
Autophagy Promotes the Survival of Adipose Mesenchymal Stem/Stromal Cells and Enhances Their Therapeutic Effects in Cisplatin-Induced Liver Injury via Modulating TGF-β1/Smad and PI3K/AKT Signaling Pathways

 , , , , ,
, , , , ,
Abstract
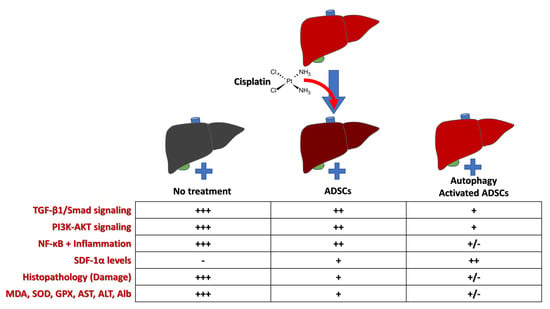
:
1. Introduction
2. Materials and Methods
2.1. Study Animals
2.2. Experimental Design
2.3. Isolation and Culture of Adipose-Derived MSCs (ADSCs)
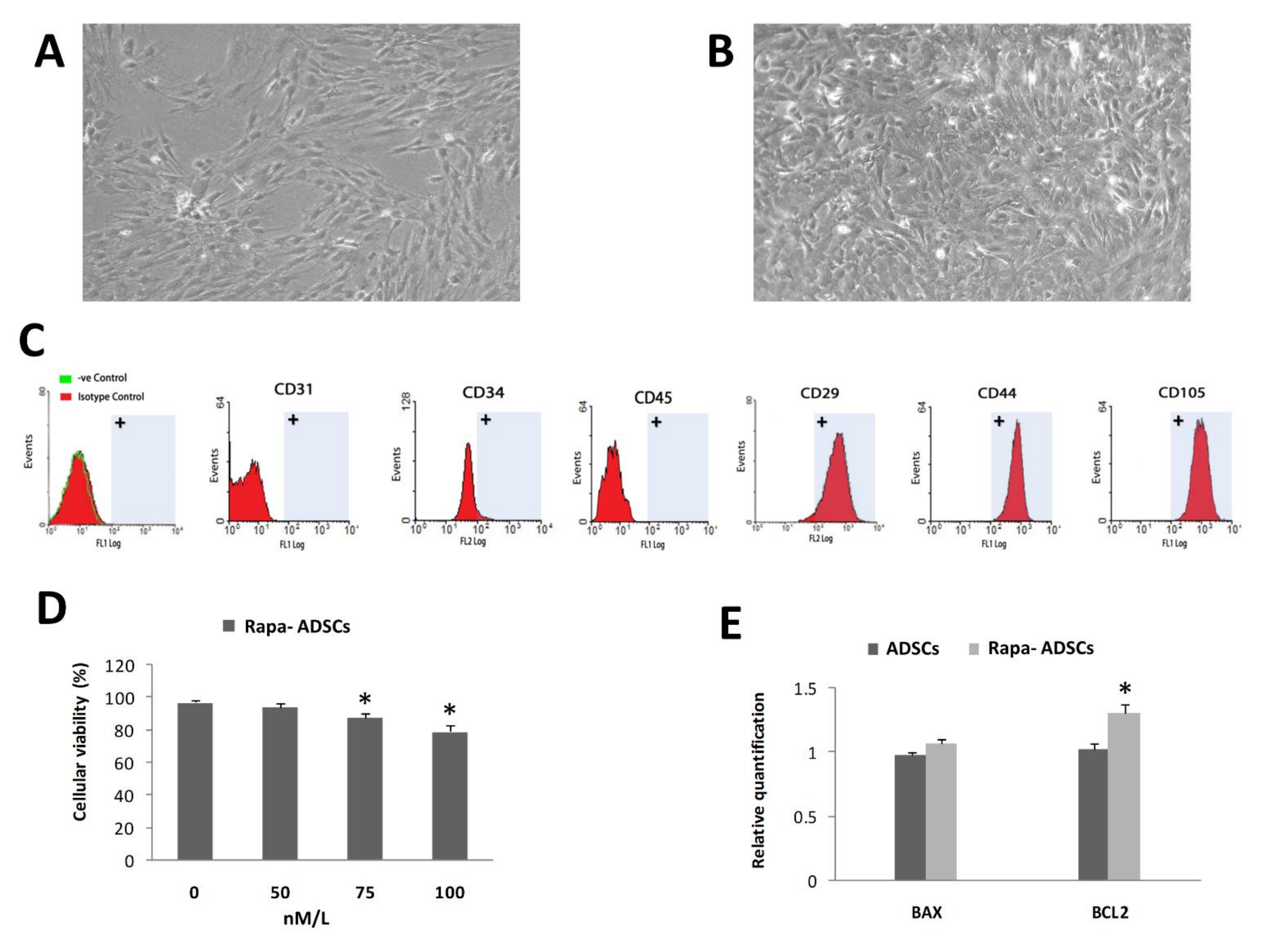
2.4. Characterization of Adipose MSCs by Flow Cytometry
2.5. Rapamycin Preconditioning of ADSCs
2.6. Assessment of Cell Viability by MTT Assay
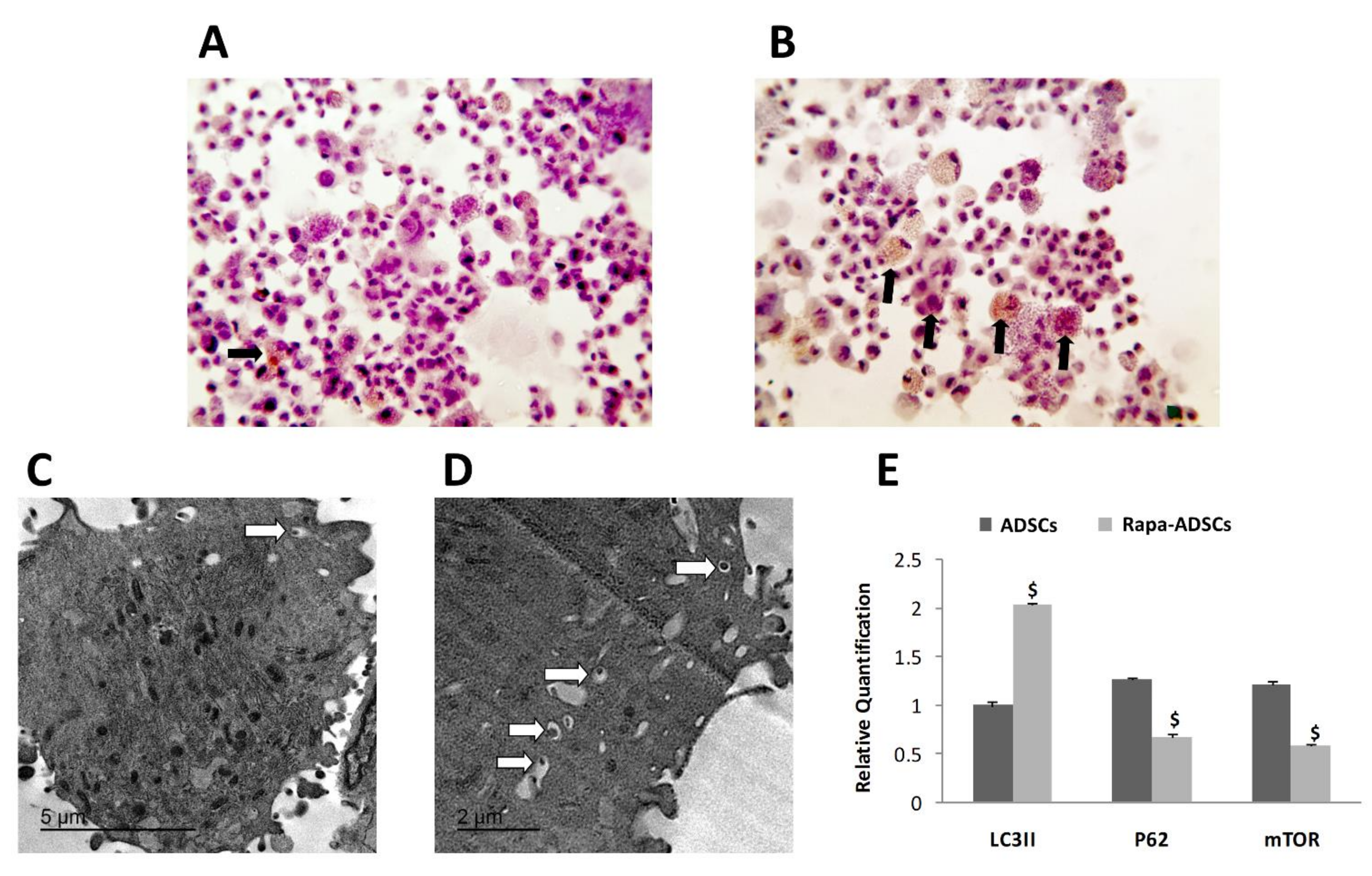
2.7. Evaluation of Autophagic Activity
2.7.1. Transmission Electron Microscopy (TEM)
2.7.2. Immunostaining of Autophagosomes
2.7.3. Gene Expression
2.8. Biochemical Analyses
2.9. Evaluation of Liver Oxidative Stress and Antioxidant Parameters
2.10. Histopathological Studies
2.11. Immunostaining
2.12. Gene Expression Analysis
2.13. Statistical Analysis
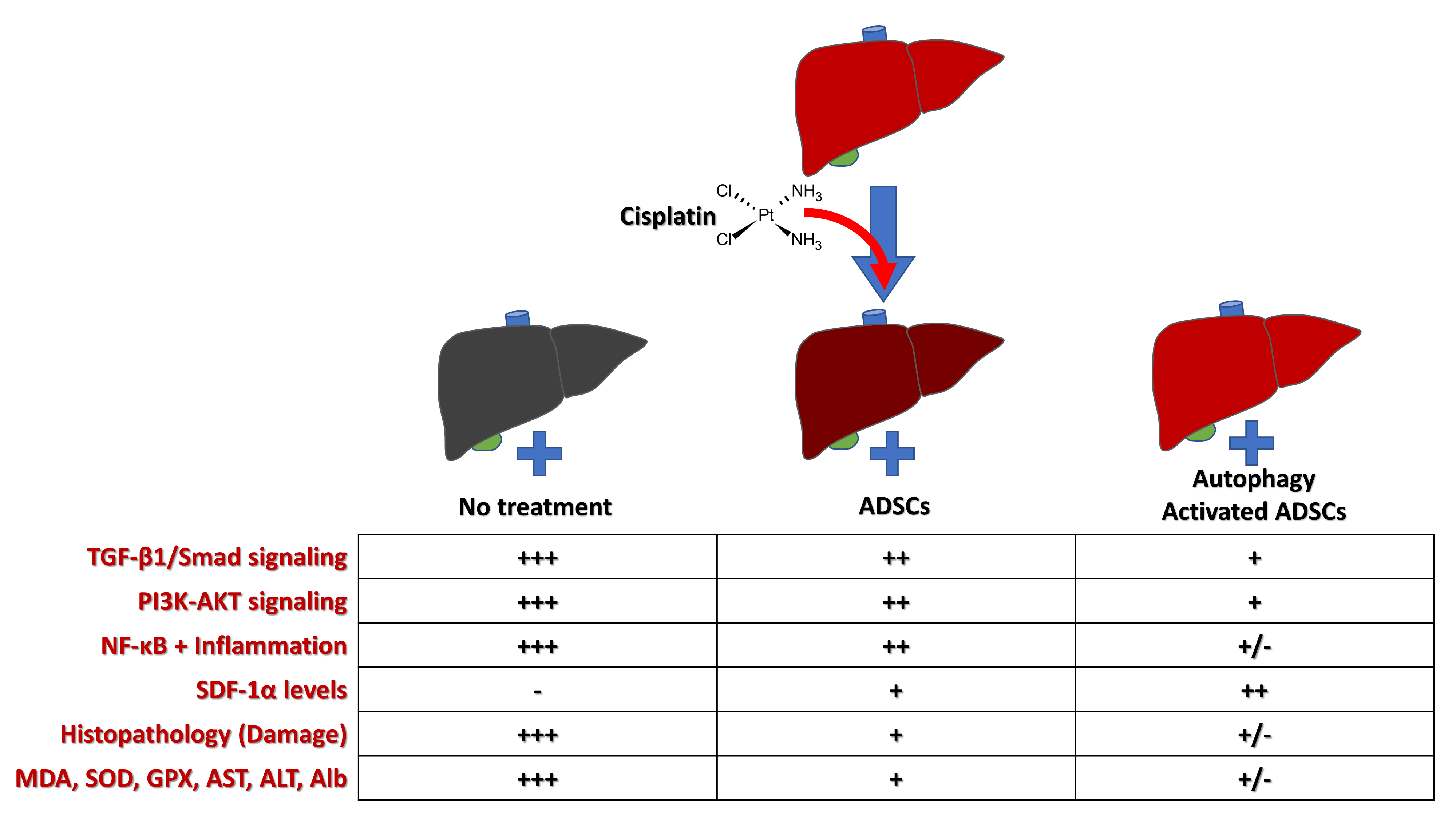
3. Results
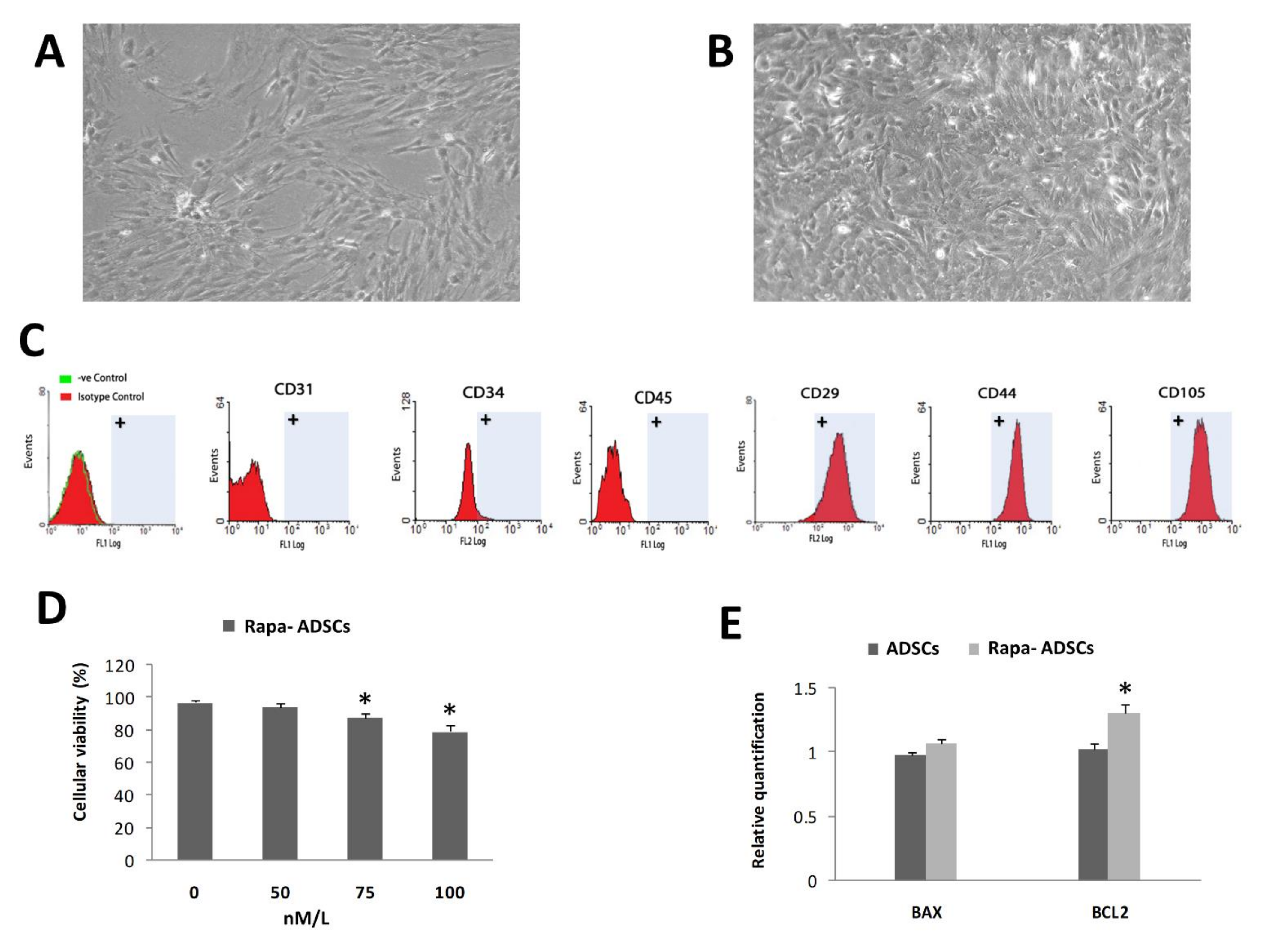
3.1. Characterization of Isolated ADSCs
3.2. Effect of Rapamycin Preconditioning on Viability of Cultured ADSCs
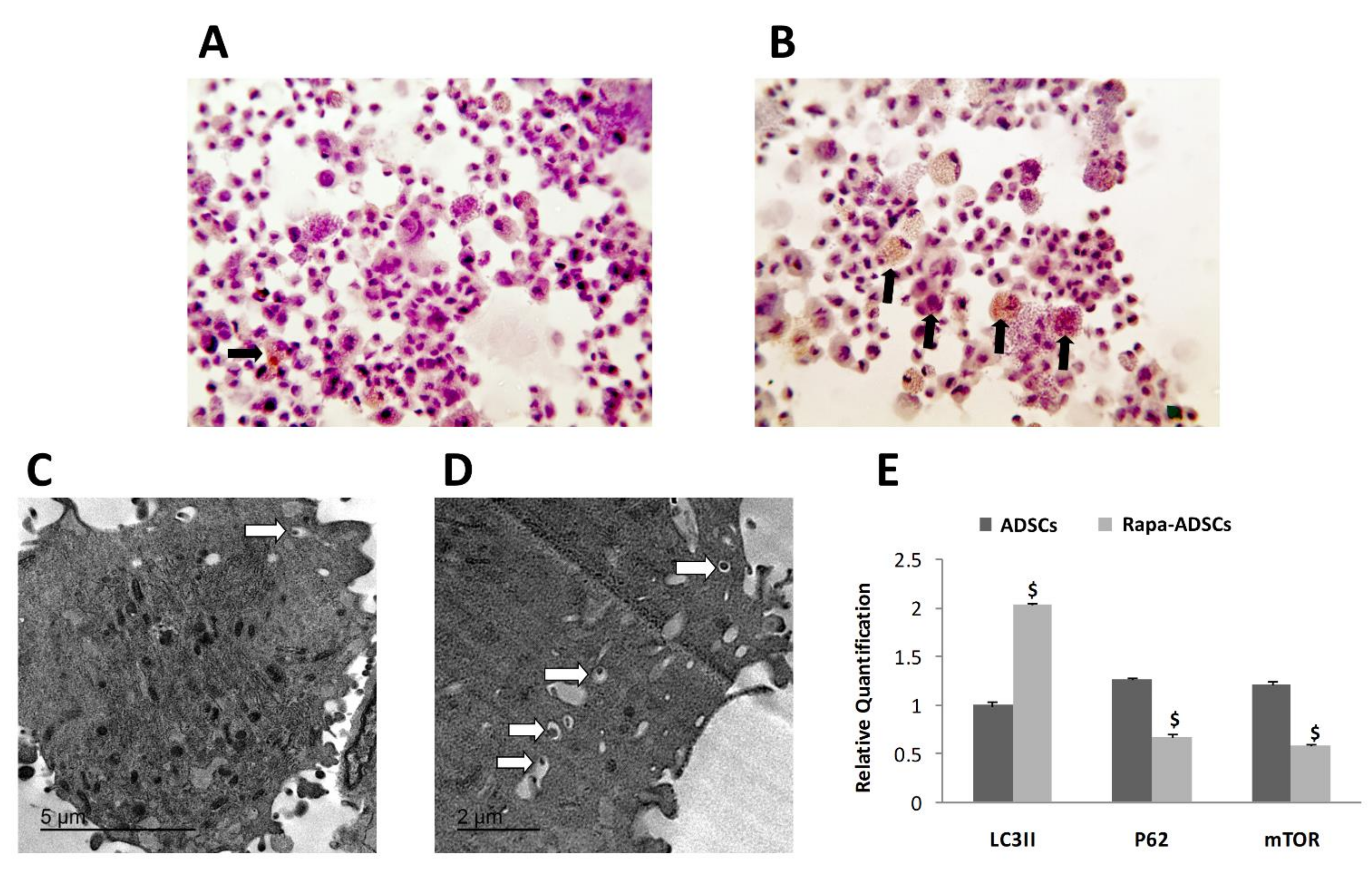
3.3. Effect of Rapamycin on Autophagy of ADSCs
3.4. Detection of Administered ADSCs in the Liver of Recipient Animals
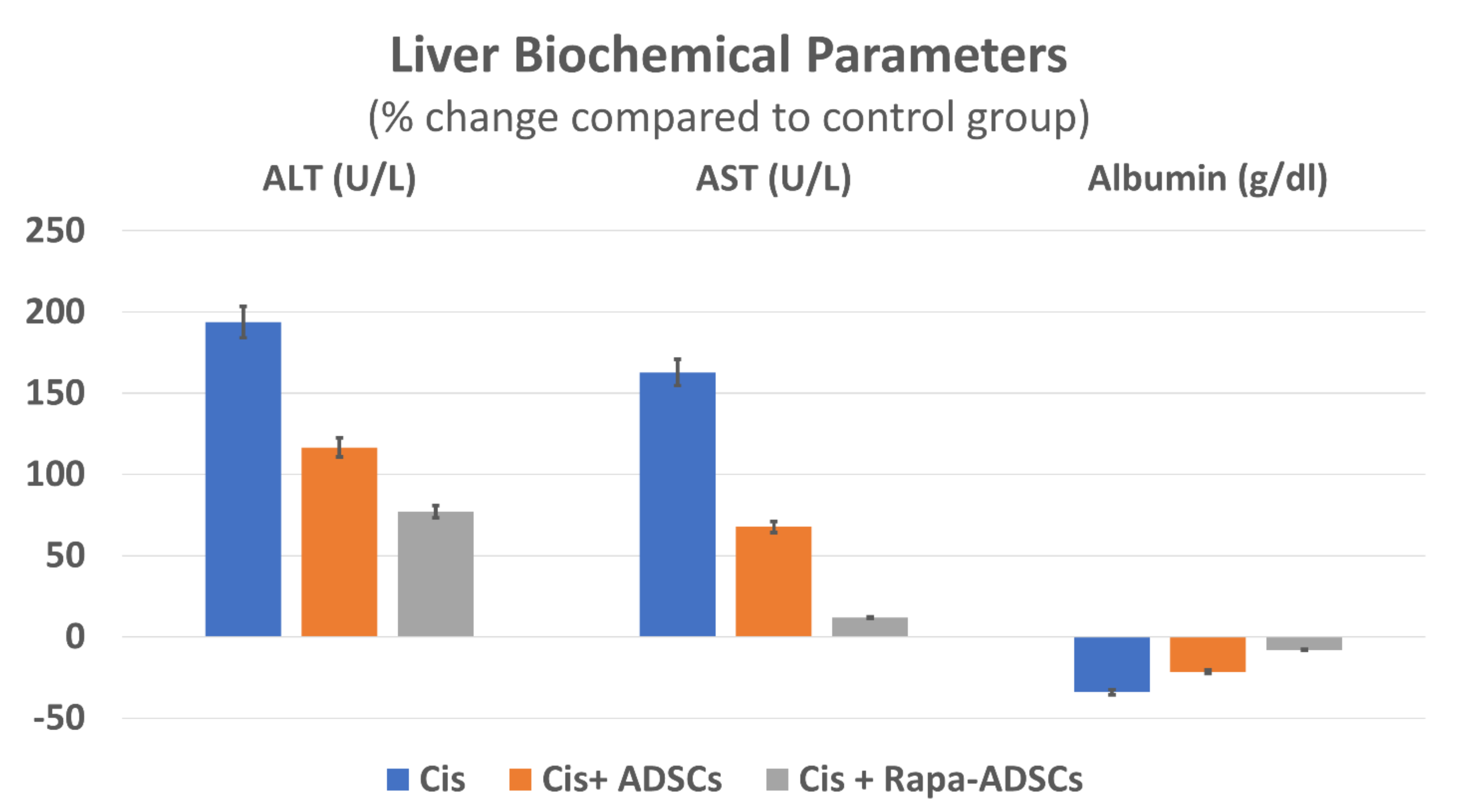
3.5. Effects of Rapamycin-Preconditioned ADSCs on Liver Function
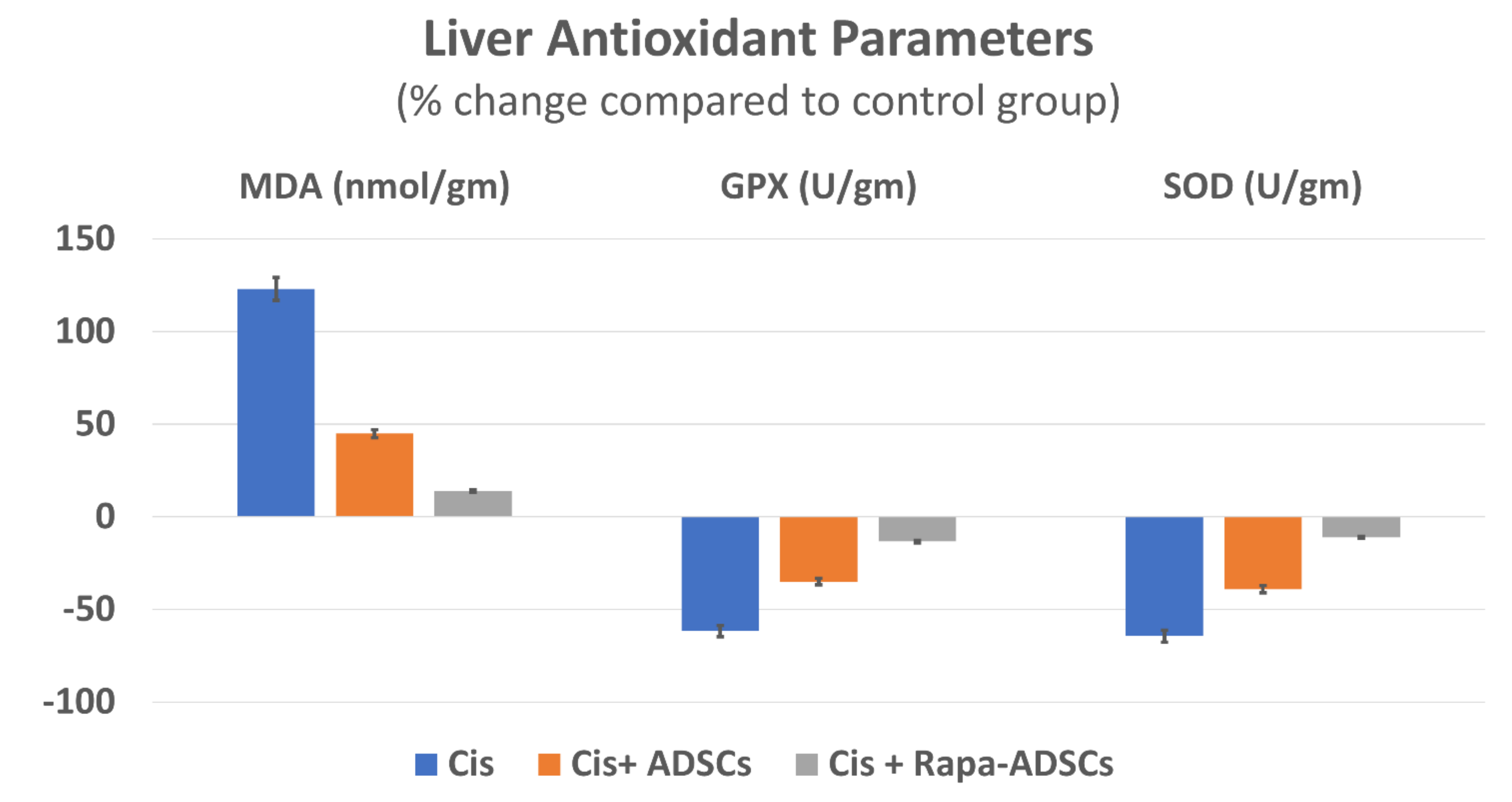
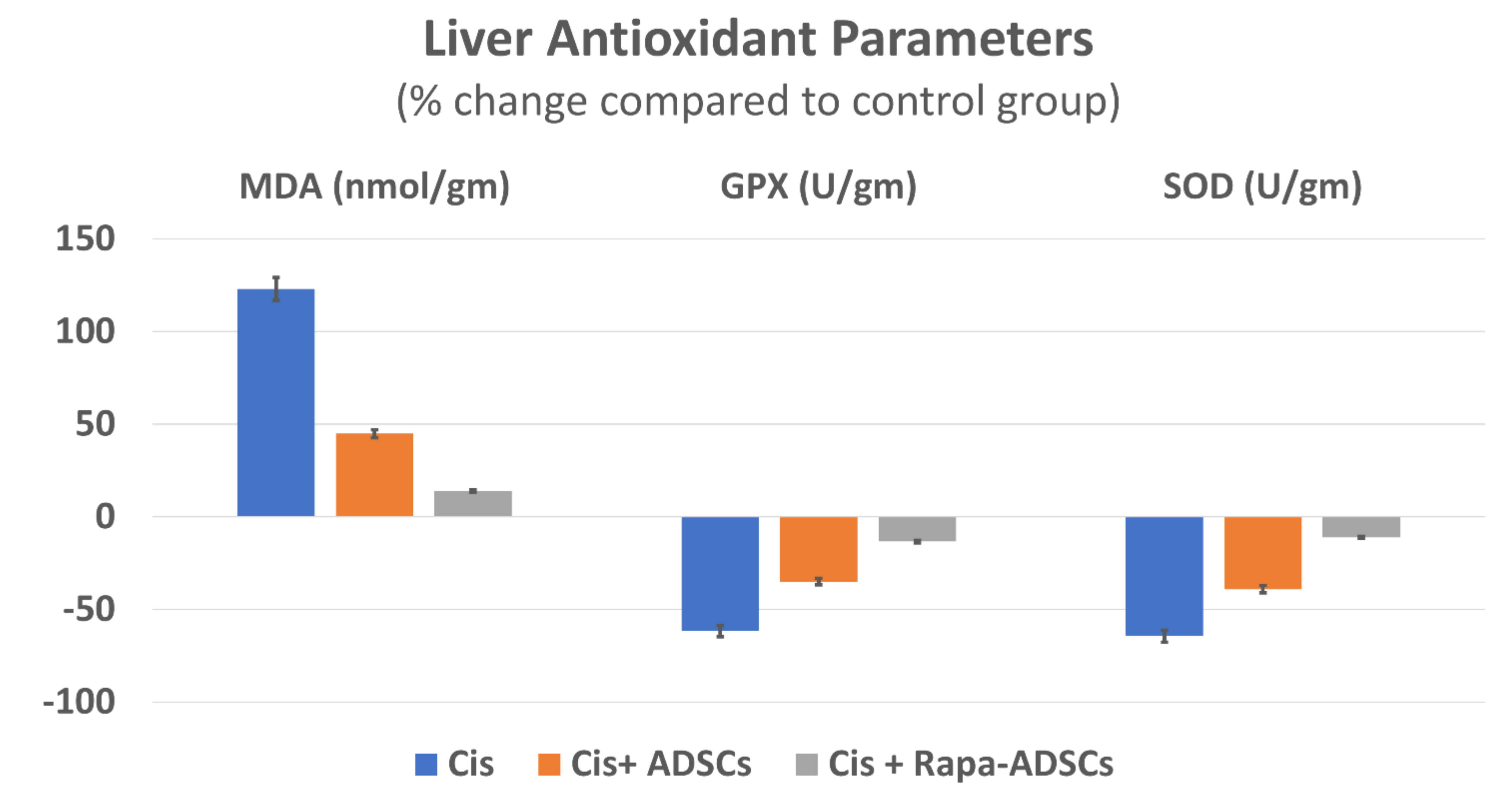
3.6. Effects of Rapamycin-Preconditioned ADSCs on Liver Antioxidant Profile
3.7. Effects of Rapamycin-Preconditioned ADSCs on Liver Histopathological Picture
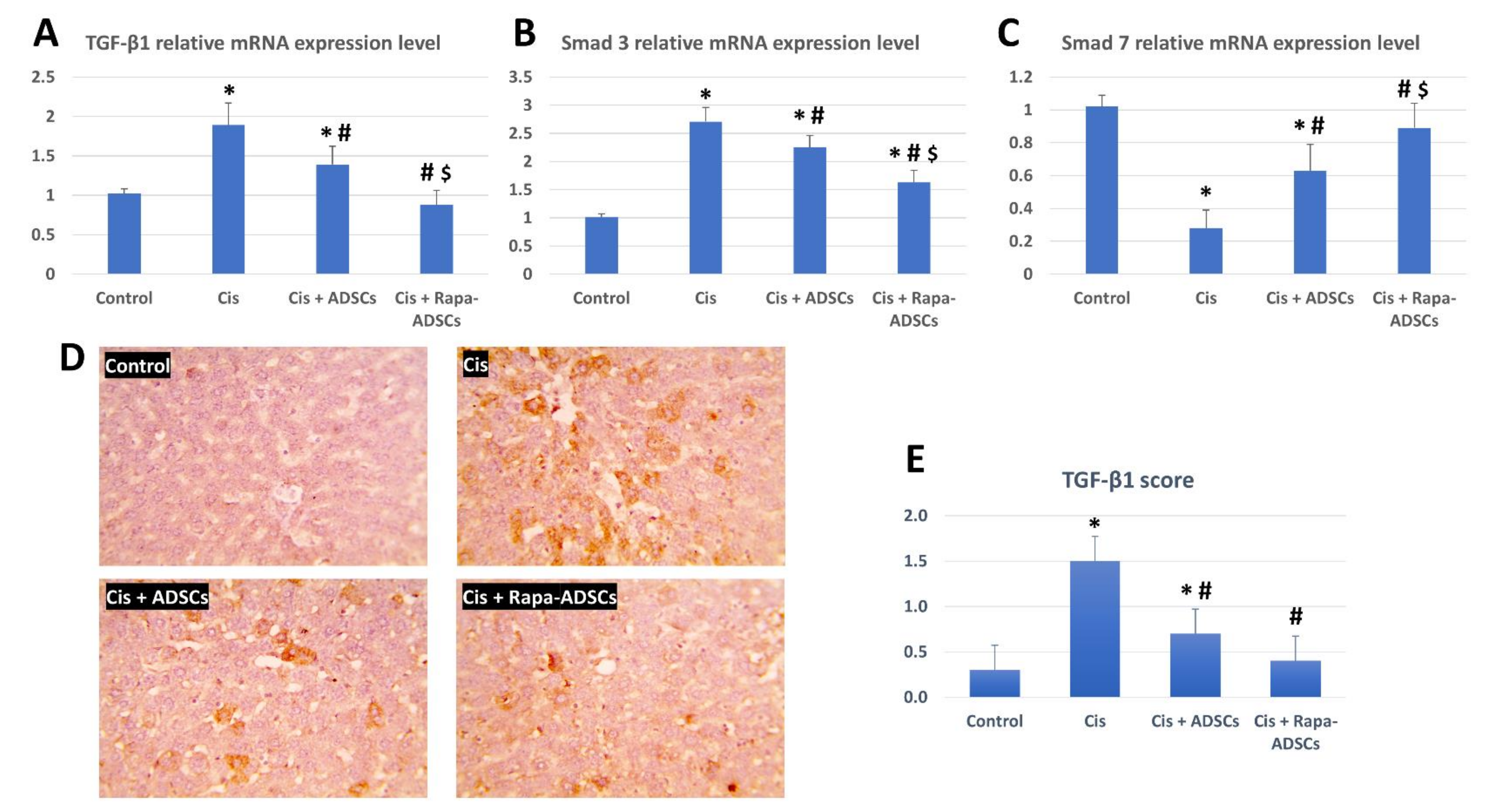
3.8. Effects of Rapamycin-Preconditioned ADSCs on Liver TGF-β1/Smad Signaling Pathway
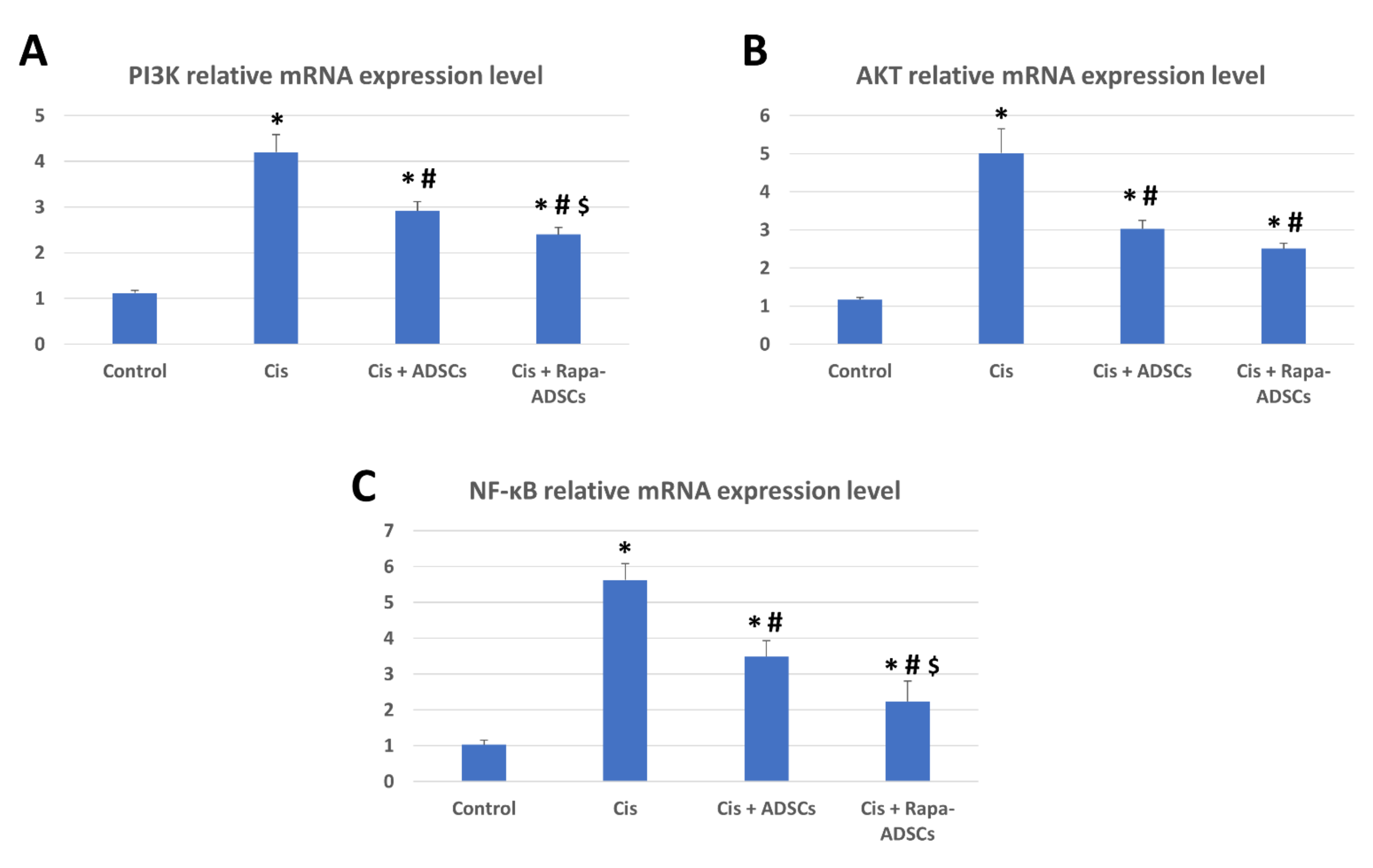
3.9. Effects of Rapamycin-Preconditioned ADSCs on Liver PI3K-AKT Signaling Pathway and NF-κB Gene Expression
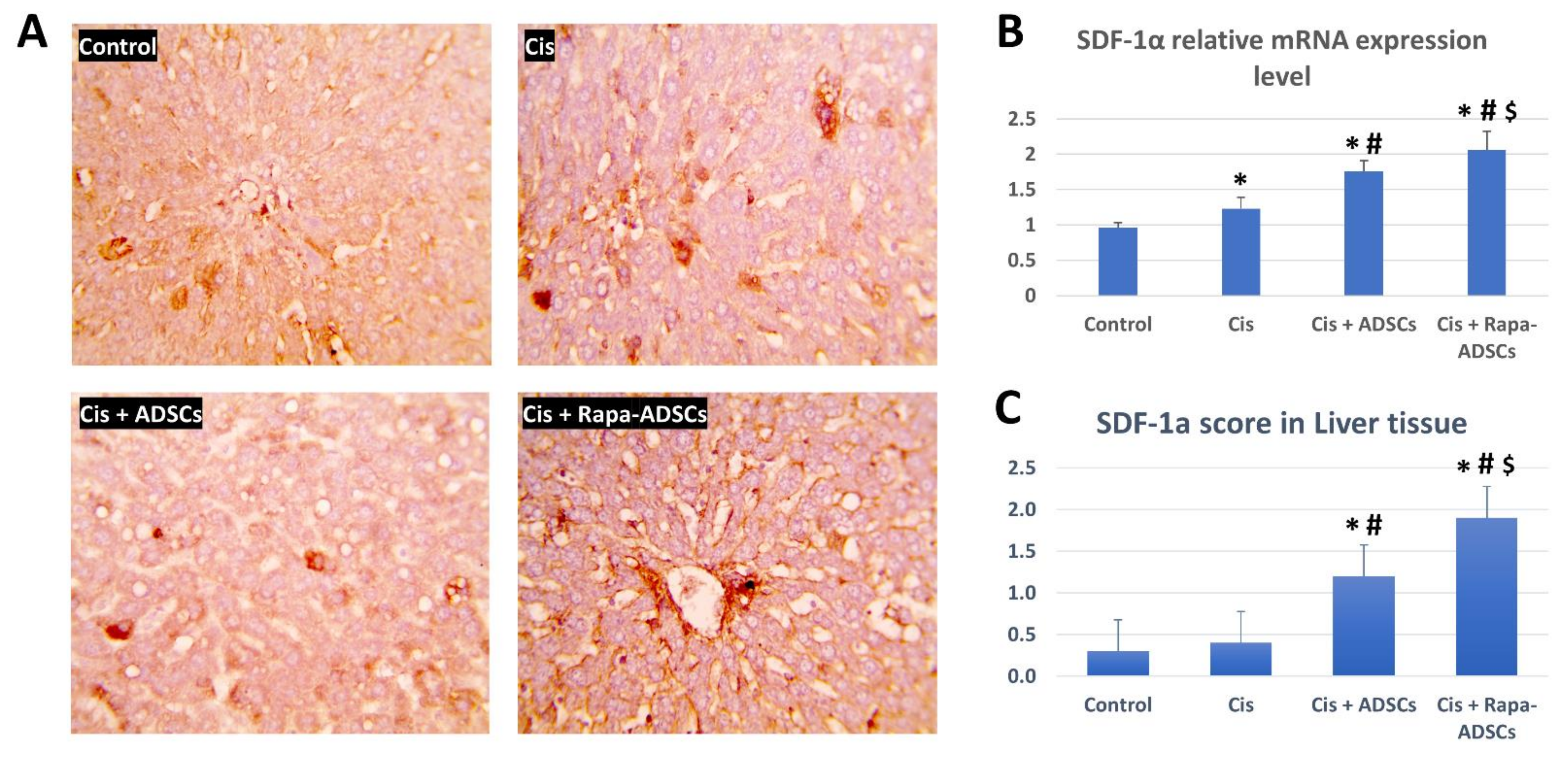
3.10. Effects of Rapamycin-Preconditioned ADSCs on Liver SDF-1α Gene Expression and Levels of SDF-1α Protein in the Liver
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Di Maio, M.; Basch, E.; Bryce, J.; Perrone, F. Patient-reported outcomes in the evaluation of toxicity of anticancer treatments. Nat. Rev. Clin. Oncol. 2016, 13, 319–325. [Google Scholar] [CrossRef]
- Liao, Y.; Lu, X.; Lu, C.; Li, G.; Jin, Y.; Tang, H. Selection of agents for prevention of cisplatin-induced hepatotoxicity. Pharmacol. Res. 2008, 57, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, N.A.G.; Rodrigues, M.A.C.; Martins, N.M.; Dos Santos, A.C. Cisplatin-induced nephrotoxicity and targets of nephroprotection: An update. Arch. Toxicol. 2012, 86, 1233–1250. [Google Scholar] [CrossRef]
- Dos Santos, N.A.G.; Ferreira, R.S.; dos Santos, A.C. Overview of cisplatin-induced neurotoxicity and ototoxicity, and the protective agents. Food Chem. Toxicol. 2020, 136, 111079. [Google Scholar] [CrossRef] [PubMed]
- Pratibha, R.; Sameer, R.; Rataboli, P.V.; Bhiwgade, D.A.; Dhume, C.Y. Enzymatic studies of cisplatin induced oxidative stress in hepatic tissue of rats. Eur. J. Pharmacol. 2006, 532, 290–293. [Google Scholar] [CrossRef] [PubMed]
- Deavall, D.G.; Martin, E.A.; Horner, J.M.; Roberts, R. Drug-induced oxidative stress and toxicity. J. Toxicol. 2012, 2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Fraser, M.; Abedini, M.R.; Bai, T.; Tsang, B.K. Regulation of apoptosis-inducing factor-mediated, cisplatin-induced apoptosis by Akt. Br. J. Cancer 2008, 98, 803–808. [Google Scholar] [CrossRef]
- Faubel, S.; Lewis, E.C.; Reznikov, L.; Ljubanovic, D.; Hoke, T.S.; Somerset, H.; Oh, D.J.; Lu, L.; Klein, C.L.; Dinarello, C.A.; et al. Cisplatin-induced acute renal failure is associated with an increase in the cytokines interleukin (IL)-1β, IL-18, IL-6, and neutrophil infiltration in the kidney. J. Pharmacol. Exp. Ther. 2007, 322, 8–15. [Google Scholar] [CrossRef]
- LiverTox: Clinical and Research Information on Drug-Induced Liver Injury (Internet). Bethesda (MD): National Institute of Diabetes and Digestive and Kidney Diseases. 2012; Cisplatin. Updated 15 September 2020. Available online: https://www.ncbi.nlm.nih.gov/books/NBK548160/ (accessed on 13 September 2021).
- Maor, Y.; Malnick, S. Liver Injury Induced by Anticancer Chemotherapy and Radiation Therapy. Int. J. Hepatol. 2013, 2013, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.; Lin, Y.; Huang, X.; Wu, S.; Wei, J.; Yang, C. Oxaliplatin aggravates hepatic oxidative stress, inflammation and fibrosis in a non-alcoholic fatty liver disease mouse model. Int. J. Mol. Med. 2019, 43, 2398–2408. [Google Scholar] [CrossRef] [PubMed]
- Le Blanc, K.; Pittenger, M.F. Mesenchymal stem cells: Progress toward promise. Cytotherapy 2005, 7, 36–45. [Google Scholar] [CrossRef]
- Shi, Y.; Hu, G.; Su, J.; Li, W.; Chen, Q.; Shou, P.; Xu, C.; Chen, X.; Huang, Y.; Zhu, Z.; et al. Mesenchymal stem cells: A new strategy for immunosuppression and tissue repair. Cell Res. 2010, 20, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Chen, X.; Wang, W.E.; Zeng, C. How to Improve the Survival of Transplanted Mesenchymal Stem Cell in Ischemic Heart? Stem Cells Int. 2016, 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Preda, M.B.; Neculachi, C.A.; Fenyo, I.M.; Vacaru, A.-M.; Publik, M.A.; Simionescu, M.; Burlacu, A. Short lifespan of syngeneic transplanted MSC is a consequence of in vivo apoptosis and immune cell recruitment in mice. Cell Death Dis. 2021, 12, 566. [Google Scholar] [CrossRef]
- Jakovljevic, J.; Harrell, C.R.; Fellabaum, C.; Arsenijevic, A.; Jovicic, N.; Volarevic, V. Modulation of autophagy as new approach in mesenchymal stem cell-based therapy. Biomed. Pharmacother. 2018, 104, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef]
- Murrow, L.; Debnath, J. Autophagy as a stress-response and quality-control mechanism: Implications for cell injury and human disease. Annu. Rev. Pathol. Mech. Dis. 2013, 8, 105–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, B.; Kroemer, G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef] [Green Version]
- Guan, J.L.; Simon, A.K.; Prescott, M.; Menendez, J.A.; Liu, F.; Wang, F.; Wang, C.; Wolvetang, E.; Vazquez-Martin, A.; Zhang, J. Autophagy in stem cells. Autophagy 2013, 9, 830–849. [Google Scholar] [CrossRef]
- Chen, X.; He, Y.; Lu, F. Autophagy in stem cell biology: A perspective on stem cell self-renewal and differentiation. Stem Cells Int. 2018, 2018. [Google Scholar] [CrossRef] [PubMed]
- García-Prat, L.; Martínez-Vicente, M.; Perdiguero, E.; Ortet, L.; Rodríguez-Ubreva, J.; Rebollo, E.; Ruiz-Bonilla, V.; Gutarra, S.; Ballestar, E.; Serrano, A.L.; et al. Autophagy maintains stemness by preventing senescence. Nature 2016, 529, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.W.; Moon, S.J.; Park, M.J.; Kim, B.M.; Kim, E.K.; Lee, S.H.; Lee, E.J.; Chung, B.H.; Yang, C.W.; Cho, M. La optimization of adipose tissue-derived mesenchymal stem cells by rapamycin in a murine model of acute graft-versus-host disease. Stem Cell Res. Ther. 2015, 6, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.-H.; Wang, Y.-L.; Wang, H.-J.; Wu, J.-H.; Tan, Y.-Z. Rapamycin-Preactivated Autophagy Enhances Survival and Differentiation of Mesenchymal Stem Cells After Transplantation into Infarcted Myocardium. Stem Cell Rev. Rep. 2020, 16, 344–356. [Google Scholar] [CrossRef] [Green Version]
- National Research Council (US); Committee for the Update of the Guide for the Care and Use of Laboratory Animals. Guide for the Care and Use of Laboratory Animals, 8th ed.; National Academies Press: Washington, DC, USA, 2011. [Google Scholar]
- Seto, Y.; Okazaki, F.; Horikawa, K.; Zhang, J.; Sasaki, H.; To, H. Influence of dosing times on cisplatin-induced peripheral neuropathy in rats. BMC Cancer 2016, 16, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Bunnell, B.A.; Flaat, M.; Gagliardi, C.; Patel, B.; Ripoll, C. Adipose-derived stem cells: Isolation, expansion and differentiation. Methods 2008, 45, 115–120. [Google Scholar] [CrossRef] [Green Version]
- Estes, B.T.; Diekman, B.O.; Gimble, J.M.; Guilak, F. Isolation of adipose-derived stem cells and their induction to a chondrogenic phenotype. Nat. Protoc. 2010, 5, 1294–1311. [Google Scholar] [CrossRef] [Green Version]
- Alzahrani, F.A.; El-Magd, M.A.; Abdelfattah-Hassan, A.; Saleh, A.A.; Saadeldin, I.M.; El-Shetry, E.S.; Badawy, A.A.; Alkarim, S. Potential Effect of Exosomes Derived from Cancer Stem Cells and MSCs on Progression of DEN-Induced HCC in Rats. Stem Cells Int. 2018, 2018, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Abdelfattah-Hassan, A.; Shalaby, S.I.; Khater, S.I.; El-Shetry, E.S.; Abd El Fadil, H.; Elsayed, S.A. Panax ginseng is superior to vitamin E as a hepatoprotector against cyclophosphamide-induced liver damage. Complement. Ther. Med. 2019, 46, 95–102. [Google Scholar] [CrossRef]
- Ahmed, M.M.; Hussein, M.M.A. Neurotoxic effects of silver nanoparticles and the protective role of rutin. Biomed. Pharmacother. 2017, 90, 731–739. [Google Scholar] [CrossRef]
- Hussein, M.M.A.; Mahfouz, M.K. Effect of resveratrol and rosuvastatin on experimental diabetic nephropathy in rats. Biomed. Pharmacother. 2016, 82, 685–692. [Google Scholar] [CrossRef]
- Matsuzawa, T.; Nomura, M.; Unno, T. Clinical Pathology Reference Ranges of Laboratory Animals. J. Vet. Med. Sci. 1993, 55, 351–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzaccara, C.; Labruna, G.; Cito, G.; Scarfò, M.; de Felice, M.; Pastore, L.; Sacchetti, L. Age-related reference intervals of the main biochemical and hematological parameters in C57BL/6J, 129SV/EV and C3H/HeJ mouse strains. PLoS ONE 2008, 3, e3772. [Google Scholar] [CrossRef]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef] [PubMed]
- Vadlamudi, R.K.; Joung, I.; Strominger, J.L.; Shin, J. p62, a phosphotyrosine-independent ligand of the SH2 domain of p56(lck), belongs to a new class of ubiquitin-binding proteins. J. Biol. Chem. 1996, 271, 20235–20237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rea, S.L.; Walsh, J.P.; Layfield, R.; Ratajczak, T.; Xu, J. New insights into the role of sequestosome 1/p62 mutant proteins in the pathogenesis of paget’s disease of bone. Endocr. Rev. 2013, 34, 501–524. [Google Scholar] [CrossRef] [Green Version]
- Clausen, T.H.; Lamark, T.; Isakson, P.; Finley, K.; Larsen, K.B.; Brech, A.; Øvervatn, A.; Stenmark, H.; Bjørkøy, G.; Simonsen, A.; et al. p62/SQSTM1 and ALFY interact to facilitate the formation of p62 bodies/ALIS and their degradation by autophagy. Autophagy 2010, 6, 330–344. [Google Scholar] [CrossRef] [Green Version]
- Benjamin, D.; Colombi, M.; Moroni, C.; Hall, M.N. Rapamycin passes the torch: A new generation of mTOR inhibitors. Nat. Rev. Drug Discov. 2011, 10, 868–880. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Yang, Y.J.; Wang, H.; Dong, Q.T.; Wang, T.J.; Qian, H.Y.; Xu, H. Autophagy activation: A novel mechanism of atorvastatin to protect mesenchymal stem cells from hypoxia and serum deprivation via AMP-activated protein kinase/mammalian target of rapamycin pathway. Stem Cells Dev. 2012, 21, 1321–1332. [Google Scholar] [CrossRef] [Green Version]
- Yang, R.; Ouyang, Y.; Li, W.; Wang, P.; Deng, H.; Song, B.; Hou, J.; Chen, Z.; Xie, Z.; Liu, Z.; et al. Autophagy Plays a Protective Role in Tumor Necrosis Factor-α-Induced Apoptosis of Bone Marrow-Derived Mesenchymal Stem Cells. Stem Cells Dev. 2016, 25, 788–797. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Zhao, L.; Wu, D.; Li, L. Modulating autophagy in mesenchymal stem cells effectively protects against hypoxia- or ischemia-induced injury. Stem Cell Res. Ther. 2019, 10, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef]
- Watabe, T.; Miyazono, K. Roles of TGF-β family signaling in stem cell renewal and differentiation. Cell Res. 2009, 19, 103–115. [Google Scholar] [CrossRef]
- Majumdar, A.; Curley, S.A.; Wu, X.; Brown, P.; Hwang, J.P.; Shetty, K.; Yao, Z.X.; He, A.R.; Li, S.; Katz, L.; et al. Hepatic stem cells and transforming growth factor β in hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 530–538. [Google Scholar] [CrossRef] [Green Version]
- Dooley, S.; Ten Dijke, P. TGF-β in progression of liver disease. Cell Tissue Res. 2012, 347, 245–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabregat, I.; Moreno-Càceres, J.; Sánchez, A.; Dooley, S.; Dewidar, B.; Giannelli, G.; ten Dijke, P. TGF-β signalling and liver disease. FEBS J. 2016, 283, 2219–2232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leask, A.; Abraham, D.J. TGF-β signaling and the fibrotic response. FASEB J. 2004, 18, 816–827. [Google Scholar] [CrossRef] [PubMed]
- Niu, L.; Cui, X.; Qi, Y.; Xie, D.; Wu, Q.; Chen, X.; Ge, J.; Liu, Z. Involvement of TGF-β1/Smad3 Signaling in Carbon Tetrachloride-Induced Acute Liver Injury in Mice. PLoS ONE 2016, 11, 156090. [Google Scholar] [CrossRef]
- Higgins, S.P.; Tang, Y.; Higgins, C.E.; Mian, B.; Zhang, W.; Czekay, R.P.; Samarakoon, R.; Conti, D.J.; Higgins, P.J. TGF-β1/p53 signaling in renal fibrogenesis. Cell. Signal. 2018, 43, 1–10. [Google Scholar] [CrossRef]
- Breitkopf, K.; Haas, S.; Wiercinska, E.; Singer, M.V.; Dooley, S. Anti-TGF-β strategies for the treatment of chronic liver disease. In Alcoholism: Clinical and Experimental Research; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2005; Volume 29. [Google Scholar]
- Park, S.-A.; Kim, M.-J.; Park, S.-Y.; Kim, J.-S.; Lee, S.-J.; Woo, H.A.; Kim, D.-K.; Nam, J.-S.; Sheen, Y.Y. EW-7197 inhibits hepatic, renal, and pulmonary fibrosis by blocking TGF-β/Smad and ROS signaling. Cell. Mol. Life Sci. 2015, 72, 2023–2039. [Google Scholar] [CrossRef] [PubMed]
- Giannelli, G.; Mikulits, W.; Dooley, S.; Fabregat, I.; Moustakas, A.; ten Dijke, P.; Portincasa, P.; Winter, P.; Janssen, R.; Leporatti, S.; et al. The rationale for targeting TGF-β in chronic liver diseases. Eur. J. Clin. Invest. 2016, 46, 349–361. [Google Scholar] [CrossRef] [Green Version]
- Porta, C.; Paglino, C.; Mosca, A. Targeting PI3K/Akt/mTOR signaling in cancer. Front. Oncol. 2014, 4, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.T.; Tan, H.L.; Huang, Q.; Ong, C.N.; Shen, H.M. Activation of the PI3K-Akt-mTOR signaling pathway promotes necrotic cell death via suppression of autophagy. Autophagy 2009, 5, 824–834. [Google Scholar] [CrossRef] [Green Version]
- Lei, Y.; Wang, Q.-L.; Shen, L.; Tao, Y.Y.; Liu, C.H. MicroRNA-101 suppresses liver fibrosis by downregulating PI3K/Akt/mTOR signaling pathway. Clin. Res. Hepatol. Gastroenterol. 2019, 43, 575–584. [Google Scholar] [CrossRef]
- Dong, Z.; Li, S.; Wang, X.; Si, L.; Ma, R.; Bao, L.; Bo, A. LncRNA GAS5 restrains CCL4-induced hepatic fibrosis by targeting miR-23a through the PTEN/PI3K/Akt signaling pathway. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 316, G539–G550. [Google Scholar] [CrossRef]
- Fu, J.; Wu, B.; Zhong, S.; Deng, W.; Lin, F. miR-29a-3p suppresses hepatic fibrosis pathogenesis by modulating hepatic stellate cell proliferation via targeting PIK3R3 gene expression. Biochem. Biophys. Res. Commun. 2020, 529, 922–929. [Google Scholar] [CrossRef]
- Mi, X.-J.; Hou, J.-G.; Wang, Z.; Han, Y.; Ren, S.; Hu, J.-N.; Chen, C.; Li, W. The protective effects of maltol on cisplatin-induced nephrotoxicity through the AMPK-mediated PI3K/Akt and p53 signaling pathways. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Pellegrino, R.; Calvisi, D.F.; Neumann, O.; Kolluru, V.; Wesely, J.; Chen, X.; Wang, C.; Wuestefeld, T.; Ladu, S.; Elgohary, N.; et al. EEF1A2 inactivates p53 by way of PI3K/AKT/mTOR-dependent stabilization of MDM4 in hepatocellular carcinoma. Hepatology 2014, 59, 1886–1899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.M.; Song, T.J.; Xiao, J.H.; Huang, Z.H.; Li, Y.; Lin, T.Y. Tripchlorolide induces autophagy in lung cancer cells by inhibiting the PI3K/AKT/mTOR pathway and improves cisplatin sensitivity in A549/DDP cells. Oncotarget 2017, 8, 63911–63922. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.; Pu, J.; Zhou, X.L.; Ning, Y.Y.; Bai, C. Silencing long Non-Coding RNA ROR improves sensitivity of Non-Small-Cell lung cancer to cisplatin resistance by inhibiting PI3K/Akt/mTOR signaling pathway. Tumor Biol. 2017, 39. [Google Scholar] [CrossRef] [Green Version]
- Lu, R.; Zhao, G.; Yang, Y.; Jiang, Z.; Cai, J.; Hu, H. Inhibition of CD133 Overcomes Cisplatin Resistance Through Inhibiting PI3K/AKT/mTOR Signaling Pathway and Autophagy in CD133-Positive Gastric Cancer Cells. Technol. Cancer Res. Treat. 2019, 18. [Google Scholar] [CrossRef] [Green Version]
- Kou, Y.; Tong, B.; Wu, W.; Liao, X.; Zhao, M. Berberine Improves Chemo-Sensitivity to Cisplatin by Enhancing Cell Apoptosis and Repressing PI3K/AKT/mTOR Signaling Pathway in Gastric Cancer. Front. Pharmacol. 2020, 11, 616251–616252. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhao, J.; Yan, T.; Mu, J.; Lin, Y.; Chen, J.; Deng, H.; Meng, X. Cyanidin-3-O-glucoside and cisplatin inhibit proliferation and downregulate the PI3K/AKT/mTOR pathway in cervical cancer cells. J. Food Sci. 2021. [Google Scholar] [CrossRef]
- Wang, S.; Zhu, M.; Wang, Q.; Hou, Y.; Li, L.; Weng, H.; Zhao, Y.; Chen, D.; Ding, H.; Guo, J.; et al. Alpha-fetoprotein inhibits autophagy to promote malignant behaviour in hepatocellular carcinoma cells by activating PI3K/AKT/mTOR signalling. Cell Death Dis. 2018, 9, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Yan, H.; Xu, Z.; Yang, B.; Luo, P.; He, Q. Molecular basis for class side effects associated with PI3K/AKT/mTOR pathway inhibitors. Expert Opin. Drug Metab. Toxicol. 2019, 15, 767–774. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Greten, F.R. NF-κB: Linking inflammation and immunity to cancer development and progression. Nat. Rev. Immunol. 2005, 5, 749–759. [Google Scholar] [CrossRef]
- Taniguchi, K.; Karin, M. NF-B, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef] [PubMed]
- Luedde, T.; Schwabe, R.F. NF-κB in the liver-linking injury, fibrosis and hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 108–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, G.; Iimuro, Y.; Seki, E.; Hirano, T.; Kaneda, Y.; Fujimoto, J. Selective inactivation of NF-κB in the liver using NF-κB decoy suppresses CCl4-induced liver injury and fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 293, 631–639. [Google Scholar] [CrossRef]
- Ma, H.; Liu, X.; Zhang, M.; Niu, J. Liver sinusoidal endothelial cells are implicated in multiple fibrotic mechanisms. Mol. Biol. Rep. 2021, 48, 2803–2815. [Google Scholar] [CrossRef] [PubMed]
- Liepelt, A.; Tacke, F. Stromal cell-derived factor-1 (SDF-1) as a target in liver diseases. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 311, G203–G209. [Google Scholar] [CrossRef] [Green Version]
- Otaka, F.; Ito, Y.; Nakamoto, S.; Nishizawa, N.; Hyodo, T.; Hosono, K.; Majima, M.; Koizumi, W.; Amano, H. Macrophages contribute to liver repair after monocrotaline-induced liver injury via SDF-1/CXCR4. Exp. Ther. Med. 2021, 22, 1–11. [Google Scholar] [CrossRef]
- Hatch, H.M.; Zheng, D.; Jorgensen, M.L.; Petersen, B.E. SDF-1α/CXCR4: A mechanism for hepatic oval cell activation and bone marrow stem cell recruitment to the injured liver of rats. Cloning Stem Cells 2002, 4, 339–351. [Google Scholar] [CrossRef]
- Toksoy, A.; Müller, V.; Gillitzer, R.; Goebeler, M. Biphasic expression of stromal cell-derived factor-1 during human wound healing. Br. J. Dermatol. 2007, 157, 1148–1154. [Google Scholar] [CrossRef] [PubMed]
- Tögel, F.; Isaac, J.; Hu, Z.; Weiss, K.; Westenfelder, C. Renal SDF-1 signals mobilization and homing of CXCR4-positive cells to the kidney after ischemic injury. Kidney Int. 2005, 67, 1772–1784. [Google Scholar] [CrossRef] [Green Version]
- Imitola, J.; Raddassi, K.; Park, K.I.; Mueller, F.J.; Nieto, M.; Teng, Y.D.; Frenkel, D.; Li, J.; Sidman, R.L.; Walsh, C.A.; et al. Directed migration of neural stem cells to sites of CNS injury by the stromal cell-derived factor 1α/CXC chemokine receptor 4 pathway. Proc. Natl. Acad. Sci. USA 2004, 101, 18117–18122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Deng, Y.; Zhou, G.Q. SDF-1α/CXCR4-mediated migration of systemically transplanted bone marrow stromal cells towards ischemic brain lesion in a rat model. Brain Res. 2008, 1195, 104–112. [Google Scholar] [CrossRef]
- Kucia, M.; Reca, R.; Miekus, K.; Wanzeck, J.; Wojakowski, W.; Janowska-Wieczorek, A.; Ratajczak, J.; Ratajczak, M.Z. Trafficking of Normal Stem Cells and Metastasis of Cancer Stem Cells Involve Similar Mechanisms: Pivotal Role of the SDF-1-CXCR4 Axis. Stem Cells 2005, 23, 879–894. [Google Scholar] [CrossRef]
- Ruart, M.; Chavarria, L.; Campreciós, G.; Suárez-Herrera, N.; Montironi, C.; Guixé-Muntet, S.; Bosch, J.; Friedman, S.L.; Garcia-Pagán, J.C.; Hernández-Gea, V. Impaired endothelial autophagy promotes liver fibrosis by aggravating the oxidative stress response during acute liver injury. J. Hepatol. 2019, 70, 458–469. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Sequence (5′-3′) | Accession No. |
|---|---|---|
| LC3 | F- CCAGGAGGAAGAAGGCTTGG R- GAGTGGAAGATGTCCGGCTC | NM_022867.2 |
| P62 | F- TGCTCCATCAGAGGATCCCA R- TTTCTGCAGAGGTGGGTGTC | NM_175843.4 |
| mTOR | F- TTGTGTCCTGCTGGCTG AAC R- GCTCTTTGTAGTGTAGTGCTTTGG | NM_019906.2 |
| BAX | F: GGCGATGAACTGGACAACAA R: CAAAGTAGAAAAGGGCAACC | NM_017059.2 |
| BCL2 | F: GGTGAACTGGGGGAGGATTG R: GCATGCTGGGGCCATATAGT | NM_016993.1 |
| NF-κB | F- GGACAGCACCACCTACGATG R- CTGGATCACTTCAATGGCCTC | NM_001276711.1 |
| TGFβ1 | F- CACTCCCGTGGCTTCTAGTG R- GGACTGGCGAGCCTTAGTTT | NM_021578.2 |
| SDF1α | F- GAGCCATGTCGCCAGAGCCAAC R- CACACCTCTCACATCTTGAGCCTCT | NM_001033882.1 |
| PI3K | F- TCTCCGTAGCGGGGCACCAG R- AACCAGCCAATATCTTCTGG | XM_017590649.2 |
| AKT | F- GAGGAGGAGACGATGGACTTC R- GGCATAGTAGCGACCTGTGG | NM_033230.3 |
| Smad3 | F- AGGGCTTTGAGGCTGTCTACC R- ACCCGATCCCTTTACTCCCA | NM_013095.3 |
| Smad7 | F- GGGGGAACGAATTATCTGGC R- CGCCATCCACTTCCCTTGT | NM_030858.2 |
| Sry | F- TGGGACTGGTGACAATTGTC R- GAGTACAGGTGTGCAGCTCT | NM_012772 |
| GAPDH | F- AGACAGCCGCATCTTCTTGT R- TTCCCATTCTCAGCCTTGAC | NM_017008.4 |
| Group | ALT (U/L) | AST (U/L) | Albumin (g/dL) |
|---|---|---|---|
| Control | 48.16 ± 3.06 | 102 ± 14.71 | 3.35 ± 0.46 |
| Cis | 141.17 ± 8.56 * | 268.67 ± 54.7 * | 2.21 ± 0.61 * |
| Cis + ADSCs | 104.33 ± 10.17 *,# | 171 ± 71.35 *,# | 2.63 ± 0.61 *,# |
| Cis + Rapa-ADSCs | 85.83 ± 10.57 *,#,$ | 114.67 ± 25.14 #,$ | 3.08 ± 0.39 #,$ |
| Group | MDA (nmol/g) | GPX (U/g) | SOD (U/g) |
|---|---|---|---|
| Control | 87.38 ± 17.86 | 120 ± 8.07 | 197.2 ± 10.87 |
| Cis | 194.3 ± 16.55 * | 46.48 ± 17.94 * | 70.89 ± 15.34 * |
| Cis + ADSCs | 126.86 ± 15.83 *,# | 78.96 ± 16.76 *,# | 120.15 ± 27.82 *,# |
| Cis + Rapa-ADSCs | 99.7 ± 15.36 #,$ | 104.4 ± 20.64 # | 175.48 ± 14.88 #,$ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El Nashar, E.M.; Alghamdi, M.A.; Alasmari, W.A.; Hussein, M.M.A.; Hamza, E.; Taha, R.I.; Ahmed, M.M.; Al-Khater, K.M.; Abdelfattah-Hassan, A. Autophagy Promotes the Survival of Adipose Mesenchymal Stem/Stromal Cells and Enhances Their Therapeutic Effects in Cisplatin-Induced Liver Injury via Modulating TGF-β1/Smad and PI3K/AKT Signaling Pathways. Cells 2021, 10, 2475. https://doi.org/10.3390/cells10092475
El Nashar EM, Alghamdi MA, Alasmari WA, Hussein MMA, Hamza E, Taha RI, Ahmed MM, Al-Khater KM, Abdelfattah-Hassan A. Autophagy Promotes the Survival of Adipose Mesenchymal Stem/Stromal Cells and Enhances Their Therapeutic Effects in Cisplatin-Induced Liver Injury via Modulating TGF-β1/Smad and PI3K/AKT Signaling Pathways. Cells. 2021; 10(9):2475. https://doi.org/10.3390/cells10092475
Chicago/Turabian StyleEl Nashar, Eman Mohamad, Mansour Abdullah Alghamdi, Wardah Abdullah Alasmari, Mohamed M. A. Hussein, Eman Hamza, Reham Ismail Taha, Mona M. Ahmed, Khulood Mohammed Al-Khater, and Ahmed Abdelfattah-Hassan. 2021. "Autophagy Promotes the Survival of Adipose Mesenchymal Stem/Stromal Cells and Enhances Their Therapeutic Effects in Cisplatin-Induced Liver Injury via Modulating TGF-β1/Smad and PI3K/AKT Signaling Pathways" Cells 10, no. 9: 2475. https://doi.org/10.3390/cells10092475
APA StyleEl Nashar, E. M., Alghamdi, M. A., Alasmari, W. A., Hussein, M. M. A., Hamza, E., Taha, R. I., Ahmed, M. M., Al-Khater, K. M., & Abdelfattah-Hassan, A. (2021). Autophagy Promotes the Survival of Adipose Mesenchymal Stem/Stromal Cells and Enhances Their Therapeutic Effects in Cisplatin-Induced Liver Injury via Modulating TGF-β1/Smad and PI3K/AKT Signaling Pathways. Cells, 10(9), 2475. https://doi.org/10.3390/cells10092475