
The Interdependency and Co-Regulation of the Vitamin D and Cholesterol Metabolism


 ,
, 
Abstract
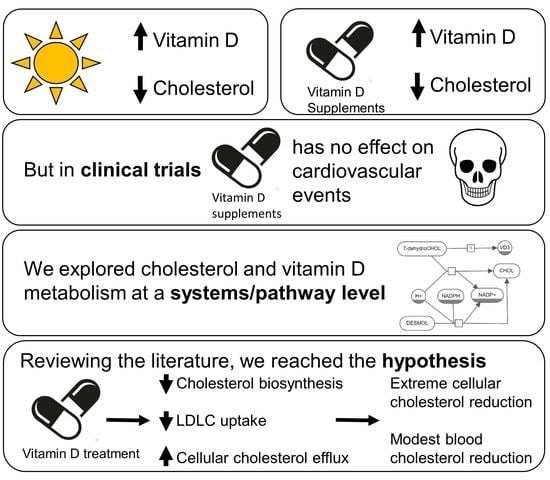
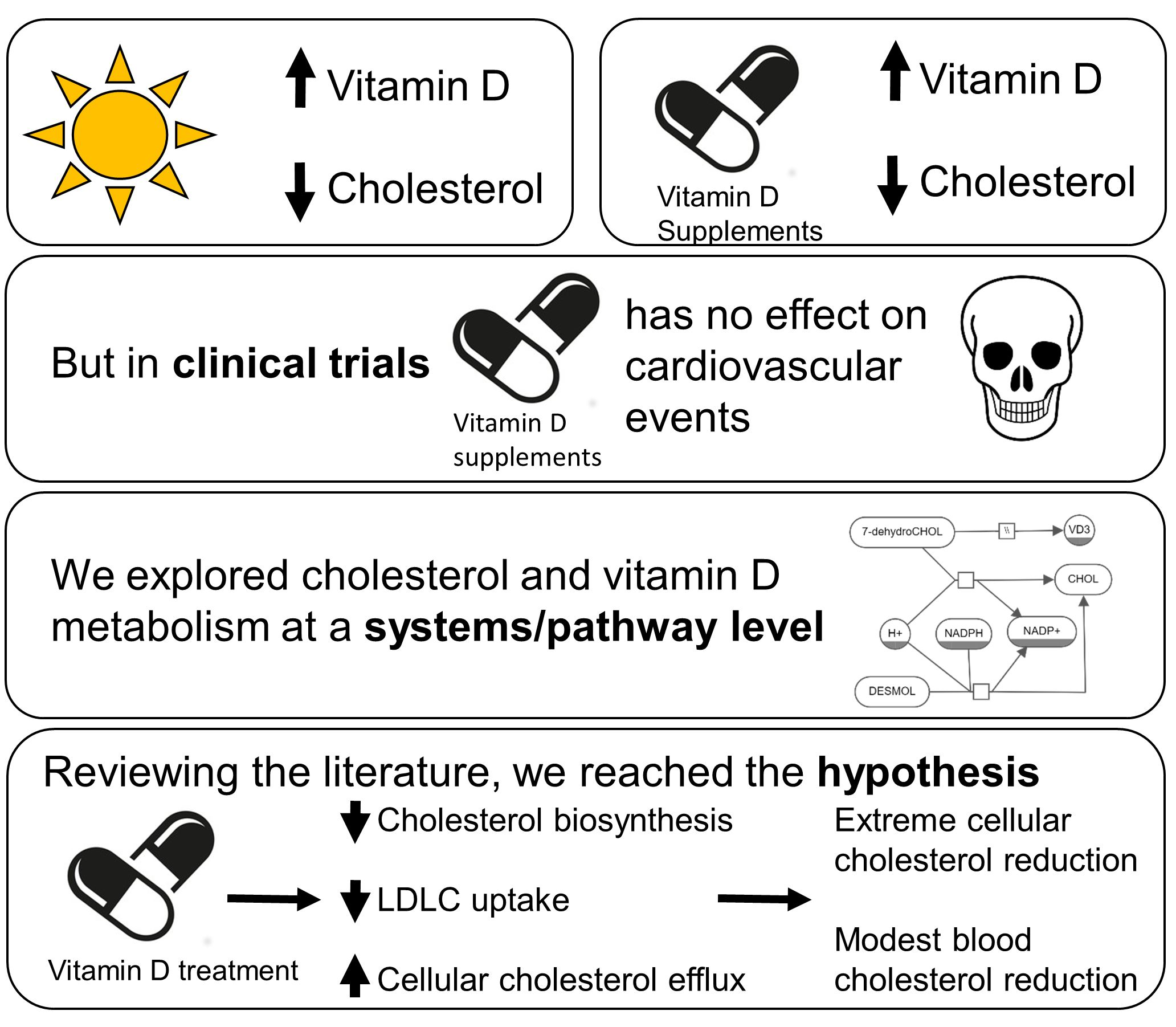{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Cholesterol in Health and Disease
3. Vitamin D in Health and Disease
4. Computational Modelling of Vitamin D and Cholesterol Metabolism
5. A Bidirectional Relationship between Cholesterol and Vitamin D Metabolisms
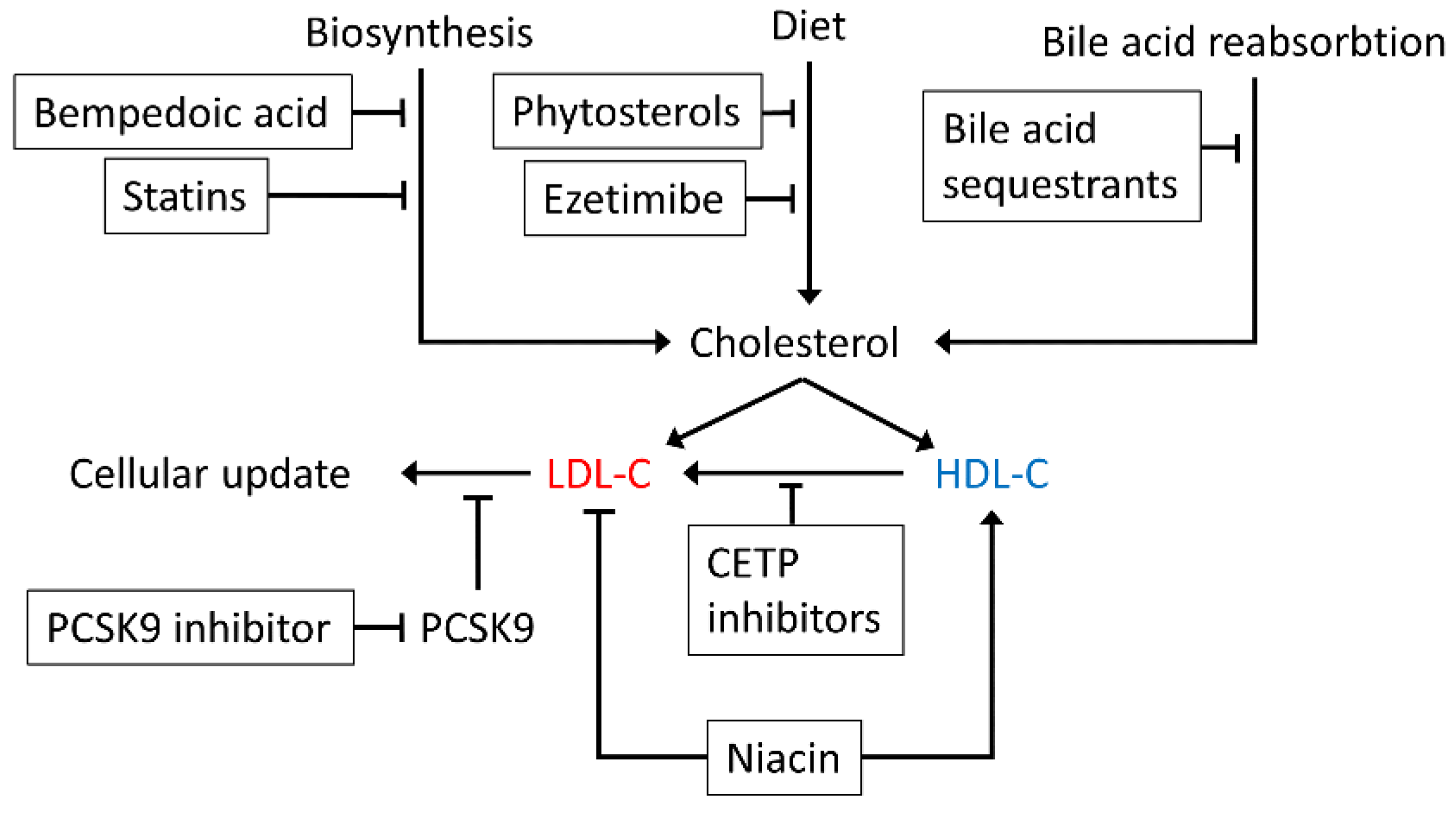
6. The Effect of Statins
7. Feedback from Vitamin D Metabolites
8. DHCR7 and Smith–Lemli–Opitz Syndrome
9. Variants and Mutations
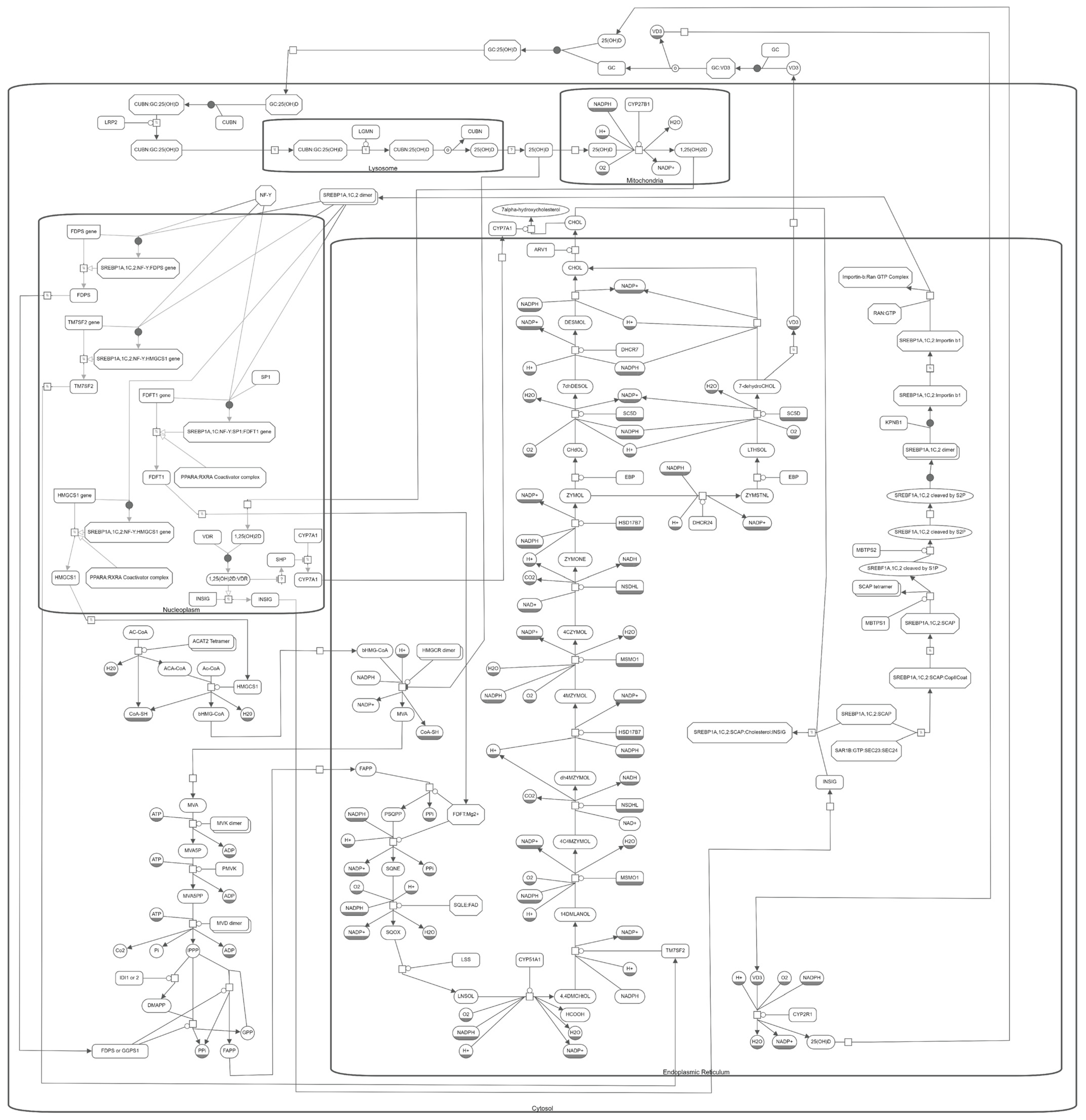
10. The Molecular Pathway of Vitamin D and Cholesterol Metabolism
11. Discussion
12. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Bullamore, J.R.; Wilkinson, R.; Gallagher, J.C.; Nordin, B.E.; Marshall, D.H. Effect of age on calcium absorption. Lancet 1970, 296, 535–537. [Google Scholar] [CrossRef]
- Jones, G. Metabolism and biomarkers of Vitamin D. Scand. J. Clin. Lab. Investig. 2012, 72, 7–13. [Google Scholar]
- Christodoulou, S.; Goula, T.; Ververidis, A.; Drosos, G. Vitamin D and bone disease. BioMed Res. Int. 2013, 2013, 396541. [Google Scholar] [CrossRef]
- Reid, I.R.; Bolland, M.J. Skeletal and Nonskeletal Effects of Vitamin D: Is Vitamin D a Tonic for Bone and Other Tissues? Springer: London, UK, 2014; pp. 2347–2357. [Google Scholar]
- Jeon, S.-M.; Shin, E.-A. Exploring vitamin D metabolism and function in cancer. Exp. Mol. Med. 2018, 50, 1–14. [Google Scholar] [CrossRef]
- Jolliffe, D.A.; Stefanidis, C.; Wang, Z.; Kermani, N.Z.; Dimitrov, V.; White, J.H.; McDonough, J.; Janssens, W.; Pfeffer, P.; Griffiths, C.J.; et al. Vitamin D metabolism is dysregulated in asthma and chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2020, 202, 371–382. [Google Scholar] [CrossRef]
- Buondonno, I.; Rovera, G.; Sassi, F.; Rigoni, M.M.; Lomater, C.; Parisi, S.; Pellerito, R.; Isaia, G.C.; D’Amelio, P. Vitamin D and immunomodulation in early rheumatoid arthritis: A randomized double-blind placebo-controlled study. PLoS ONE 2017, 12, e0178463. [Google Scholar] [CrossRef] [PubMed]
- Treiber, G.; Prietl, B.; Fröhlich-Reiterer, E.; Lechner, E.; Ribitsch, A.; Fritsch, M.; Rami-Merhar, B.; Steigleder-Schweiger, C.; Graninger, W.; Borkenstein, M.; et al. Cholecalciferol supplementation improves suppressive capacity of regulatory T-cells in young patients with new-onset type 1 diabetes mellitus—A randomized clinical trial. Clin. Immunol. 2015, 161, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Sassi, F.; Tamone, C.; D’Amelio, P. Vitamin D: Nutrient, hormone, and immunomodulator. Nutrients 2018, 10, 1656. [Google Scholar] [CrossRef]
- DeLuca, H.F. Evolution of our understanding of vitamin D. Nutr. Rev. 2008, 66, S73–S87. [Google Scholar] [CrossRef] [PubMed]
- Gil, A.; Plaza-Diaz, J.; Mesa, M.D. Vitamin D: Classic and novel actions. Ann. Nutr. Metab. 2018, 72, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Zidovetzki, R.; Levitan, I. Use of cyclodextrins to manipulate plasma membrane cholesterol content: Evidence, misconceptions and control strategies. Biochim. Biophys. Acta Biomembr. 2007, 1768, 1311–1324. [Google Scholar] [CrossRef] [PubMed]
- Mazein, A.; Watterson, S.; Hsieh, W.Y.; Griffiths, W.J.; Ghazal, P. A comprehensive machine-readable view of the mammalian cho-lesterol biosynthesis pathway. Biochem. Pharmacol. 2013, 86, 56–66. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, Q. Cholesterol metabolism and homeostasis in the brain. Protein Cell 2015, 6, 254–264. [Google Scholar] [CrossRef] [PubMed]
- Henderson, R.; O’Kane, M.; McGilligan, V.; Watterson, S. The genetics and screening of familial hypercholesterolaemia. J. Biomed. Sci. 2016, 23, 39. [Google Scholar] [CrossRef] [PubMed]
- Parton, A.; McGilligan, V.; Chemaly, M.; O’Kane, M.; Watterson, S. New models of atherosclerosis and multi-drug therapeutic interventions. Bioinformatics 2018, 35, 2449–2457. [Google Scholar] [CrossRef]
- Robertson, K.A.; Hsieh, W.Y.; Forster, T.; Blanc, M.; Lu, H.; Crick, P.J.; Yutuc, E.; Watterson, S.; Martin, K.; Griffiths, S.J.; et al. An interferon regulated MicroRNA provides broad cell-intrinsic antiviral immunity through multihit host-directed targeting of the sterol pathway. PLoS Biol. 2016, 14, e1002364. [Google Scholar] [CrossRef]
- Blanc, M.; Hsieh, W.Y.; Robertson, K.A.; Watterson, S.; Shui, G.; Lacaze, P.; Khondoker, M.; Dickinson, P.; Sing, G.; Rodríguez-Martín, S.; et al. Host defense against viral infection involves interferon mediated down-regulation of sterol biosynthesis. PLoS Biol. 2011, 9, e1000598. [Google Scholar] [CrossRef]
- Portincasa, P.; Moschetta, A.; Palasciano, G. Cholesterol gallstone disease. Lancet 2006, 368, 230–239. [Google Scholar] [CrossRef]
- Yvan-Charvet, L.; Bonacina, F.; Guinamard, R.R.; Norata, G.D. Immunometabolic function of cholesterol in cardiovascular disease and beyond. Cardiovasc. Res. 2019, 115, 1393–1407. [Google Scholar] [CrossRef]
- Skaaby, T. The relationship of vitamin D status to risk of cardiovascular disease and mortality. Dan. Med. J. 2015, 62, B5008. [Google Scholar]
- Mozos, I.; Marginean, O. Links between Vitamin D deficiency and cardiovascular diseases. BioMed Res. Int. 2015, 2015, 109275. [Google Scholar] [CrossRef] [PubMed]
- Lupton, J.R.; Faridi, K.F.; Martin, S.S.; Sharma, S.; Kulkarni, K.; Jones, S.R. Deficient serum 25-hydroxyvitamin D is associated with an atherogenic lipid profile: The very large database of lipids (VLDL-3) study. J. Clin. Lipidol. 2016, 10, 72–81. [Google Scholar] [CrossRef]
- Zittermann, A.; Trummer, C.; Theiler-Schwetz, V.; Lerchbaum, E.; März, W.; Pilz, S. Vitamin D and cardiovascular disease: An updated narrative review. Int. J. Mol. Sci. 2021, 22, 2896. [Google Scholar] [CrossRef]
- Zhang, Y.; Fang, F.; Tang, J.; Jia, L.; Feng, Y.; Xu, P.; Faramand, A. Association between vitamin D supplementation and mortality: Systematic review and meta-analysis. BMJ 2019, 366, l4673. [Google Scholar] [CrossRef] [PubMed]
- Jorde, R.; Figenschau, Y.; Hutchinson, M.; Emaus, N.; Grimnes, G. High serum 25-hydroxyvitamin D concentrations are associated with a favorable serum lipid profile. Eur. J. Clin. Nutr. 2010, 64, 1457–1464. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, K.; Islam, N.; Azam, I.; Mehboobali, N.; Iqbal, M.P. Lack of association of statin use with Vitamin D levels in a hospital based population of type 2 diabetes mellitus patients. Pak. J. Med. Sci. 2018, 34, 204–208. [Google Scholar] [CrossRef]
- Khayznikov, M.; Hemachrandra, K.; Pandit, R.; Kumar, A.; Wang, P.; GLueck, C.J. Statin intolerance because of myalgia, myositis, myopathy, or myonecrosis can in most cases be safely resolved by vitamin D supplementation. N. Am. J. Med. Sci. 2015, 7, 86–93. [Google Scholar] [CrossRef]
- Schwartz, J.B. Effects of Vitamin D supplementation in atorvastatin-treated patients: A new drug interaction with an unexpected consequence. Clin. Pharmacol. Ther. 2008, 85, 198–203. [Google Scholar] [CrossRef]
- Mc Auley, M.T.; Wilkinson, D.J.; Jones, J.J.L.; Kirkwood, T.B.L. A whole-body mathematical model of cholesterol metabolism and its age-associated dysregulation. BMC Syst. Biol. 2012, 6, 130. [Google Scholar] [CrossRef]
- Veldurthy, V.; Wei, R.; Oz, L.; Dhawan, P.; Jeon, Y.H.; Christakos, S. Vitamin D, calcium homeostasis and aging. Bone Res. 2016, 4, 16041. [Google Scholar] [CrossRef]
- Cohen, D.E. Balancing cholesterol synthesis and absorption in the gastrointestinal tract. J. Clin. Lipidol. 2008, 2, S1–S3. [Google Scholar] [CrossRef]
- Dietschy, J.M.; Turley, S.D.; Spady, D.K. Role of liver in the maintenance of cholesterol and low density lipoprotein homeostasis in different animal species, including humans. J. Lipid Res. 1993, 34, 1637–1659. [Google Scholar] [CrossRef]
- Nagashima, S.; Yagyu, H.; Tozawa, R.; Tazoe, F.; Takahashi, M.; Kitamine, T.; Yamamuro, D.; Sakai, K.; Sekiya, M.; Okazaki, H.; et al. Plasma cholesterol-lowering and transient liver dysfunction in mice lacking squalene synthase in the liver. J. Lipid Res. 2015, 56, 998–1005. [Google Scholar] [CrossRef] [PubMed]
- Nakano, T.; Inoue, I.; Murakoshi, T. A newly integrated model for intestinal cholesterol absorption and efflux reappraises how plant sterol intake reduces circulating cholesterol levels. Nutrition 2019, 11, 310. [Google Scholar] [CrossRef]
- Chemaly, M.; McGilligan, V.; Gibson, M.; Clauss, M.; Watterson, S.; Alexander, H.D.; Bjourson, A.J.; Peace, A. Role of tumour necrosis factor alpha converting enzyme (TACE/ADAM17) and associated proteins in coronary artery disease and cardiac events. Arch. Cardiovasc. Dis. 2017, 110, 700–711. [Google Scholar] [CrossRef]
- Libby, P. Changing concepts of atherogenesis. J. Intern. Med. 2000, 247, 349–358. [Google Scholar] [CrossRef]
- Collet, J.P.; Thiele, H.; Barbato, E.; Barthélémy, O.; Bauersachs, J.; Bhatt, D.L.; Dendale, P.; Dorobantu, M.; Edvardsen, T.; Folliguet, T.; et al. 2020 ESC Guidelines for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation: The task force for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation of the European Society of Cardiology (ESC). Eur. Heart J. 2020, 42, 1289–1367. [Google Scholar]
- Lu, H.; Talbot, S.; Robertson, K.A.; Watterson, S.; Forster, T.; Roy, D.; Ghazal, P. Rapid proteasomal elimination of 3-hydroxy-3-methylglutaryl-CoA reductase by interferon-γ in primary macrophages requires endogenous 25-hydroxycholesterol synthesis. Steroids 2015, 99, 219–229. [Google Scholar] [CrossRef]
- Nissen, S.E.; Tuzcu, E.M.; Schoenhagen, P.; Crowe, T.; Sasiela, W.J.; Tsai, J.; Orazem, J.; Magorien, R.D.; O’Shaughnessy, C.; Ganz, P. Statin therapy, LDL cholesterol, C-reactive protein, and coronary artery disease. N. Engl. J. Med. 2005, 352, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Watterson, S.; Guerriero, M.L.; Blanc, M.; Mazein, A.; Loewe, L.; Robertson, K.A.; Gibbs, H.; Shui, G.; Wenk, M.R.; Hillston, J.; et al. A model of flux regulation in the cholesterol biosynthesis pathway: Immune mediated graduated flux reduction versus statin-like led stepped flux reduction. Biochimie 2013, 95, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Costa, J.; Borges, M.; David, C.; Carneiro, A.V. Efficacy of lipid lowering drug treatment for diabetic and non-diabetic patients: Metaanalysis of randomised controlled trials. BMJ 2006, 332, 1115–1124. [Google Scholar] [CrossRef]
- Baigent, C. Cholesterol Treatment Trialists’(CTT) Collaborators: Efficacy and safety of cholesterol-lowering treatment: Pro-spective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet 2005, 366, 1267–1278. [Google Scholar]
- Michalska-Kasiczak, M.; Sahebkar, A.; Mikhailidis, D.P.; Rysz, J.; Muntner, P.; Toth, P.P.; Jones, S.R.; Rizzo, M.; Hovingh, G.K.; Farnier, M.; et al. Analysis of vitamin D levels in patients with and without statin-associated myalgia—A systematic review and meta-analysis of 7 studies with 2420 patients. Int. J. Cardiol. 2015, 178, 111–116. [Google Scholar] [CrossRef]
- Riche, K.D.; Arnall, J.; Rieser, K.; East, H.E.; Riche, D.M. Impact of vitamin D status on statin-induced myopathy. J. Clin. Transl. Endocrinol. 2016, 6, 56–59. [Google Scholar] [CrossRef] [PubMed]
- Golomb, B.A.; Evans, M.A. Statin adverse effects. Am. J. Cardiovasc. Drugs. 2008, 8, 373–418. [Google Scholar] [CrossRef] [PubMed]
- Abd, T.T.; Jacobson, T. Statin-induced myopathy: A review and update. Expert Opin. Drug Saf. 2011, 10, 373–387. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-W.; Kang, H.-J.; Jhon, M.; Kim, J.-W.; Lee, J.-Y.; Walker, A.; Agustini, B.; Kim, J.-M.; Berk, M. Statins and inflammation: New therapeutic opportunities in psychiatry. Front. Psychiatry 2019, 10, 103. [Google Scholar] [CrossRef]
- Greenwood, J.; Steinman, L.; Zamvil, S.S. Statin therapy and autoimmune disease: From protein prenylation to immunomodulation. Nat. Rev. Immunol. 2006, 6, 358–370. [Google Scholar] [CrossRef]
- Henriksbo, B.D.; Tamrakar, A.K.; Phulka, J.S.; Barra, N.G.; Schertzer, J.D. Statins activate the NLRP3 inflammasome and impair insulin signaling via p38 and mTOR. Am. J. Physiol. Metab. 2020, 319, E110–E116. [Google Scholar] [CrossRef]
- Koushki, K.; Shahbaz, S.K.; Mashayekhi, K.; Sadeghi, M.; Zayeri, Z.D.; Taba, M.Y.; Banach, M.; Al-Rasadi, K.; Johnston, T.P.; Sahebkar, A. Anti-inflammatory action of statins in cardiovascular disease: The role of inflammasome and toll-like receptor pathways. Clin. Rev. Allergy Immunol. 2020, 60, 175–199. [Google Scholar] [CrossRef]
- Satoh, M.; Tabuchi, T.; Itoh, T.; Nakamura, M. NLRP3 inflammasome activation in coronary artery disease: Results from prospective and randomized study of treatment with atorvastatin or rosuvastatin. Clin. Sci. 2013, 126, 233–241. [Google Scholar] [CrossRef]
- Nutescu, E.A.; Shapiro, N.L. Ezetimibe: A selective cholesterol absorption inhibitor. J. Hum. Pharmacol. Drug Ther. 2003, 23, 1463–1474. [Google Scholar] [CrossRef] [PubMed]
- Ostlund, R.E. Phytosterols, cholesterol absorption and healthy diets. Lipids 2007, 42, 41–45. [Google Scholar] [CrossRef]
- Insull, W. Clinical utility of bile acid sequestrants in the treatment of dyslipidemia: A scientific review. South. Med. J. 2006, 99, 257–274. [Google Scholar] [CrossRef] [PubMed]
- Ganji, S.H.; Kamanna, V.S.; Kashyap, M.L. Niacin and cholesterol: Role in cardiovascular disease (Review). J. Nutr. Biochem. 2003, 14, 298–305. [Google Scholar] [CrossRef]
- Seidah, N.G.; Awan, Z.; Chrétien, M.; Mbikay, M. PCSK9: A key modulator of cardiovascular health. Circ. Res. 2014, 114, 1022–1036. [Google Scholar] [CrossRef] [PubMed]
- Greig, S.L.; Deeks, E.D. Alirocumab: A review in hypercholesterolemia. Am. J. Cardiovasc. Drugs 2016, 16, 141–152. [Google Scholar] [CrossRef]
- Dadu, R.T.; Ballantyne, C.M. Lipid lowering with PCSK9 inhibitors. Nat. Rev. Cardiol. 2014, 11, 563–575. [Google Scholar] [CrossRef]
- Lee, J.; Lee, Y.; Kwon, N.; Ryu, K. Old target, but new drug: 2nd generation cetp inhibitor, CKD-508. Atherosclerosis 2020, 315, e258. [Google Scholar] [CrossRef]
- Chen, C.; Sun, R.; Sun, Y.; Chen, X.; Li, F.; Wen, X.; Yuan, H.; Chen, D. Synthesis, biological evaluation and SAR studies of ursolic acid 3β-ester derivatives as novel CETP inhibitors. Bioorg. Med. Chem. Lett. 2020, 30, 126824. [Google Scholar] [CrossRef]
- Laufs, U.; Banach, M.; Mancini, G.B.J.; Gaudet, D.; Bloedon, L.T.; Sterling, L.R.; Kelly, S.; Stroes, E.S.G. Efficacy and safety of bempedoic acid in patients with hypercholesterolemia and statin intolerance. J. Am. Hear. Assoc. 2019, 8, e011662. [Google Scholar] [CrossRef]
- Futema, M.; Plagnol, V.; Li, K.; Whittall, A.R.; Neil, H.A.W.; Seed, M.; Bertolini, S.; Calandra, S.; Descamps, O.S.; Graham, C.; et al. Whole exome sequencing of familial hypercholesterolaemia patients negative for LDLR/APOB/PCSK9 mutations. J. Med. Genet. 2014, 51, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Dharmayat, K.I.; Stevens, C.A.; Sharabiani, M.T.; Jones, R.S.; Watts, G.F.; Genest, J.; Ray, K.K.; Vallejo-Vaz, A.J. Prevalence of familial hypercholesterolemia among the general population and patients with atherosclerotic cardiovascular disease: A systematic review and meta-analysis. Circulation 2020, 141, 1742–1759. [Google Scholar] [CrossRef] [PubMed]
- Carroll, M.D.; Lacher, D.A.; Sorlie, P.D.; Cleeman, J.I.; Gordon, D.J.; Wolz, M.; Grundy, S.M.; Johnson, C.L. Trends in serum lipids and lipoproteins of adults, 1960–2002. JAMA 2005, 294, 1773–1781. [Google Scholar] [CrossRef]
- Félix-Redondo, F.J.; Grau, M.; Fernandez-Berges, D. Cholesterol and cardiovascular disease in the elderly. Facts and gaps. Aging Dis. 2013, 4, 154–169. [Google Scholar] [PubMed]
- Morgan, A.E.; Mc Auley, M.T. Cholesterol homeostasis: An in silico investigation into how aging disrupts its key hepatic regulatory mechanisms. Biology 2020, 9, 314. [Google Scholar] [CrossRef] [PubMed]
- Morgan, A.; Mooney, K.M.; Wilkinson, S.J.; Pickles, N.; Mc Auley, M.T. Mathematically modelling the dynamics of cholesterol metabolism and ageing. Biosystem 2016, 145, 19–32. [Google Scholar] [CrossRef]
- Morgan, A.; Mooney, K.; Wilkinson, S.; Pickles, N.; Mc Auley, M. Cholesterol metabolism: A review of how ageing disrupts the biological mechanisms responsible for its regulation. Ageing Res. Rev. 2016, 27, 108–124. [Google Scholar] [CrossRef]
- Chyou, P.H.; Eaker, E.D. Serum cholesterol concentrations and all-cause mortality in older people. Age Ageing 2000, 29, 69–74. [Google Scholar] [CrossRef]
- Weverling-Rijnsburger, A.W.E.; Jonkers, I.J.A.M.; van Exel, E.; Gussekloo, J.; Westendorp, R.G.J. High-density vs low-density lipoprotein cholesterol as the risk factor for coronary artery disease and stroke in old age. Arch. Intern. Med. 2003, 163, 1549–1554. [Google Scholar] [CrossRef]
- Ogami, M.; Ikura, Y.; Ohsawa, M.; Matsuo, T.; Kayo, S.; Yoshimi, N.; Hai, E.; Shirai, N.; Ehara, S.; Komatsu, R.; et al. Telomere shortening in human coronary artery diseases. Arter. Thromb. Vasc. Biol. 2004, 24, 546–550. [Google Scholar] [CrossRef] [PubMed]
- Yegorov, Y.; Poznyak, A.; Nikiforov, N.; Starodubova, A.; Orekhov, A. Role of telomeres shortening in atherogenesis: An overview. Cells 2021, 10, 395. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.A.; Stolzing, A. The role of lipid metabolism in aging, lifespan regulation, and age related disease. Aging Cell 2019, 18, e13048. [Google Scholar] [CrossRef]
- Duan, L.-P.; Wang, H.H.; Ohashi, A.; Wang, D.Q.-H. Role of intestinal sterol transporters Abcg5, Abcg8, and Npc1l1 in cholesterol absorption in mice: Gender and age effects. Am. J. Physiol. Liver Physiol. 2006, 290, G269–G276. [Google Scholar] [CrossRef] [PubMed]
- Field, P.A.; Gibbons, G.F. Decreased hepatic expression of the low-density lipoprotein (LDL) receptor and LDL receptor-related protein in aging rats is associated with delayed clearance of chylomicrons from the circulation. Metabolism 2000, 49, 492–498. [Google Scholar] [CrossRef]
- Millar, J.S.; Lichtenstein, A.H.; Cuchel, M.; Dolnikowski, G.; Hachey, D.L.; Cohn, J.S.; Schaefer, E.J. Impact of age on the metabolism of VLDL, IDL, and LDL apolipoprotein B-100 in men. J. Lipid Res. 1995, 36, 1155–1167. [Google Scholar] [CrossRef]
- Zhang, Y.; Ma, K.L.; Ruan, X.Z.; Liu, B.C. Dysregulation of the low-density lipoprotein receptor pathway is involved in lipid disor-der-mediated organ injury. Int. J. Biol. Sci. 2016, 12, 569. [Google Scholar] [CrossRef] [PubMed]
- Venkataraman, K.; Khurana, S.; Tai, T.C. Oxidative stress in aging-matters of the heart and mind. Int. J. Mol. Sci. 2013, 14, 17897–17925. [Google Scholar] [CrossRef]
- Christakos, S.; Ajibade, D.V.; Dhawan, P.; Fechner, A.J.; Mady, L.J. Vitamin D: Metabolism. Endocrinol. Metab. Clin. 2010, 39, 243–253. [Google Scholar] [CrossRef]
- Christakos, S.; Dhawan, P.; Verstuyf, A.; Verlinden, L.; Carmeliet, G. Vitamin D: Metabolism, molecular mechanism of action, and pleiotropic effects. Physiol. Rev. 2016, 96, 365–408. [Google Scholar] [CrossRef] [PubMed]
- Bouillon, R.; Carmeliet, G. Vitamin D insufficiency: Definition, diagnosis and management. Best Pract. Res. Clin. Endocrinol. Metab. 2018, 32, 669–684. [Google Scholar] [CrossRef]
- Pludowski, P.; Holick, M.F.; Grant, W.B.; Konstantynowicz, J.; Mascarenhas, M.R.; Haq, A.; Povoroznyuk, V.; Balatska, N.; Barbosa, A.P.; Karonova, T.; et al. Vitamin D supplementation guidelines. J. Steroid Biochem. Mol. Biol. 2018, 175, 125–135. [Google Scholar] [CrossRef]
- Zmijewski, M.A.; Carlberg, C. Vitamin D receptor(s): In the nucleus but also at membranes? Exp. Dermatol. 2020, 29, 876–884. [Google Scholar] [CrossRef]
- Mousa, A.; Misso, M.; Teede, H.; Scragg, R.; De Courten, B. Effect of vitamin D supplementation on inflammation: Protocol for a systematic review. BMJ Open 2016, 6, e010804. [Google Scholar] [CrossRef] [PubMed]
- Rao, Z.; Chen, X.; Wu, J.; Xiao, M.; Zhang, J.; Wang, B.; Fang, L.; Zhang, H.; Wang, X.; Yang, S.; et al. Vitamin D receptor inhibits NLRP3 activation by impeding its BRCC3-mediated deubiquitination. Front. Immunol. 2019, 10, 2783. [Google Scholar] [CrossRef]
- Edwards, M.; Cole, Z.; Harvey, N.; Cooper, C. The global epidemiology of vitamin D status. J. Aging Res. 2014, 3, 148–158. [Google Scholar] [CrossRef]
- Cashman, K.D.; van den Heuvel, E.G.; Schoemaker, R.J.; Prévéraud, D.P.; Macdonald, H.M.; Arcot, J. 25-Hydroxyvitamin D as a biomarker of vitamin D status and its modeling to inform strategies for prevention of vitamin D deficiency within the population. Int. Rev. J. 2017, 8, 947–957. [Google Scholar] [CrossRef]
- Cashman, K.D.; Dowling, K.G.; Škrabáková, Z.; Gonzalez-Gross, M.; Valtueña, J.; de Henauw, S.; Moreno, L.; Damsgaard, C.T.; Kim, F.M.; Molgaard, C. Vitamin D deficiency in Europe: Pandemic? Am. J. Clin. Nutr. 2016, 103, 1033–1044. [Google Scholar] [CrossRef] [PubMed]
- Holick, M.; Binkley, N.C.; Bischoff-Ferrari, H.; Gordon, C.M.; Hanley, D.A.; Heaney, R.P.; Murad, M.H.; Weaver, C.M. Evaluation, treatment, and prevention of Vitamin D deficiency: An endocrine society clinical practice guideline. J. Clin. Endocrinol. Metab. 2011, 96, 1911–1930. [Google Scholar] [CrossRef] [PubMed]
- Dawson-Hughes, B.; Heaney, R.P.; Holick, M.; Lips, P.; Meunier, P.J.; Vieth, R. Estimates of optimal vitamin D status. Osteoporos. Int. 2005, 16, 713–716. [Google Scholar] [CrossRef] [PubMed]
- Henry, H.L.; Bouillon, R.; Norman, A.W.; Gallagher, J.C.; Lips, P.; Heaney, R.P.; Vieth, R.; Pettifor, J.; Dawson-Hughes, B.; Lamberg-Allardt, C.; et al. 14th Vitamin D Workshop consensus on vitamin D nutritional guidelines. J. Steroid Biochem. Mol. Biol. 2010, 121, 4–6. [Google Scholar] [CrossRef]
- Sempos, C.T.; Vesper, H.W.; Phinney, K.W.; Thienpont, L.M.; Coates, P.M. Vitamin D status as an international issue: National surveys and the problem of standardization. Scand. J. Clin. Lab. Investig. 2012, 7, 243. [Google Scholar] [CrossRef]
- Binkley, N.; Sempos, C.T.; VDSP. Standardizing vitamin D assays: The way forward. J. Bone Miner. Res. 2014, 29, 1709–1714. [Google Scholar] [CrossRef] [PubMed]
- Sempos, C.T.; Heijboer, A.C.; Bikle, D.D.; Bollerslev, J.; Bouillon, R.; Brannon, P.M.; DeLuca, H.F.; Jones, G.; Munns, C.F.; Bilezikian, J.P.; et al. Vitamin D assays and the definition of hypovitaminosis D: Results from the First International Conference on Controversies in Vitamin D. Br. J. Clin. Pharmacol. 2018, 84, 2194–2207. [Google Scholar] [CrossRef] [PubMed]
- SACN Scientific Advisory Committee on Nutition. Vitamin D and Health; Public Health England: London, UK, 2016.
- EFSA Panel on Dietetic Products, Nutrition and Allergies (NDA). Dietary reference values for vitamin D. EFSA J. 2016, 14, e04547. [Google Scholar] [CrossRef]
- Pilz, S.; März, W.; Cashman, K.D.; Kiely, M.E.; Whiting, S.J.; Holick, M.F.; Grant, W.B.; Pludowski, P.; Hiligsmann, M.; Trummer, C.; et al. Rationale and plan for vitamin D food fortification: A review and guidance paper. Front. Endocrinol. 2018, 9, 373. [Google Scholar] [CrossRef]
- Lips, P. Vitamin D deficiency and secondary hyperparathyroidism in the elderly: Consequences for bone loss and fractures and therapeutic implications. Endocr. Rev. 2001, 22, 477–501. [Google Scholar] [CrossRef]
- Oudshoorn, C.; Van Der Cammen, T.J.M.; McMurdo, M.E.T.; Van Leeuwen, J.P.T.M.; Colin, E.M. Ageing and vitamin D deficiency: Effects on calcium homeostasis and considerations for vitamin D supplementation. Br. J. Nutr. 2009, 101, 1597–1606. [Google Scholar] [CrossRef]
- MacLaughlin, J.; Holick, M.F. Aging decreases the capacity of human skin to produce vitamin D3. J. Clin. Investig. 1985, 76, 1536–1538. [Google Scholar] [CrossRef]
- Duque, G.; El Abdaimi, K.; Macoritto, M.; Miller, M.M.; Kremer, R. Estrogens (E2) regulate expression and response of 1,25-dihydroxyvitamin D3 receptors in bone cells: Changes with aging and hormone deprivation. Biochem. Biophys. Res. Commun. 2002, 299, 446–454. [Google Scholar] [CrossRef]
- Bischoff-Ferrari, H.; Borchers, M.; Gudat, F.; Dürmüller, U.; Stähelin, H.B.; Dick, W. Vitamin D receptor expression in human muscle tissue decreases with age. J. Bone Miner. Res. 2004, 19, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Stein, M.S.; Wark, J.D.; Scherer, S.C.; Walton, S.L.; Chick, P.; Di Carlantonio, M.; Zajac, J.D.; Flicker, L. Falls relate to vitamin D and parathyroid hormone in an Australian nursing home and hostel. J. Am. Geriatr. Soc. 1999, 47, 1195–1201. [Google Scholar] [CrossRef]
- Bischoff, H.A.; Stähelin, H.B.; Tyndall, A.; Theiler, R. Relationship between muscle strength and vitamin D metabolites: Are there therapeutic possibilities in the elderly? Z. Rheumatol. 2000, 59, I39–I41. [Google Scholar] [CrossRef]
- Tzotzas, T.; Papadopoulou, F.G.; Tziomalos, K.; Karras, S.; Gastaris, K.; Perros, P.; Krassas, G.E. Rising serum 25-hydroxy-vitamin D levels after weight loss in obese women correlate with improvement in insulin resistance. J. Clin. Endocrinol. Metab. 2010, 95, 4251–4257. [Google Scholar] [CrossRef]
- Kim, D. The role of Vitamin D in thyroid diseases. Int. J. Mol. Sci. 2017, 18, 1949. [Google Scholar] [CrossRef]
- Scragg, R.; Stewart, A.W.; Waayer, D.; Lawes, C.M.M.; Toop, L.; Sluyter, J. Effect of monthly high-dose vitamin D supplementation on cardiovascular disease in the vitamin D assessment study: A randomized clinical trial. JAMA Cardiol. 2017, 2, 608–616. [Google Scholar] [CrossRef]
- Dziedzic, E.A.; Gąsior, J.S.; Pawłowski, M.; Wodejko-Kucharska, B.; Saniewski, T.; Marcisz, A.; Dąbrowski, M.J. Vitamin D level is associated with severity of coronary artery atherosclerosis and incidence of acute coronary syndromes in non-diabetic cardiac patients. Arch. Med Sci. 2019, 15, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Wilson, L.R.; Tripkovic, L.; Hart, K.H.; Lanham-New, S.A. Vitamin D deficiency as a public health issue: Using vitamin D2or vitamin D3in future fortification strategies. Proc. Nutr. Soc. 2017, 76, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Prietl, B.; Treiber, G.; Pieber, T.R.; Amrein, K. Vitamin D and immune function. Nutrients 2013, 5, 2502–2521. [Google Scholar] [CrossRef]
- Bjelakovic, G.; Gluud, L.L.; Nikolova, D.; Whitfield, K.; Wetterslev, J.; Simonetti, R.G.; Bjelakovic, M.; Gluud, C. Vitamin D supplementation for prevention of mortality in adults. Cochrane Database Syst. Rev. 2014, 1. [Google Scholar] [CrossRef]
- Petkovich, M.; Bishop, C. Extended Release Calcifediol in Renal Disease, Vitamin D; Academic Press: Cambridge, MA, USA, 2018; pp. 667–678. [Google Scholar]
- Zmuda, J.M.; Cauley, J.; Ferrell, R.E. Molecular epidemiology of vitamin D receptor gene variants. Epidemiol. Rev. 2000, 22, 203–217. [Google Scholar] [CrossRef]
- Valdivielso, J.M.; Fernandez, E. Vitamin D receptor polymorphisms and diseases. Clin. Chim. Acta 2006, 371, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Slater, N.A.; Rager, M.L.; Havrda, D.E.; Harralson, A.F. Genetic variation in CYP2R1 and GC genes associated with vitamin D de-ficiency status. J. Pharm. Pract. 2017, 30, 31–36. [Google Scholar] [CrossRef] [PubMed]
- McAuley, M.T.; Proctor, C.J.; Corfe, B.M.; Cuskelly, G.J.; Mooney, K.M. Nutrition research and the impact of computational systems biology. J. Comput. Sci. Syst. Biol. 2013, 6, 271–285. [Google Scholar]
- Wilkinson, D.J. Stochastic modelling for quantitative description of heterogeneous biological systems. Nat. Rev. Genet. 2009, 10, 122–133. [Google Scholar] [CrossRef]
- Polynikis, A.; Hogan, S.; di Bernardo, M. Comparing different ODE modelling approaches for gene regulatory networks. J. Theor. Biol. 2009, 261, 511–530. [Google Scholar] [CrossRef] [PubMed]
- Klann, M.; Koeppl, H. Spatial simulations in systems biology: From molecules to cells. Int. J. Mol. Sci. 2012, 13, 7798–7827. [Google Scholar] [CrossRef]
- Watterson, S.; Ghazal, P. Use of logic theory in understanding regulatory pathway signaling in response to infection. Futur. Microbiol. 2010, 5, 163–176. [Google Scholar] [CrossRef] [PubMed]
- Dunn, J.F. Computer simulation of Vitamin D transport. Ann. N. Y. Acad. Sci. 1988, 538, 69–76. [Google Scholar] [CrossRef]
- Chun, R.F.; Peercy, B.E.; Adams, J.; Hewison, M. Vitamin D binding protein and monocyte response to 25-Hydroxyvitamin D and 1,25-Dihydroxyvitamin D: Analysis by mathematical modeling. PLoS ONE 2012, 7, e30773. [Google Scholar] [CrossRef]
- Peterson, M.C.; Riggs, M.M. A physiologically based mathematical model of integrated calcium homeostasis and bone remodeling. Bone 2010, 46, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Raposo, J.F.; Sobrinho, L.G.; Ferreira, H.G. A minimal mathematical model of calcium homeostasis. J. Clin. Endocrinol. Metab. 2002, 87, 4330–4340. [Google Scholar] [CrossRef]
- Foissac, F.; Treluyer, J.-M.; Souberbielle, J.-C.; Rostane, H.; Urien, S.; Viard, J.-P. Vitamin D3 supplementation scheme in HIV-infected patients based upon pharmacokinetic modelling of 25-hydroxycholecalciferol. Br. J. Clin. Pharmacol. 2013, 75, 1312–1320. [Google Scholar] [CrossRef] [PubMed]
- Chelliah, V.; Juty, N.; Ajmera, I.; Ali, R.; Dumousseau, M.; Glonț, M.; Hucka, M.; Jalowicki, G.; Keating, S.; Knight-Schrijver, V.; et al. BioModels: Ten-year anniversary. Nucleic Acids Res. 2014, 43, D542–D548. [Google Scholar] [CrossRef]
- Benson, H.E.; Watterson, S.; Sharman, J.L.; Mpamhanga, C.P.; Parton, A.; Southan, C.; Harmar, A.J.; Ghazal, P. Is systems pharmacology ready to impact upon therapy development? A study on the cholesterol biosynthesis pathway. Br. J. Pharma. 2017, 174, 4362–4382. [Google Scholar] [CrossRef] [PubMed]
- Pool, F.; Currie, R.; Sweby, P.K.; Salazar, J.D.; Tindall, M.J. A mathematical model of the mevalonate cholesterol biosynthesis pathway. J. Theor. Biol. 2018, 443, 157–176. [Google Scholar] [CrossRef]
- Pool, F.; Sweby, P.K.; Tindall, M.J. An integrated mathematical model of cellular cholesterol biosynthesis and lipoprotein metabolism. Processes 2018, 6, 134. [Google Scholar] [CrossRef]
- Bhattacharya, B.S.; Sweby, P.K.; Minihane, A.-M.; Jackson, K.; Tindall, M.J. A mathematical model of the sterol regulatory element binding protein 2 cholesterol biosynthesis pathway. J. Theor. Biol. 2014, 349, 150–162. [Google Scholar] [CrossRef] [PubMed]
- El Khatib, N.; Génieys, S.; Kazmierczak, B.; Volpert, V. Mathematical modelling of atherosclerosis as an inflammatory disease. Philos. Trans. R. Soc. Math. Phys. Eng. Sci. 2009, 367, 4877–4886. [Google Scholar] [CrossRef]
- Bulelzai, M.; Dubbeldam, J.L. Long time evolution of atherosclerotic plaques. J. Theor. Biol. 2012, 297, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Friedman, A.; Hao, W. A Mathematical model of atherosclerosis with reverse cholesterol transport and associated risk factors. Bull. Math. Biol. 2014, 77, 758–781. [Google Scholar] [CrossRef]
- Rai, V.; Agrawal, D.K. Role of vitamin D in cardiovascular diseases. Endocrinol. Metab. Clin. N. Am. 2017, 46, 1039–1059. [Google Scholar] [CrossRef]
- Skaaby, T.; Husemoen, L.L.; Pisinger, C.; Jørgensen, T.; Thuesen, B.H.; Fenger, M.; Linneberg, A. Vitamin D status and changes in car-diovascular risk factors: A prospective study of a general population. Cardiology 2012, 123, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Dibaba, D.T. Effect of vitamin D supplementation on serum lipid profiles: A systematic review and meta-analysis. Nutr. Rev. 2019, 77, 890–902. [Google Scholar] [CrossRef] [PubMed]
- Skaaby, T.; Husemoen, L.L.; Pisinger, C.; Jørgensen, T.; Thuesen, B.H.; Fenger, M.; Linneberg, A. Vitamin D status and incident cardi-ovascular disease and all-cause mortality: A general population study. Endocrine 2013, 43, 618–625. [Google Scholar] [CrossRef] [PubMed]
- Jorde, R.; Figenschau, Y.; Emaus, N.; Hutchinson, M.; Grimnes, G. Serum 25-Hydroxyvitamin D levels are strongly related to systolic blood pressure but do not predict future hypertension. Hypertension 2010, 55, 792–798. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Glenn, D.J.; Ni, W.; Grigsby, C.; Olsen, K.; Nishimoto, M.; Law, C.S.; Gardner, D.G. Expression of the Vitamin D receptor is increased in the hypertrophic heart. Hypertension 2008, 52, 1106–1112. [Google Scholar] [CrossRef]
- Norman, P.; Powell, J. Vitamin D and cardiovascular disease. Circ. Res. 2014, 114, 379–393. [Google Scholar] [CrossRef]
- Riek, A.E.; Oh, J.; Bernal-Mizrachi, C. 1,25(OH)2 vitamin D suppresses macrophage migration and reverses atherogenic cholesterol metabolism in type 2 diabetic patients. J. Steroid Biochem. Mol. Biol. 2013, 136, 309–312. [Google Scholar] [CrossRef]
- Playford, M.P.; Dey, A.K.; Zierold, C.; Joshi, A.A.; Blocki, F.; Bonelli, F.; Rodante, J.A.; Harrington, C.L.; Rivers, J.P.; Elnabawi, Y.A.; et al. Serum active 1,25 (OH) 2D, but not inactive 25 (OH) D vitamin D levels are associated with cardiometabolic and cardio-vascular disease risk in psoriasis. Atherosclerosis 2019, 289, 44–50. [Google Scholar] [CrossRef]
- Mohania, D.; Chandel, S.; Kumar, P.; Verma, V.; Digvijay, K.; Tripathi, D.; Choudhury, K.; Mitten, S.K.; Shah, D. Ultraviolet radiations: Skin defense-damage mechanism. Ultrav. Light Hum. Health Dis. Environ. 2017, 996, 71–87. [Google Scholar] [CrossRef]
- Maxwell, J.D. Seasonal variation in vitamin D. Proc. Nutr. Soc. 1994, 53, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Wacker, M.; Holick, M.F. Sunlight and vitamin D: A global perspective for health. Dermatoendocrinology 2013, 5, 51–108. [Google Scholar] [CrossRef] [PubMed]
- Pereira, L.A.; Luz, F.B.; Carneiro, C.M.M.D.O.; Xavier, A.L.R.; Kanaan, S.; Miot, H.A. Evaluation of vitamin D plasma levels after mild exposure to the sun with photoprotection. An. Bras. Dermatol. 2019, 94, 56–61. [Google Scholar] [CrossRef]
- Nikooyeh, B.; Abdollahi, Z.; Hajifaraji., M.; Alavi-Majd, H.; Salehi, F.; Yarparvar, A.H.; Neyestani, T.R. Healthy changes in some cardiometabolic risk factors accompany the higher summertime serum 25-hydroxyvitamin D concentrations in Iranian children: National Food and Nutrition Surveillance. Public Health Nutr. 2018, 21, 2013–2021. [Google Scholar] [CrossRef]
- Huotari, A.; Herzig, K.-H. International Journal of Circumpolar Health Vitamin D and living in northern latitudes, an endemic risk area for vitamin D deficiency. Circumpolar Health 2008, 67, 164–178. [Google Scholar] [CrossRef]
- Grimes, D.S.; Hindle, E.; Dyer, T. Sunlight, cholesterol and coronary heart disease. QJM Int. J. Med. 1996, 89, 579–590. [Google Scholar] [CrossRef]
- Liu, Y.; Brook, R.D.; Liu, X.; Byrd, J.B. Abstract P300: Countries’ geographic latitude and their populations’ cholesterol and blood pressure. Hypertension 2018, 72, 300. [Google Scholar] [CrossRef]
- Scragg, R. Seasonality of cardiovascular disease mortality and the possible protective effect of ultra-violet radiation. Int. J. Epidemiol. 1981, 10, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Collins, R.; Reith, C.; Emberson, J.; Armitage, J.; Baigent, C.; Blackwell, L.; Blumenthal, R.; Danesh, J.; Smith, G.D.; DeMets, D.; et al. Interpretation of the evidence for the efficacy and safety of statin therapy. Lancet 2016, 388, 2532–2561. [Google Scholar] [CrossRef]
- Heart Protection Study Collaborative Group. MRC/BHF heart protection study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: A randomised placebocontrolled trial. Lancet 2002, 360, 7–22. [Google Scholar] [CrossRef]
- Pinal-Fernandez, I.; Casal-Dominguez, M.; Mammen, A.L. Statins: Pros and cons. Med. Clínica 2018, 150, 398–402. [Google Scholar] [CrossRef]
- Gupta, A.; Thompson, P.D. The relationship of vitamin D deficiency to statin myopathy. Atherosclerosis 2011, 215, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Turner, R.M.; Pirmohamed, M. Statin-related myotoxicity: A comprehensive review of pharmacokinetic, pharmacogenomic and muscle components. J. Clin. Med. 2019, 9, 22. [Google Scholar] [CrossRef]
- Qin, X.F.; Zhao, L.S.; Chen, W.R.; Wang, H. Effects of vitamin D on plasma lipid profiles in statin-treated patients with hypercho-lesterolemia: A randomized placebo-controlled trial. Clin. Nutr. 2015, 34, 201–206. [Google Scholar] [CrossRef]
- Aloia, J.F.; Li-Ng, M.; Pollack, S. Statins and vitamin D. Am. J. Cardiol. 2007, 8, 1329. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Castrillón, J.L.; Vega, G.; Abad, L.; Sanz, A.; Chaves, J.; Hernandez, G.; Dueñas, A. Effects of atorvastatin on Vitamin D levels in patients with acute ischemic heart disease. Am. J. Cardiol. 2007, 99, 903–905. [Google Scholar] [CrossRef] [PubMed]
- Sahebkar, A.; Reiner, Ž.; Simental-Mendía, L.; Ferretti, G.; Della Corte, C.; Nobili, V. Impact of statin therapy on plasma vitamin D levels: A systematic review and meta-analysis. Curr. Pharm. Des. 2017, 23, 861–869. [Google Scholar] [CrossRef]
- Thummel, K.E.; Brimer, C.; Yasuda, K.; Thottassery, J.; Senn, T.; Lin, Y.; Ishizuka, H.; Kharasch, E.; Schuetz, J.; Schuetz, E. Transcriptional control of intestinal cytochrome P-4503A by 1α, 25-dihydroxy vitamin D3. Mol. Pharmacol. 2001, 60, 1399–1406. [Google Scholar] [CrossRef]
- Drocourt, L.; Ourlin, J.-C.; Pascussi, J.M.; Maurel, P.; Vilarem, M.-J. Expression of CYP3A4, CYP2B6, and CYP2C9 is regulated by the Vitamin D receptor pathway in primary human hepatocytes. J. Biol. Chem. 2002, 277, 25125–25132. [Google Scholar] [CrossRef]
- Pérez-Castrillón, J.L.; Manteca, L.A.; Vega, G.; Montes, J.D.P.; De Luis, D.; Laita, A.D. Vitamin D levels and lipid response to atorvastatin. Int. J. Endocrinol. 2009, 2010, 320721. [Google Scholar] [CrossRef]
- Brown, M.S.; Goldstein, J.L. The SREBP pathway: Regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 1997, 89, 331–340. [Google Scholar] [CrossRef]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Investig. 2002, 109, 1125–1131. [Google Scholar] [CrossRef]
- Bengoechea-Alonso, M.T.; Ericsson, J. SREBP in signal transduction: Cholesterol metabolism and beyond. Curr. Opin. Cell Biol. 2007, 19, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; He, Y.; Lin, S.; Hao, L.; Ye, Y.; Lv, L.; Sun, Z.; Fan, H.; Shi, Z.; Li, J.; et al. Increase of circulating cholesterol in vitamin D deficiency is linked to reduced vitamin D receptor activity via the Insig-2/SREBP-2 pathway. Mol. Nutr. Food Res. 2015, 60, 798–809. [Google Scholar] [CrossRef]
- Lee, S.; Lee, D.-K.; Choi, E.; Lee, J.W. Identification of a functional vitamin D response element in the murine insig-2 promoter and its potential role in the differentiation of 3T3-L1 preadipocytes. Mol. Endocrinol. 2005, 19, 399–408. [Google Scholar] [CrossRef][Green Version]
- Quach, H.P.; Dzekic, T.; Bukuroshi, P.; Pang, K.S. Potencies of vitamin D analogs, 1α-hydroxyvitamin D3, 1α-hydroxyvitamin D2 and 25-hydroxyvitamin D3, in lowering cholesterol in hypercholesterolemic mice in vivo. Biopharm. Drug Dispos. 2018, 39, 196–204. [Google Scholar] [CrossRef]
- Defay, R.; Astruc, M.E.; Roussillon, S.; Descomps, B.; de Paulet, A.C. DNA synthesis and 3-hydroxy-3-methylglutaryl CoA reductase activity in PHA stimulated human lymphocytes: A comparative study of the inhibitory effects of some oxysterols with special reference to side chain hydroxylated derivatives. Biochem. Biophys. Res. Commun. 1982, 106, 362–372. [Google Scholar] [CrossRef]
- Chow, E.C.; Magomedova, L.; Quach, H.P.; Patel, R.; Durk, M.R.; Fan, J.; Maeng, H.-J.; Irondi, K.; Anakk, S.; Moore, D.D.; et al. Vitamin D receptor activation down-regulates the small heterodimer partner and increases CYP7A1 to lower cholesterol. Gastroenterology 2014, 146, 1048.e7–1059.e7. [Google Scholar] [CrossRef]
- Chambers, K.F.; Day, P.E.; Aboufarrag, H.T.; Kroon, P.A. Polyphenol effects on cholesterol metabolism via bile acid biosynthesis, CYP7A1: A Review. Nutrients 2019, 11, 2588. [Google Scholar] [CrossRef]
- Prabhu, A.; Luu, W.; Li, D.; Sharpe, L.; Brown, A.J. DHCR7: A vital enzyme switch between cholesterol and vitamin D production. Prog. Lipid Res. 2016, 64, 138–151. [Google Scholar] [CrossRef]
- Prabhu, A.; Luu, W.; Sharpe, L.; Brown, A.J. Cholesterol-mediated degradation of 7-Dehydrocholesterol reductase switches the balance from cholesterol to vitamin D synthesis. J. Biol. Chem. 2016, 291, 8363–8373. [Google Scholar] [CrossRef]
- Cross, J.L.; Iben, J.; Simpson, C.L.; Thurm, A.; Swedo, S.; Tierney, E.; Bailey-Wilson, J.E.; Biesecker, L.G.; Porter, F.D.; Wassif, C.A. Determination of the allelic frequency in Smith-Lemli-Opitz syndrome by analysis of massively parallel sequencing data sets. Clin. Genet. 2015, 87, 570–575. [Google Scholar] [CrossRef]
- Honda, M.; Tint, G.S.; Honda, A.; Nguyen, L.B.; Chen, T.S.; Shefer, S. 7-Dehydrocholesterol down-regulates cholesterol biosynthesis in cultured Smith-Lemli-Opitz syndrome skin fibroblasts. J. Lipid Res. 1998, 39, 647–657. [Google Scholar] [CrossRef]
- Lamberson, C.R.; Muchalski, H.; McDuffee, K.B.; Tallman, K.A.; Xu, L.; Porter, N.A. Propagation rate constants for the peroxidation of sterols on the biosynthetic pathway to cholesterol. Chem. Phys. Lipids 2017, 207, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Movassaghi, M.; Bianconi, S.; Feinn, R.; Wassif, C.A.; Porter, F.D. Vitamin D levels in Smith-Lemli-Opitz syndrome. Am. J. Med. Genet. 2017, 173, 2577–2583. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.J.; Zhang, F.; Richards, J.B.; Kestenbaum, B.; Van Meurs, J.B.; Berry, D.; Kiel, D.P.; Streeten, E.A.; Ohlsson, C.; Koller, D.L.; et al. Common genetic determinants of vitamin D insufficiency: A genome-wide association study. Lancet 2010, 376, 180–188. [Google Scholar] [CrossRef]
- Ahn, J.; Yu, K.; Stolzenberg-Solomon, R.; Simon, K.C.; McCullough, M.L.; Gallicchio, L.; Jacobs, E.J.; Ascherio, A.; Helzlsouer, K.; Jacobs, K.; et al. Genome-wide association study of circulating vitamin D levels. Hum. Mol. Genet. 2010, 19, 2739–2745. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Chitipiralla, S.; Brown, G.R.; Chen, C.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; Kaur, K.; Liu, C.; et al. ClinVar: Improvements to accessing data. Nucleic Acids Res. 2020, 48, D835–D844. [Google Scholar] [CrossRef] [PubMed]
- Andresen, B.S.; Christensen, E.; Corydon, T.J.; Bross, P.; Pilgaard, B.; Wanders, R.J.; Ruiter, J.P.; Simonsen, H.; Winter, V.; Knudsen, I.; et al. Isolated 2-methylbutyrylglycinuria caused by short/branched-chain acyl-CoA dehydrogenase deficiency: Identification of a new enzyme defect, resolution of its molecular basis, and evidence for distinct acyl-CoA dehydrogenases in iso-leucine and valine metabolism. Am. J. Hum. Genet. 2000, 67, 1095–1103. [Google Scholar] [PubMed]
- Gibson, K.M.; Burlingame, T.G.; Hogema, B.; Jakobs, C.; Schutgens, R.B.H.; Millington, D.; Roe, C.R.; Roe, D.S.; Sweetman, L.; Steiner, R.; et al. 2-Methylbutyryl-coenzyme a dehydrogenase deficiency: A new inborn error of L-isoleucine metabolism. Pediatric Res. 2000, 47, 830–833. [Google Scholar] [CrossRef][Green Version]
- Le Novere, N.; Hucka, M.; Mi, H.; Moodie, S.; Schreiber, F.; Sorokin, A.; Demir, E.; Wegner, K.; Aladjem, M.I.; Wimalaratne, S.M.; et al. The systems biology graphical notation. Nat. Biotech. 2009, 27, 735–741. [Google Scholar] [CrossRef]
- Van Iersel, M.P.; Villéger, A.C.; Czauderna, T.; Boyd, S.E.; Bergmann, F.T.; Luna, A.; Demir, E.; Sorokin, A.; Dogrusoz, U.; Matsuoka, Y.; et al. Software support for SBGN maps: SBGN-ML and LibSBGN. Bioinformatics 2012, 28, 2016–2021. [Google Scholar] [CrossRef]
- Fabregat, A.; Sidiropoulos, K.; Viteri, G.; Marin-Garcia, P.; Ping, P.; Stein, L.; D’Eustachio, P.; Hermjakob, H. Reactome diagram viewer: Data structures and strategies to boost performance. Bioinformatics 2017, 34, 1208–1214. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S.; Maitra, A. Possible mechanisms of interaction between statins and vitamin D. Qjm. Int. J. Med. 2012, 105, 487–491. [Google Scholar] [CrossRef] [PubMed]
- Nikolic, D.; Banach, M.; Chianetta, R.; Luzzu, L.M.; Stoian, A.P.; Diaconu, C.C.; Citarrella, R.; Montalto, G.; Rizzo, M. An overview of statin-induced myopathy and perspectives for the future. Expert Opin. Drug Saf. 2020, 19, 601–615. [Google Scholar] [CrossRef] [PubMed]
- Banach, M.; Serban, C.; Ursoniu, S.; Rysz, J.; Muntner, P.; Toth, P.P.; Jones, S.R.; Rizzo, M.; Glasser, S.; Watts, G.; et al. Statin therapy and plasma coenzyme Q10 concentrations—A systematic review and meta-analysis of placebo-controlled trials. Pharmacol. Res. 2015, 99, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.L.; DeBose-Boyd, R.A.; Brown, M.S. Protein sensors for membrane sterols. Cell 2006, 124, 35–46. [Google Scholar] [CrossRef]
- Munir, M.T.; Ponce, C.; Santos, J.M.; Sufian, H.B.; Al-Harrasi, A.; Gollahon, L.S.; Hussain, F.; Rahman, S.M. VD3 and LXR agonist (T0901317) combination demonstrated greater potency in inhibiting cholesterol accumulation and inducing apoptosis via ABCA1-CHOP-BCL-2 cascade in MCF-7 breast cancer cells. Mol. Biol. Rep. 2020, 47, 7771–7782. [Google Scholar] [CrossRef]
- Lisse, T.S.; Chun, R.F.; Rieger, S.; Adams, J.S.; Hewison, M. Vitamin D activation of functionally distinct regulatory miRNAs in primary human osteoblasts. J. Bone Miner. Res. 2013, 28, 1478–1488. [Google Scholar] [CrossRef]
- Decourt, C.; Janin, A.; Moindrot, M.; Chatron, N.; Nony, S.; Muntaner, M.; Dumont, S.; Divry, E.; Dauchet, L.; Meirhaeghe, A.; et al. PCSK9 post-transcriptional regulation: Role of a 3’ UTR microRNA-binding site variant in linkage disequilibrium with c. 1420G. Atherosclerosis 2020, 314, 63–70. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Warren, T.; McAllister, R.; Morgan, A.; Rai, T.S.; McGilligan, V.; Ennis, M.; Page, C.; Kelly, C.; Peace, A.; Corfe, B.M.; et al. The Interdependency and Co-Regulation of the Vitamin D and Cholesterol Metabolism. Cells 2021, 10, 2007. https://doi.org/10.3390/cells10082007
Warren T, McAllister R, Morgan A, Rai TS, McGilligan V, Ennis M, Page C, Kelly C, Peace A, Corfe BM, et al. The Interdependency and Co-Regulation of the Vitamin D and Cholesterol Metabolism. Cells. 2021; 10(8):2007. https://doi.org/10.3390/cells10082007
Chicago/Turabian StyleWarren, Tara, Roisin McAllister, Amy Morgan, Taranjit Singh Rai, Victoria McGilligan, Matthew Ennis, Christopher Page, Catriona Kelly, Aaron Peace, Bernard M. Corfe, and et al. 2021. "The Interdependency and Co-Regulation of the Vitamin D and Cholesterol Metabolism" Cells 10, no. 8: 2007. https://doi.org/10.3390/cells10082007
APA StyleWarren, T., McAllister, R., Morgan, A., Rai, T. S., McGilligan, V., Ennis, M., Page, C., Kelly, C., Peace, A., Corfe, B. M., Mc Auley, M., & Watterson, S. (2021). The Interdependency and Co-Regulation of the Vitamin D and Cholesterol Metabolism. Cells, 10(8), 2007. https://doi.org/10.3390/cells10082007