
Transcriptome and Methylome Analysis Reveal Complex Cross-Talks between Thyroid Hormone and Glucocorticoid Signaling at Xenopus Metamorphosis

 , , , and
, , , and 
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Methods
2.1. Animal Care
2.2. Whole Tadpole Hormonal Treatments
2.3. Organotypic Tail Culture
2.4. RNA Isolation and Measure of Gene Expression
2.5. RNA-Seq Data Processing
2.6. Clustering
- -
- This is a gene-level transformation, with few constraints on the actual number of genes within each cluster. This differs from the widely used k-mean clustering, which tends to produce clusters with similar number of genes. It is clear that there is no reason a priori to impose that each type of biological response should be constrained in term of the number of genes.
- -
- It has better control of false negatives because it does not call multiple times for statistics with limited power, as calling multiple times for differential analysis performed with few biological replicates (n = 3, as the current standard suggests) would. Thus, statistics may deviate slightly from the differential analysis.
- -
- The biological response of genes can be compared without the confounding effect of their expression level, which range over five orders of magnitude.
2.7. Signaling and Metabolic Network
2.8. PPI Network
2.9. Prediction of Nuclear Receptors Binding Sites
2.10. MethylCap-Seq and Identification of Differentially Methylated Regions (DMRs)
3. Results
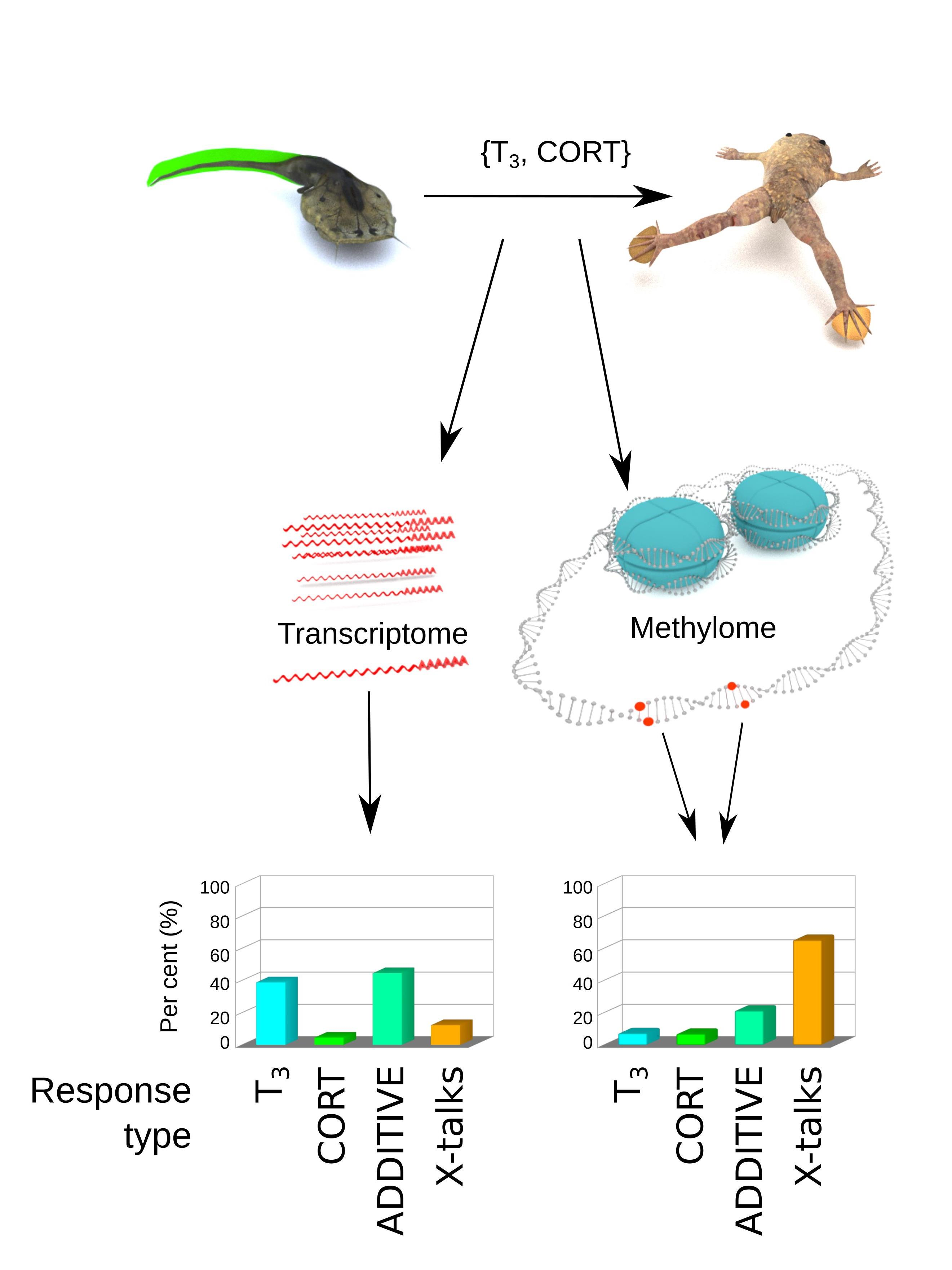
3.1. Experimental System to Address Tailfin Regression
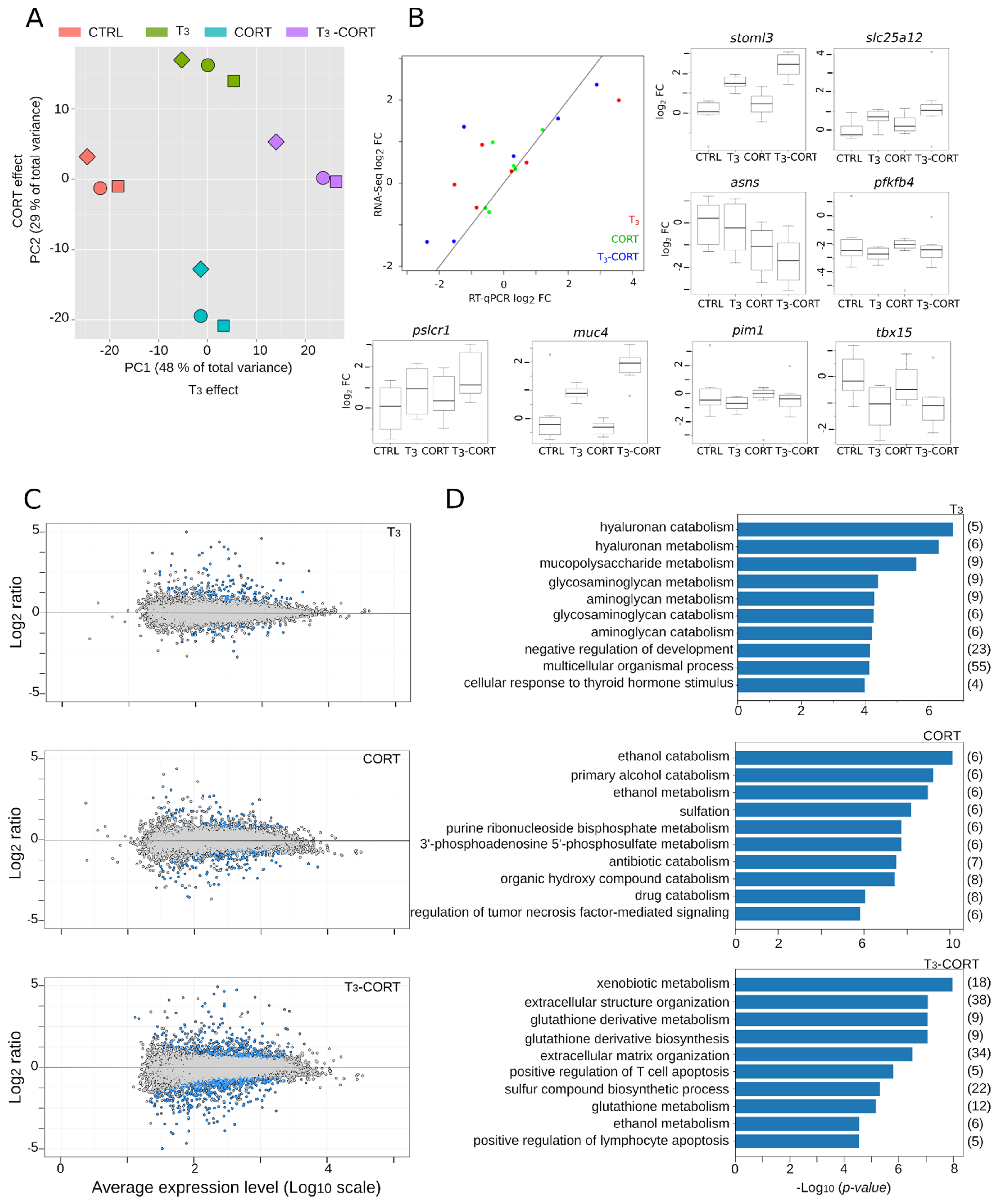
3.2. Standard Analysis Suggests Additive T3 and CORT Effects
3.3. Cross-Talks Do Exist, and They Only Represent a Fraction of Transcriptional Responses
- -
- Most of CORT-regulated genes are also regulated by T3 in tailfin.
- -
- We provide a new list of CORT-only responsive genes, which are notoriously difficult to isolate [70].
- -
- Qualitatively, the effects of T3 and CORT co-treatment are not limited to the cumulative response from each hormone individually. There is a large palette of biological responses involving complex interactions between pathways (X-talks).
- -
- At the transcriptome level, transcriptional responses are dominated by the T3 effect and ADDITIVE, and although X-talks do exist, they correspond to ~10% of differentially regulated genes.
3.4. Understanding Molecular Phenotypes: From Lists of DE Genes to System Biology
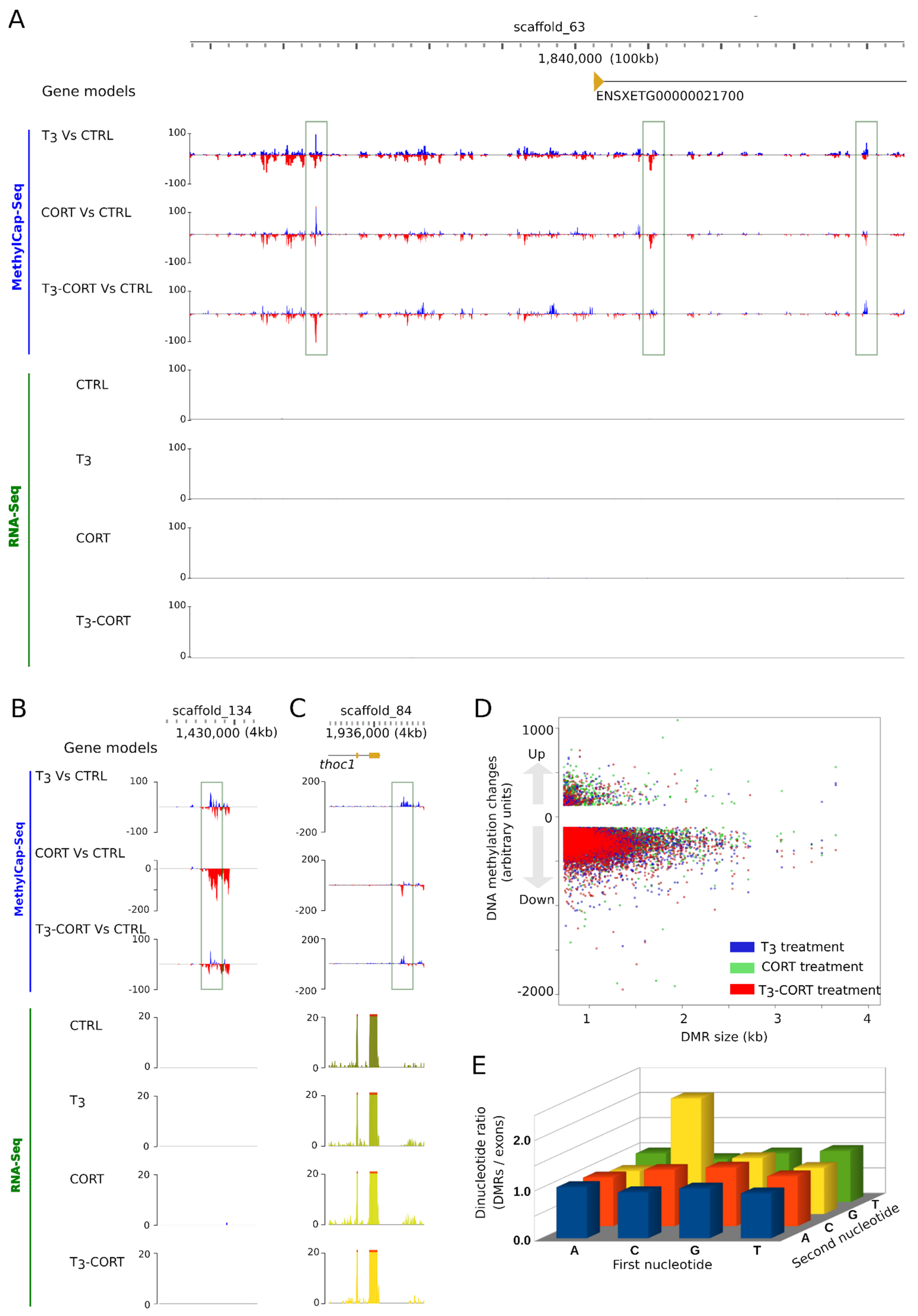
3.5. T3 and CORT Induced Massive and Complex Changes of DNA Methylation
3.6. The Cross-Talks-Like Complex Regulations Drive the Majority of DNA Methylation Changes
3.7. DMRs Are Located Far from Genes
4. Discussion
4.1. TH and GC X-Talks: A Large Repertoire of Transcriptional Regulations
4.2. X-Talks: An Unexpected Large Impact on DNA Methylation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Crespi, E.J.; Denver, R.J. Ancient Origins of Human Developmental Plasticity. Am. J. Hum. Biol. 2005, 17, 44–54. [Google Scholar] [CrossRef]
- Zoeller, R.T.; Tan, S.W.; Tyl, R.W. General Background on the Hypothalamic-Pituitary-Thyroid (HPT) Axis. Crit. Rev. Toxicol. 2007, 37, 11–53. [Google Scholar] [CrossRef]
- Wada, H. Glucocorticoids: Mediators of Vertebrate Ontogenetic Transitions. Gen. Comp. Endocrinol. 2008, 156, 441–453. [Google Scholar] [CrossRef] [PubMed]
- Buchholz, D.R. More Similar than You Think: Frog Metamorphosis as a Model of Human Perinatal Endocrinology. Dev. Biol. 2015, 408, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Bianco, A.C.; Dumitrescu, A.; Gereben, B.; Ribeiro, M.O.; Fonseca, T.L.; Fernandes, G.W.; Bocco, B.M.L.C. Paradigms of Dynamic Control of Thyroid Hormone Signaling. Endocr. Rev. 2019, 40, 1000–1047. [Google Scholar] [CrossRef]
- Bassett, J.H.D.; Williams, G.R. Role of Thyroid Hormones in Skeletal Development and Bone Maintenance. Endocr. Rev. 2016, 37, 135–187. [Google Scholar] [CrossRef]
- Rovet, J.F. The Role of Thyroid Hormones for Brain Development and Cognitive Function. Paediatr. Thyroidol. 2014, 26, 26–43. [Google Scholar]
- Liu, Y.-Y.; Brent, G.A. Thyroid Hormone and the Brain: Mechanisms of Action in Development and Role in Protection and Promotion of Recovery after Brain Injury. Pharmacol. Ther. 2018, 186, 176–185. [Google Scholar] [CrossRef]
- Ritter, M.J.; Amano, I.; Hollenberg, A.N. Thyroid Hormone Signaling and the Liver. Hepatology 2020, 72, 742–752. [Google Scholar] [CrossRef] [PubMed]
- Bloise, F.F.; Cordeiro, A.; Ortiga-Carvalho, T.M. Role of Thyroid Hormone in Skeletal Muscle Physiology. J. Endocrinol. 2018, 236, R57–R68. [Google Scholar] [CrossRef]
- Juszczak, G.R.; Stankiewicz, A.M. Glucocorticoids, Genes and Brain Function. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2018, 82, 136–168. [Google Scholar] [CrossRef]
- Liu, B.; Zhang, T.-N.; Knight, J.K.; Goodwin, J.E. The Glucocorticoid Receptor in Cardiovascular Health and Disease. Cells 2019, 8, 1227. [Google Scholar] [CrossRef] [PubMed]
- Whirledge, S.; DeFranco, D.B. Glucocorticoid Signaling in Health and Disease: Insights from Tissue-Specific GR Knockout Mice. Endocrinology 2018, 159, 46–64. [Google Scholar] [CrossRef]
- Robson, H.; Siebler, T.; Shalet, S.M.; Williams, G.R. Interactions between GH, IGF-I, Glucocorticoids, and Thyroid Hormones during Skeletal Growth. Pediatr. Res. 2002, 52, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Miranda, A.; Sousa, N. Maternal Hormonal Milieu Influence on Fetal Brain Development. Brain Behav. 2018, 8, e00920. [Google Scholar] [CrossRef] [PubMed]
- Bagamasbad, P.D.; Espina, J.E.C.; Knoedler, J.R.; Subramani, A.; Harden, A.J.; Denver, R.J. Coordinated Transcriptional Regulation by Thyroid Hormone and Glucocorticoid Interaction in Adult Mouse Hippocampus-Derived Neuronal Cells. PLoS ONE 2019, 14, e0220378. [Google Scholar] [CrossRef]
- Uyama, N.; Geerts, A.; Reynaert, H. Neural Connections between the Hypothalamus and the Liver. Anat. Rec. 2004, 280A, 808–820. [Google Scholar] [CrossRef]
- Timmermans, S.; Souffriau, J.; Libert, C. A General Introduction to Glucocorticoid Biology. Front. Immunol. 2019, 10, 1545. [Google Scholar] [CrossRef]
- Muthusami, S.; Vidya, B.; Shankar, E.M.; Vadivelu, J.; Ramachandran, I.; Stanley, J.A.; Selvamurugan, N. The Functional Significance of Endocrine-Immune Interactions in Health and Disease. Curr. Protein Pept. Sci. 2020, 21, 52–65. [Google Scholar] [CrossRef]
- Lazar, M.A. Maturing of the Nuclear Receptor Family. J. Clin. Investig. 2017, 127, 1123–1125. [Google Scholar] [CrossRef]
- Fullwood, M.J.; Liu, M.H.; Pan, Y.F.; Liu, J.; Xu, H.; Mohamed, Y.B.; Orlov, Y.L.; Velkov, S.; Ho, A.; Mei, P.H.; et al. An Oestrogen-Receptor-Alpha-Bound Human Chromatin Interactome. Nature 2009, 462, 58–64. [Google Scholar] [CrossRef]
- Bernal, J. Thyroid Hormone Regulated Genes in Cerebral Cortex Development. J. Endocrinol. 2017, 232, R83–R97. [Google Scholar] [CrossRef]
- Lamont, K.R.; Tindall, D.J. Androgen Regulation of Gene Expression. Adv. Cancer Res. 2010, 107, 137–162. [Google Scholar] [PubMed]
- Hu, F.; Knoedler, J.R.; Denver, R.J. A Mechanism to Enhance Cellular Responsivity to Hormone Action: Krüppel-Like Factor 9 Promotes Thyroid Hormone Receptor-β Autoinduction During Postembryonic Brain Development. Endocrinology 2016, 157, 1683–1693. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Buaas, F.W.; Kirsh, A.L.; Sharma, M.; McLean, D.J.; Morris, J.L.; Griswold, M.D.; de Rooij, D.G.; Braun, R.E. Plzf Is Required in Adult Male Germ Cells for Stem Cell Self-Renewal. Nat. Genet. 2004, 36, 647–652. [Google Scholar] [CrossRef] [PubMed]
- De Bosscher, K.; Desmet, S.J.; Clarisse, D.; Estébanez-Perpiña, E.; Brunsveld, L. Nuclear Receptor Crosstalk—Defining the Mechanisms for Therapeutic Innovation. Nat. Rev. Endocrinol. 2020, 16, 363–377. [Google Scholar] [CrossRef]
- Nilsson, O.; Marino, R.; De Luca, F.; Phillip, M.; Baron, J. Endocrine Regulation of the Growth Plate. Horm. Res. Paediatr. 2005, 64, 157–165. [Google Scholar] [CrossRef]
- Ycaza Herrera, A.; Mather, M. Actions and Interactions of Estradiol and Glucocorticoids in Cognition and the Brain: Implications for Aging Women. Neurosci. Biobehav. Rev. 2015, 55, 36–52. [Google Scholar] [CrossRef]
- Rabelo, E.M.; Baker, B.S.; Tata, J.R. Interplay between Thyroid Hormone and Estrogen in Modulating Expression of Their Receptor and Vitellogenin Genes during Xenopus Metamorphosis. Mech. Dev. 1994, 45, 49–57. [Google Scholar] [CrossRef]
- Davis, P.J.; Lin, H.-Y.; Mousa, S.A.; Luidens, M.K.; Hercbergs, A.A.; Wehling, M.; Davis, F.B. Overlapping Nongenomic and Genomic Actions of Thyroid Hormone and Steroids. Steroids 2011, 76, 829–833. [Google Scholar] [CrossRef][Green Version]
- Duarte-Guterman, P.; Navarro-Martín, L.; Trudeau, V.L. Mechanisms of Crosstalk between Endocrine Systems: Regulation of Sex Steroid Hormone Synthesis and Action by Thyroid Hormones. Gen. Comp. Endocrinol. 2014, 203, 69–85. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, D.; Padmanabhan, V.; Sargis, R.M. Stress, Sex, and Sugar: Glucocorticoids and Sex-Steroid Crosstalk in the Sex-Specific Misprogramming of Metabolism. J. Endocr. Soc. 2020, 4, bvaa087. [Google Scholar] [CrossRef]
- Samarasinghe, R.A.; Witchell, S.F.; DeFranco, D.B. Cooperativity and Complementarity: Synergies in Non-Classical and Classical Glucocorticoid Signaling. Cell Cycle 2012, 11, 2819–2827. [Google Scholar] [CrossRef] [PubMed]
- Weikum, E.R.; Knuesel, M.T.; Ortlund, E.A.; Yamamoto, K.R. Glucocorticoid Receptor Control of Transcription: Precision and Plasticity via Allostery. Nat. Rev. Mol. Cell Biol. 2017, 18, 159–174. [Google Scholar] [CrossRef] [PubMed]
- Davis, P.J.; Goglia, F.; Leonard, J.L. Nongenomic Actions of Thyroid Hormone. Nat. Rev. Endocrinol. 2016, 12, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Flamant, F.; Cheng, S.-Y.; Hollenberg, A.N.; Moeller, L.C.; Samarut, J.; Wondisford, F.E.; Yen, P.M.; Refetoff, S. Thyroid Hormone Signaling Pathways: Time for a More Precise Nomenclature. Endocrinology 2017, 158, 2052–2057. [Google Scholar] [CrossRef]
- Stahn, C.; Buttgereit, F. Genomic and Nongenomic Effects of Glucocorticoids. Nat. Clin. Pract. Rheumatol. 2008, 4, 525–533. [Google Scholar] [CrossRef]
- Martin, N.P.; Marron Fernandez de Velasco, E.; Mizuno, F.; Scappini, E.L.; Gloss, B.; Erxleben, C.; Williams, J.G.; Stapleton, H.M.; Gentile, S.; Armstrong, D.L. A Rapid Cytoplasmic Mechanism for PI3 Kinase Regulation by the Nuclear Thyroid Hormone Receptor, TRβ, and Genetic Evidence for Its Role in the Maturation of Mouse Hippocampal Synapses In Vivo. Endocrinology 2014, 155, 3713–3724. [Google Scholar] [CrossRef]
- Arancibia, S.; Benítez, D.; Núñez, L.E.; Jewell, C.M.; Langjahr, P.; Candia, E.; Zapata-Torres, G.; Cidlowski, J.A.; González, M.-J.; Hermoso, M.A. Phosphatidylinositol 3-Kinase Interacts with the Glucocorticoid Receptor upon TLR2 Activation. J. Cell. Mol. Med. 2011, 15, 339–349. [Google Scholar] [CrossRef]
- Sachs, L.M.; Buchholz, D.R. Insufficiency of Thyroid Hormone in Frog Metamorphosis and the Role of Glucocorticoids. Front. Endocrinol. 2019, 10, 287. [Google Scholar] [CrossRef]
- Shi, Y.-B. Amphibian Metamorphosis: From Morphology to Molecular Biology; Wiley-Liss: New York, NY, USA, 2000; ISBN 0471244759. [Google Scholar]
- Bonett, R.M.; Hoopfer, E.D.; Denver, R.J. Molecular Mechanisms of Corticosteroid Synergy with Thyroid Hormone during Tadpole Metamorphosis. Gen. Comp. Endocrinol. 2010, 168, 209–219. [Google Scholar] [CrossRef]
- Kulkarni, S.S.; Denver, R.J.; Gomez-Mestre, I.; Buchholz, D.R. Genetic Accommodation via Modified Endocrine Signalling Explains Phenotypic Divergence among Spadefoot Toad Species. Nat. Commun. 2017, 8, 993. [Google Scholar] [CrossRef]
- Kulkarni, S.S.; Buchholz, D.R. Corticosteroid Signaling in Frog Metamorphosis. Gen. Comp. Endocrinol. 2014, 203, 225–231. [Google Scholar] [CrossRef]
- Denver, R.J. Stress Hormones Mediate Environment-Genotype Interactions during Amphibian Development. Gen. Comp. Endocrinol. 2009, 164, 20–31. [Google Scholar] [CrossRef]
- Sterner, Z.R.; Shewade, L.H.; Mertz, K.M.; Sturgeon, S.M.; Buchholz, D.R. Glucocorticoid Receptor Is Required for Survival through Metamorphosis in the Frog Xenopus Tropicalis. Gen. Comp. Endocrinol. 2020, 291, 113419. [Google Scholar] [CrossRef]
- Tribondeau, A.; Sachs, L.M.; Buisine, N. Are Paedomorphs Actual Larvae? Dev. Dyn. 2021, 250, 779–787. [Google Scholar] [CrossRef]
- Ulisse, S.; Iwamuro, S.; Tata, J.R. Differential Responses to Ligands of Overexpressed Thyroid Hormone and Retinoid X Receptors in a Xenopus Cell Line and in Vivo. Mol. Cell. Endocrinol. 1997, 126, 17–24. [Google Scholar] [CrossRef]
- Krain, L.P.; Denver, R.J. Developmental Expression and Hormonal Regulation of Glucocorticoid and Thyroid Hormone Receptors during Metamorphosis in Xenopus Laevis. J. Endocrinol. 2004, 181, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Galton, V.A. Mechanisms Underlying the Acceleration of Thyroid Hormone-Induced Tadpole Metamorphosis by Corticosterone*. Endocrinology 1990, 127, 2997–3002. [Google Scholar] [CrossRef] [PubMed]
- Kühn, E.R.; Geris, K.L.; van der Geyten, S.; Mol, K.A.; Darras, V.M. Inhibition and Activation of the Thyroidal Axis by the Adrenal Axis in Vertebrates. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 1998, 120, 169–174. [Google Scholar] [CrossRef]
- Kulkarni, S.S.; Buchholz, D.R. Beyond Synergy: Corticosterone and Thyroid Hormone Have Numerous Interaction Effects on Gene Regulation in Xenopus Tropicalis Tadpoles. Endocrinology 2012, 153, 5309–5324. [Google Scholar] [CrossRef] [PubMed]
- Nieuwkoop, P.D.; Faber, J. Normal Table of Xenopus Laevis (Daudin): A Systematic and Chronological Survey of the Development from the Fertilized Egg Till the End of Metamorphosis, 1st ed.; North-Holland Pub. Co: Amsterdam, The Netherlands, 1956; ISBN 0815318960. [Google Scholar]
- Kerdivel, G.; Blugeon, C.; Fund, C.; Rigolet, M.; Sachs, L.M.; Buisine, N. Opposite T3 Response of ACTG1–FOS Subnetwork Differentiate Tailfin Fate in Xenopus Tadpole and Post-Hatching Axolotl. Front. Endocrinol. 2019, 10, 194. [Google Scholar] [CrossRef] [PubMed]
- Andersen, C.L.; Jensen, J.L.; Ørntoft, T.F. Normalization of Real-Time Quantitative Reverse Transcription-PCR Data: A Model-Based Variance Estimation Approach to Identify Genes Suited for Normalization, Applied to Bladder and Colon Cancer Data Sets. Cancer Res. 2004, 64, 5245–5250. [Google Scholar] [CrossRef] [PubMed]
- Hellsten, U.; Harland, R.M.; Gilchrist, M.J.; Hendrix, D.; Jurka, J.; Kapitonov, V.; Ovcharenko, I.; Putnam, N.H.; Shu, S.; Taher, L.; et al. The Genome of the Western Clawed Frog Xenopus Tropicalis. Science 2010, 328, 633–636. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and Memory-Efficient Alignment of Short DNA Sequences to the Human Genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef]
- Buisine, N.; Ruan, X.; Bilesimo, P.; Grimaldi, A.; Alfama, G.; Ariyaratne, P.; Mulawadi, F.; Chen, J.; Sung, W.-K.; Liu, E.T.; et al. Xenopus Tropicalis Genome Re-Scaffolding and Re-Annotation Reach the Resolution Required for In Vivo ChIA-PET Analysis. PLoS ONE 2015, 10, e0137526. [Google Scholar] [CrossRef]
- Anders, S.; McCarthy, D.J.; Chen, Y.; Okoniewski, M.; Smyth, G.K.; Huber, W.; Robinson, M.D. Count-Based Differential Expression Analysis of RNA Sequencing Data Using R and Bioconductor. Nat. Protoc. 2013, 8, 1765–1786. [Google Scholar] [CrossRef]
- Eden, E.; Navon, R.; Steinfeld, I.; Lipson, D.; Yakhini, Z. GOrilla: A Tool for Discovery and Visualization of Enriched GO Terms in Ranked Gene Lists. BMC Bioinform. 2009, 10, 48. [Google Scholar] [CrossRef]
- Winterhalter, C.; Widera, P.; Krasnogor, N. JEPETTO: A Cytoscape Plugin for Gene Set Enrichment and Topological Analysis Based on Interaction Networks. Bioinformatics 2014, 30, 1029–1030. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Oughtred, R.; Rust, J.; Chang, C.; Breitkreutz, B.; Stark, C.; Willems, A.; Boucher, L.; Leung, G.; Kolas, N.; Zhang, F.; et al. The BioGRID Database: A Comprehensive Biomedical Resource of Curated Protein, Genetic, and Chemical Interactions. Protein Sci. 2021, 30, 187–200. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a Reference Resource for Gene and Protein Annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef]
- Margolis, R.N.; Evans, R.M.; O’Malley, B.W.; NURSA Atlas Consortium. The Nuclear Receptor Signaling Atlas: Development of a Functional Atlas of Nuclear Receptors. Mol. Endocrinol. 2005, 19, 2433–2436. [Google Scholar] [CrossRef] [PubMed]
- Becnel, L.B.; Darlington, Y.F.; Ochsner, S.A.; Easton-Marks, J.R.; Watkins, C.M.; McOwiti, A.; Kankanamge, W.H.; Wise, M.W.; DeHart, M.; Margolis, R.N.; et al. Nuclear Receptor Signaling Atlas: Opening Access to the Biology of Nuclear Receptor Signaling Pathways. PLoS ONE 2015, 10, e0135615. [Google Scholar] [CrossRef] [PubMed]
- Sandelin, A.; Wasserman, W.W. Prediction of Nuclear Hormone Receptor Response Elements. Mol. Endocrinol. 2005, 19, 595–606. [Google Scholar] [CrossRef]
- Buels, R.; Yao, E.; Diesh, C.M.; Hayes, R.D.; Munoz-Torres, M.; Helt, G.; Goodstein, D.M.; Elsik, C.G.; Lewis, S.E.; Stein, L.; et al. JBrowse: A Dynamic Web Platform for Genome Visualization and Analysis. Genome Biol. 2016, 17, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Bilesimo, P.; Jolivet, P.; Alfama, G.; Buisine, N.; Le Mevel, S.; Havis, E.; Demeneix, B.A.; Sachs, L.M. Specific Histone Lysine 4 Methylation Patterns Define TR-Binding Capacity and Differentiate Direct T3 Responses. Mol. Endocrinol. 2011, 25, 225–237. [Google Scholar] [CrossRef]
- Schneider, K.A.; Shewade, L.H.; Buisine, N.; Sachs, L.M.; Buchholz, D.R. A Novel Stress Hormone Response Gene in Tadpoles of Xenopus Tropicalis. Gen. Comp. Endocrinol. 2018, 260, 107–114. [Google Scholar] [CrossRef]
- Ma’ayan, A. Insights into the Organization of Biochemical Regulatory Networks Using Graph Theory Analyses. J. Biol. Chem. 2009, 284, 5451–5455. [Google Scholar] [CrossRef]
- Tata, J.R.; Kawahara, A.; Baker, B.S. Prolactin Inhibits Both Thyroid Hormone-Induced Morphogenesis and Cell Death in Cultured Amphibian Larval Tissues. Dev. Biol. 1991, 146, 72–80. [Google Scholar] [CrossRef]
- Tata, J.R. Amphibian Metamorphosis as a Model for Studying the Developmental Actions of Thyroid Hormone. Biochimie 1999, 81, 359–366. [Google Scholar] [CrossRef]
- Zhu, X.; Gerstein, M.; Snyder, M. Getting Connected: Analysis and Principles of Biological Networks. Genes Dev. 2007, 21, 1010–1024. [Google Scholar] [CrossRef] [PubMed]
- Charitou, T.; Bryan, K.; Lynn, D.J. Using Biological Networks to Integrate, Visualize and Analyze Genomics Data. Genet. Sel. Evol. 2016, 48, 27. [Google Scholar] [CrossRef]
- Kyono, Y.; Raj, S.; Sifuentes, C.J.; Buisine, N.; Sachs, L.; Denver, R.J. DNA Methylation Dynamics Underlie Metamorphic Gene Regulation Programs in Xenopus Tadpole Brain. Dev. Biol. 2020, 462, 180–196. [Google Scholar] [CrossRef]
- Meijsing, S.H.; Pufall, M.A.; So, A.Y.; Bates, D.L.; Chen, L.; Yamamoto, K.R. DNA Binding Site Sequence Directs Glucocorticoid Receptor Structure and Activity. Science 2009, 324, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Wahlström, G.M.; Sjöberg, M.; Andersson, M.; Nordström, K.; Vennström, B. Binding Characteristics of the Thyroid Hormone Receptor Homo- and Heterodimers to Consensus AGGTCA Repeat Motifs. Mol. Endocrinol. 1992, 6, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- Shabtai, Y.; Nagaraj, N.K.; Batmanov, K.; Cho, Y.-W.; Guan, Y.; Jiang, C.; Remsberg, J.; Forrest, D.; Lazar, M.A. A Coregulator Shift, Rather than the Canonical Switch, Underlies Thyroid Hormone Action in the Liver. Genes Dev. 2021, 35, 367–378. [Google Scholar] [CrossRef]
- Ayers, S.; Switnicki, M.P.; Angajala, A.; Lammel, J.; Arumanayagam, A.S.; Webb, P. Genome-Wide Binding Patterns of Thyroid Hormone Receptor Beta. PLoS ONE 2014, 9, e81186. [Google Scholar] [CrossRef] [PubMed]
- Lizcano, F.; Rodríguez, J.S. Thyroid Hormone Therapy Modulates Hypothalamo-Pituitary-Adrenal Axis. Endocr. J. 2011, 58, 137–142. [Google Scholar] [CrossRef]
- Johnson, E.O.; Kamilaris, T.C.; Calogero, A.E.; Gold, P.W.; Chrousos, G.P. Experimentally-Induced Hyperthyroidism Is Associated with Activation of the Rat Hypothalamic–Pituitary–Adrenal Axis. Eur. J. Endocrinol. 2005, 153, 177–185. [Google Scholar] [CrossRef][Green Version]
- Darras, V.M.; Van der Geyten, S.; Cox, C.; Segers, I.B.; De Groef, B.; Kühn, E.R. Effects of Dexamethasone Treatment on Iodothyronine Deiodinase Activities and on Metamorphosis-Related Morphological Changes in the Axolotl (Ambystoma Mexicanum). Gen. Comp. Endocrinol. 2002, 127, 157–164. [Google Scholar] [CrossRef]
- Erwin, D.H.; Davidson, E.H. The Evolution of Hierarchical Gene Regulatory Networks. Nat. Rev. Genet. 2009, 10, 141–148. [Google Scholar] [CrossRef]
- Grimaldi, A.; Buisine, N.; Miller, T.; Shi, Y.-B.; Sachs, L.M. Mechanisms of Thyroid Hormone Receptor Action during Development: Lessons from Amphibian Studies. Biochim. Biophys. Acta 2012, 1830, 3882–3892. [Google Scholar] [CrossRef] [PubMed]
- Bonett, R.M.; Hu, F.; Bagamasbad, P.; Denver, R.J. Stressor and Glucocorticoid-Dependent Induction of the Immediate Early Gene Kruppel-like Factor 9: Implications for Neural Development and Plasticity. Endocrinology 2009, 150, 1757–1765. [Google Scholar] [CrossRef] [PubMed]
- Bagamasbad, P.; Ziera, T.; Borden, S.A.; Bonett, R.M.; Rozeboom, A.M.; Seasholtz, A.; Denver, R.J. Molecular Basis for Glucocorticoid Induction of the Kruppel-like Factor 9 Gene in Hippocampal Neurons. Endocrinology 2012, 153, 5334–5345. [Google Scholar] [CrossRef] [PubMed]
- Raj, S.; Kyono, Y.; Sifuentes, C.J.; Arellanes-Licea, E.d.C.; Subramani, A.; Denver, R.J. Thyroid Hormone Induces DNA Demethylation in Xenopus Tadpole Brain. Endocrinology 2020, 161, bqaa155. [Google Scholar] [CrossRef]
- Denver, R.J.; Pavgi, S.; Shi, Y.B. Thyroid Hormone-Dependent Gene Expression Program for Xenopus Neural Development. J. Biol. Chem. 1997, 272, 8179–8188. [Google Scholar] [CrossRef]
- Leloup, J.; Buscaglia, M. Triiodothyronine, Hormone of Amphibian Metamorphosis. Comptes Rendus Hebd. Des Seances L Acad. Des Sci. Ser. D 1977, 284, 2261–2263. [Google Scholar]
- Kyono, Y.; Sachs, L.M.; Bilesimo, P.; Wen, L.; Denver, R.J. Developmental and Thyroid Hormone Regulation of the DNA Methyltransferase 3a Gene in Xenopus Tadpoles. Endocrinology 2016, 157, 4961–4972. [Google Scholar] [CrossRef]
- Kyono, Y.; Subramani, A.; Ramadoss, P.; Hollenberg, A.N.; Bonett, R.M.; Denver, R.J. Liganded Thyroid Hormone Receptors Transactivate the DNA Methyltransferase 3a Gene in Mouse Neuronal Cells. Endocrinology 2016, 157, 3647–3657. [Google Scholar] [CrossRef]
- Guan, W.; Guyot, R.; Samarut, J.; Flamant, F.; Wong, J.; Gauthier, K.C. Methylcytosine Dioxygenase TET3 Interacts with Thyroid Hormone Nuclear Receptors and Stabilizes Their Association to Chromatin. Proc. Natl. Acad. Sci. USA 2017, 114, 8229–8234. [Google Scholar] [CrossRef]
- Barrera-Hernandez, G.; Zhan, Q.; Wong, R.; Cheng, S.-Y. Thyroid Hormone Receptor Is a Negative Regulator in P53-Mediated Signaling Pathways. DNA Cell Biol. 1998, 17, 743–750. [Google Scholar] [CrossRef]
- Bungay, A.; Selden, C.; Brown, D.; Malik, R.; Hubank, M.; Hodgson, H. Microarray Analysis of Mitogenic Effects of T3 on the Rat Liver. J. Gastroenterol. Hepatol. 2008, 23, 1926–1933. [Google Scholar] [CrossRef]
- Fu, L.; Li, C.; Na, W.; Shi, Y.-B. Thyroid Hormone Activates Xenopus MBD3 Gene via an Intronic TRE in Vivo. Front. Biosci. 2020, 25, 437–451. [Google Scholar]
- Mukhopadhyay, D.; Plateroti, M.; Anant, S.; Nassir, F.; Samarut, J.; Davidson, N.O. Thyroid Hormone Regulates Hepatic Triglyceride Mobilization and Apolipoprotein B Messenger Ribonucleic Acid Editing in a Murine Model of Congenital Hypothyroidism. Endocrinology 2003, 144, 711–719. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Crudo, A.; Petropoulos, S.; Suderman, M.; Moisiadis, V.G.; Kostaki, A.; Hallett, M.; Szyf, M.; Matthews, S.G. Effects of Antenatal Synthetic Glucocorticoid on Glucocorticoid Receptor Binding, DNA Methylation, and Genome-Wide MRNA Levels in the Fetal Male Hippocampus. Endocrinology 2013, 154, 4170–4181. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-J.; Chen, Y.-L.; Lee, S.-C. Coactivator TIF1β Interacts with Transcription Factor C/EBPβ and Glucocorticoid Receptor to Induce A1-Acid Glycoprotein Gene Expression. Mol. Cell. Biol. 1998, 18, 5880–5887. [Google Scholar] [CrossRef]
- Hoffman, J.A.; Trotter, K.W.; Ward, J.M.; Archer, T.K. BRG1 Governs Glucocorticoid Receptor Interactions with Chromatin and Pioneer Factors across the Genome. eLife 2018, 7, e35073. [Google Scholar] [CrossRef] [PubMed]
- Vettorazzi, S.; Nalbantoglu, D.; Gebhardt, J.C.M.; Tuckermann, J. A Guide to Changing Paradigms of Glucocorticoid Receptor Function—A Model System for Genome Regulation and Physiology. FEBS J. 2021, febs.16100. [Google Scholar] [CrossRef]
- Quatrini, L.; Ugolini, S. New Insights into the Cell- and Tissue-Specificity of Glucocorticoid Actions. Cell. Mol. Immunol. 2021, 18, 269–278. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buisine, N.; Grimaldi, A.; Jonchere, V.; Rigolet, M.; Blugeon, C.; Hamroune, J.; Sachs, L.M. Transcriptome and Methylome Analysis Reveal Complex Cross-Talks between Thyroid Hormone and Glucocorticoid Signaling at Xenopus Metamorphosis. Cells 2021, 10, 2375. https://doi.org/10.3390/cells10092375
Buisine N, Grimaldi A, Jonchere V, Rigolet M, Blugeon C, Hamroune J, Sachs LM. Transcriptome and Methylome Analysis Reveal Complex Cross-Talks between Thyroid Hormone and Glucocorticoid Signaling at Xenopus Metamorphosis. Cells. 2021; 10(9):2375. https://doi.org/10.3390/cells10092375
Chicago/Turabian StyleBuisine, Nicolas, Alexis Grimaldi, Vincent Jonchere, Muriel Rigolet, Corinne Blugeon, Juliette Hamroune, and Laurent M. Sachs. 2021. "Transcriptome and Methylome Analysis Reveal Complex Cross-Talks between Thyroid Hormone and Glucocorticoid Signaling at Xenopus Metamorphosis" Cells 10, no. 9: 2375. https://doi.org/10.3390/cells10092375
APA StyleBuisine, N., Grimaldi, A., Jonchere, V., Rigolet, M., Blugeon, C., Hamroune, J., & Sachs, L. M. (2021). Transcriptome and Methylome Analysis Reveal Complex Cross-Talks between Thyroid Hormone and Glucocorticoid Signaling at Xenopus Metamorphosis. Cells, 10(9), 2375. https://doi.org/10.3390/cells10092375