ach, T.; Huisingh, C.; McGwin, G., Jr.; Messinger, J.D.; Zhang, T.; Bentley, M.J.; Gutierrez, D.B.; Ablonczy, Z.; Smith, R.T.; Sloan, K.R.; et al. Quantitative autofluorescence and cell density maps of the human retinal pigment epithelium. Investig. Ophthalmol. Vis. Sci. 2014, 55, 4832–4841. [Google Scholar] [CrossRef] [Green Version]
- Del Priore, L.V.; Kuo, Y.H.; Tezel, T.H. Age-related changes in human RPE cell density and apoptosis proportion in situ. Investig. Ophthalmol. Vis. Sci. 2002, 43, 3312–3318. [Google Scholar]
- Steinberg, J.S.; Fleckenstein, M.; Holz, F.G.; Schmitz-Valckenberg, S. Foveal sparing of reticular drusen in eyes with early and intermediate age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2015, 56, 4267–4274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sene, A.; Khan, A.A.; Cox, D.; Nakamura, R.E.; Santeford, A.; Kim, B.M.; Sidhu, R.; Onken, M.D.; Harbour, J.W.; Hagbi-Levi, S.; et al. Impaired cholesterol efflux in senescent macrophages promotes age-related macular degeneration. Cell Metab. 2013, 17, 549–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lafosse, E.; Wolffsohn, J.S.; Talens-Estarelles, C.; García-Lázaro, S. Presbyopia and the aging eye: Existing refractive approaches and their potential impact on dry eye signs and symptoms. Cont. Lens Anterior Eye 2020, 43, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.D.; Johnson, L.V.; Herrmann, R.; Farsiu, S.; Smith, S.G.; Groelle, M.; Mace, B.E.; Sullivan, P.; Jamison, J.A.; Kelly, U.; et al. Anti-amyloid therapy protects against retinal pigmented epithelium damage and vision loss in a model of age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2011, 108, E279–E287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruban, J.; Glotin, A.L.; Dinet, V.; Chalour, N.; Sennlaub, F.; Jonet, L.; An, N.; Faussat, A.M.; Mascarelli, F. Amyloid-beta(1-42) alters structure and function of retinal pigmented epithelial cells. Aging Cell. 2009, 8, 162–177. [Google Scholar] [CrossRef] [PubMed]
- Kolb, H.; Fernandez, E.; Nelson, R. Webvision: The Organization of the Retina and Visual System [Internet]; University of Utah Health Sciences Center: Salt Lake City, UT, USA, 1995. [Google Scholar]
- Freund, K.B.; Yannuzzi, L.A.; Sorenson, J.A. Age-related macular degeneration and choroidal neovascularization. Am. J. Ophthalmol. 1993, 115, 786–791. [Google Scholar] [CrossRef]
- Cunha-Vaz, J.; Bernardes, R.; Lobo, C. Blood-retinal barrier. Eur. J. Ophthalmol. 2011, 21 (Suppl. S6), S3–S9. [Google Scholar] [CrossRef]
- Campbell, M.; Humphries, P. The blood-retina barrier: Tight junctions and barrier modulation. Adv. Exp. Med. Biol. 2012, 763, 70–84. [Google Scholar] [PubMed]
- Fogli, S.; Del Re, M.; Rofi, E.; Posarelli, C.; Figus, M.; Danesi, R. Clinical pharmacology of intravitreal anti-VEGF drugs. Eye 2018, 32, 1010–1020. [Google Scholar] [CrossRef] [Green Version]
- Beatty, S.; Koh, H.; Phil, M.; Henson, D.; Boulton, M. The role of oxidative stress in the pathogenesis of age-related macular degeneration. Surv. Ophthalmol. 2000, 45, 115–134. [Google Scholar] [CrossRef] [Green Version]
- Nashine, S.; Cohen, P.; Chwa, M.; Lu, S.; Nesburn, A.B.; Kuppermann, B.D.; Kenney, M.C. Humanin G (HNG) protects age-related macular degeneration (AMD) transmitochondrial ARPE-19 cybrids from mitochondrial and cellular damage. Cell Death Dis. 2017, 8, e2951. [Google Scholar] [CrossRef] [PubMed]
- Nashine, S.; Cohen, P.; Nesburn, A.B.; Kuppermann, B.D.; Kenney, M.C. Characterizing the protective effects of SHLP2, a mitochondrial-derived peptide, in macular degeneration. Sci. Rep. 2018, 8, 15175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abokyi, S.; To, C.H.; Lam, T.T.; Tse, D.Y. Central role of oxidative stress in age-related macular degeneration: Evidence from a review of the molecular mechanisms and animal models. Oxid. Med. Cell. Longev. 2020, 2020, 7901270. [Google Scholar] [CrossRef] [Green Version]
- Yildirim, Z.; Ucgun, N.I.; Yildirim, F. The role of oxidative stress and antioxidants in the pathogenesis of age-related macular degeneration. Clinics 2011, 66, 743–746. [Google Scholar] [PubMed]
- Sparrow, J.R. Vitamin A-aldehyde adducts: AMD risk and targeted therapeutics. Proc. Natl. Acad. Sci. USA 2016, 113, 4564–4569. [Google Scholar] [CrossRef] [Green Version]
- Bosch-Morell, F.; Villagrasa, V.; Ortega, T.; Acero, N.; Muñoz-Mingarro, D.; González-Rosende, M.E.; Castillo, E.; Sanahuja, M.A.; Soriano, P. Medicinal plants and natural products as neuroprotective agents in age-related macular degeneration. Neural Regen Res. 2020, 15, 2207–2216. [Google Scholar]
- Hirota, K.; Matsui, M.; Iwata, S.; Nishiyama, A.; Mori, K.; Yodoi, J. AP-1 transcriptional activity is regulated by a direct association between thioredoxin and Ref-1. Proc. Natl. Acad. Sci. USA 1997, 94, 3633–3638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, Z.; Basi, D.; Li, Q.; Mariash, A.; Xia, Y.F.; Geng, J.G.; Kao, E.; Hall, J.L. Loss of redox factor 1 decreases NF-kappaB activity and increases susceptibility of endothelial cells to apoptosis. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 96–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sardar Pasha, S.P.B.; Sishtla, K.; Sulaiman, R.S.; Park, B.; Shetty, T.; Shah, F.; Fishel, M.L.; Wikel, J.H.; Kelley, M.R.; Corson, T.W. Ref-1/APE1 inhibition with novel small molecules blocks ocular neovascularization. J. Pharmacol. Exp. Ther. 2018, 367, 108–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Liu, X.; Zhou, T.; Kelley, M.R.; Edwards, P.A.; Gao, H.; Qiao, X. Suppression of choroidal neovascularization through inhibition of APE1/Ref-1 redox activity. Investig. Ophthalmol. Vis. Sci. 2014, 55, 4461–4469. [Google Scholar] [CrossRef]
- Silvestri, G.; Williams, M.A.; McAuley, C.; Oakes, K.; Sillery, E.; Henderson, D.C.; Ferguson, S.; Silvestri, V.; Muldrew, K.A. Drusen prevalence and pigmentary changes in Caucasians aged 18–54 years. Eye 2012, 26, 1357–1362. [Google Scholar] [CrossRef]
- Abdelsalam, A.; Del Priore, L.; Zarbin, M.A. Drusen in age-related macular degeneration: Pathogenesis, natural course, and laser photocoagulation-induced regression. Surv. Ophthalmol. 1999, 44, 1–29. [Google Scholar] [CrossRef]
- Crabb, J.W.; Miyagi, M.; Gu, X.; Shadrach, K.; West, K.A.; Sakaguchi, H.; Kamei, M.; Hasan, A.; Yan, L.; Rayborn, M.E.; et al. Drusen proteome analysis: An approach to the etiology of age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2002, 99, 14682–14687. [Google Scholar] [CrossRef] [Green Version]
- Anderson, D.H.; Mullins, R.F.; Hageman, G.S.; Johnson, L.V. A role for local inflammation in the formation of drusen in the aging eye. Am. J. Ophthalmol. 2002, 134, 411–431. [Google Scholar] [CrossRef]
- Kurji, K.H.; Cui, J.Z.; Lin, T.; Harriman, D.; Prasad, S.S.; Kojic, L.; Matsubara, J.A. Microarray analysis identifies changes in inflammatory gene expression in response to amyloid-beta stimulation of cultured human retinal pigment epithelial cells. Investig. Ophthalmol. Vis. Sci. 2010, 51, 1151–1163. [Google Scholar] [CrossRef]
- Sandgren, O. Ocular amyloidosis, with special reference to the hereditary forms with vitreous involvement. Surv. Ophthalmol 1995, 40, 173–196. [Google Scholar] [CrossRef]
- Teng, J.; Takei, Y.; Harada, A.; Nakata, T.; Chen, J.; Hirokawa, N. Synergistic effects of MAP2 and MAP1B knockout in neuronal migration, dendritic outgrowth, and microtubule organization. J. Cell. Biol. 2001, 155, 65–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luheshi, L.M.; Tartaglia, G.G.; Brorsson, A.C.; Pawar, A.P.; Watson, I.E.; Chiti, F.; Vendruscolo, M.; Lomas, D.A.; Dobson, C.M.; Crowther, D.C. Systematic in vivo analysis of the intrinsic determinants of amyloid Beta pathogenicity. PLoS Biol. 2007, 5, e290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Zhu, C.; Xu, Y.; Liu, B.; Wang, M.; Wu, K. Development and expression of amyloid-beta peptide 42 in retinal ganglion cells in rats. Anat. Rec. 2011, 294, 1401–1405. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Ohno-Matsui, K.; Ichinose, S.; Sato, T.; Iwata, N.; Saido, T.C.; Hisatomi, T.; Mochizuki, M.; Morita, I. The potential role of amyloid beta in the pathogenesis of age-related macular degeneration. J. Clin. Investig. 2005, 115, 2793–2800. [Google Scholar] [CrossRef] [PubMed]
- Ward, R.V.; Jennings, K.H.; Jepras, R.; Neville, W.; Owen, D.E.; Hawkins, J.; Christie, G.; Davis, J.B.; George, A.; Karran, E.H.; et al. Fractionation and characterization of oligomeric, protofibrillar and fibrillar forms of beta-amyloid peptide. Biochem. J. 2000, 348, 137–144. [Google Scholar] [CrossRef]
- Sabate, R.; Estelrich, J. Evidence of the existence of micelles in the fibrillogenesis of beta-amyloid peptide. J. Phys. Chem. B 2005, 109, 11027–11032. [Google Scholar] [CrossRef]
- Prasad, T.; Zhu, P.; Verma, A.; Chakrabarty, P.; Rosario, A.M.; Golde, T.E.; Li, Q. Amyloid β peptides overexpression in retinal pigment epithelial cells via AAV-mediated gene transfer mimics AMD-like pathology in mice. Sci. Rep. 2017, 7, 3222. [Google Scholar] [CrossRef]
- Gravius, A.; Klein, K.; Levin, L.; Lagreze, W.; Wegener, N.; Danysz, W. Topical administration of MRZ-99030, a β-amyloid aggregation modulator, protects retinal ganglion cells and axons in a rodent model of glaucoma. Invest. Ophthalmol. Vis. Sci. 2013, 54, 2625. [Google Scholar]
- Parsons, C.G.; Ruitenberg, M.; Freitag, C.E.; Sroka-Saidi, K.; Russ, H.; Rammes, G. MRZ-99030—A novel modulator of Aβ aggregation: I—Mechanism of action (MoA) underlying the potential neuroprotective treatment of Alzheimer’s disease, glaucoma and age-related macular degeneration (AMD). Neuropharmacology 2015, 92, 158–169. [Google Scholar] [CrossRef]
- Frydman-Marom, A.; Rechter, M.; Shefler, I.; Bram, Y.; Shalev, D.E.; Gazit, E. Cognitive-performance recovery of Alzheimer’s disease model mice by modulation of early soluble amyloidal assemblies. Angew. Chem. Int. Ed. Engl. 2009, 48, 1981–1986. [Google Scholar] [CrossRef]
- Rammes, G.; Parsons, C.G. The Aβ aggregation modulator MRZ-99030 prevents and even reverses synaptotoxic effects of Aβ1-42 on LTP even following serial dilution to a 500:1 stoichiometric excess of Aβ1-42, suggesting a beneficial prion-like seeding mechanism. Neuropharmacology 2020, 179, 108267. [Google Scholar] [CrossRef]
- Adler, R.; Landa, K.B.; Manthorpe, M.; Varon, S. Cholinergic neuronotrophic factors: Intraocular distribution of trophic activity for ciliary neurons. Science 1979, 204, 1434–1436. [Google Scholar] [CrossRef] [Green Version]
- LaVail, M.M.; Yasumura, D.; Matthes, M.T.; Lau-Villacorta, C.; Unoki, K.; Sung, C.H.; Steinberg, R.H. Protection of mouse photoreceptors by survival factors in retinal degenerations. Investig. Ophthalmol. Vis. Sci. 1998, 39, 592–602. [Google Scholar]
- Li, R.; Wen, R.; Banzon, T.; Maminishkis, A.; Miller, S.S. CNTF mediates neurotrophic factor secretion and fluid absorption in human retinal pigment epithelium. PLoS ONE 2011, 6, e23148. [Google Scholar] [CrossRef] [Green Version]
- Koh, S.W. Ciliary neurotrophic factor released by corneal endothelium surviving oxidative stress ex vivo. Investig. Ophthalmol. Vis. Sci. 2002, 43, 2887–2896. [Google Scholar]
- Sendtner, M.; Götz, R.; Holtmann, B.; Thoenen, H. Endogenous ciliary neurotrophic factor is a lesion factor for axotomized motoneurons in adult mice. J. Neurosci. 1997, 17, 6999–7006. [Google Scholar] [CrossRef]
- ALS CNTF Treatment Study Group. A double-blind placebo-controlled clinical trial of subcutaneous recombinant human ciliary neurotrophic factor (rHCNTF) in amyotrophic lateral sclerosis ALS CNTF Treatment Study Group. Neurology 1996, 46, 1244–1249. [Google Scholar] [CrossRef] [PubMed]
- Dittrich, F.; Thoenen, H.; Sendtner, M. Ciliary neurotrophic factor: Pharmacokinetics and acute-phase response in rat. Ann. Neurol. 1994, 35, 151–163. [Google Scholar] [CrossRef] [Green Version]
- Maysinger, D.; Krieglstein, K.; Filipovic-Grcic, J.; Sendtner, M.; Unsicker, K.; Richardson, P. Microencapsulated ciliary neurotrophic factor: Physical properties and biological activities. Exp. Neurol. 1996, 138, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Kauper, K.; McGovern, C.; Sherman, S.; Heatherton, P.; Rapoza, R.; Stabila, P.; Dean, B.; Lee, A.; Borges, S.; Bouchard, B.; et al. Two-year intraocular delivery of ciliary neurotrophic factor by encapsulated cell technology implants in patients with chronic retinal degenerative diseases. Investig. Ophthalmol. Vis. Sci. 2012, 53, 7484–7491. [Google Scholar] [CrossRef] [PubMed]
- Bokoch, G.M. Biology of the Rap proteins, members of the ras superfamily of GTP-binding proteins. Biochem. J. 1993, 289, 17–24. [Google Scholar] [CrossRef] [Green Version]
- Zwartkruis, F.J.; Bos, J.L. Ras and Rap1: Two highly related small GTPases with distinct function. Exp. Cell Res. 1999, 253, 157–165. [Google Scholar] [CrossRef]
- Wittchen, E.S.; Worthylake, R.A.; Kelly, P.; Casey, P.J.; Quilliam, L.A.; Burridge, K. Rap1 GTPase inhibits leukocyte transmigration by promoting endothelial barrier function. J. Biol. Chem. 2005, 280, 11675–11682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wittchen, E.S.; Hartnett, M.E. The small GTPase Rap1 is a novel regulator of RPE cell barrier function. Investig. Ophthalmol. Vis. Sci. 2011, 52, 7455–7463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Yan, J.; De, P.; Chang, H.C.; Yamauchi, A.; Christopherson, K.W., 2nd; Paranavitana, N.C.; Peng, X.; Kim, C.; Munugalavadla, V.; et al. Rap1a null mice have altered myeloid cell functions suggesting distinct roles for the closely related Rap1a and 1b proteins. J. Immunol. 2007, 179, 8322–8331. [Google Scholar] [CrossRef] [Green Version]
- Chrzanowska-Wodnicka, M.; White, G.C., 2nd; Quilliam, L.A.; Whitehead, K.J. Small GTPase Rap1 is essential for mouse development and formation of functional vasculature. PLoS ONE 2015, 10, e0145689. [Google Scholar]
- Yan, J.; Li, F.; Ingram, D.A.; Quilliam, L.A. Rap1a is a key regulator of fibroblast growth factor 2-induced angiogenesis and together with Rap1b controls human endothelial cell functions. Mol. Cell Biol. 2008, 28, 5803–5810. [Google Scholar] [CrossRef] [Green Version]
- Pizon, V.; Lerosey, I.; Chardin, P.; Tavitian, A. Nucleotide sequence of a human cDNA encoding a ras-related protein (rap1B). Nucleic Acids Res. 1988, 16, 7719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmona, G.; Göttig, S.; Orlandi, A.; Scheele, J.; Bäuerle, T.; Jugold, M.; Kiessling, F.; Henschler, R.; Zeiher, A.M.; Dimmeler, S.; et al. Role of the small GTPase Rap1 for integrin activity regulation in endothelial cells and angiogenesis. Blood 2009, 113, 488–497. [Google Scholar] [CrossRef]
- Fukuhra, S.; Sakurai, A.; Yamagishi, A.; Sako, K.; Mochizuki, N. Vascular endothelial cadherin-mediated cell-cell adhesion regulated by a small GTPase, Rap1. J. Biochem. Mol. Biol. 2006, 39, 132–139. [Google Scholar] [CrossRef]
- Duchniewicz, M.; Zemojtel, T.; Kolanczyk, M.; Grossmann, S.; Scheele, J.S.; Zwartkruis, F.J. Rap1A-deficient T and B cells show impaired integrin-mediated cell adhesion. Mol. Cell. Biol. 2006, 26, 643–653. [Google Scholar] [CrossRef] [Green Version]
- Price, L.S.; Hajdo-Milasinovic, A.; Zhao, J.; Zwartkruis, F.J.; Collard, J.G.; Bos, J.L. Rap1 regulates E-cadherin-mediated cell-cell adhesion. J. Biol. Chem. 2004, 279, 35127–35132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hogan, C.; Serpente, N.; Cogram, P.; Hosking, C.R.; Bialucha, C.U.; Feller, S.M.; Braga, V.M.; Birchmeier, W.; Fujita, Y. Rap1 regulates the formation of E-cadherin-based cell-cell contacts. Mol. Cell. Biol. 2004, 24, 6690–6700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Rooij, J.; Rehmann, H.; van Triest, M.; Cool, R.H.; Wittinghofer, A.; Bos, J.L. Mechanism of regulation of the Epac family of cAMP-dependent RapGEFs. J. Biol. Chem. 2000, 275, 20829–20836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawasaki, H.; Springett, G.M.; Mochizuki, N.; Toki, S.; Nakaya, M.; Matsuda, M.; Housman, D.E.; Graybiel, A.M. A family of cAMP-binding proteins that directly activate Rap1. Science 1998, 282, 2275–2279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos, C.J.; Lin, C.; Liu, X.; Antonetti, D.A. The EPAC-Rap1 pathway prevents and reverses cytokine-induced retinal vascular permeability. J. Biol. Chem. 2018, 293, 717–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birukova, A.A.; Fu, P.; Xing, J.; Birukov, K.G. Rap1 mediates protective effects of iloprost against ventilator-induced lung injury. J. Appl. Physiol. (1985) 2009, 107, 1900–1910. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Zhang, R.; Wang, C.; Wang, X.; Xu, M.; Ma, J.; Shang, Q. Activation of the small GTPase Rap1 inhibits Choroidal neovascularization by regulating cell junctions and ROS generation in rats. Curr. Eye Res. 2018, 43, 934–940. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Han, X.; Bretz, C.A.; Becker, S.; Gambhir, D.; Smith, G.W.; Samulski, R.J.; Wittchen, E.S.; Quilliam, L.A.; Chrzanowska-Wodnicka, M.; et al. Retinal pigment epithelial cell expression of active Rap 1a by scAAV2 inhibits choroidal neovascularization. Mol. Ther. Methods Clin. Dev. 2016, 3, 16056. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Fotheringham, L.; Wittchen, E.S.; Hartnett, M.E. Rap1 GTPase inhibits tumor necrosis factor-α-induced choroidal endothelial migration via NADPH oxidase- and NF-κB-dependent activation of Rac1. Am. J. Pathol. 2015, 185, 3316–3325. [Google Scholar] [CrossRef] [Green Version]
- Kauppinen, A.; Paterno, J.J.; Blasiak, J.; Salminen, A.; Kaarniranta, K. Inflammation and its role in age-related macular degeneration. Cell. Mol. Life Sci. 2016, 73, 1765–1786. [Google Scholar] [CrossRef] [Green Version]
- Nashine, S.; Chwa, M.; Kazemian, M.; Thaker, K.; Lu, S.; Nesburn, A.; Kuppermann, B.D.; Kenney, M.C. Differential expression of complement markers in normal and AMD transmitochondrial Cybrids. PLoS ONE 2016, 11, e0159828. [Google Scholar] [CrossRef] [Green Version]
- Schoenberger, S.D.; Kim, S.J.; Sheng, J.; Rezaei, K.A.; Lalezary, M.; Cherney, E. Increased prostaglandin E2 (PGE2) levels in proliferative diabetic retinopathy, and correlation with VEGF and inflammatory cytokines. Investig. Ophthalmol. Vis. Sci. 2012, 53, 5906–5911. [Google Scholar] [CrossRef] [Green Version]
- Qazi, Y.; Maddula, S.; Ambati, B.K. Mediators of ocular angiogenesis. J. Genet. 2009, 88, 495–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chin, M.S.; Nagineni, C.N.; Hooper, L.C.; Detrick, B.; Hooks, J.J. Cyclooxygenase-2 gene expression and regulation in human retinal pigment epithelial cells. Investig. Ophthalmol. Vis. Sci. 2001, 42, 2338–2346. [Google Scholar]
- Wilkinson-Berka, J.L.; Alousis, N.S.; Kelly, D.J.; Gilbert, R.E. COX-2 inhibition and retinal angiogenesis in a mouse model of retinopathy of prematurity. Investig. Ophthalmol. Vis. Sci. 2003, 44, 974–979. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.Z.; Cai, N.; Liu, N.N. Celecoxib down-regulates the hypoxia-induced expression of HIF-1α and VEGF through the PI3K/AKT pathway in retinal pigment epithelial cells. Cell. Physiol. Biochem. 2017, 44, 1640–1650. [Google Scholar] [CrossRef] [Green Version]
- Amrite, A.C.; Kompella, U.B. Celecoxib inhibits proliferation of retinal pigment epithelial and choroid-retinal endothelial cells by a cyclooxygenase-2-independent mechanism. J. Pharmacol. Exp. Ther. 2008, 324, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Amrite, A.C.; Ayalasomayajula, S.P.; Cheruvu, N.P.; Kompella, U.B. Single periocular injection of celecoxib-PLGA microparticles inhibits diabetes-induced elevations in retinal PGE2, VEGF, and vascular leakage. Investig. Ophthalmol. Vis. Sci. 2006, 47, 1149–1160. [Google Scholar] [CrossRef] [Green Version]
- Ayalasomayajula, S.P.; Kompella, U.B. Celecoxib, a selective cyclooxygenase-2 inhibitor, inhibits retinal vascular endothelial growth factor expression and vascular leakage in a streptozotocin-induced diabetic rat model. Eur. J. Pharmacol. 2003, 458, 283–289. [Google Scholar] [CrossRef]
- Ayalasomayajula, S.P.; Kompella, U.B. Subconjunctivally administered celecoxib-PLGA microparticles sustain retinal drug levels and alleviate diabetes-induced oxidative stress in a rat model. Eur. J. Pharmacol. 2005, 511, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Hill, B.G.; Benavides, G.A.; Lancaster, J.R., Jr.; Ballinger, S.; Dell’Italia, L.; Jianhua, Z.; Darley-Usmar, V.M. Integration of cellular bioenergetics with mitochondrial quality control and autophagy. Biol. Chem. 2012, 393, 1485–1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.J.; Toma, H.; Shah, R.; Kompella, U.B.; Vooturi, S.K.; Sheng, J. The safety, pharmacokinetics, and efficacy of intraocular celecoxib. Investig. Ophthalmol. Vis. Sci. 2014, 55, 1409–1418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seedher, N.; Bhatia, S. Solubility enhancement of Cox-2 inhibitors using various solvent systems. AAPS PharmSciTech. 2003, 4, E33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terluk, M.R.; Kapphahn, R.J.; Soukup, L.M.; Gong, H.; Gallardo, C.; Montezuma, S.R.; Ferrington, D.A. Investigating mitochondria as a target for treating age-related macular degeneration. J. Neurosci. 2015, 35, 7304–7311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikon, N.; Ryan, R.O. Cardiolipin and mitochondrial cristae organization. Biochim. Biophys. Acta Biomembr. 2017, 1859, 1156–1163. [Google Scholar] [CrossRef]
- Paradies, G.; Paradies, V.; De Benedictis, V.; Ruggiero, F.M.; Petrosillo, G. Functional role of cardiolipin in mitochondrial bioenergetics. Biochim. Biophys. Acta 2014, 1837, 408–417. [Google Scholar] [CrossRef] [Green Version]
- Dudek, J. Role of cardiolipin in mitochondrial signaling pathways. Front. Cell. Dev. Biol. 2017, 5, 90. [Google Scholar] [CrossRef] [Green Version]
- Batandier, C.; Leverve, X.; Fontaine, E. Opening of the mitochondrial permeability transition pore induces reactive oxygen species production at the level of the respiratory chain complex I. J. Biol. Chem. 2004, 279, 17197–17204. [Google Scholar] [CrossRef] [Green Version]
- Szeto, H.H. Cell-permeable, mitochondrial-targeted, peptide antioxidants. AAPS J. 2006, 8, E277–E283. [Google Scholar] [CrossRef]
- Wu, X.; Pang, Y.; Zhang, Z.; Li, X.; Wang, C.; Lei, Y.; Li, A.; Yu, L.; Ye, J. Mitochondria-targeted antioxidant peptide SS-31 mediates neuroprotection in a rat experimental glaucoma model. Acta Biochim. Biophys. Sin. 2019, 51, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Liu, B.; Gao, Q.; Zhuo, Y.; Ge, J. Mitochondria-targeted peptide MTP-131 alleviates mitochondrial dysfunction and oxidative damage in human trabecular meshwork cells. Investig. Ophthalmol. Vis. Sci. 2011, 52, 7027–7037. [Google Scholar] [CrossRef] [PubMed]
- Allingham, M.J.; Mettu, P.S.; Cousins, S.W. Elamipretide, a Mitochondrial-Targeted Drug, for the Treatment of Vision Loss in Dry AMD with High Risk Drusen: Results of the Phase 1 ReCLAIM Study. Invest. Ophthalmol. Vis. Sci. 2019, 60, 361. [Google Scholar]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nashine, S. Potential Therapeutic Candidates for Age-Related Macular Degeneration (AMD). Cells 2021, 10, 2483. https://doi.org/10.3390/cells10092483
Nashine S. Potential Therapeutic Candidates for Age-Related Macular Degeneration (AMD). Cells. 2021; 10(9):2483. https://doi.org/10.3390/cells10092483
Chicago/Turabian StyleNashine, Sonali. 2021. "Potential Therapeutic Candidates for Age-Related Macular Degeneration (AMD)" Cells 10, no. 9: 2483. https://doi.org/10.3390/cells10092483
APA StyleNashine, S. (2021). Potential Therapeutic Candidates for Age-Related Macular Degeneration (AMD). Cells, 10(9), 2483. https://doi.org/10.3390/cells10092483