
Detection of Novel Potential Regulators of Stem Cell Differentiation and Cardiogenesis through Combined Genome-Wide Profiling of Protein-Coding Transcripts and microRNAs



Abstract
:1. Introduction
2. Materials and Methods
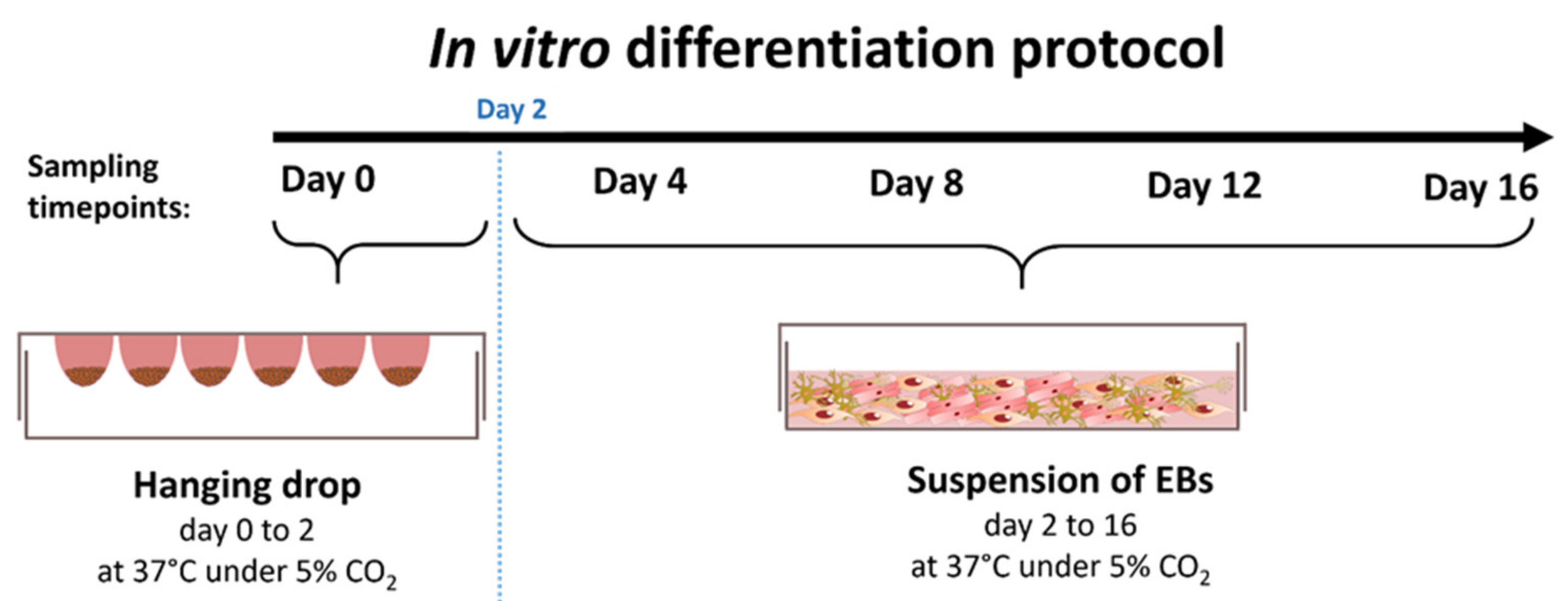
2.1. Culturing and Differentiation Assays of ESCs
2.2. Microarray Data Processing and Data Quality Assessment
2.3. Determining miRNA Peak Patterns
2.4. Functional Enrichment Analysis
2.5. Integrative Analysis of Expression Data and Potential Regulatory Interactions
3. Results
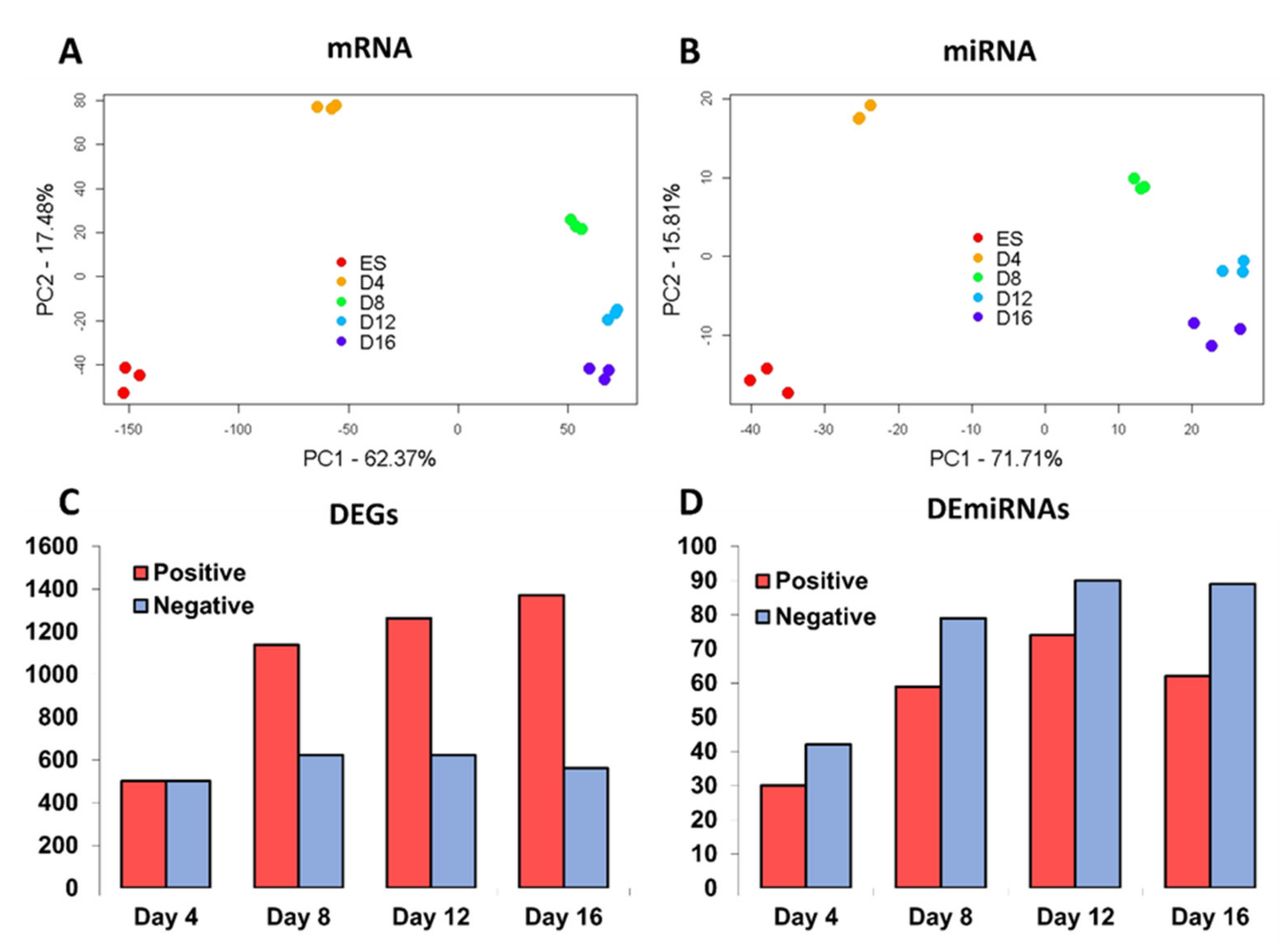
3.1. Differential Gene Expression during In Vitro Differentiation of ESCs
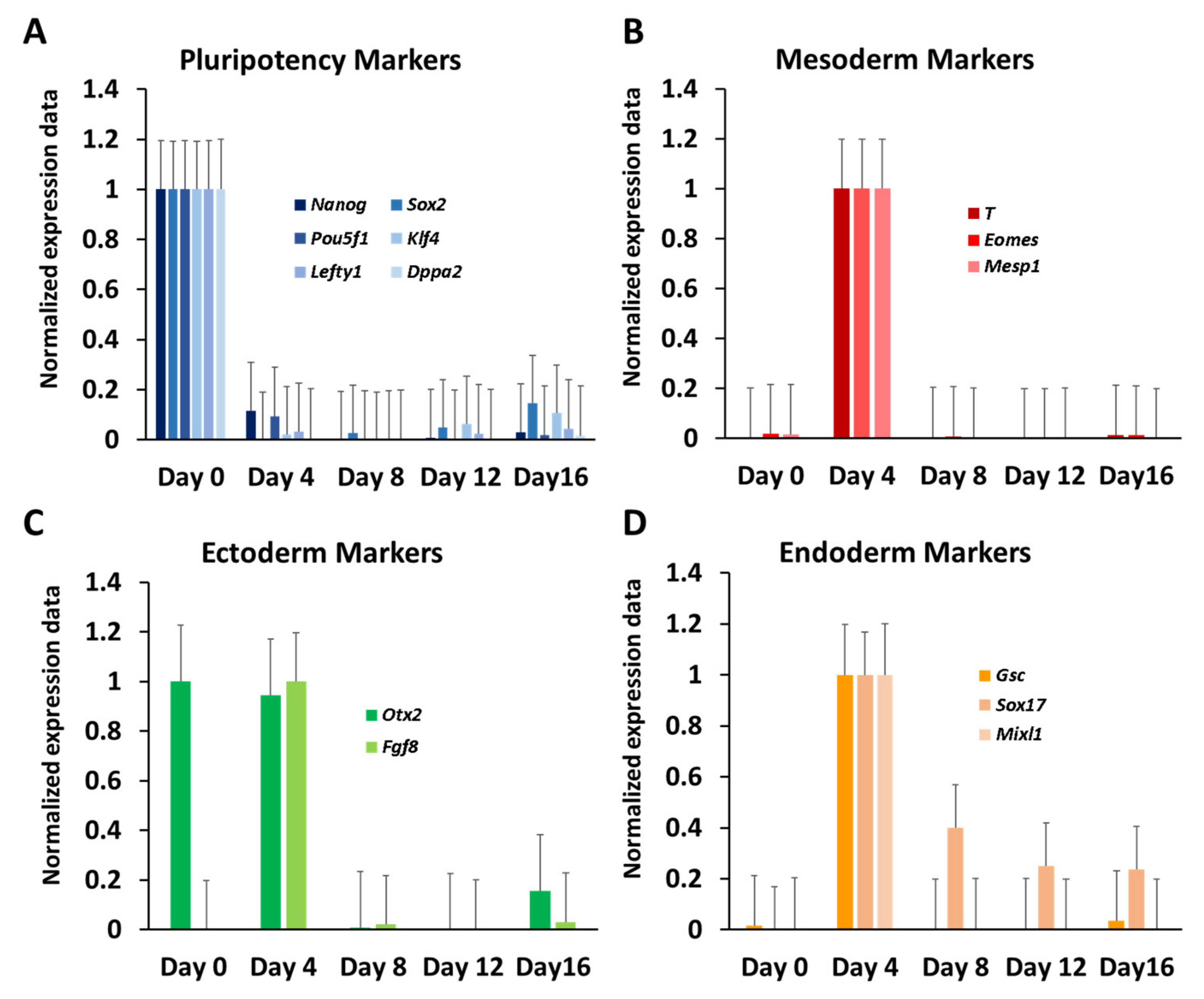
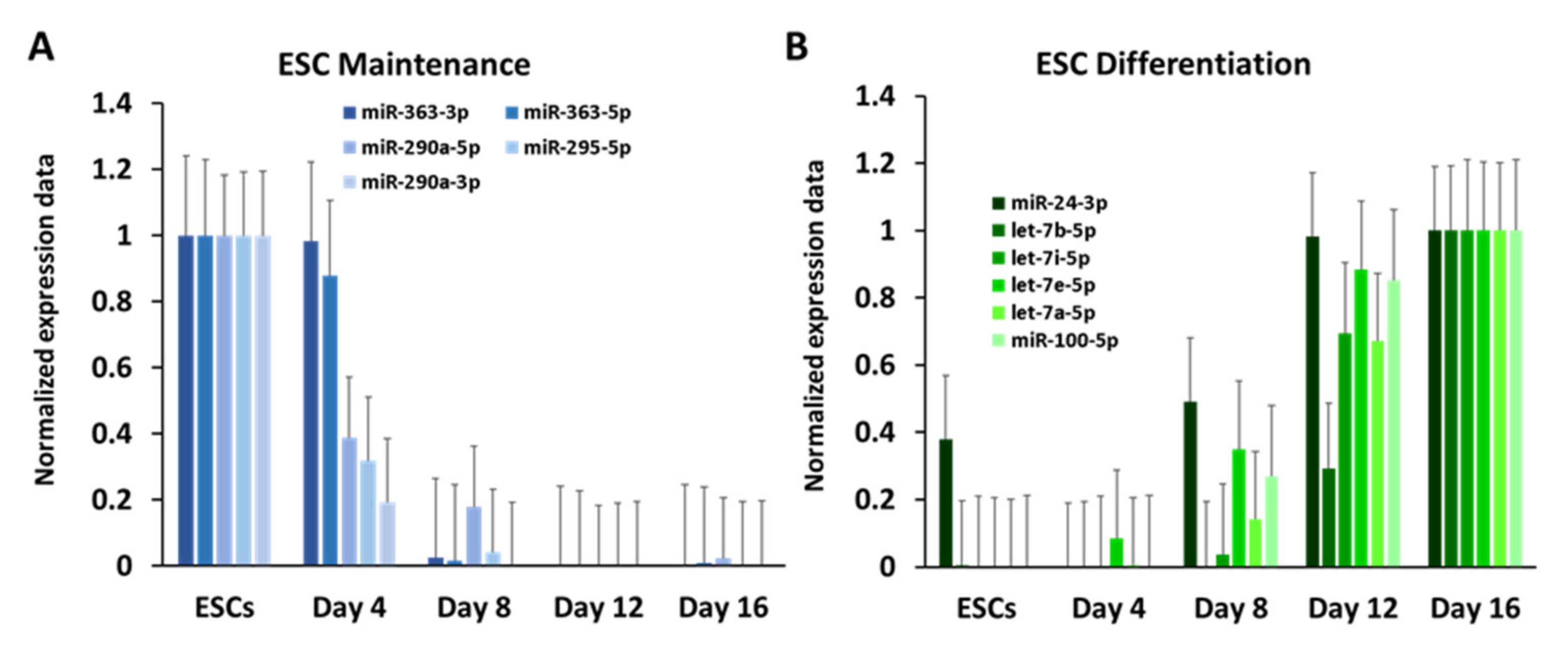
3.2. Dynamic Expression of Marker Genes and miRNAs
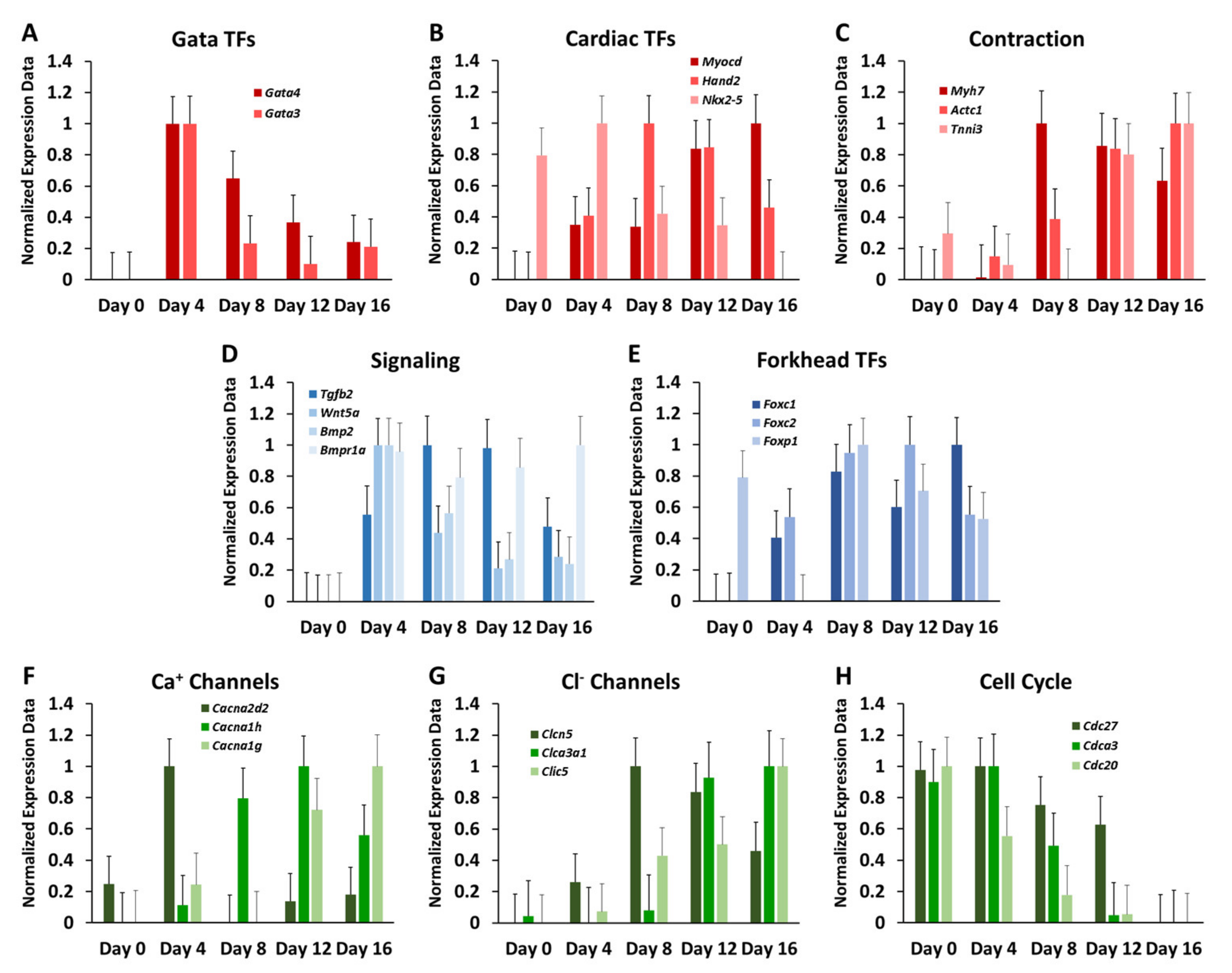
3.3. Elucidation of a Cardiogenic Signature in Embryonic Bodies
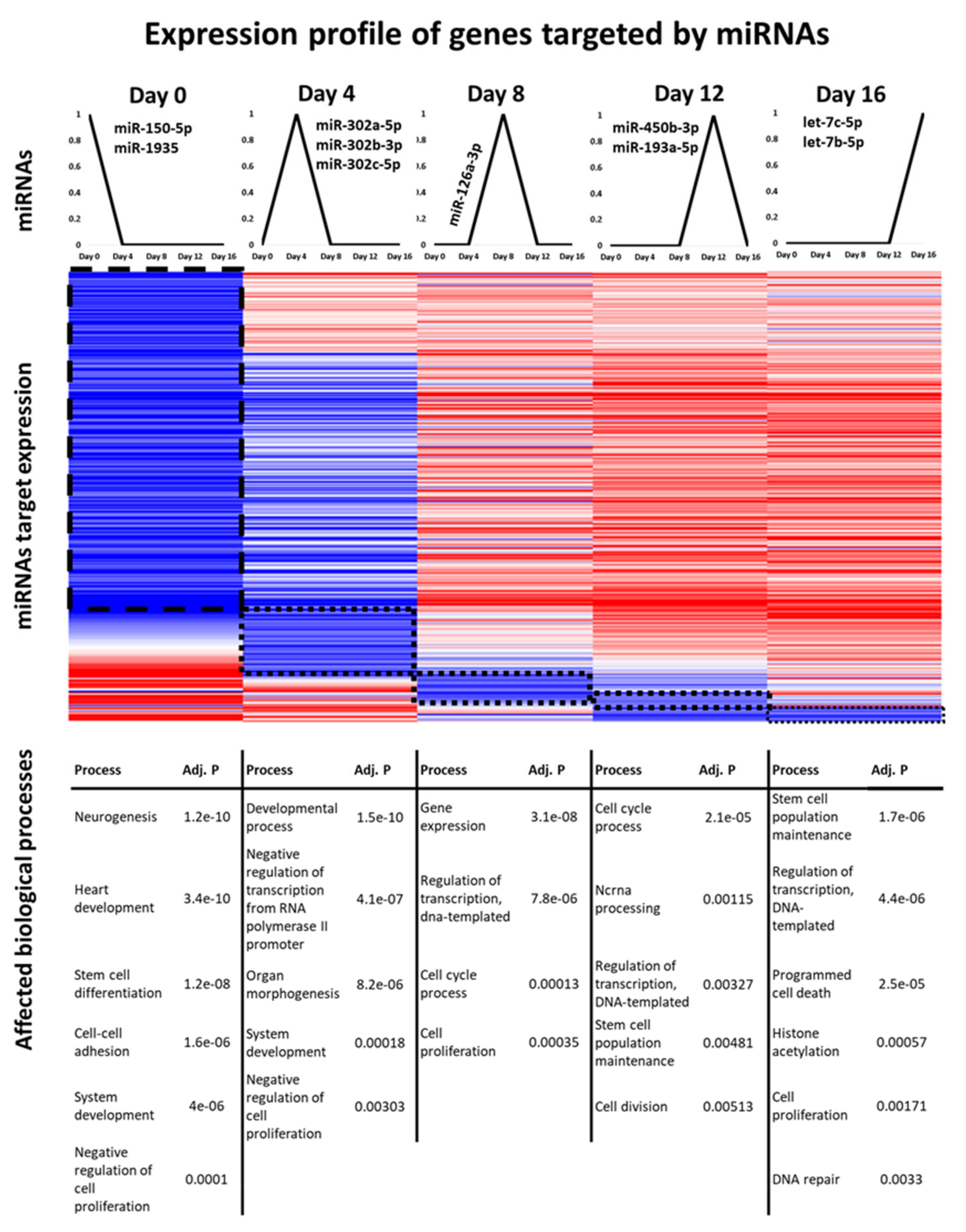
3.4. Identification of miRNAs That Might Serve as Genetic Switches
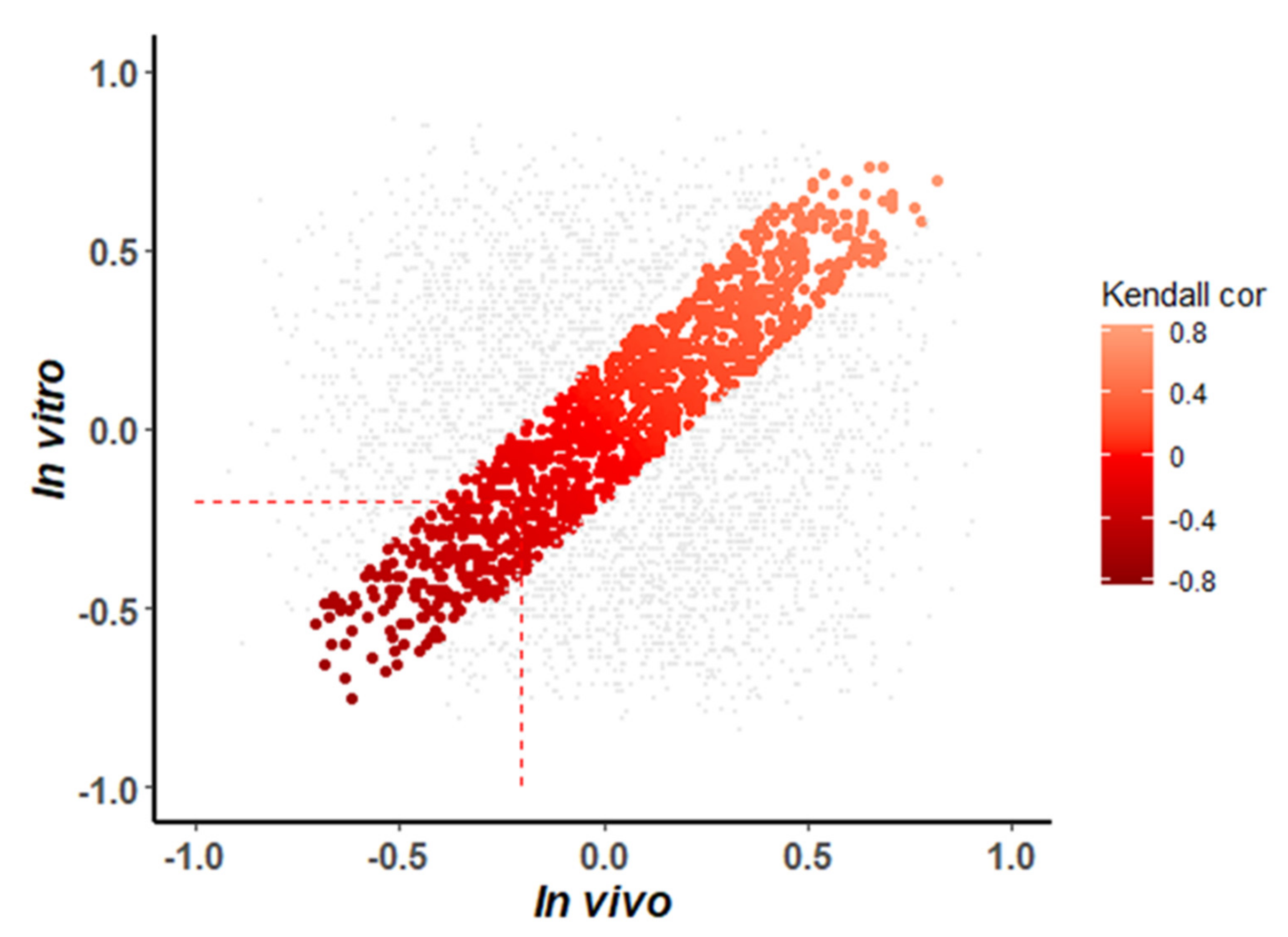
3.5. Detection of Shared In Vitro and In Vivo miRNA-mRNA Interactions
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Keller, G. In vitro differentiation of embryonic stem cells. Curr. Opin. Cell Biol. 1995, 7, 862–869. [Google Scholar] [CrossRef]
- Lancaster, M.; Knoblich, J.A. Organogenesis in a dish: Modeling development and disease using organoid technologies. Science 2014, 345, 1247125. [Google Scholar] [CrossRef] [PubMed]
- Lei, I.; Bu, L.; Wang, Z. Derivation of cardiac progenitor cells from embryonic stem cells. J. Vis. Exp. 2015, 95, 52047. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, J.A.; Doss, M.X.; Winkler, J.; Wagh, V.; Hescheler, J.; Kolde, R.; Vilo, J.; Schulz, H.; Sachinidis, A. Gene Expression Signatures Defining Fundamental Biological Processes in Pluripotent, Early, and Late Differentiated Embryonic Stem Cells. Stem Cells Dev. 2012, 21, 2471–2484. [Google Scholar] [CrossRef] [PubMed]
- Niwa, H. How is pluripotency determined and maintained? Development 2007, 134, 635–646. [Google Scholar] [CrossRef]
- Chen, X.; Xu, H.; Yuan, P.; Fang, F.; Huss, M.; Vega, V.B.; Wong, E.; Orlov, Y.; Zhang, W.; Jiang, J.; et al. Integration of External Signaling Pathways with the Core Transcriptional Network in Embryonic Stem Cells. Cell 2008, 133, 1106–1117. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-M.; Kuang, S.; Xiong, X.; Gao, T.; Liu, C.; Guo, A.-Y. Transcription factor and microRNA co-regulatory loops: Important regulatory motifs in biological processes and diseases. Brief. Bioinform. 2015, 16, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Carthew, R.W.; Sontheimer, E.J. Origins and Mechanisms of miRNAs and siRNAs. Cell 2009, 136, 642–655. [Google Scholar] [CrossRef]
- Hadjimichael, C.; Nikolaou, C.; Papamatheakis, J.; Kretsovali, A. MicroRNAs for Fine-Tuning of Mouse Embryonic Stem Cell Fate Decision through Regulation of TGF-β Signaling. Stem Cell Rep. 2016, 6, 292–301. [Google Scholar] [CrossRef]
- Katz, M.G.; Fargnoli, A.S.; Kendle, A.P.; Hajjar, R.J.; Bridges, C.R. The role of microRNAs in cardiac development and regenerative capacity. Am. J. Physiol. Circ. Physiol. 2016, 310, H528–H541. [Google Scholar] [CrossRef]
- Kuppusamy, K.; Sperber, H.; Baker, H.; Ruohola-Baker, H. MicroRNA regulation and role in stem cell maintenance, cardiac differentiation and hypertrophy. Curr. Mol. Med. 2013, 13, 757–764. [Google Scholar] [CrossRef]
- Li, N.; Long, B.; Han, W.; Yuan, S.; Wang, K. microRNAs: Important regulators of stem cells. Stem Cell Res. Ther. 2017, 8, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, J.; Ruohola-Baker, H. Regulation of Stem Cell Populations by microRNAs. Adv. Exp. Med. Biol. 2013, 786, 329–351. [Google Scholar] [CrossRef] [PubMed]
- Sabour, D.; Machado, R.S.; Pinto, J.P.; Rohani, S.; Sahito, R.G.; Hescheler, J.; Futschik, M.E.; Sachinidis, A. Parallel Genome-wide Profiling of Coding and Non-coding RNAs to Identify Novel Regulatory Elements in Embryonic and Maturated Heart. Mol. Ther. Nucleic Acids 2018, 12, 158–173. [Google Scholar] [CrossRef] [PubMed]
- Bai, F.; Lim, C.H.; Jia, J.; Santostefano, K.; Simmons, C.; Kasahara, H.; Wu, W.; Terada, N.; Jin, S. Directed Differentiation of Embryonic Stem Cells into Cardiomyocytes by Bacterial Injection of Defined Transcription Factors. Sci. Rep. 2015, 5, 15014. [Google Scholar] [CrossRef]
- Van Vliet, P.; Wu, S.M.; Zaffran, S.; Puceat, M. Early cardiac development: A view from stem cells to embryos. Cardiovasc. Res. 2012, 96, 352–362. [Google Scholar] [CrossRef]
- Sabour, D.; Srinivasan, S.P.; Rohani, S.; Wagh, V.; Gaspar, J.A.; Panek, D.; Ardestani, M.A.; Doss, M.X.; Riet, N.; Abken, H.; et al. STRIP2 Is Indispensable for the Onset of Embryonic Stem Cell Differentiation. Mol. Ther. Methods Clin. Dev. 2017, 5, 116–129. [Google Scholar] [CrossRef]
- Gissel, C.; Voolstra, C.; Doss, M.X.; Koehler, C.I.; Winkler, J.; Hescheler, J.; Sachinidis, A. An optimized embryonic stem cell model for consistent gene expression and developmental studies. A fundamental study. Thromb. Haemost. 2005, 94, 719–727. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Carlson, M. org.Mm.eg.db: Genome Wide Annotation for Mouse. 2014. Available online: https://bioconductor.org/packages/release/data/annotation/html/org.Mm.eg.db.html (accessed on 12 September 2021).
- Falcon, S.; Gentleman, R. Using GOstats to test gene lists for GO term association. Bioinformatics 2006, 23, 257–258. [Google Scholar] [CrossRef]
- Betel, D.; Wilson, M.; Gabow, A.; Marks, D.S.; Sander, C. The microRNA.org resource: Targets and expression. Nucleic Acids Res. 2008, 36, D149–D153. [Google Scholar] [CrossRef]
- Kertesz, M.; Iovino, N.; Unnerstall, U.; Gaul, U.; Segal, E. The role of site accessibility in microRNA target recognition. Nat. Genet. 2007, 39, 1278–1284. [Google Scholar] [CrossRef]
- Wong, N.; Wang, X. miRDB: An online resource for microRNA target prediction and functional annotations. Nucleic Acids Res. 2015, 43, D146–D152. [Google Scholar] [CrossRef]
- Agarwal, V.; Bell, G.W.; Nam, J.-W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015, 4, e05005. [Google Scholar] [CrossRef]
- Chou, C.-H.; Chang, N.-W.; Shrestha, S.; Hsu, S.-D.; Lin, Y.-L.; Lee, W.-H.; Yang, C.-D.; Hong, H.-C.; Wei, T.-Y.; Tu, S.-J.; et al. miRTarBase 2016: Updates to the experimentally validated miRNA-target interactions database. Nucleic Acids Res. 2016, 44, D239–D247. [Google Scholar] [CrossRef] [PubMed]
- Kashyap, V.; Rezende, N.C.; Scotland, K.B.; Shaffer, S.M.; Persson, J.; Gudas, L.J.; Mongan, N. Regulation of Stem Cell Pluripotency and Differentiation Involves a Mutual Regulatory Circuit of the Nanog, OCT4, and SOX2 Pluripotency Transcription Factors with Polycomb Repressive Complexes and Stem Cell microRNAs. Stem Cells Dev. 2009, 18, 1093–1108. [Google Scholar] [CrossRef] [PubMed]
- Masui, S.; Nakatake, Y.; Toyooka, Y.; Shimosato, D.; Yagi, R.; Takahashi, K.; Okochi, H.; Okuda, A.; Matoba, R.; Sharov, A.A.; et al. Pluripotency governed by Sox2 via regulation of Oct3/4 expression in mouse embryonic stem cells. Nature 2007, 9, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Macatee, T.L.; Hammond, B.P.; Arenkiel, B.; Francis, L.; Frank, D.U.; Moon, A.M. Ablation of specific expression domains reveals discrete functions of ectoderm- and endoderm-derived FGF8 during cardiovascular and pharyngeal development. Development 2003, 130, 6361–6374. [Google Scholar] [CrossRef]
- Pankratz, M.T.; Li, X.-J.; LaVaute, T.M.; Lyons, E.A.; Chen, X.; Zhang, S.-C. Directed Neural Differentiation of Human Embryonic Stem Cells via an Obligated Primitive Anterior Stage. Stem Cells 2007, 25, 1511–1520. [Google Scholar] [CrossRef] [PubMed]
- Faial, T.; Bernardo, A.S.; Mendjan, S.; Diamanti, E.; Ortmann, D.; Gentsch, G.E.; Mascetti, V.L.; Trotter, M.; Smith, J.; Pedersen, R.A. Brachyury and SMAD signalling collaboratively orchestrate distinct mesoderm and endoderm gene regulatory networks in differentiating human embryonic stem cells. Development 2015, 142, 2121–2135. [Google Scholar] [CrossRef]
- Kitajima, S.; Takagi, A.; Inoue, T.; Saga, Y. MesP1 and MesP2 are essential for the development of cardiac mesoderm. Development 2000, 127, 3215–3226. [Google Scholar] [CrossRef]
- Kim, P.T.W.; Ong, C.J. Differentiation of Definitive Endoderm from Mouse Embryonic Stem Cells. Results Probl. Cell Differ. 2012, 55, 303–319. [Google Scholar] [CrossRef]
- Ma, Y.; Yao, N.; Liu, G.; Dong, L.; Liu, Y.; Zhang, M.; Wang, F.; Wang, B.; Wei, X.; Dong, H.; et al. Functional screen reveals essential roles of miR-27a/24 in differentiation of embryonic stem cells. EMBO J. 2015, 34, 361–378. [Google Scholar] [CrossRef]
- Schulman, B.R.M.; Esquela-Kerscher, A.; Slack, F.J. Reciprocal expression oflin-41and the microRNAslet-7andmir-125during mouse embryogenesis. Dev. Dyn. 2005, 234, 1046–1054. [Google Scholar] [CrossRef]
- Wulczyn, F.G.; Smirnova, L.; Rybak-Wolf, A.; Brandt, C.; Kwidzinski, E.; Ninnemann, O.; Strehle, M.; Seiler, A.; Schumacher, S.; Nitsch, R. Post-transcriptional regulation of the let-7 microRNA during neural cell specification. FASEB J. 2006, 21, 415–426. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Han, S.; Kwon, C.S.; Lee, D. Biogenesis and regulation of the let-7 miRNAs and their functional implications. Protein Cell 2016, 7, 100–113. [Google Scholar] [CrossRef]
- Tarantino, C.; Paolella, G.; Cozzuto, L.; Minopoli, G.; Pastore, L.; Parisi, S.; Russo, T. miRNA 34a, 100, and 137 modulate differentiation of mouse embryonic stem cells. FASEB J. 2010, 24, 3255–3263. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yang, P. In Vitro Differentiation of Mouse Embryonic Stem (mES) Cells Using the Hanging Drop Method. J. Vis. Exp. 2008, e825. [Google Scholar] [CrossRef] [PubMed]
- Miano, J.M. Myocardin in biology and disease. J. Biomed. Res. 2015, 29, 3–19. [Google Scholar] [CrossRef]
- England, J.; Loughna, S. Heavy and light roles: Myosin in the morphogenesis of the heart. Cell. Mol. Life Sci. 2012, 70, 1221–1239. [Google Scholar] [CrossRef]
- Sakaki-Yumoto, M.; Katsuno, Y.; Derynck, R. TGF-β family signaling in stem cells. Biochim. Biophys. Acta BBA Gen. Subj. 2013, 1830, 2280–2296. [Google Scholar] [CrossRef] [PubMed]
- Andersen, P.; Tampakakis, E.; Jimenez, D.V.; Kannan, S.; Miyamoto, M.; Shin, H.K.; Saberi, A.; Murphy, S.; Sulistio, E.; Chelko, S.; et al. Precardiac organoids form two heart fields via Bmp/Wnt signaling. Nat. Commun. 2018, 9, 3140. [Google Scholar] [CrossRef] [PubMed]
- Papanicolaou, K.N.; Izumiya, Y.; Walsh, K. Forkhead Transcription Factors and Cardiovascular Biology. Circ. Res. 2008, 102, 16–31. [Google Scholar] [CrossRef] [PubMed]
- Kume, T.; Jiang, H.; Topczewska, J.M.; Hogan, B.L. The murine winged helix transcription factors, Foxc1 and Foxc2, are both required for cardiovascular development and somitogenesis. Genes Dev. 2001, 15, 2470–2482. [Google Scholar] [CrossRef] [PubMed]
- Seo, S.; Kume, T. Forkhead transcription factors, Foxc1 and Foxc2, are required for the morphogenesis of the cardiac outflow tract. Dev. Biol. 2006, 296, 421–436. [Google Scholar] [CrossRef] [PubMed]
- Leishman, E.; Howard, J.M.; Garcia, G.E.; Miao, Q.; Ku, A.T.; Dekker, J.D.; Tucker, H.; Nguyen, H. Foxp1 maintains hair follicle stem cell quiescence through regulation of Fgf18. Development 2013, 140, 3809–3818. [Google Scholar] [CrossRef]
- Wang, B.; Weidenfeld, J.; Lu, M.M.; Maika, S.; Kuziel, W.A.; Morrisey, E.E.; Tucker, P.W. Foxp1 regulates cardiac outflow tract, endocardial cushion morphogenesis and myocyte proliferation and maturation. Development 2004, 131, 4477–4487. [Google Scholar] [CrossRef] [PubMed]
- Ponnalagu, D.; Rao, S.G.; Farber, J.; Xin, W.; Hussain, A.T.; Shah, K.; Tanda, S.; Berryman, M.; Edwards, J.C.; Singh, H. Molecular identity of cardiac mitochondrial chloride intracellular channel proteins. Mitochondrion 2016, 27, 6–14. [Google Scholar] [CrossRef]
- Bao, M.-H.; Feng, X.; Zhang, Y.-W.; Lou, X.-Y.; Cheng, Y.; Zhou, H. Let-7 in Cardiovascular Diseases, Heart Development and Cardiovascular Differentiation from Stem Cells. Int. J. Mol. Sci. 2013, 14, 23086–23102. [Google Scholar] [CrossRef]
- The Gene Ontology Consortium Expansion of the Gene Ontology knowledgebase and resources. Nucleic Acids Res. 2017, 45, D331–D338. [CrossRef] [PubMed]
- Dorfeshan, P.; Novin, M.G.; Salehi, M.; Farifteh, F. Expression of miR-302 in human embryo derived from in-vitro matured oocyte. Int. J. Reprod. Biomed. 2019, 17, 405–412. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Xu, J. miR-126: A Potential Biomarker for Coronary Artery Disease? Cardiology 2019, 142, 2–3. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Xu, R.; Huang, J.; Yao, Y.; Pan, X.; Chen, Z.; Ma, G. Insulin-like growth factor-1-mediated regulation of miR-193a expression promotes the migration and proliferation of c-kit-positive mouse cardiac stem cells. Stem Cell Res. Ther. 2018, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Lambers, E.; Arnone, B.; Fatima, A.; Qin, G.; Wasserstrom, J.A.; Kume, T. Foxc1 Regulates Early Cardiomyogenesis and Functional Properties of Embryonic Stem Cell Derived Cardiomyocytes. Stem Cells 2016, 34, 1487–1500. [Google Scholar] [CrossRef]
- Chu, W.; Li, X.; Li, C.; Wan, L.; Shi, H.; Song, X.; Liu, X.; Chen, X.; Zhang, C.; Shan, H.; et al. TGFBR3, a potential negative regulator of TGF-β signaling, protects cardiac fibroblasts from hypoxia-induced apoptosis. J. Cell. Physiol. 2011, 226, 2586–2594. [Google Scholar] [CrossRef]
- Ieda, M. Key Regulators of Cardiovascular Differentiation and Regeneration: Harnessing the Potential of Direct Reprogramming to Treat Heart Failure. J. Card. Fail. 2020, 26, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Wan, G.-X.; Cheng, L.; Qin, H.-L.; Zhang, Y.-Z.; Wang, L.-Y.; Zhang, Y.-G. MiR-15b-5p is Involved in Doxorubicin-Induced Cardiotoxicity via Inhibiting Bmpr1a Signal in H9c2 Cardiomyocyte. Cardiovasc. Toxicol. 2018, 19, 264–275. [Google Scholar] [CrossRef]
- Cai, C.-L.; Liang, X.; Shi, Y.; Chu, P.-H.; Pfaff, S.L.; Chen, J.; Evans, S. Isl1 Identifies a Cardiac Progenitor Population that Proliferates Prior to Differentiation and Contributes a Majority of Cells to the Heart. Dev. Cell 2003, 5, 877–889. [Google Scholar] [CrossRef]
- Li, X.; Zhu, Y.; Zhang, H.; Ma, G.; Wu, G.; Xiang, A.; Shi, X.; Yang, G.S.; Sun, S. MicroRNA-106a-5p Inhibited C2C12 Myogenesis via Targeting PIK3R1 and Modulating the PI3K/AKT Signaling. Genes 2018, 9, 333. [Google Scholar] [CrossRef] [PubMed]
- Winnier, G.E.; Kume, T.; Deng, K.; Rogers, R.; Bundy, J.; Raines, C.; Walter, M.A.; Hogan, B.L.M.; Conway, S.J. Roles for the Winged Helix Transcription Factors MF1 and MFH1 in Cardiovascular Development Revealed by Nonallelic Noncomplementation of Null Alleles. Dev. Biol. 1999, 213, 418–431. [Google Scholar] [CrossRef]
- Gabdoulline, R.; Kaisers, W.; Gaspar, A.; Meganathan, K.; Jesudoss, M.X.D.; Jagtap, S.; Hescheler, J.; Sachinidis, A.; Schwender, H. Differences in the Early Development of Human and Mouse Embryonic Stem Cells. PLoS ONE 2015, 10, e0140803. [Google Scholar] [CrossRef] [PubMed]
- Jayawardena, T.M.; Egemnazarov, B.; Finch, E.A.; Zhang, L.; Payne, J.A.; Pandya, K.; Zhang, Z.; Rosenberg, P.; Mirotsou, M.; Dzau, V.J. MicroRNA-Mediated In Vitro and In Vivo Direct Reprogramming of Cardiac Fibroblasts to Cardiomyocytes. Circ. Res. 2012, 110, 1465–1473. [Google Scholar] [CrossRef]
- Jayawardena, T.M.; Finch, E.A.; Zhang, L.; Zhang, H.; Hodgkinson, C.P.; Pratt, R.E.; Rosenberg, P.; Mirotsou, M.; Dzau, V.J. MicroRNA Induced Cardiac Reprogramming In Vivo. Circ. Res. 2015, 116, 418–424. [Google Scholar] [CrossRef]
- Kuppusamy, K.T.; Jones, D.C.; Sperber, H.; Madan, A.; Fischer, K.A.; Rodriguez, M.L.; Pabon, L.; Zhu, W.-Z.; Tulloch, N.L.; Yang, X.; et al. Let-7 family of microRNA is required for maturation and adult-like metabolism in stem cell-derived cardiomyocytes. Proc. Natl. Acad. Sci. USA 2015, 112, E2785–E2794. [Google Scholar] [CrossRef]
- Gabisonia, K.; Prosdocimo, G.; Aquaro, G.D.; Carlucci, L.; Zentilin, L.; Secco, I.; Ali, H.; Braga, L.; Gorgodze, N.; Bernini, F.; et al. MicroRNA therapy stimulates uncontrolled cardiac repair after myocardial infarction in pigs. Nature 2019, 569, 418–422. [Google Scholar] [CrossRef] [PubMed]
- Beissert, T.; Perkovic, M.; Vogel, A.; Erbar, S.; Walzer, K.C.; Hempel, T.; Brill, S.; Haefner, E.; Becker, R.; Türeci, Ö.; et al. A Trans-amplifying RNA Vaccine Strategy for Induction of Potent Protective Immunity. Mol. Ther. 2020, 28, 119–128. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miRID | Count | Gene Symbol | PMID |
|---|---|---|---|
| mmu-miR-27a-3p | 7 | Robo1, Gata3, Zfp36l1, Flrt2, Mef2c, Slc8a1, Bmpr1a | 28293796 |
| mmu-miR-34a-5p | 7 | Pofut1, Fat4, Tab2, Maml1, Angpt1, Pdgfra, Kdm2a | 30474870 |
| mmu-miR-680 | 6 | Ctnnb1, Zfp36l1, Foxc1, Gnaq, Zmiz1, Tgfbr3 | |
| mmu-let-7b-3p | 6 | Sfrp2, Lmo4, Vegfa, Hectd1, Acvr1, Frs2 | 27072074 |
| mmu-miR-23b-3p | 5 | Has2, Pcsk5, Adam19, Whsc1, Nox4 | 31103821 |
| mmu-miR-705 | 5 | Ncam1, Sh3pxd2b, Kdm2a, Casp7, Gpc3 | - |
| mmu-miR-23a-3p | 5 | Isl1, Sox11, Robo1, Aldh1a2, Med12 | 31130720 |
| mmu-miR-762 | 5 | Ncam1, Pdpn, Tgfbr1, Zfp36l1, Nedd4 | |
| mmu-miR-27b-3p | 4 | Smad4, Pdgfra, Ccm2, Tgfbr3 | 27072074 |
| mmu-miR-20b-5p | 4 | Heg1, Pkd1, Nrp2, Tgfbr2 | 25898012 |
| mmu-miR-150-5p | 4 | Efnb2, Prkar1a, Trps1, Erbb2 | 31122130 |
| mmu-miR-152-3p | 4 | Dicer1, Ube4b, Gys1, Sos1 | - |
| mmu-miR-3473a | 4 | Vegfa, Gab1, Camk2d, Mdm4 | - |
| mmu-miR-466f-3p | 4 | Pitx2, Wisp1, Zfpm2, Kif3a | - |
| mmu-miR-106b-5p | 4 | Pkd1, Heg1, Tgfbr2, Grhl2 | - |
| mmu-miR-148a-3p | 3 | Dicer1, Ube4b, Adam19 | 25630970 |
| mmu-miR-30c-5p | 3 | Nfatc3, Adam19, Bcor | 30279543 |
| mmu-miR-19b-3p | 3 | Mef2a, Tgfbr2, Sgcb | 29664809 |
| mmu-miR-3472 | 3 | Sema3c, Tmem100, Foxc1 | - |
| mmu-miR-106a-5p | 3 | Heg1, Pkd1, Tgfbr2 | 30004470 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Machado, R.; Sachinidis, A.; Futschik, M.E. Detection of Novel Potential Regulators of Stem Cell Differentiation and Cardiogenesis through Combined Genome-Wide Profiling of Protein-Coding Transcripts and microRNAs. Cells 2021, 10, 2477. https://doi.org/10.3390/cells10092477
Machado R, Sachinidis A, Futschik ME. Detection of Novel Potential Regulators of Stem Cell Differentiation and Cardiogenesis through Combined Genome-Wide Profiling of Protein-Coding Transcripts and microRNAs. Cells. 2021; 10(9):2477. https://doi.org/10.3390/cells10092477
Chicago/Turabian StyleMachado, Rui, Agapios Sachinidis, and Matthias E. Futschik. 2021. "Detection of Novel Potential Regulators of Stem Cell Differentiation and Cardiogenesis through Combined Genome-Wide Profiling of Protein-Coding Transcripts and microRNAs" Cells 10, no. 9: 2477. https://doi.org/10.3390/cells10092477
APA StyleMachado, R., Sachinidis, A., & Futschik, M. E. (2021). Detection of Novel Potential Regulators of Stem Cell Differentiation and Cardiogenesis through Combined Genome-Wide Profiling of Protein-Coding Transcripts and microRNAs. Cells, 10(9), 2477. https://doi.org/10.3390/cells10092477