
The Landscape of IFN/ISG Signaling in HIV-1-Infected Macrophages and Its Possible Role in the HIV-1 Latency


Abstract
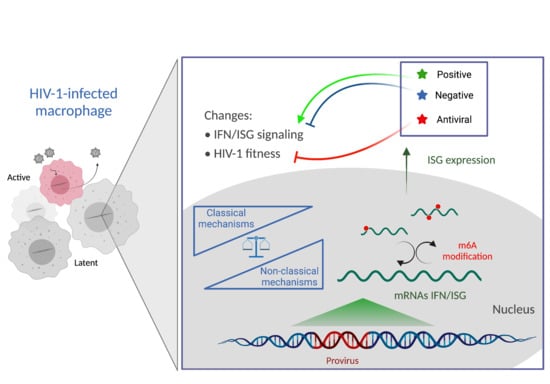
1. Current Status
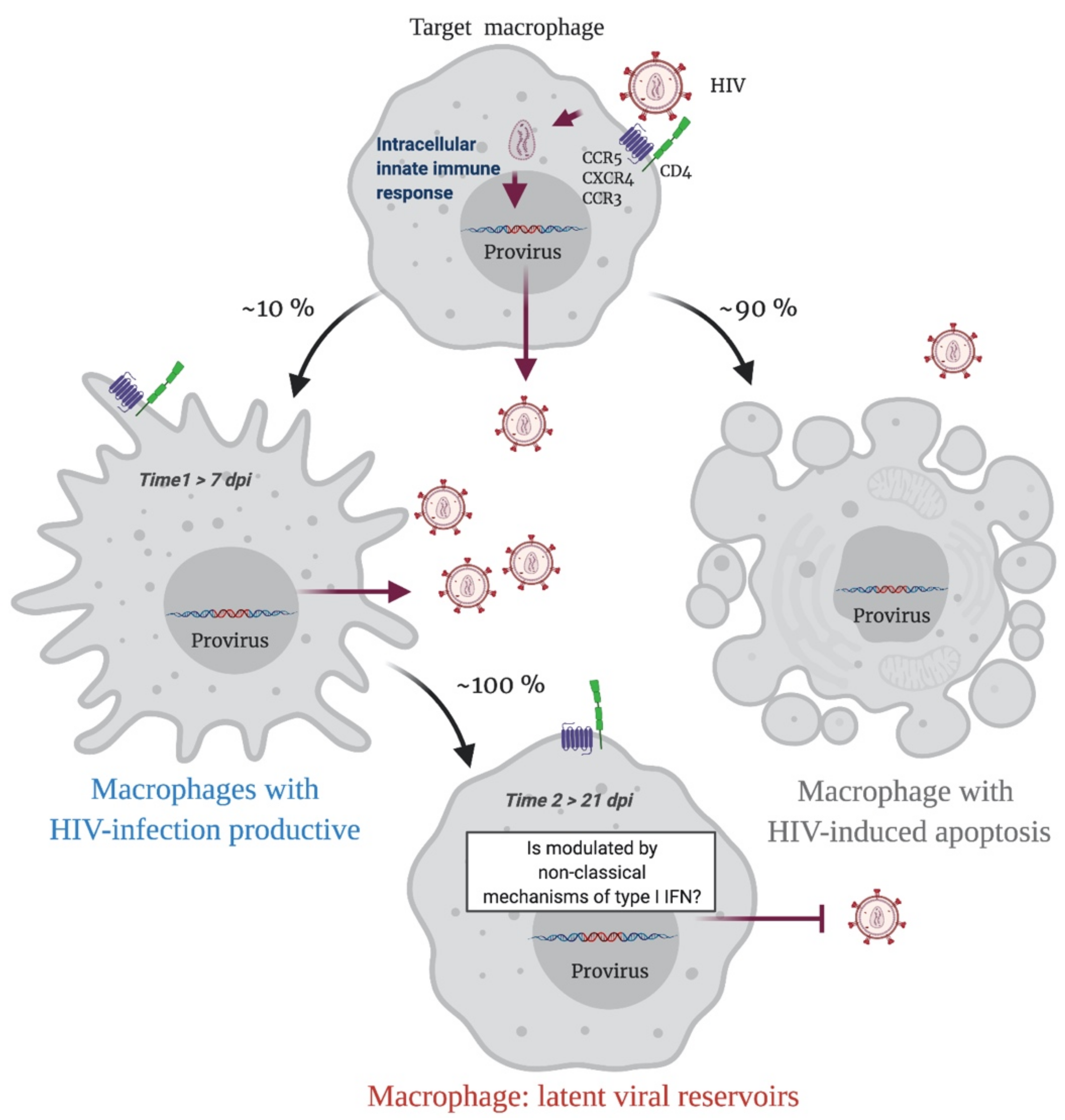
2. Macrophages as HIV-1 Reservoirs
3. Intracellular Innate Immune Response in HIV-1-Infected Macrophages
3.1. The IFN/ISG Signaling Network and HIV-1
- Antiviral effectors: There is a plethora of HIV-1-induced antiviral effectors. For example, some molecules with anti-HIV-1 activity, such as the apolipoprotein B mRNA editing complex 3 (APOBEC3) family of proteins, act by binding to HIV-1 RNA and inducing hypermutation in the newly synthesized HIV-1 DNA early in the viral cycle, which restricts provirus replication [51,67]. Members of the tripartite motif (TRIM) family of proteins, such as TRIM5α, enhance the fragmentation of viral cores, preventing HIV-1 cDNA synthesis [57,68]. Sterile alpha motif and histidine–aspartate domain-containing protein 1 (SAMHD1) can restrict viral replication by reducing the number of nucleotides available for viral DNA synthesis [69,70]. Some members of the dynamin GTPase superfamily, such as myxovirus resistance 2 (Mx2), prevent the nuclear import and integration of viral DNA [57,71] while tetherin inhibits the release of the virus [51,72].
- Positive regulators of IFN signaling: These include molecules such as IFN regulatory factor 3 (IRF3) [73], 1, 2, and 7 [74]; cyclic GMP-AMP synthase (cGAS) [75]; melanoma differentiation-associated gene 5 (MDA5) [76]; and RIG-1 [77]. These proteins act as sensors, second messengers, or effector molecules and contribute to the antiviral response. Some lentiviruses, including HIV-1, can induce the production of several positive regulators of IFN signaling, such as IRF1, IRF2, IRF7, cGAS, MDA5, RIG-1, and IFN-γ-inducible protein 16 (IFI16), which confer protection against infection in a species- and cell-type-dependent manner [78].
- Negative regulators of IFN signaling: These include suppressor of cytokine signaling (SOCS) proteins, which inhibit JAK/STAT signaling [79], or ubiquitin-specific peptidase 18 (USP18) [80], which induces a state of desensitization in the target cell, thereby rendering the cell refractory to IFN stimulation [56]. HIV-1 infection can reportedly induce SOCS1, which, in turn, can affect the innate and adaptive immunity responses [81]. Another study revealed that, in CD4+ T cells of HIV-infected patients, SOCS1/3 mRNA levels were upregulated, whereas their protein levels were downregulated, which may explain the lack of attenuation of the JAK/STAT pathway [82]. Similarly, it was proposed that the reduced viability of memory CD4+ T cells induced by type I IFN during HIV-infection is USP18/protein kinase B (AKT)/phosphatase and tensin homolog (PTEN)-dependent [83]. In macrophages and dendritic cells, USP18 can promote HIV-1 replication by enhancing reverse transcription through the downregulation of the expression of p21 (a cyclin-dependent kinase inhibitor), which correlates with the antiviral-inactive form of SAMHD1 [84].
3.2. The Induction of IFN and ISG Expression in HIV-Infected Macrophages
4. The Relevance of the IFN/ISG Response in HIV-1-Infected Cells
- Type I IFN responses are thought to be the main selective pressure for the emergence of HIV-1 genotypes (transmitted/founder [T/F] variants) that initiate the infection process in humans. Different groups have reported that the T/F virus is more resistant to Type I IFN compared with the virus present during the chronic phase of infection [51,109,110]. T/F viruses can usually infect CD4+ T lymphocytes but not macrophages. However, the virus can eventually infect macrophages when they express an envelope with a high affinity for CD4. Nevertheless, the molecular mechanisms underlying the IFN-induced restriction of HIV-1 infection and how the virus evolves its tropism to macrophages remain unknown [39,111].
- Lentiviruses, and HIV-1 in particular, have developed accessory proteins and several strategies to counter ISG activity [70]. Viral proteins include viral infectivity factor (Vif), which inhibits the antiviral factor APOBEC3 [67]; viral protein X (Vpx), which inhibits SAMHD1 [69,112]; and viral protein unique (Vpu); which inhibits tetherin [113].
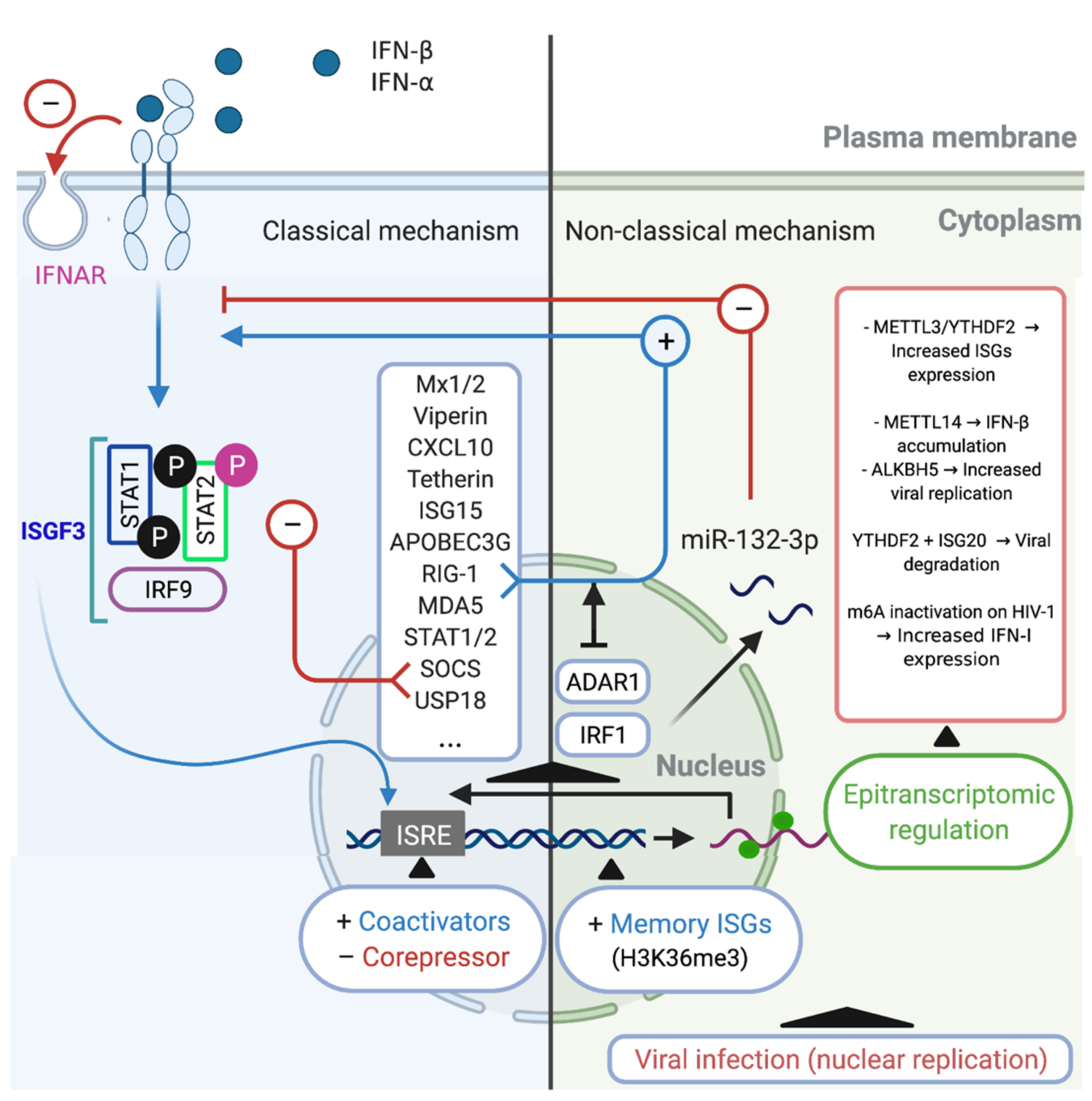
5. The Regulation of the IFN/ISG Signaling Network in HIV-1-Infected Cells
5.1. Classical Mechanism
5.2. Non-Classical Mechanisms
5.3. Alternative Mechanism: Epitranscriptomic Regulation
6. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Nature. Nature.com, 30 November 2018. Available online: https://www.nature.com/collections/mghkkdjlgx (accessed on 18 June 2019).
- UNAIDS. unaids.org, 1 December 2019. Available online: https://www.unaids.org/en/resources/fact-sheet (accessed on 7 April 2020).
- Zicari, S.; Sessa, L.; Cotugno, N.; Ruggiero, A.; Morrocchi, E.; Concato, C.; Rocca, S.; Zangari, P.; Manno, E.C.; Palma, P. Immune Activation, Inflammation, and Non-AIDS Co-Morbidities in HIV-Infected Patients under Long-Term ART. Viruses 2019, 11, 200. [Google Scholar] [CrossRef]
- Veenstra, M.; León-Rivera, R.; Li, M.; Gama, L.; Clements, J.E.; Berman, J.W. Mechanisms of CNS Viral Seeding by HIV + CD14 + CD16 + Monocytes: Establishment and Reseeding of Viral Reservoirs Contributing to HIV-Associated Neurocognitive Disorders. MBio 2017, 8, e01280-17. [Google Scholar] [CrossRef]
- Tirumuru, N.; Zhao, B.; Lu, W.; Lu, Z.; He, C.; Wu, L. N6-methyladenosine of HIV-1 RNA regulates viral infection and HIV-1 Gag protein expression. ELife 2016, 5, 1–20. [Google Scholar] [CrossRef]
- Davey, R.T.; Bhat, N.; Yoder, C.; Chun, T.-W.; Metcalf, J.A.; Dewar, R.; Natarajan, V.; Lempicki, R.; Adelsberger, J.W.; Miller, K.D.; et al. HIV-1 and T cell dynamics after interruption of highly active antiretroviral therapy (HAART) in patients with a history of sustained viral suppression. Proc. Natl. Acad. Sci. USA 1999, 96, 15109–15114. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo-Redondo, R.; Fryer, H.R.; Bedford, T.; Kim, E.Y.; Archer, J.; Pond, S.L.K.; Chung, Y.S.; Penugonda, S.; Chipman, J.G.; Fletcher, C.V.; et al. Persistent HIV-1 replication maintains the tissue reservoir during therapy. Nature 2016, 530, 51–56. [Google Scholar] [CrossRef]
- Nowlin, B.T.; Wang, J.; Schafer, J.L.; Autissier, P.; Burdo, T.H.; Williams, K.C. Monocyte subsets exhibit transcriptional plasticity and a shared response to interferon in SIV-infected rhesus macaques. J. Leukoc. Biol. 2018, 103, 141–155. [Google Scholar] [CrossRef] [PubMed]
- Burdo, T.H. Editor’s Commentary for Special Issue: “The Role of Macrophages in HIV Persistence”. J. Neuroimmune Pharmacol. 2019, 14, 2–5. [Google Scholar] [CrossRef] [PubMed]
- Doranz, B.J.; Rucker, J.; Yi, Y.; Smyth, R.J.; Samson, M.; Peiper, S.C.; Parmentier, M.; Collman, R.G.; Doms, R.W. A Dual-Tropic Primary HIV-1 Isolate That Uses Fusin and the β-Chemokine Receptors CKR-5, CKR-3, and CKR-2b as Fusion Cofactors. Cell 1996, 85, 1149–1158. [Google Scholar] [CrossRef]
- Feng, Y.; Broder, C.C.; Kennedy, P.E.; Berger, E.A. HIV-1 Entry Cofactor: Functional cDNA Cloning of a Seven-Transmembrane, G Protein-Coupled Receptor. Science 1996, 272, 872–877. [Google Scholar] [CrossRef]
- Stevenson, M. HIV-1 pathogenesis. Nat. Med. 2003, 9, 853–860. [Google Scholar] [CrossRef]
- Henderson, L.; Nath, A. Astrocytes as an HIV Reservoir: Mechanism of HIV Infection. Curr. HIV Res. 2016, 14, 373–381. [Google Scholar] [CrossRef]
- Burdo, T.H.; Soulas, C.; Orzechowski, K.; Button, J.; Krishnan, A.; Sugimoto, C.; Alvarez, X.; Kuroda, M.J.; Williams, K.C. Increased Monocyte Turnover from Bone Marrow Correlates with Severity of SIV Encephalitis and CD163 Levels in Plasma. PLoS Pathog. 2010, 6, e1000842. [Google Scholar] [CrossRef]
- Witwer, K.; Gama, L.; Li, M.; Bartizal, C.M.; Queen, S.E.; Varrone, J.J.; Brice, A.K.; Graham, D.R.; Tarwater, P.M.; Mankowski, J.L.; et al. Coordinated Regulation of SIV Replication and Immune Responses in the CNS. PLoS ONE 2009, 4, e8129. [Google Scholar] [CrossRef]
- Valcour, V.; Chalermchai, T.; Sailasuta, N.; Marovich, M.A.; Lerdlum, S.; Suttichom, D.; Suwanwela, N.C.; Jagodzinski, L.L.; Michael, N.L.; Spudich, S.; et al. Central Nervous System Viral Invasion and Inflammation During Acute HIV Infection. J. Infect. Dis. 2012, 206, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Portilla, I.; Reus, S.; León, R.; Hofstadt, C.V.-D.; Sánchez, J.; López, N.; Boix, V.; Merino, E.; Portilla, J. Neurocognitive Impairment in Well-Controlled HIV-Infected Patients: A Cross-Sectional Study. AIDS Res. Hum. Retrovir. 2019, 35, 634–641. [Google Scholar] [CrossRef]
- Buckner, C.M.; Calderon, T.M.; Willams, D.W.; Belbin, T.J.; Berman, J.W. Characterization of monocyte maturation/differentiation that facilitates their transmigration across the blood–brain barrier and infection by HIV: Implications for NeuroAIDS. Cell Immunol. 2011, 267, 109–123. [Google Scholar] [CrossRef]
- Kanmogne, G.D.; Schall, K.; Leibhart, J.; Knipe, B.; Gendelman, H.E.; Persidsky, Y. HIV-1 gp120 Compromises Blood–Brain Barrier Integrity and Enhance Monocyte Migration across Blood–Brain Barrier: Implication for Viral Neuropathogenesis. Br. J. Pharmacol. 2006, 27, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Nath, A.; Conant, K.; Chen, P.; Scott, C.; Major, E.O. Transient Exposure to HIV-1 Tat Protein Results in Cytokine Production in Macrophages and Astrocytes. J. Biol. Chem. 1999, 274, 17098–17102. [Google Scholar] [CrossRef]
- Duette, G.; Gerber, P.P.; Rubione, J.; Perez, P.S.; Landay, A.L.; Crowe, S.M.; Liao, Z.; Witwer, K.W.; Holgado, M.P.; Salido, J.; et al. Induction of HIF-1α by HIV-1 Infection in CD4 + T Cells Promotes Viral Replication and Drives Extracellular Vesicle-Mediated Inflammation. MBio 2018, 9, e00757-18. [Google Scholar] [CrossRef]
- Akiyama, H.; Miller, C.; Ettinger, C.R.; Belkina, A.C.; Snyder-Cappione, J.E.; Gummuluru, S. HIV-1 intron-containing RNA expression induces innate immune activation and T cell dysfunction. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef]
- Bruzzesi, E.; Sereti, I. Residual Immune Activation and Latency. Curr. Top. Microbiol. Immunol. 2018, 417, 157–180. [Google Scholar] [CrossRef] [PubMed]
- Duffau, P.; Wittkop, L.; Lazaro, E.; Le Marec, F.; Cognet, C.; Blanco, P.; Moreau, J.-F.; Dauchy, F.-A.; Cazanave, C.; Vandenhende, M.-A.; et al. Association of immune-activation and senescence markers with non-AIDS-defining comorbidities in HIV-suppressed patients. AIDS 2015, 29, 2099–2108. [Google Scholar] [CrossRef]
- Brady, M.T.; Oleske, J.; Williams, P.L.; Elgie, C.; Mofenson, L.; Dankner, W.M.; Van Dyke, R.B. Declines in Mortality Rates and Changes in Causes of Death in HIV-1-Infected Children During the HAART Era. JAIDS J. Acquir. Immune Defic. Syndr. 2010, 53, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Stacey, A.R.; Norris, P.J.; Qin, L.; Haygreen, E.A.; Taylor, E.; Heitman, J.; Lebedeva, M.; DeCamp, A.; Li, D.; Grove, D.; et al. Induction of a Striking Systemic Cytokine Cascade prior to Peak Viremia in Acute Human Immunodeficiency Virus Type 1 Infection, in Contrast to More Modest and Delayed Responses in Acute Hepatitis B and C Virus Infections. J. Virol. 2009, 83, 3719–3733. [Google Scholar] [CrossRef]
- Freeman, M.L.; Shive, C.L.; Nguyen, T.P.; Younes, S.-A.; Panigrahi, S.; Lederman, M.M. Cytokines and T-Cell Homeostasis in HIV Infection. J. Infect. Dis. 2016, 214, S51–S57. [Google Scholar] [CrossRef]
- Prevedel, L.; Ruel, N.; Castellano, P.; Smith, C.; Malik, S.; Villeux, C.; Bomsel, M.; Morgello, S.; Eugenin, E.A. Identification, Localization, and Quantification of HIV Reservoirs Using Microscopy. Curr. Protoc. Cell Biol. 2018, 82, e64. [Google Scholar] [CrossRef]
- Ruelas, D.S.; Greene, W.C. An Integrated Overview of HIV-1 Latency. Cell 2013, 155, 519–529. [Google Scholar] [CrossRef]
- Abreu, C.; Shirk, E.N.; Queen, S.E.; Mankowski, J.L.; Gama, L.; Clements, J.E. A Quantitative Approach to SIV Functional Latency in Brain Macrophages. J. Neuroimmune Pharmacol. 2018, 14, 23–32. [Google Scholar] [CrossRef]
- Sengupta, S.; Siliciano, R.F. Targeting the Latent Reservoir for HIV-1. Immunity 2018, 48, 872–895. [Google Scholar] [CrossRef]
- Siliciano, R.F.; Greene, W.C. HIV Latency. Cold Spring Harb. Perspect. Med. 2011, 1, a007096. [Google Scholar] [CrossRef] [PubMed]
- Cribbs, S.K.; Lennox, J.; Caliendo, A.M.; Brown, L.A.; Guidot, D.M. Healthy HIV-1-Infected Individuals on Highly Active Antiretroviral Therapy Harbor HIV-1 in Their Alveolar Macrophages. AIDS Res. Hum. Retrovir. 2015, 31, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Zalar, A.; Figueroa, M.I.; Ruibal-Ares, B.; Baré, P.; Cahn, P.; Bracco, M.M.D.E.D.; Belmonte, L. Macrophage HIV-1 infection in duodenal tissue of patients on long term HAART. Antivir. Res. 2010, 87, 269–271. [Google Scholar] [CrossRef]
- Josefsson, L.; von Stockenstrom, S.; Faria, N.R.; Sinclair, E.; Bacchetti, P.; Killian, M.; Epling, L.; Stanga, L.L.; Ho, T.; Lemey, P.; et al. The HIV-1 reservoir in eight patients on long-term suppressive antiretroviral therapy is stable with few genetic changes over time. Proc. Natl. Acad. Sci. USA 2013, 110, E4987–E4996. [Google Scholar] [CrossRef] [PubMed]
- Churchill, M.J.; Gorry, P.R.; Cowley, D.; Lal, L.; Sonza, S.; Purcell, D.F.J.; Thompson, K.A.; Gabuzda, D.; McArthur, J.C.; Pardo, C.A.; et al. Use of laser capture microdissection to detect integrated HIV-1 DNA in macrophages and astrocytes from autopsy brain tissues. J. Neurovirol. 2006, 12, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Calantone, N.; Wu, F.; Klase, Z.; Deleage, C.; Perkins, M.; Matsuda, K.; Thompson, E.A.; Ortiz, A.M.; Vinton, C.L.; Ourmanov, I.; et al. Tissue Myeloid Cells in SIV-Infected Primates Acquire Viral DNA through Phagocytosis of Infected T Cells. Immunity 2014, 41, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Baxter, A.; Russell, R.A.; Duncan, C.; Moore, R.; Willberg, C.; Pablos, J.L.; Finzi, A.; Kaufmann, D.E.; Ochsenbauer, C.; Kappes, J.C.; et al. Macrophage Infection via Selective Capture of HIV-1-Infected CD4+ T Cells. Cell Host Microbe 2014, 16, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Andrade, V.M.; Stevenson, M. Host and Viral Factors Influencing Interplay between the Macrophage and HIV-1. J. Neuroimmune Pharmacol. 2019, 14, 33–43. [Google Scholar] [CrossRef]
- Castellano, P.; Prevedel, L.; Eugenin, E.A. HIV-infected macrophages and microglia that survive acute infection become viral reservoirs by a mechanism involving Bim. Sci. Rep. 2017, 7, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Tay, T.L.; Mai, D.; Dautzenberg, J.; Fernandez-Klett, F.; Lin, G.; Sagar; Datta, M.; Drougard, A.; Stempfl, T.; Ardura-Fabregat, A.; et al. A new fate mapping system reveals context-dependent random or clonal expansion of microglia. Nat. Neurosci. 2017, 20, 793–803. [Google Scholar] [CrossRef] [PubMed]
- Thompson, K.A.; Cherry, C.L.; Bell, J.E.; McLean, C.A. Brain Cell Reservoirs of Latent Virus in Presymptomatic HIV-Infected Individuals. Am. J. Pathol. 2011, 179, 1623–1629. [Google Scholar] [CrossRef]
- Kierdorf, K.; Masuda, T.; Jordão, M.J.C.; Prinz, M. Macrophages at CNS interfaces: Ontogeny and function in health and disease. Nat. Rev. Neurosci. 2019, 20, 547–562. [Google Scholar] [CrossRef] [PubMed]
- Albright, A.V.; Shieh, J.T.; O’Connor, M.J.; González-Scarano, F. Characterization of cultured microglia that can be infected by HIV-1. J. Neurovirol. 2000, 6 (Suppl. S1), S53–S60. [Google Scholar] [PubMed]
- Honeycutt, J.B.; Wahl, A.; Baker, C.; Spagnuolo, R.A.; Foster, J.; Zakharova, O.; Wietgrefe, S.; Caro-Vegas, C.; Madden, V.; Sharpe, G.; et al. HIV persistence in tissue macrophages of humanized myeloid-only mice during antiretroviral therapy. Nat. Med. 2016, 23, 638–643. [Google Scholar] [CrossRef] [PubMed]
- Honeycutt, J.B.; Wahl, A.; Baker, C.; Spagnuolo, R.A.; Foster, J.; Zakharova, O.; Wietgrefe, S.; Caro-Vegas, C.; Madden, V.; Sharpe, G.; et al. Macrophages sustain HIV replication in vivo independently of T cells. J. Clin. Investig. 2017, 126, 1353–1366. [Google Scholar] [CrossRef]
- Ko, A.; Kang, G.; Hattler, J.B.; Galadima, H.; Zhang, J.; Li, Q.; Kim, W.-K. Macrophages but not Astrocytes Harbor HIV DNA in the Brains of HIV-1-Infected Aviremic Individuals on Suppressive Antiretroviral Therapy. J. Neuroimmune Pharmacol. 2019, 14, 110–119. [Google Scholar] [CrossRef]
- Castellano, P.; Prevedel, L.; Valdebenito, S.; Eugenin, E.A. HIV infection and latency induce a unique metabolic signature in human macrophages. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef]
- Garcia-Mesa, Y.; Jay, T.R.; Checkley, M.A.; Luttge, B.; Dobrowolski, C.; Valadkhan, S.; Landreth, G.E.; Karn, J.; Alvarez-Carbonell, D. Immortalization of primary microglia: A new platform to study HIV regulation in the central nervous system. J. Neurovirol. 2017, 23, 47–66. [Google Scholar] [CrossRef]
- Alvarez-Carbonell, D.; Ye, F.; Ramanath, N.; Garcia-Mesa, Y.; Knapp, P.E.; Hauser, K.F.; Karn, J. Cross-talk between microglia and neurons regulates HIV latency. PLoS Pathog. 2019, 15, e1008249. [Google Scholar] [CrossRef]
- Sumner, R.P.; Thorne, L.G.; Fink, D.L.; Khan, H.; Milne, R.S.; Towers, G.J. Are Evolution and the Intracellular Innate Immune System Key Determinants in HIV Transmission? Front. Immunol. 2017, 8, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Vermeire, J.; Roesch, F.; Sauter, D.; Rua, R.; Hotter, D.; Van Nuffel, A.; Vanderstraeten, H.; Naessens, E.; Iannucci, V.; Landi, A.; et al. HIV Triggers a cGAS-Dependent, Vpu- and Vpr-Regulated Type I Interferon Response in CD4+ T Cells. Cell Rep. 2016, 17, 413–424. [Google Scholar] [CrossRef]
- Aso, H.; Ito, J.; Koyanagi, Y.; Sato, K. Comparative Description of the Expression Profile of Interferon-Stimulated Genes in Multiple Cell Lineages Targeted by HIV-1 Infection. Front. Microbiol. 2019, 10, 429. [Google Scholar] [CrossRef]
- Isaacs, A.; Lindenmann, J. Virus interference. I. The interferon. Proc. R. Soc. Lond. Ser. B Boil Sci. 1957, 147, 258–267. [Google Scholar] [CrossRef]
- Shirazi, Y.; Pitha, P.M. Alpha interferon inhibits early stages of the human immunodeficiency virus type 1 replication cycle. J. Virol. 1992, 66, 1321–1328. [Google Scholar] [CrossRef]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-Stimulated Genes: A Complex Web of Host Defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef] [PubMed]
- Doyle, T.; Goujon, C.; Malim, M.H. HIV-1 and interferons: Who’s interfering with whom? Nat. Rev. Genet. 2015, 13, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Rustagi, A.; Gale, M. Innate Antiviral Immune Signaling, Viral Evasion and Modulation by HIV-1. J. Mol. Biol. 2014, 426, 1161–1177. [Google Scholar] [CrossRef] [PubMed]
- Jiménez, V.C.; Booiman, T.; De Taeye, S.W.; Van Dort, K.A.; Rits, M.A.N.; Hamann, J.; Kootstra, N.A. Differential expression of HIV-1 interfering factors in monocyte-derived macrophages stimulated with polarizing cytokines or interferons. Sci. Rep. 2012, 2, 763. [Google Scholar] [CrossRef]
- Liu, M.-Q.; Zhao, M.; Kong, W.-H.; Tang, L.; Wang, F.; Zhu, Z.-R.; Wang, X.; Qiu, H.-Y.; Zhou, D.-J.; Wang, X.; et al. Combination antiretroviral therapy (cART) restores HIV-1 infection-mediated impairment of JAK-STAT signaling pathway. Oncotarget 2017, 8, 22524–22533. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef]
- Meier, A.; Alter, G.; Frahm, N.; Sidhu, H.; Li, B.; Bagchi, A.; Teigen, N.; Streeck, H.; Stellbrink, H.-J.; Hellman, J.; et al. MyD88-Dependent Immune Activation Mediated by Human Immunodeficiency Virus Type 1-Encoded Toll-Like Receptor Ligands. J. Virol. 2007, 81, 8180–8191. [Google Scholar] [CrossRef]
- Berg, R.K.; Melchjorsen, J.; Rintahaka, J.; Diget, E.; Søby, S.; Horan, K.A.; Gorelick, R.J.; Matikainen, S.; Larsen, C.S.; Østergaard, L.; et al. Genomic HIV RNA Induces Innate Immune Responses through RIG-I-Dependent Sensing of Secondary-Structured RNA. PLoS ONE 2012, 7, e29291. [Google Scholar] [CrossRef]
- Solis, M.; Nakhaei, P.; Jalalirad, M.; Lacoste, J.; Douville, R.; Arguello, M.; Zhao, T.; Laughrea, M.; Wainberg, M.A.; Hiscott, J. RIG-I-Mediated Antiviral Signaling Is Inhibited in HIV-1 Infection by a Protease-Mediated Sequestration of RIG-I. J. Virol. 2010, 85, 1224–1236. [Google Scholar] [CrossRef] [PubMed]
- Qiu, L.; Wang, T.; Tang, Q.; Li, G.; Wu, P.; Chen, K. Long Non-coding RNAs: Regulators of Viral Infection and the Interferon Antiviral Response. Front. Microbiol. 2018, 9, 1621. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.; Kessler, D.S.; Pine, R.; Darnell, J.E. Cytoplasmic activation of ISGF3, the positive regulator of interferon-alpha-stimulated transcription, reconstituted in vitro. Genes Dev. 1989, 3, 1362–1371. [Google Scholar] [CrossRef] [PubMed]
- Sheehy, A.M.; Gaddis, N.; Choi, J.D.; Malim, M. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nat. Cell Biol. 2002, 418, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Stremlau, M.; Owens, C.M.; Perron, M.J.; Kiessling, M.; Autissier, P.; Sodroski, J. The cytoplasmic body component TRIM5α restricts HIV-1 infection in Old World monkeys. Nat. Cell Biol. 2004, 427, 848–853. [Google Scholar] [CrossRef]
- Goldstone, D.; Ennis-Adeniran, V.; Hedden, J.J.; Groom, H.C.T.; Rice, G.; Christodoulou, E.; Walker, P.A.; Kelly, G.; Haire, L.F.; Yap, M.W.; et al. HIV-1 restriction factor SAMHD1 is a deoxynucleoside triphosphate triphosphohydrolase. Nat. Cell Biol. 2011, 480, 379–382. [Google Scholar] [CrossRef]
- Merindol, N. Restriction Factors in HIV-1 Disease Progression. Curr. HIV Res. 2015, 13, 448–461. [Google Scholar] [CrossRef] [PubMed]
- Goujon, C.; Moncorge, O.; Bauby, H.; Doyle, T.; Ward, C.C.; Schaller, T.; Hue, S.; Barclay, W.; Schulz, R.; Malim, M.H. Human MX2 is an interferon-induced post-entry inhibitor of HIV-1 infection. Nat. Cell Biol. 2013, 502, 559–562. [Google Scholar] [CrossRef]
- Galao, R.; Le Tortorec, A.; Pickering, S.; Kueck, T.; Neil, S.J. Innate Sensing of HIV-1 Assembly by Tetherin Induces NFκB-Dependent Proinflammatory Responses. Cell Host Microbe 2012, 12, 633–644. [Google Scholar] [CrossRef]
- Lin, R.; Heylbroeck, C.; Pitha, P.M.; Hiscott, J. Virus-Dependent Phosphorylation of the IRF-3 Transcription Factor Regulates Nuclear Translocation, Transactivation Potential, and Proteasome-Mediated Degradation. Mol. Cell. Biol. 1998, 18, 2986–2996. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.; Hiscott, J.; Pitha, P.M. The growing family of interferon regulatory factors. Cytokine Growth Factor Rev. 1997, 8, 293–312. [Google Scholar] [CrossRef]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP Synthase Is a Cytosolic DNA Sensor That Activates the Type I Interferon Pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.-C.; Gopalkrishnan, R.V.; Wu, Q.; Jankowsky, E.; Pyle, A.M.; Fisher, P.B. mda-5: An interferon-inducible putative RNA helicase with double-stranded RNA-dependent ATPase activity and melanoma growth-suppressive properties. Proc. Natl. Acad. Sci. USA 2002, 99, 637–642. [Google Scholar] [CrossRef] [PubMed]
- Ishii, K.; Coban, C.; Kato, H.; Takahashi, K.; Torii, Y.; Takeshita, F.; Ludwig, H.; Sutter, G.; Suzuki, K.; Hemmi, H.; et al. A Toll-like receptor–independent antiviral response induced by double-stranded B-form DNA. Nat. Immunol. 2005, 7, 40–48. [Google Scholar] [CrossRef]
- Kane, M.; Zang, T.M.; Rihn, S.; Zhang, F.; Kueck, T.; Alim, M.; Schoggins, J.; Rice, C.M.; Wilson, S.J.; Bieniasz, P.D. Identification of Interferon-Stimulated Genes with Antiretroviral Activity. Cell Host Microbe 2016, 20, 392–405. [Google Scholar] [CrossRef]
- Dalpke, A.H.; Opper, S.; Zimmermann, S.; Heeg, K. Suppressors of Cytokine Signaling (SOCS)-1 and SOCS-3 Are Induced by CpG-DNA and Modulate Cytokine Responses in APCs. J. Immunol. 2001, 166, 7082–7089. [Google Scholar] [CrossRef]
- François-Newton, V.; Almeida, G.; Payelle-Brogard, B.; Monneron, D.; Pichard-Garcia, L.; Piehler, J.; Pellegrini, S.; Uzé, G. USP18-Based Negative Feedback Control Is Induced by Type I and Type III Interferons and Specifically Inactivates Interferon α Response. PLoS ONE 2011, 6, e22200. [Google Scholar] [CrossRef]
- Yadav, A.; Fitzgerald, P.; Sajadi, M.M.; Gilliam, B.; Lafferty, M.K.; Redfield, R.; Reid, W. Increased expression of suppressor of cytokine signaling-1 (SOCS-1): A mechanism for dysregulated T helper-1 responses in HIV-1 disease. Virology 2009, 385, 126–133. [Google Scholar] [CrossRef][Green Version]
- Miller, R.C.; Schlaepfer, E.; Baenziger, S.; Crameri, R.; Zeller, S.; Byland, R.; Audigé, A.; Nadal, D.; Speck, R.F. HIV interferes with SOCS-1 and -3 expression levels driving immune activation. Eur. J. Immunol. 2011, 41, 1058–1069. [Google Scholar] [CrossRef]
- Dagenais-Lussier, X.; Loucif, H.; Cadorel, H.; Blumberger, J.; Isnard, S.; Bego, M.G.; Cohen, É.A.; Routy, J.-P.; Van Grevenynghe, J.; The Montreal Primary Infection Study Group. USP18 is a significant driver of memory CD4 T-cell reduced viability caused by type I IFN signaling during primary HIV-1 infection. PLoS Pathog. 2019, 15, e1008060. [Google Scholar] [CrossRef]
- Kuffour, E.O.; Schott, K.; Vasudevan, A.A.J.; Holler, J.; Schulz, W.; Lang, P.A.; Lang, K.; Kim, B.; Häussinger, D.; König, R.; et al. USP18 (UBP43) Abrogates p21-Mediated Inhibition of HIV-1. J. Virol. 2018, 92, e00592-18. [Google Scholar] [CrossRef] [PubMed]
- Hubel, P.; Urban, C.; Bergant, V.; Schneider, W.M.; Knauer, B.; Stukalov, A.; Scaturro, P.; Mann, A.; Brunotte, L.; Hoffmann, H.H.; et al. A protein-interaction network of interferon-stimulated genes extends the innate immune system landscape. Nat. Immunol. 2019, 20, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Tokarev, A.; Suarez, M.; Kwan, W.; Fitzpatrick, K.; Singh, R.; Guatelli, J. Stimulation of NF- B Activity by the HIV Restriction Factor BST. J. Virol. 2012, 87, 2046–2057. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Zhang, X.; Feng, Y.; Zhang, H.; Wang, X.; Zheng, Y.; Qiao, W.; Liu, X. Structural and Functional Study of Apoptosis-linked Gene-2·Heme-binding Protein 2 Interactions in HIV-1 Production. J. Biol. Chem. 2016, 291, 26670–26685. [Google Scholar] [CrossRef] [PubMed]
- Goffinet, C. Cellular Antiviral Factors that Target Particle Infectivity of HIV-1. Curr. HIV Res. 2016, 14, 211–216. [Google Scholar] [CrossRef][Green Version]
- Jakobsen, M.R.; Bak, R.O.; Andersen, A.; Berg, R.K.; Jensen, S.B.; Jin, T.; Laustsen, A.; Hansen, K.; Østergaard, L.; Fitzgerald, K.A.; et al. PNAS Plus: From the Cover: IFI16 senses DNA forms of the lentiviral replication cycle and controls HIV-1 replication. Proc. Natl. Acad. Sci. USA 2013, 110, E4571–E4580. [Google Scholar] [CrossRef]
- Soper, A.; Kimura, I.; Nagaoka, S.; Konno, Y.; Yamamoto, K.; Koyanagi, Y.; Sato, K. Type I Interferon Responses by HIV-1 Infection: Association with Disease Progression and Control. Front. Immunol. 2018, 8, 1823. [Google Scholar] [CrossRef]
- Perry, A.K.; Chow, E.; Goodnough, J.B.; Yeh, W.-C.; Cheng, G. Differential Requirement for TANK-binding Kinase-1 in Type I Interferon Responses to Toll-like Receptor Activation and Viral Infection. J. Exp. Med. 2004, 199, 1651–1658. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.H.; Murti, A.; Pfeffer, S.R.; Basu, L.; Kim, J.G.; Pfeffer, L. IFNalpha/beta promotes cell survival by activating NF-kappa B. Proc. Natl. Acad. Sci. USA 2000, 97, 13631–13636. [Google Scholar] [CrossRef]
- McCauley, S.M.; Kim, K.; Nowosielska, A.; Dauphin, A.; Yurkovetskiy, L.; Diehl, W.E.; Luban, J. Intron-containing RNA from the HIV-1 provirus activates type I interferon and inflammatory cytokines. Nat. Commun. 2018, 9, 1–10. [Google Scholar] [CrossRef]
- Decalf, J.; Desdouits, M.; Rodrigues, V.; Gobert, F.-X.; Gentili, M.; Marques-Ladeira, S.; Chamontin, C.; Mougel, M.; de Alencar, B.; Benaroch, P. Sensing of HIV-1 Entry Triggers a Type I Interferon Response in Human Primary Macrophages. J. Virol. 2017, 91, e00147-17. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Langer, S.; Zhang, Z.; Herbert, K.M.; Yoh, S.; König, R.; Chanda, S.K. Sensor Sensibility—HIV-1 and the Innate Immune Response. Cells 2020, 9, 254. [Google Scholar] [CrossRef] [PubMed]
- Utay, N.S.; Douek, D.C. Interferons and HIV Infection: The Good, the Bad, and the Ugly. Pathog. Immun. 2016, 1, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Rasaiyaah, J.; Tan, C.P.; Fletcher, A.J.; Price, A.J.; Blondeau, C.; Hilditch, L.; Jacques, D.; Selwood, D.; James, L.C.; Noursadeghi, M.; et al. HIV-1 evades innate immune recognition through specific cofactor recruitment. Nat. Cell Biol. 2013, 503, 402–405. [Google Scholar] [CrossRef]
- Wang, B.; Kang, W.; Zuo, J.; Kang, W.; Sun, Y. The Significance of Type-I Interferons in the Pathogenesis and Therapy of Human Immunodeficiency Virus 1 Infection. Front. Immunol. 2017, 8, 1–12. [Google Scholar] [CrossRef]
- Hardy, G.A.D.; Sieg, S.; Rodriguez, B.; Anthony, D.; Asaad, R.; Jiang, W.; Mudd, J.; Schacker, T.; Funderburg, N.; Pilch-Cooper, H.A.; et al. Interferon-α Is the Primary Plasma Type-I IFN in HIV-1 Infection and Correlates with Immune Activation and Disease Markers. PLoS ONE 2013, 8, e56527. [Google Scholar] [CrossRef]
- Bosinger, S.E.; Li, Q.; Gordon, S.N.; Klatt, N.R.; Duan, L.; Xu, L.; Francella, N.; Sidahmed, A.; Smith, A.J.; Cramer, E.M.; et al. Global genomic analysis reveals rapid control of a robust innate response in SIV-infected sooty mangabeys. J. Clin. Investig. 2009, 119, 3556–3572. [Google Scholar] [CrossRef] [PubMed]
- Nganou-Makamdop, K.; Douek, D.C. Manipulating the Interferon Signaling Pathway: Implications for HIV Infection. Virol. Sin. 2019, 34, 192–196. [Google Scholar] [CrossRef]
- Machmach, K.; Leal, M.; Gras, C.; Viciana, P.; Genebat, M.; Franco, E.; Boufassa, F.; Lambotte, O.; Herbeuval, J.P.; Ruiz-Mateos, E. Plasmacytoid Dendritic Cells Reduce HIV Production in Elite Controllers. J. Virol. 2012, 86, 4245–4252. [Google Scholar] [CrossRef]
- Granier, C.; Battivelli, E.; Lécuroux, C.; Venet, A.; Lambotte, O.; Schmitt-Boulanger, M.; Delaugerre, C.; Molina, J.-M.; Chakrabarti, L.; Clavel, F.; et al. Pressure from TRIM5α Contributes to Control of HIV-1 Replication by Individuals Expressing Protective HLA-B Alleles. J. Virol. 2013, 87, 10368–10380. [Google Scholar] [CrossRef]
- Van Der Sluis, R.M.; Zerbato, J.M.; Rhodes, J.W.; Pascoe, R.D.; Solomon, A.; Kumar, N.A.; Dantanarayana, A.I.; Tennakoon, S.; Dufloo, J.; McMahon, J.; et al. Diverse effects of interferon alpha on the establishment and reversal of HIV latency. PLoS Pathog. 2020, 16, e1008151. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Wu, J.; Wu, Y.-T.; Du, F.; Aroh, C.; Yan, N.; Sun, L.; Chen, Z.J. Cyclic GMP-AMP Synthase Is an Innate Immune Sensor of HIV and Other Retroviruses. Science 2013, 341, 903–906. [Google Scholar] [CrossRef]
- Ringeard, M.; Marchand, V.; Decroly, E.; Motorin, Y.; Yamina, B. FTSJ3 is an RNA 2′-O-methyltransferase recruited by HIV to avoid innate immune sensing. Nature 2019, 565, 500–504. [Google Scholar] [CrossRef]
- Wang, M.Q.; Huang, Y.L.; Huang, J.; Zheng, J.L.; Qian, G.X. RIG-I detects HIV-1 infection and mediates type I interferon response in human macrophages from patients with HIV-1-associated neurocognitive disorders. Genet. Mol. Res. 2015, 14, 13799–13811. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, H.; Liu, M.-Q.; Li, J.-L.; Zhou, R.-H.; Zhou, Y.; Wang, Y.-Z.; Zhou, W.; Ho, W.-Z. IFN-λ Inhibits Drug-Resistant HIV Infection of Macrophages. Front. Immunol. 2017, 8, 1–8. [Google Scholar] [CrossRef]
- Parrish, N.F.; Gao, F.; Li, H.; Giorgi, E.E.; Barbian, H.J.; Parrish, E.H.; Zajic, L.; Iyer, S.S.; Decker, J.M.; Kumar, A.; et al. Phenotypic properties of transmitted founder HIV-1. Proc. Natl. Acad. Sci. USA 2013, 110, 6626–6633. [Google Scholar] [CrossRef]
- Iyer, S.S.; Bibollet-Ruche, F.; Sherrill-Mix, S.; Learn, G.H.; Plenderleith, L.; Smith, A.G.; Barbian, H.J.; Russell, R.M.; Gondim, M.V.P.; Bahari, C.Y.; et al. Resistance to type 1 interferons is a major determinant of HIV-1 transmission fitness. Proc. Natl. Acad. Sci. USA 2017, 114, E590–E599. [Google Scholar] [CrossRef] [PubMed]
- Salazar-Gonzalez, J.F.; Salazar, M.G.; Keele, B.F.; Learn, G.; Giorgi, E.E.; Li, H.; Decker, J.M.; Wang, S.; Baalwa, J.; Kraus, M.H.; et al. Genetic identity, biological phenotype, and evolutionary pathways of transmitted/founder viruses in acute and early HIV-1 infection. J. Exp. Med. 2009, 206, 1273–1289. [Google Scholar] [CrossRef] [PubMed]
- Hrecka, K.; Hao, C.; Gierszewska, M.; Swanson, S.K.; Kesik-Brodacka, M.; Srivastava, S.; Florens, L.; Washburn, M.; Skowronski, J. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nat. Cell Biol. 2011, 474, 658–661. [Google Scholar] [CrossRef]
- Neil, S.; Zang, T.; Bieniasz, P.D. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nat. Cell Biol. 2008, 451, 425–430. [Google Scholar] [CrossRef]
- Duncan, C.J.A.; Sattentau, Q.J. Viral Determinants of HIV-1 Macrophage Tropism. Viruses 2011, 3, 2255–2279. [Google Scholar] [CrossRef] [PubMed]
- Winkler, R.; Gillis, E.; Lasman, L.; Safra, M.; Geula, S.; Soyris, C.; Nachshon, A.; Tai-Schmiedel, J.; Friedman, N.; Le-Trilling, V.T.K.; et al. m6A modification controls the innate immune response to infection by targeting type I interferons. Nat. Immunol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Filipowicz, M.; Wang, X.; Yan, M.; Duong, F.H.T.; Poli, V.; Hilton, D.J.; Zhang, D.-E.; Heim, M.H. Alpha Interferon Induces Long-Lasting Refractoriness of JAK-STAT Signaling in the Mouse Liver through Induction of USP18/UBP43. Mol. Cell. Biol. 2009, 29, 4841–4851. [Google Scholar] [CrossRef] [PubMed]
- Marijanovic, Z.; Ragimbeau, J.; Van Der Heyden, J.; Uzé, G.; Pellegrini, S. Comparable potency of IFNα2 and IFNβ on immediate JAK/STAT activation but differential down-regulation of IFNAR2. Biochem. J. 2007, 407, 141–151. [Google Scholar] [CrossRef]
- Coccia, E.M.; Uzé, G.; Pellegrini, G.U. Negative Regulation of Type I Interferon Signaling: Facts and Mechanisms (Noisy-le-grand). Cell. Mol. Biol. 2006, 52, 77–87. Available online: https://pubmed.ncbi.nlm.nih.gov/16914099/ (accessed on 16 July 2021). [PubMed]
- David, M.; Chen, H.E.; Goelz, S.; Larner, A.C.; Neel, B.G. Differential regulation of the alpha/beta interferon-stimulated Jak/Stat pathway by the SH2 domain-containing tyrosine phosphatase SHPTP1. Mol. Cell. Biol. 1995, 15, 7050–7058. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Xu, L.; Su, J.; Peppelenbosch, M.; Pan, Q. Transcriptional Regulation of Antiviral Interferon-Stimulated Genes. Trends Microbiol. 2017, 25, 573–584. [Google Scholar] [CrossRef]
- Bluyssen, H.; Muzaffar, R.; Vlieststra, R.J.; van der Made, A.C.; Leung, S.; Stark, G.R.; Kerr, I.M.; Trapman, J.; Levy, D. Combinatorial association and abundance of components of interferon-stimulated gene factor 3 dictate the selectivity of interferon responses. Proc. Natl. Acad. Sci. USA 1995, 92, 5645–5649. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Cui, K.; Murray, D.M.; Ling, C.; Xue, Y.; Gerstein, A.; Parsons, R.; Zhao, K.; Wang, W. PBAF chromatin-remodeling complex requires a novel specificity subunit, BAF200, to regulate expression of selective interferon-responsive genes. Genes Dev. 2005, 19, 1662–1667. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.J.; Zhao, Y.; Chait, B.T.; Lathem, W.; Ritzi, M.; Knippers, R.; Darnell, J.E. Ser727-dependent recruitment of MCM5 by Stat1α in IFN-γ-induced transcriptional activation. EMBO J. 1998, 17, 6963–6971. [Google Scholar] [CrossRef]
- Kadota, S.; Nagata, K. Silencing of IFN-stimulated gene transcription is regulated by histone H1 and its chaperone TAF-I. Nucleic Acids Res. 2014, 42, 7642–7653. [Google Scholar] [CrossRef]
- Mostafavi, S.; Yoshida, H.; Moodley, D.; LeBoité, H.; Rothamel, K.; Raj, T.; Ye, C.J.; Chevrier, N.; Zhang, S.-Y.; Feng, T.; et al. Parsing the Interferon Transcriptional Network and Its Disease Associations. Cell 2016, 164, 564–578. [Google Scholar] [CrossRef] [PubMed]
- Rubio, R.M.; Depledge, D.P.; Bianco, C.; Thompson, L.; Mohr, I. RNA m6A modification enzymes shape innate responses to DNA by regulating interferon β. Genes Dev. 2018, 32, 1472–1484. [Google Scholar] [CrossRef]
- Imam, H.; Kim, G.-W.; Mir, S.A.; Khan, M.; Siddiqui, A. Interferon-stimulated gene 20 (ISG20) selectively degrades N6-methyladenosine modified Hepatitis B Virus transcripts. PLoS Pathog. 2020, 16, e1008338. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Kumar, S.; Espada, C.E.; Tirumuru, N.; Cahill, M.P.; Hu, L.; He, C.; Wu, L. N6-methyladenosine modification of HIV-1 RNA suppresses type-I interferon induction in differentiated monocytic cells and primary macrophages. PLoS Pathog. 2021, 17, e1009421. [Google Scholar] [CrossRef]
- Pujantell, M.; Riveira-Muñoz, E.; Badia, R.; Castellví, M.; Garcia-Vidal, E.; Sirera, G.; Puig, T.; Ramirez, C.; Clotet, B.; Esté, J.A.; et al. RNA editing by ADAR1 regulates innate and antiviral immune functions in primary macrophages. Sci. Rep. 2017, 7, 13339. [Google Scholar] [CrossRef] [PubMed]
- George, C.X.; Samuel, C.E. Human RNA-specific adenosine deaminase ADAR1 transcripts possess alternative exon 1 structures that initiate from different promoters, one constitutively active and the other interferon inducible. Proc. Natl. Acad. Sci. USA 1999, 96, 4621–4626. [Google Scholar] [CrossRef]
- Patterson, J.B.; Samuel, C.E. Expression and regulation by interferon of a double-stranded-RNA-specific adenosine deaminase from human cells: Evidence for two forms of the deaminase. Mol. Cell. Biol. 1995, 15, 5376–5388. [Google Scholar] [CrossRef]
- Cuadrado, E.; Booiman, T.; Van Hamme, J.L.; Jansen, M.H.; Van Dort, K.A.; Vanderver, A.; Rice, G.I.; Crow, Y.J.; Kootstra, N.A.; Kuijpers, T.W. ADAR1 Facilitates HIV-1 Replication in Primary CD4+ T Cells. PLoS ONE 2015, 10, e0143613. [Google Scholar] [CrossRef]
- Yang, C.H.; Yue, J.; Fan, M.; Pfeffer, L.M. IFN Induces miR-21 through a Signal Transducer and Activator of Transcription 3–Dependent Pathway as a Suppressive Negative Feedback on IFN-Induced Apoptosis. Cancer Res. 2010, 70, 8108–8116. [Google Scholar] [CrossRef]
- Gomez, J.A.; Wapinski, O.L.; Yang, Y.W.; Bureau, J.-F.; Gopinath, S.; Monack, D.M.; Chang, H.Y.; Brahic, M.; Kirkegaard, K. The NeST Long ncRNA Controls Microbial Susceptibility and Epigenetic Activation of the Interferon-γ Locus. Cell 2013, 152, 743–754. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Lin, X.; Yang, X.; Lu, G.; Zhang, Q.; Zhang, C. MicroRNA-132-3p suppresses type I IFN response through targeting IRF1 to facilitate H1N1 influenza A virus infection. Biosci. Rep. 2019, 39, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Wang, F.; Argyris, E.; Chen, K.; Liang, Z.; Tian, H.; Huang, W.; Squires, K.; Verlinghieri, G.; Zhang, H. Cellular microRNAs contribute to HIV-1 latency in resting primary CD4+ T lymphocytes. Nat. Med. 2007, 13, 1241–1247. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhou, R.; Liu, Y.; Guo, L.; Wang, X.; Hu, W.; Ho, W. HIV infection suppresses TLR3 activation-mediated antiviral immunity in microglia and macrophages. Immunology 2020, 160, 269–279. [Google Scholar] [CrossRef]
- Martinez-Nunez, R.T.; Louafi, F.; Friedmann, P.S.; Sanchez-Elsner, T. MicroRNA-155 Modulates the Pathogen Binding Ability of Dendritic Cells (DCs) by Down-regulation of DC-specific Intercellular Adhesion Molecule-3 Grabbing Non-integrin (DC-SIGN). J. Biol. Chem. 2009, 284, 16334–16342. [Google Scholar] [CrossRef]
- Swaminathan, G.; Rossi, F.; Sierra, L.-J.; Gupta, A.; Navas-Martin, S.; Martín-García, J. A Role for microRNA-155 Modulation in the Anti-HIV-1 Effects of Toll-Like Receptor 3 Stimulation in Macrophages. PLoS Pathog. 2012, 8, e1002937. [Google Scholar] [CrossRef] [PubMed]
- Ruelas, D.S.; Chan, J.K.; Oh, E.; Heidersbach, A.J.; Hebbeler, A.M.; Chavez, L.; Verdin, E.; Rape, M.; Greene, W.C. MicroRNA-155 Reinforces HIV Latency. J. Biol. Chem. 2015, 290, 13736–13748. [Google Scholar] [CrossRef]
- Wang, X.; Ye, L.; Zhou, Y.; Liu, M.-Q.; Zhou, D.-J.; Ho, W.-Z. Inhibition of Anti-HIV MicroRNA Expression: A Mechanism for Opioid-Mediated Enhancement of HIV Infection of Monocytes. Am. J. Pathol. 2011, 178, 41–47. [Google Scholar] [CrossRef]
- Zhang, Q.; Cao, X. Epigenetic regulation of the innate immune response to infection. Nat. Rev. Immunol. 2019, 19, 417–432. [Google Scholar] [CrossRef]
- Kamada, R.; Yang, W.; Zhang, Y.; Patel, M.C.; Yang, Y.; Ouda, R.; Dey, A.; Wakabayashi, Y.; Sakaguchi, K.; Fujita, T.; et al. Interferon stimulation creates chromatin marks and establishes transcriptional memory. Proc. Natl. Acad. Sci. USA 2018, 115, E9162–E9171. [Google Scholar] [CrossRef]
- Pereira-Montecinos, C.; Valiente-Echeverria, F.; Soto-Rifo, R. Epitranscriptomic regulation of viral replication. Biochim. Biophys. Acta-Bioenerg. 2017, 1860, 460–471. [Google Scholar] [CrossRef] [PubMed]
- Oriol-Tordera, B.; Berdasco, M.; Llano, A.; Mothe, B.; Gálvez, C.; Martinez-Picado, J.; Carrillo, J.; Blanco, J.; Duran-Castells, C.; Ganoza, C.; et al. Methylation regulation of Antiviral host factors, Interferon Stimulated Genes (ISGs) and T-cell responses associated with natural HIV control. PLoS Pathog. 2020, 16, e1008678. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.; Calis, J.J.; Wu, X.; Sun, T.; Yu, Y.; Sarbanes, S.L.; Thi, V.L.D.; Shilvock, A.; Hoffmann, H.-H.; Rosenberg, B.; et al. Human ADAR1 Prevents Endogenous RNA from Triggering Translational Shutdown. Cell 2018, 172, 811–824.e14. [Google Scholar] [CrossRef]
- Fu, Y.; Dominissini, D.; Rechavi, G.; He, C. Gene expression regulation mediated through reversible m6A RNA methylation. Nat. Rev. Genet. 2014, 15, 293–306. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Xiong, X.; Yi, C. Epitranscriptome sequencing technologies: Decoding RNA modifications. Nat. Methods 2016, 14, 23–31. [Google Scholar] [CrossRef]
- Kennedy, E.M.; Courtney, D.G.; Tsai, K.; Cullen, B.R. Viral Epitranscriptomics. J. Virol. 2017, 91, e02263-16. [Google Scholar] [CrossRef] [PubMed]
- Brocard, M.; Ruggieri, A.; Locker, N. m6A RNA methylation, a new hallmark in virus-host interactions. J. Gen. Virol. 2017, 98, 2207–2214. [Google Scholar] [CrossRef] [PubMed]
- Horn, S.R.G.-V.; Sarnow, P. Making the Mark: The Role of Adenosine Modifications in the Life Cycle of RNA Viruses. Cell Host Microbe 2017, 21, 661–669. [Google Scholar] [CrossRef]
- Zhao, B.; Roundtree, I.A.; He, C. Post-transcriptional gene regulation by mRNA modifications. Nat. Rev. Mol. Cell Biol. 2017, 18, 31–42. [Google Scholar] [CrossRef]
- Wang, X.; Zhao, B.; Roundtree, I.A.; Lu, Z.; Han, D.; Ma, H.; Weng, X.; Chen, K.; Shi, H.; He, C. N6-methyladenosine Modulates Messenger RNA Translation Efficiency. Cell 2015, 161, 1388–1399. [Google Scholar] [CrossRef] [PubMed]
- Batista, P.J.; Molinie, B.; Wang, J.; Qu, K.; Zhang, J.; Li, L.; Bouley, D.M.; Lujan, E.; Haddad, B.; Daneshvar, K.; et al. m6A RNA Modification Controls Cell Fate Transition in Mammalian Embryonic Stem Cells. Cell Stem Cell 2014, 15, 707–719. [Google Scholar] [CrossRef]
- Schwartz, S.; Mumbach, M.; Jovanovic, M.; Wang, T.; Maciag, K.; Bushkin, G.G.; Mertins, P.; Ter-Ovanesyan, D.; Habib, N.; Cacchiarelli, D.; et al. Perturbation of m6A Writers Reveals Two Distinct Classes of mRNA Methylation at Internal and 5′ Sites. Cell Rep. 2014, 8, 284–296. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Fu, Y.; Zhao, X.; Dai, Q.; Zheng, G.; Yang, Y.; Yi, C.; Lindahl, T.; Pan, T.; Yang, Y.-G.; et al. Erratum: Corrigendum: N6-Methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 2012, 8, 1008. [Google Scholar] [CrossRef]
- Zheng, G.; Dahl, J.A.; Niu, Y.; Fedorcsak, P.; Huang, C.-M.; Li, C.J.; Vågbø, C.B.; Shi, Y.; Wang, W.-L.; Song, S.-H.; et al. ALKBH5 Is a Mammalian RNA Demethylase that Impacts RNA Metabolism and Mouse Fertility. Mol. Cell 2013, 49, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.; Roundtree, I.A.; Wang, P.; Wang, X.; Wang, L.; Sun, C.; Tian, Y.; Li, J.; He, C.; Xu, Y. Crystal structure of the YTH domain of YTHDF2 reveals mechanism for recognition of N6-methyladenosine. Cell Res. 2014, 24, 1493–1496. [Google Scholar] [CrossRef] [PubMed]
- Liao, S.; Sun, H.; Xu, C. YTH Domain: A Family of N 6 -methyladenosine (m 6 A) Readers. Genom. Proteom. Bioinform. 2018, 16, 99–107. [Google Scholar] [CrossRef]
- Gokhale, N.S.; McIntyre, A.B.R.; Mattocks, M.D.; Holley, C.L.; Lazear, H.M.; Mason, C.E.; Horner, S.M. Altered m6A Modification of Specific Cellular Transcripts Affects Flaviviridae Infection. Mol. Cell 2020, 77, 542–555.e8. [Google Scholar] [CrossRef]
- Huangfu, N.; Zheng, W.; Xu, Z.; Wang, S.; Wang, Y.; Cheng, J.; Li, Z.; Cheng, K.; Zhang, S.; Chen, X.; et al. RBM4 regulates M1 macrophages polarization through targeting STAT1-mediated glycolysis. Int. Immunopharmacol. 2020, 83, 106432. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, Z.; Tang, H.; Shen, Y.; Gong, Z.; Xie, N.; Zhang, X.; Wang, W.; Kong, W.; Zhou, Y.; et al. The N6-methyladenosine (m6A)-forming enzyme METTL3 facilitates M1 macrophage polarization through the methylation of STAT1 mRNA. Am. J. Physiol. Physiol. 2019, 317, C762–C775. [Google Scholar] [CrossRef]
- Lichinchi, G.; Gao, S.; Saletore, Y.; Gonzalez, G.M.; Bansal, V.; Wang, Y.; Mason, C.E.; Rana, T.M. Dynamics of the human and viral m6A RNA methylomes during HIV-1 infection of T cells. Nat. Microbiol. 2016, 1, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Riquelme-Barrios, S.; Pereira-Montecinos, C.; Valiente-Echeverría, F.; Rifo, R.S. Emerging Roles of N6-Methyladenosine on HIV-1 RNA Metabolism and Viral Replication. Front. Microbiol. 2018, 9, 576. [Google Scholar] [CrossRef] [PubMed]
- Jurczyszak, D.; Zhang, W.; Terry, S.N.; Kehrer, T.; González, M.C.B.; McGregor, E.; Mulder, L.C.F.; Eckwahl, M.J.; Pan, T.; Simon, V. HIV protease cleaves the antiviral m6A reader protein YTHDF3 in the viral particle. PLoS Pathog. 2020, 16, e1008305. [Google Scholar] [CrossRef]
- Kennedy, E.M.; Bogerd, H.P.; Kornepati, A.V.R.; Kang, D.; Ghoshal, D.; Marshall, J.B.; Poling, B.C.; Tsai, K.; Gokhale, N.S.; Horner, S.M.; et al. Posttranscriptional m 6 A Editing of HIV-1 mRNAs Enhances Viral Gene Expression. Cell Host Microbe 2016, 19, 675–685. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Cell Type | IFNs | ISGs | Other Cytokines and Costimulatory or Activation Molecules | Reference |
|---|---|---|---|---|
| THP1, Primary human MDMs | IFN-β | TREX1, CXCL10 | CXCL10 | [105] |
| Primary human MDMs THP1/MDM Murine macrophages | IFN-β IFN-λ1 | CXCL10, IFI16 | IL-6, TNF-α, IL-1β | [89] |
| THP1, Primary human MDMs | IFN-β IFN-α2 IFN-α4 | ISG56, ISG15, APOBEC3G | CXCL10, IL-6, IL-12α | [64] |
| Primary human MDMs | IFN-α IFN-β | No reported | No reported | [106] |
| (HIV-1)-infected macrophages from patients with HAND | IFN-α2 IFN-α1 IFN-β | RIG-1, MDA5 | No reported | [107] |
| Primary human MDMs | IFN-β1 | ISG15 | CD86, HLA-DR | [93] |
| Primary human MDMs | IFN-β IFN-α2 | CD169/Siglec1, CXCL10 | CD160, CXCL10, MCP-1, IL-15, VEGF | [22] |
| Primary human MDMs | IFN-λs | Mx2, Tetherin | No reported | [108] |
| Primary human MDMs | IFN-β | >17 ISGs upregulated, i.e., Mx1, Viperin, ISG15, CXCL10, TNFSF10 (or TRAIL) | CXCL10 | [94] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rojas, M.; Luz-Crawford, P.; Soto-Rifo, R.; Reyes-Cerpa, S.; Toro-Ascuy, D. The Landscape of IFN/ISG Signaling in HIV-1-Infected Macrophages and Its Possible Role in the HIV-1 Latency. Cells 2021, 10, 2378. https://doi.org/10.3390/cells10092378
Rojas M, Luz-Crawford P, Soto-Rifo R, Reyes-Cerpa S, Toro-Ascuy D. The Landscape of IFN/ISG Signaling in HIV-1-Infected Macrophages and Its Possible Role in the HIV-1 Latency. Cells. 2021; 10(9):2378. https://doi.org/10.3390/cells10092378
Chicago/Turabian StyleRojas, Masyelly, Patricia Luz-Crawford, Ricardo Soto-Rifo, Sebastián Reyes-Cerpa, and Daniela Toro-Ascuy. 2021. "The Landscape of IFN/ISG Signaling in HIV-1-Infected Macrophages and Its Possible Role in the HIV-1 Latency" Cells 10, no. 9: 2378. https://doi.org/10.3390/cells10092378
APA StyleRojas, M., Luz-Crawford, P., Soto-Rifo, R., Reyes-Cerpa, S., & Toro-Ascuy, D. (2021). The Landscape of IFN/ISG Signaling in HIV-1-Infected Macrophages and Its Possible Role in the HIV-1 Latency. Cells, 10(9), 2378. https://doi.org/10.3390/cells10092378