
The Immune Privilege of Cancer Stem Cells: A Key to Understanding Tumor Immune Escape and Therapy Failure


Abstract
1. Introduction
2. The Immune Privileged Status of CSCs

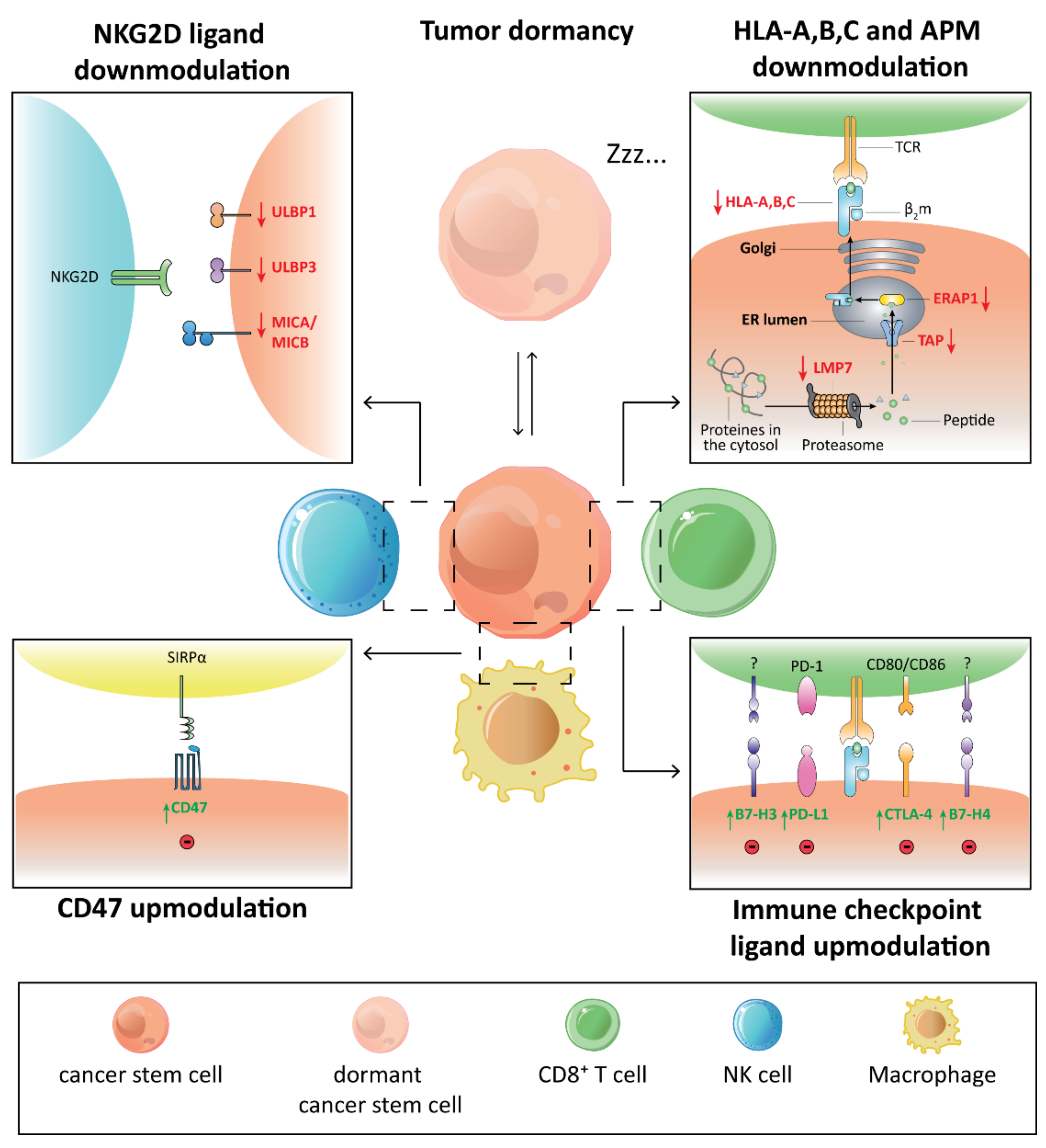{kind=link}
{kind=link}
{kind=link}
| Marker | Tumor type | Rfs |
|---|---|---|
| Surface markers | ||
| CD44 molecule | Lung, breast, gastric, liver, colorectal | [15,20] |
| Plasminogen sctivator, urokinase receptor (PLAUR, CD87) | Lung | [16] |
| Thy-1 cell surface antigen (THY1, CD90) | Lung, breast, gastric, liver | [17] |
| Prominin 1 (PROM1, CD133) | Lung, breast, gastric, liver, colorectal | [16] |
| Activated leukocyte cell adhesion molecule (ALCAM, CD166) | Lung, breast, gastric, liver, colorectal | [18] |
| Epithelial cell adhesion molecule (EpCAM) | Lung, colorectal | [19] |
| CD24 molecule (CD24) | Lung, breast, gastric, liver, colorectal | [20] |
| Integrin subunit beta 1 (ITGB1, CD29) | Breast | [21] |
| ITGA6 integrin subunit alpha 6 (ITGA6, CD49f) | Breast | [21] |
| Integrin subunit beta 3 (ITGB3, CD61) | Breast | [22] |
| CD70 molecule (CD70) | Breast | [23] |
| C-X-C motif chemokine receptor 4 (CXCR4) | Breast, gastric | [24] |
| Leucine rich repeat containing G protein-coupled receptor 5 (LGR5) | Breast, gastric, glioma, colorectal | [25] |
| Protein C receptor (PROCR) | Breast | [26] |
| Leucine rich repeat and Ig domain containing 2 (LINGO2) | Gastric | [27] |
| CD33 molecule (CD33) | AML, CML | [28,29] |
| Interleukin 3 receptor subunit alpha (IL3RA, CD123) | AML, CML | [28,29] |
| C-type lectin domain family 12 member A (CLEC12A, CCL1) | AML | [28] |
| HAVCR2 hepatitis A virus cellular receptor 2 (TIM3) | AML | [28] |
| Interleukin 2 receptor subunit alpha (IL2RA, CD25) | CML | [29] |
| Dipeptidyl peptidase 4 (DPP4, CD26) | CML | [29] |
| KIT proto-oncogene, receptor tyrosine kinase (KIT, CD117) | CML | [29] |
| CD36 molecule (CD36) | CML | [29] |
| Interleukin 1 receptor accessory protein (IL1RAP) | CML | [29] |
| Intracellular markers and pathways | ||
| Aldehyde dehydrogenase (ALDH) | Lung, breast, gastric, colorectal, AML | [20] |
| Nanog homeobox (NANOG) | Lung, breast, gastric, liver, colorectal, AML | [33] |
| POU class 5 homeobox 1 (POU5F1, Oct-3/4) | Lung, breast, gastric, liver, colorectal, AML | [34] |
| SRY-box transcription factor 2 (SOX2) | Breast, gastric, liver, colorectal, AML | [35] |
| BMI proto-oncogene, polycomb ring finger (BMI1) | Breast | [36] |
| Wnt/β-Catenin signaling | Breast, liver, CML | [38] |
| JAK/STAT signaling | CML | [37] |
| FOXO signaling | CML | [29] |
| Hedgehog/Smo/Gli2 signaling | CML | [38] |
| Notch signaling | Breast, liver | [38] |
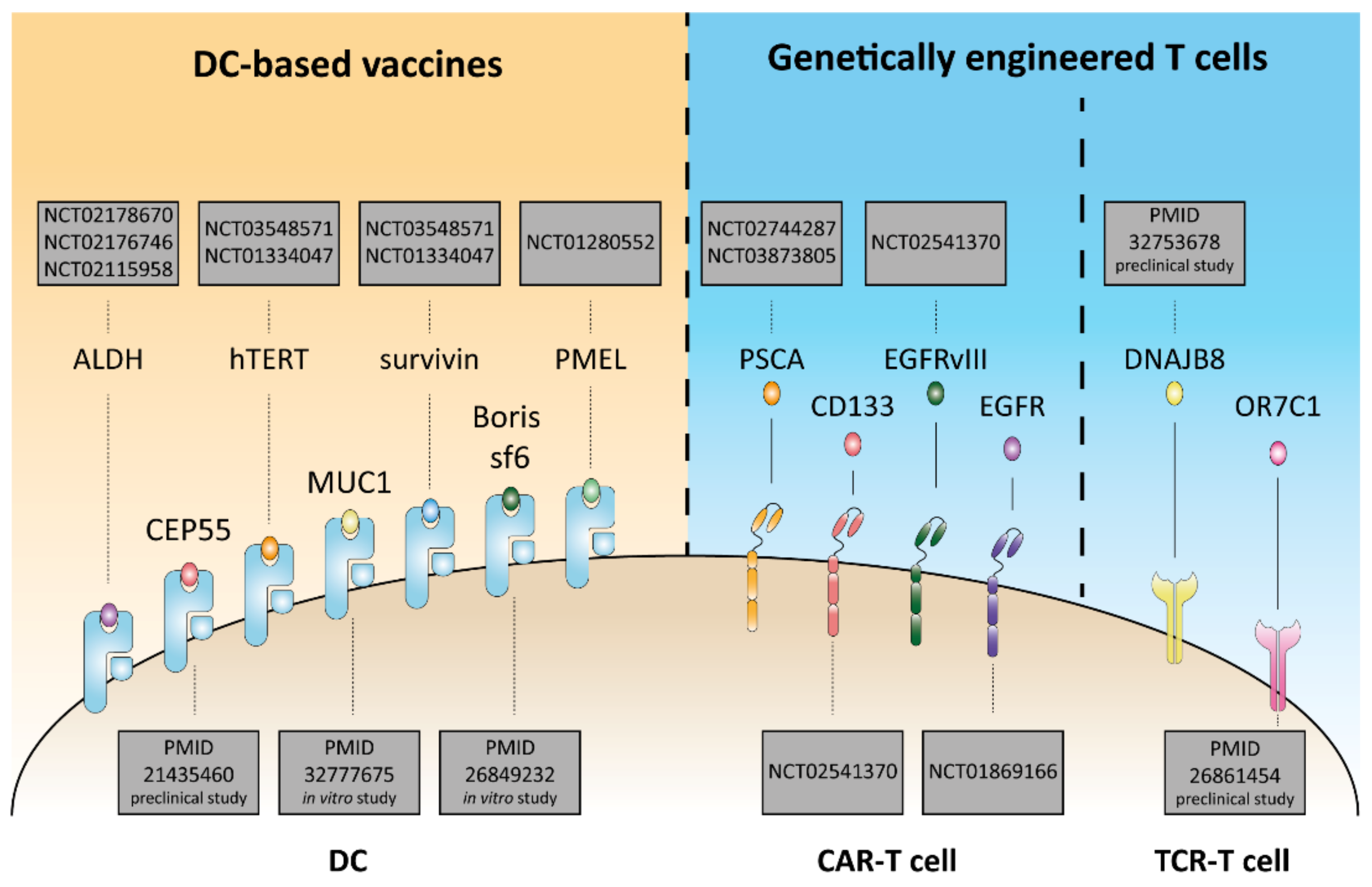
3. Immunomodulatory Traits of CSCs and Their Immune Context
4. Immunotherapeutic Strategies against CSCs
5. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef]
- Dean, M.; Fojo, T.; Bates, S. Tumour stem cells and drug resistance. Nat. Rev. Cancer 2005, 5, 275–284. [Google Scholar] [CrossRef]
- Valent, P.; Bonnet, D.; De Maria, R.; Lapidot, T.; Copland, M.; Melo, J.V.; Chomienne, C.; Ishikawa, F.; Schuringa, J.J.; Stassi, G.; et al. Cancer stem cell definitions and terminology: The devil is in the details. Nat. Rev. Cancer 2012, 12, 767–775. [Google Scholar] [CrossRef]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Xia, P. Surface markers of cancer stem cells in solid tumors. Curr. Stem Cell Res. Ther. 2014, 9, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Al-Othman, N.; Alhendi, A.; Ihbaisha, M.; Barahmeh, M.; Alqaraleh, M.; Al-Momany, B.Z. Role of CD44 in breast cancer. Breast Dis. 2020, 39, 1–13. [Google Scholar] [CrossRef]
- Clevers, H. The cancer stem cell: Premises, promises and challenges. Nat. Med. 2011, 17, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Boyle, S.E.; Fedele, C.G.; Corbin, V.; Wybacz, E.; Szeto, P.; Lewin, J.; Young, R.J.; Wong, A.; Fuller, R.; Spillane, J.; et al. CD271 Expression on Patient Melanoma Cells Is Unstable and Unlinked to Tumorigenicity. Cancer Res. 2016, 76, 3965–3977. [Google Scholar] [CrossRef] [PubMed]
- Jeter, C.R.; Yang, T.; Wang, J.; Chao, H.P.; Tang, D.G. Concise Review: NANOG in Cancer Stem Cells and Tumor Development: An Update and Outstanding Questions. Stem Cells 2015, 33, 2381–2390. [Google Scholar] [CrossRef]
- Takebe, N.; Miele, L.; Harris, P.J.; Jeong, W.; Bando, H.; Kahn, M.; Yang, S.X.; Ivy, S.P. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: Clinical update. Nat. Rev. Clin. Oncol. 2015, 12, 445–464. [Google Scholar] [CrossRef] [PubMed]
- Hill, R.; Wu, H. PTEN, stem cells, and cancer stem cells. J. Biol. Chem. 2009, 284, 11755–11759. [Google Scholar] [CrossRef] [PubMed]
- Maugeri-Sacca, M.; Zeuner, A.; De Maria, R. Therapeutic targeting of cancer stem cells. Front. Oncol. 2011, 1, 10. [Google Scholar] [CrossRef]
- Manic, G.; Sistigu, A.; Corradi, F.; Musella, M.; De Maria, R.; Vitale, I. Replication stress response in cancer stem cells as a target for chemotherapy. Semin. Cancer Biol. 2018, 53, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Bhatia, R. Stem cell quiescence. Clin. Cancer Res. 2011, 17, 4936–4941. [Google Scholar] [CrossRef] [PubMed]
- Signore, M.; Ricci-Vitiani, L.; De Maria, R. Targeting apoptosis pathways in cancer stem cells. Cancer Lett. 2013, 332, 374–382. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Ma, S.; Cao, K.; Zhou, S.; Zhao, A.; Li, M.; Qian, F.; Zhu, C. Therapeutic approaches targeting cancer stem cells. J. Cancer Res. Ther. 2018, 14, 1469–1475. [Google Scholar] [CrossRef]
- Puram, S.V.; Tirosh, I.; Parikh, A.S.; Patel, A.P.; Yizhak, K.; Gillespie, S.; Rodman, C.; Luo, C.L.; Mroz, E.A.; Emerick, K.S.; et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell 2017, 171, 1611–1624 e1624. [Google Scholar] [CrossRef]
- Schatton, T.; Frank, M.H. Antitumor immunity and cancer stem cells. Ann. N. Y. Acad. Sci. 2009, 1176, 154–169. [Google Scholar] [CrossRef]
- Dahan, P.; Martinez Gala, J.; Delmas, C.; Monferran, S.; Malric, L.; Zentkowski, D.; Lubrano, V.; Toulas, C.; Cohen-Jonathan Moyal, E.; Lemarie, A. Ionizing radiations sustain glioblastoma cell dedifferentiation to a stem-like phenotype through survivin: Possible involvement in radioresistance. Cell Death Dis. 2014, 5, e1543. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Chen, I.; Shimoda, L.A.; Park, Y.; Zhang, C.; Tran, L.; Zhang, H.; Semenza, G.L. Chemotherapy-Induced Ca(2+) Release Stimulates Breast Cancer Stem Cell Enrichment. Cell Rep. 2017, 18, 1946–1957. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, L.A.; Hemann, M.T. DNA damage-mediated induction of a chemoresistant niche. Cell 2010, 143, 355–366. [Google Scholar] [CrossRef]
- Bruttel, V.S.; Wischhusen, J. Cancer stem cell immunology: Key to understanding tumorigenesis and tumor immune escape? Front. Immunol. 2014, 5, 360. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, A.; Kumar, Y. Cancer stem cells and tumor immunoediting: Putting two and two together. Expert Rev. Clin. Immunol. 2016, 12, 605–607. [Google Scholar] [CrossRef][Green Version]
- Grau, J.J.; Mesia, R.; de la Iglesia-Vicente, M.; Williams, E.S.; Taberna, M.; Caballero, M.; Larque, A.B.; de la Oliva, J.; Cordon-Cardo, C.; Domingo-Domenech, J. Enrichment of Cells with Cancer Stem Cell-Like Markers in Relapses of Chemoresistant Patients with Locally Advanced Head and Neck Squamous Cell Carcinoma. Oncology 2016, 90, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Schatton, T.; Schutte, U.; Frank, N.Y.; Zhan, Q.; Hoerning, A.; Robles, S.C.; Zhou, J.; Hodi, F.S.; Spagnoli, G.C.; Murphy, G.F.; et al. Modulation of T-cell activation by malignant melanoma initiating cells. Cancer Res. 2010, 70, 697–708. [Google Scholar] [CrossRef]
- Di Tomaso, T.; Mazzoleni, S.; Wang, E.; Sovena, G.; Clavenna, D.; Franzin, A.; Mortini, P.; Ferrone, S.; Doglioni, C.; Marincola, F.M.; et al. Immunobiological characterization of cancer stem cells isolated from glioblastoma patients. Clin. Cancer Res. 2010, 16, 800–813. [Google Scholar] [CrossRef] [PubMed]
- Morrison, B.J.; Steel, J.C.; Morris, J.C. Reduction of MHC-I expression limits T-lymphocyte-mediated killing of Cancer-initiating cells. BMC Cancer 2018, 18, 469. [Google Scholar] [CrossRef] [PubMed]
- Chikamatsu, K.; Takahashi, G.; Sakakura, K.; Ferrone, S.; Masuyama, K. Immunoregulatory properties of CD44+ cancer stem-like cells in squamous cell carcinoma of the head and neck. Head Neck 2011, 33, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Inoda, S.; Hirohashi, Y.; Torigoe, T.; Morita, R.; Takahashi, A.; Asanuma, H.; Nakatsugawa, M.; Nishizawa, S.; Tamura, Y.; Tsuruma, T.; et al. Cytotoxic T lymphocytes efficiently recognize human colon cancer stem-like cells. Am. J. Pathol. 2011, 178, 1805–1813. [Google Scholar] [CrossRef]
- Voutsadakis, I.A. Expression and function of immune ligand-receptor pairs in NK cells and cancer stem cells: Therapeutic implications. Cell Oncol. 2018, 41, 107–121. [Google Scholar] [CrossRef]
- Tallerico, R.; Todaro, M.; Di Franco, S.; Maccalli, C.; Garofalo, C.; Sottile, R.; Palmieri, C.; Tirinato, L.; Pangigadde, P.N.; La Rocca, R.; et al. Human NK cells selective targeting of colon cancer-initiating cells: A role for natural cytotoxicity receptors and MHC class I molecules. J. Immunol. 2013, 190, 2381–2390. [Google Scholar] [CrossRef]
- Pietra, G.; Manzini, C.; Vitale, M.; Balsamo, M.; Ognio, E.; Boitano, M.; Queirolo, P.; Moretta, L.; Mingari, M.C. Natural killer cells kill human melanoma cells with characteristics of cancer stem cells. Int. Immunol. 2009, 21, 793–801. [Google Scholar] [CrossRef]
- Castriconi, R.; Daga, A.; Dondero, A.; Zona, G.; Poliani, P.L.; Melotti, A.; Griffero, F.; Marubbi, D.; Spaziante, R.; Bellora, F.; et al. NK cells recognize and kill human glioblastoma cells with stem cell-like properties. J. Immunol. 2009, 182, 3530–3539. [Google Scholar] [CrossRef] [PubMed]
- Tseng, H.C.; Arasteh, A.; Paranjpe, A.; Teruel, A.; Yang, W.; Behel, A.; Alva, J.A.; Walter, G.; Head, C.; Ishikawa, T.O.; et al. Increased lysis of stem cells but not their differentiated cells by natural killer cells; de-differentiation or reprogramming activates NK cells. PLoS ONE 2010, 5, e11590. [Google Scholar] [CrossRef] [PubMed]
- Paczulla, A.M.; Rothfelder, K.; Raffel, S.; Konantz, M.; Steinbacher, J.; Wang, H.; Tandler, C.; Mbarga, M.; Schaefer, T.; Falcone, M.; et al. Publisher Correction: Absence of NKG2D ligands defines leukaemia stem cells and mediates their immune evasion. Nature 2019, 572, E19. [Google Scholar] [CrossRef]
- Wang, B.; Wang, Q.; Wang, Z.; Jiang, J.; Yu, S.C.; Ping, Y.F.; Yang, J.; Xu, S.L.; Ye, X.Z.; Xu, C.; et al. Metastatic consequences of immune escape from NK cell cytotoxicity by human breast cancer stem cells. Cancer Res. 2014, 74, 5746–5757. [Google Scholar] [CrossRef]
- Logtenberg, M.E.W.; Scheeren, F.A.; Schumacher, T.N. The CD47-SIRPalpha Immune Checkpoint. Immunity 2020, 52, 742–752. [Google Scholar] [CrossRef]
- Brown, E.J.; Frazier, W.A. Integrin-associated protein (CD47) and its ligands. Trends Cell Biol. 2001, 11, 130–135. [Google Scholar] [CrossRef]
- Barclay, A.N.; Brown, M.H. The SIRP family of receptors and immune regulation. Nat. Rev. Immunol. 2006, 6, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Cioffi, M.; Trabulo, S.; Hidalgo, M.; Costello, E.; Greenhalf, W.; Erkan, M.; Kleeff, J.; Sainz, B., Jr.; Heeschen, C. Inhibition of CD47 Effectively Targets Pancreatic Cancer Stem Cells via Dual Mechanisms. Clin. Cancer Res. 2015, 21, 2325–2337. [Google Scholar] [CrossRef] [PubMed]
- Majeti, R.; Chao, M.P.; Alizadeh, A.A.; Pang, W.W.; Jaiswal, S.; Gibbs, K.D., Jr.; van Rooijen, N.; Weissman, I.L. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell 2009, 138, 286–299. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.K.; Cheung, V.C.; Lu, P.; Lau, E.Y.; Ma, S.; Tang, K.H.; Tong, M.; Lo, J.; Ng, I.O. Blockade of CD47-mediated cathepsin S/protease-activated receptor 2 signaling provides a therapeutic target for hepatocellular carcinoma. Hepatology 2014, 60, 179–191. [Google Scholar] [CrossRef]
- Alvarado, A.G.; Thiagarajan, P.S.; Mulkearns-Hubert, E.E.; Silver, D.J.; Hale, J.S.; Alban, T.J.; Turaga, S.M.; Jarrar, A.; Reizes, O.; Longworth, M.S.; et al. Glioblastoma Cancer Stem Cells Evade Innate Immune Suppression of Self-Renewal through Reduced TLR4 Expression. Cell Stem Cell 2017, 20, 450–461 e454. [Google Scholar] [CrossRef]
- Marotta, L.L.; Almendro, V.; Marusyk, A.; Shipitsin, M.; Schemme, J.; Walker, S.R.; Bloushtain-Qimron, N.; Kim, J.J.; Choudhury, S.A.; Maruyama, R.; et al. The JAK2/STAT3 signaling pathway is required for growth of CD44(+)CD24(-) stem cell-like breast cancer cells in human tumors. J. Clin. Invest. 2011, 121, 2723–2735. [Google Scholar] [CrossRef] [PubMed]
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef] [PubMed]
- Quesnel, B. Tumor dormancy and immunoescape. APMIS 2008, 116, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Maccalli, C.; Rasul, K.I.; Elawad, M.; Ferrone, S. The role of cancer stem cells in the modulation of anti-tumor immune responses. Semin. Cancer Biol. 2018, 53, 189–200. [Google Scholar] [CrossRef]
- Malladi, S.; Macalinao, D.G.; Jin, X.; He, L.; Basnet, H.; Zou, Y.; de Stanchina, E.; Massague, J. Metastatic Latency and Immune Evasion through Autocrine Inhibition of WNT. Cell 2016, 165, 45–60. [Google Scholar] [CrossRef]
- Saudemont, A.; Quesnel, B. In a model of tumor dormancy, long-term persistent leukemic cells have increased B7-H1 and B7.1 expression and resist CTL-mediated lysis. Blood 2004, 104, 2124–2133. [Google Scholar] [CrossRef]
- Vitale, I.; Sistigu, A.; Manic, G.; Rudqvist, N.P.; Trajanoski, Z.; Galluzzi, L. Mutational and Antigenic Landscape in Tumor Progression and Cancer Immunotherapy. Trends Cell Biol. 2019, 29, 396–416. [Google Scholar] [CrossRef]
- Saudemont, A.; Hamrouni, A.; Marchetti, P.; Liu, J.; Jouy, N.; Hetuin, D.; Colucci, F.; Quesnel, B. Dormant tumor cells develop cross-resistance to apoptosis induced by CTLs or imatinib mesylate via methylation of suppressor of cytokine signaling 1. Cancer Res. 2007, 67, 4491–4498. [Google Scholar] [CrossRef] [PubMed]
- Plaks, V.; Kong, N.; Werb, Z. The cancer stem cell niche: How essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 2015, 16, 225–238. [Google Scholar] [CrossRef]
- Sistigu, A.; Musella, M.; Galassi, C.; Vitale, I.; De Maria, R. Tuning Cancer Fate: Tumor Microenvironment’s Role in Cancer Stem Cell Quiescence and Reawakening. Front. Immunol. 2020, 11, 2166. [Google Scholar] [CrossRef] [PubMed]
- Hinshaw, D.C.; Shevde, L.A. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res. 2019, 79, 4557–4566. [Google Scholar] [CrossRef]
- Sung, T.C.; Huang, W.L.; Ban, L.K.; Lee, H.H.; Wang, J.H.; Su, H.Y.; Jen, S.H.; Chang, Y.H.; Yang, J.M.; Higuchi, A.; et al. Enrichment of cancer-initiating cells from colon cancer cells through porous polymeric membranes by a membrane filtration method. J. Mater. Chem. B 2020, 8, 10577–10585. [Google Scholar] [CrossRef]
- Vonderheide, R.H.; Anderson, K.S.; Hahn, W.C.; Butler, M.O.; Schultze, J.L.; Nadler, L.M. Characterization of HLA-A3-restricted cytotoxic T lymphocytes reactive against the widely expressed tumor antigen telomerase. Clin. Cancer Res. 2001, 7, 3343–3348. [Google Scholar]
- Warrier, N.M.; Agarwal, P.; Kumar, P. Emerging Importance of Survivin in Stem Cells and Cancer: The Development of New Cancer Therapeutics. Stem Cell Rev. Rep. 2020, 16, 828–852. [Google Scholar] [CrossRef]
- Morita, R.; Nishizawa, S.; Torigoe, T.; Takahashi, A.; Tamura, Y.; Tsukahara, T.; Kanaseki, T.; Sokolovskaya, A.; Kochin, V.; Kondo, T.; et al. Heat shock protein DNAJB8 is a novel target for immunotherapy of colon cancer-initiating cells. Cancer Sci. 2014, 105, 389–395. [Google Scholar] [CrossRef]
- Morita, R.; Hirohashi, Y.; Torigoe, T.; Ito-Inoda, S.; Takahashi, A.; Mariya, T.; Asanuma, H.; Tamura, Y.; Tsukahara, T.; Kanaseki, T.; et al. Olfactory Receptor Family 7 Subfamily C Member 1 Is a Novel Marker of Colon Cancer-Initiating Cells and Is a Potent Target of Immunotherapy. Clin. Cancer Res. 2016, 22, 3298–3309. [Google Scholar] [CrossRef] [PubMed]
- Asano, T.; Hirohashi, Y.; Torigoe, T.; Mariya, T.; Horibe, R.; Kuroda, T.; Tabuchi, Y.; Saijo, H.; Yasuda, K.; Mizuuchi, M.; et al. Brother of the regulator of the imprinted site (BORIS) variant subfamily 6 is involved in cervical cancer stemness and can be a target of immunotherapy. Oncotarget 2016, 7, 11223–11237. [Google Scholar] [CrossRef] [PubMed]
- Engelmann, K.; Shen, H.; Finn, O.J. MCF7 side population cells with characteristics of cancer stem/progenitor cells express the tumor antigen MUC1. Cancer Res. 2008, 68, 2419–2426. [Google Scholar] [CrossRef]
- Phuphanich, S.; Wheeler, C.J.; Rudnick, J.D.; Mazer, M.; Wang, H.; Nuno, M.A.; Richardson, J.E.; Fan, X.; Ji, J.; Chu, R.M.; et al. Phase I trial of a multi-epitope-pulsed dendritic cell vaccine for patients with newly diagnosed glioblastoma. Cancer Immunol. Immunother. 2013, 62, 125–135. [Google Scholar] [CrossRef]
- Gedye, C.; Quirk, J.; Browning, J.; Svobodova, S.; John, T.; Sluka, P.; Dunbar, P.R.; Corbeil, D.; Cebon, J.; Davis, I.D. Cancer/testis antigens can be immunological targets in clonogenic CD133+ melanoma cells. Cancer Immunol. Immunother. 2009, 58, 1635–1646. [Google Scholar] [CrossRef]
- Hont, A.B.; Cruz, C.R.; Ulrey, R.; O’Brien, B.; Stanojevic, M.; Datar, A.; Albihani, S.; Saunders, D.; Hanajiri, R.; Panchapakesan, K.; et al. Immunotherapy of Relapsed and Refractory Solid Tumors With Ex Vivo Expanded Multi-Tumor Associated Antigen Specific Cytotoxic T Lymphocytes: A Phase I Study. J. Clin. Oncol. 2019, 37, 2349–2359. [Google Scholar] [CrossRef] [PubMed]
- Mennonna, D.; Maccalli, C.; Romano, M.C.; Garavaglia, C.; Capocefalo, F.; Bordoni, R.; Severgnini, M.; De Bellis, G.; Sidney, J.; Sette, A.; et al. T cell neoepitope discovery in colorectal cancer by high throughput profiling of somatic mutations in expressed genes. Gut 2017, 66, 454–463. [Google Scholar] [CrossRef] [PubMed]
- Korkaya, H.; Liu, S.; Wicha, M.S. Breast cancer stem cells, cytokine networks, and the tumor microenvironment. J. Clin. Invest. 2011, 121, 3804–3809. [Google Scholar] [CrossRef]
- Cella, M.; Sallusto, F.; Lanzavecchia, A. Origin, maturation and antigen presenting function of dendritic cells. Curr. Opin. Immunol. 1997, 9, 10–16. [Google Scholar] [CrossRef]
- Hubo, M.; Trinschek, B.; Kryczanowsky, F.; Tuettenberg, A.; Steinbrink, K.; Jonuleit, H. Costimulatory molecules on immunogenic versus tolerogenic human dendritic cells. Front. Immunol. 2013, 4, 82. [Google Scholar] [CrossRef]
- Domogalla, M.P.; Rostan, P.V.; Raker, V.K.; Steinbrink, K. Tolerance through Education: How Tolerogenic Dendritic Cells Shape Immunity. Front. Immunol. 2017, 8, 1764. [Google Scholar] [CrossRef]
- Hanke, N.; Alizadeh, D.; Katsanis, E.; Larmonier, N. Dendritic cell tumor killing activity and its potential applications in cancer immunotherapy. Crit. Rev. Immunol. 2013, 33, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Keller, A.M.; Xiao, Y.; Peperzak, V.; Naik, S.H.; Borst, J. Costimulatory ligand CD70 allows induction of CD8+ T-cell immunity by immature dendritic cells in a vaccination setting. Blood 2009, 113, 5167–5175. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef]
- Hsu, Y.L.; Chen, Y.J.; Chang, W.A.; Jian, S.F.; Fan, H.L.; Wang, J.Y.; Kuo, P.L. Interaction between Tumor-Associated Dendritic Cells and Colon Cancer Cells Contributes to Tumor Progression via CXCL1. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef]
- Condeelis, J.; Pollard, J.W. Macrophages: Obligate partners for tumor cell migration, invasion, and metastasis. Cell 2006, 124, 263–266. [Google Scholar] [CrossRef] [PubMed]
- DeNardo, D.G.; Ruffell, B. Macrophages as regulators of tumour immunity and immunotherapy. Nat. Rev. Immunol. 2019, 19, 369–382. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef] [PubMed]
- Wan, S.; Zhao, E.; Kryczek, I.; Vatan, L.; Sadovskaya, A.; Ludema, G.; Simeone, D.M.; Zou, W.; Welling, T.H. Tumor-associated macrophages produce interleukin 6 and signal via STAT3 to promote expansion of human hepatocellular carcinoma stem cells. Gastroenterology 2014, 147, 1393–1404. [Google Scholar] [CrossRef]
- Kim, S.Y.; Kang, J.W.; Song, X.; Kim, B.K.; Yoo, Y.D.; Kwon, Y.T.; Lee, Y.J. Role of the IL-6-JAK1-STAT3-Oct-4 pathway in the conversion of non-stem cancer cells into cancer stem-like cells. Cell Signal. 2013, 25, 961–969. [Google Scholar] [CrossRef]
- Sherry, M.M.; Reeves, A.; Wu, J.K.; Cochran, B.H. STAT3 is required for proliferation and maintenance of multipotency in glioblastoma stem cells. Stem Cells 2009, 27, 2383–2392. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wulfkuhle, J.; Zhang, H.; Gu, P.; Yang, Y.; Deng, J.; Margolick, J.B.; Liotta, L.A.; Petricoin, E., 3rd; Zhang, Y. Activation of the PTEN/mTOR/STAT3 pathway in breast cancer stem-like cells is required for viability and maintenance. Proc. Natl. Acad. Sci. USA 2007, 104, 16158–16163. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Yao, Q.; Liu, Y.; Du, S.; Liu, A.; Guo, Z.; Sun, A.; Ruan, J.; Chen, L.; Ye, C.; et al. IL-6-induced epithelial-mesenchymal transition promotes the generation of breast cancer stem-like cells analogous to mammosphere cultures. Int. J. Oncol. 2012, 40, 1171–1179. [Google Scholar] [CrossRef] [PubMed]
- Iliopoulos, D.; Hirsch, H.A.; Wang, G.; Struhl, K. Inducible formation of breast cancer stem cells and their dynamic equilibrium with non-stem cancer cells via IL6 secretion. Proc. Natl. Acad. Sci. USA 2011, 108, 1397–1402. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Ping, Y.F.; Zhou, W.; He, Z.C.; Chen, C.; Bian, B.S.; Zhang, L.; Chen, L.; Lan, X.; Zhang, X.C.; et al. Tumour-associated macrophages secrete pleiotrophin to promote PTPRZ1 signalling in glioblastoma stem cells for tumour growth. Nat. Commun. 2017, 8, 15080. [Google Scholar] [CrossRef] [PubMed]
- Nakano, M.; Kikushige, Y.; Miyawaki, K.; Kunisaki, Y.; Mizuno, S.; Takenaka, K.; Tamura, S.; Okumura, Y.; Ito, M.; Ariyama, H.; et al. Dedifferentiation process driven by TGF-beta signaling enhances stem cell properties in human colorectal cancer. Oncogene 2019, 38, 780–793. [Google Scholar] [CrossRef]
- Mitchem, J.B.; Brennan, D.J.; Knolhoff, B.L.; Belt, B.A.; Zhu, Y.; Sanford, D.E.; Belaygorod, L.; Carpenter, D.; Collins, L.; Piwnica-Worms, D.; et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 2013, 73, 1128–1141. [Google Scholar] [CrossRef] [PubMed]
- Condamine, T.; Ramachandran, I.; Youn, J.I.; Gabrilovich, D.I. Regulation of tumor metastasis by myeloid-derived suppressor cells. Annu. Rev. Med. 2015, 66, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016, 37, 208–220. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, H.; Mabuchi, S.; Yokoi, E.; Komura, N.; Kozasa, K.; Matsumoto, Y.; Kawano, M.; Takahashi, R.; Sasano, T.; Shimura, K.; et al. Prostaglandin E2 produced by myeloid-derived suppressive cells induces cancer stem cells in uterine cervical cancer. Oncotarget 2018, 9, 36317–36330. [Google Scholar] [CrossRef] [PubMed]
- Peng, D.; Tanikawa, T.; Li, W.; Zhao, L.; Vatan, L.; Szeliga, W.; Wan, S.; Wei, S.; Wang, Y.; Liu, Y.; et al. Myeloid-Derived Suppressor Cells Endow Stem-like Qualities to Breast Cancer Cells through IL6/STAT3 and NO/NOTCH Cross-talk Signaling. Cancer Res. 2016, 76, 3156–3165. [Google Scholar] [CrossRef]
- Cui, T.X.; Kryczek, I.; Zhao, L.; Zhao, E.; Kuick, R.; Roh, M.H.; Vatan, L.; Szeliga, W.; Mao, Y.; Thomas, D.G.; et al. Myeloid-derived suppressor cells enhance stemness of cancer cells by inducing microRNA101 and suppressing the corepressor CtBP2. Immunity 2013, 39, 611–621. [Google Scholar] [CrossRef]
- Wing, J.B.; Tanaka, A.; Sakaguchi, S. Human FOXP3(+) Regulatory T Cell Heterogeneity and Function in Autoimmunity and Cancer. Immunity 2019, 50, 302–316. [Google Scholar] [CrossRef]
- Togashi, Y.; Shitara, K.; Nishikawa, H. Regulatory T cells in cancer immunosuppression - implications for anticancer therapy. Nat. Rev. Clin. Oncol. 2019, 16, 356–371. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Wang, B.; Guan, C.; Wu, B.; Cai, C.; Wang, M.; Zhang, B.; Liu, T.; Yang, P. Foxp3+IL-17+ T cells promote development of cancer-initiating cells in colorectal cancer. J. Leukoc. Biol. 2011, 89, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Dong, X.; Qi, P.; Ye, Y.; Shen, W.; Leng, L.; Wang, L.; Li, X.; Luo, X.; Chen, Y.; et al. Sox2 Communicates with Tregs Through CCL1 to Promote the Stemness Property of Breast Cancer Cells. Stem Cells 2017, 35, 2351–2365. [Google Scholar] [CrossRef] [PubMed]
- Asadzadeh, Z.; Mohammadi, H.; Safarzadeh, E.; Hemmatzadeh, M.; Mahdian-Shakib, A.; Jadidi-Niaragh, F.; Azizi, G.; Baradaran, B. The paradox of Th17 cell functions in tumor immunity. Cell Immunol. 2017, 322, 15–25. [Google Scholar] [CrossRef]
- Chang, S.H. T helper 17 (Th17) cells and interleukin-17 (IL-17) in cancer. Arch. Pharm. Res. 2019, 42, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zoltan, M.; Riquelme, E.; Xu, H.; Sahin, I.; Castro-Pando, S.; Montiel, M.F.; Chang, K.; Jiang, Z.; Ling, J.; et al. Immune Cell Production of Interleukin 17 Induces Stem Cell Features of Pancreatic Intraepithelial Neoplasia Cells. Gastroenterology 2018, 155, 210–223 e213. [Google Scholar] [CrossRef] [PubMed]
- Reading, J.L.; Galvez-Cancino, F.; Swanton, C.; Lladser, A.; Peggs, K.S.; Quezada, S.A. The function and dysfunction of memory CD8(+) T cells in tumor immunity. Immunol. Rev. 2018, 283, 194–212. [Google Scholar] [CrossRef]
- Song, M.; Ping, Y.; Zhang, K.; Yang, L.; Li, F.; Zhang, C.; Cheng, S.; Yue, D.; Maimela, N.R.; Qu, J.; et al. Low-Dose IFNgamma Induces Tumor Cell Stemness in Tumor Microenvironment of Non-Small Cell Lung Cancer. Cancer Res. 2019, 79, 3737–3748. [Google Scholar] [CrossRef]
- Stein, R.G.; Ebert, S.; Schlahsa, L.; Scholz, C.J.; Braun, M.; Hauck, P.; Horn, E.; Monoranu, C.M.; Thiemann, V.J.; Wustrow, M.P.; et al. Cognate Nonlytic Interactions between CD8(+) T Cells and Breast Cancer Cells Induce Cancer Stem Cell-like Properties. Cancer Res. 2019, 79, 1507–1519. [Google Scholar] [CrossRef] [PubMed]
- Ravindran, S.; Rasool, S.; Maccalli, C. The Cross Talk between Cancer Stem Cells/Cancer Initiating Cells and Tumor Microenvironment: The Missing Piece of the Puzzle for the Efficient Targeting of these Cells with Immunotherapy. Cancer Microenviron 2019, 12, 133–148. [Google Scholar] [CrossRef]
- Roth, P.; Junker, M.; Tritschler, I.; Mittelbronn, M.; Dombrowski, Y.; Breit, S.N.; Tabatabai, G.; Wick, W.; Weller, M.; Wischhusen, J. GDF-15 contributes to proliferation and immune escape of malignant gliomas. Clin. Cancer Res. 2010, 16, 3851–3859. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, K.; Massague, J. TGF-beta Inhibition and Immunotherapy: Checkmate. Immunity 2018, 48, 626–628. [Google Scholar] [CrossRef] [PubMed]
- Kokubu, Y.; Tabu, K.; Muramatsu, N.; Wang, W.; Murota, Y.; Nobuhisa, I.; Jinushi, M.; Taga, T. Induction of protumoral CD11c(high) macrophages by glioma cancer stem cells through GM-CSF. Genes Cells 2016, 21, 241–251. [Google Scholar] [CrossRef]
- Yamashina, T.; Baghdadi, M.; Yoneda, A.; Kinoshita, I.; Suzu, S.; Dosaka-Akita, H.; Jinushi, M. Cancer stem-like cells derived from chemoresistant tumors have a unique capacity to prime tumorigenic myeloid cells. Cancer Res. 2014, 74, 2698–2709. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Cai, D.J.; Li, B. Ovarian cancer stem-like cells elicit the polarization of M2 macrophages. Mol. Med. Rep. 2015, 11, 4685–4693. [Google Scholar] [CrossRef]
- Jinushi, M.; Chiba, S.; Yoshiyama, H.; Masutomi, K.; Kinoshita, I.; Dosaka-Akita, H.; Yagita, H.; Takaoka, A.; Tahara, H. Tumor-associated macrophages regulate tumorigenicity and anticancer drug responses of cancer stem/initiating cells. Proc. Natl. Acad. Sci. USA 2011, 108, 12425–12430. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Ke, S.Q.; Huang, Z.; Flavahan, W.; Fang, X.; Paul, J.; Wu, L.; Sloan, A.E.; McLendon, R.E.; Li, X.; et al. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat. Cell Biol. 2015, 17, 170–182. [Google Scholar] [CrossRef]
- Guo, X.; Zhao, Y.; Yan, H.; Yang, Y.; Shen, S.; Dai, X.; Ji, X.; Ji, F.; Gong, X.G.; Li, L.; et al. Single tumor-initiating cells evade immune clearance by recruiting type II macrophages. Genes Dev. 2017, 31, 247–259. [Google Scholar] [CrossRef]
- Mirzaei, R.; Sarkar, S.; Dzikowski, L.; Rawji, K.S.; Khan, L.; Faissner, A.; Bose, P.; Yong, V.W. Brain tumor-initiating cells export tenascin-C associated with exosomes to suppress T cell activity. Oncoimmunology 2018, 7, e1478647. [Google Scholar] [CrossRef] [PubMed]
- You, Y.; Li, Y.; Li, M.; Lei, M.; Wu, M.; Qu, Y.; Yuan, Y.; Chen, T.; Jiang, H. Ovarian cancer stem cells promote tumour immune privilege and invasion via CCL5 and regulatory T cells. Clin. Exp. Immunol. 2018, 191, 60–73. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Chen, M.; Wu, P.; Chen, C.; Xu, Z.P.; Gu, W. Increased PD-L1 expression in breast and colon cancer stem cells. Clin. Exp. Pharmacol. Physiol. 2017, 44, 602–604. [Google Scholar] [CrossRef] [PubMed]
- Kikushige, Y.; Miyamoto, T.; Yuda, J.; Jabbarzadeh-Tabrizi, S.; Shima, T.; Takayanagi, S.; Niiro, H.; Yurino, A.; Miyawaki, K.; Takenaka, K.; et al. A TIM-3/Gal-9 Autocrine Stimulatory Loop Drives Self-Renewal of Human Myeloid Leukemia Stem Cells and Leukemic Progression. Cell Stem Cell 2015, 17, 341–352. [Google Scholar] [CrossRef]
- Gao, L.; Yu, S.; Zhang, X. Hypothesis: Tim-3/galectin-9, a new pathway for leukemia stem cells survival by promoting expansion of myeloid-derived suppressor cells and differentiating into tumor-associated macrophages. Cell Biochem. Biophys. 2014, 70, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Wang, X.; Jin, K.; Zhu, J.; Wang, Y.; Xiong, S.; Mao, Y.; Zhou, L. B7-H4 is preferentially expressed in non-dividing brain tumor cells and in a subset of brain tumor stem-like cells. J. Neurooncol. 2008, 89, 121–129. [Google Scholar] [CrossRef]
- Yao, Y.; Ye, H.; Qi, Z.; Mo, L.; Yue, Q.; Baral, A.; Hoon, D.S.B.; Vera, J.C.; Heiss, J.D.; Chen, C.C.; et al. B7-H4(B7x)-Mediated Cross-talk between Glioma-Initiating Cells and Macrophages via the IL6/JAK/STAT3 Pathway Lead to Poor Prognosis in Glioma Patients. Clin. Cancer Res. 2016, 22, 2778–2790. [Google Scholar] [CrossRef]
- Caputo, S.; Grioni, M.; Brambillasca, C.S.; Monno, A.; Brevi, A.; Freschi, M.; Piras, I.S.; Elia, A.R.; Pieri, V.; Baccega, T.; et al. Galectin-3 in Prostate Cancer Stem-Like Cells Is Immunosuppressive and Drives Early Metastasis. Front. Immunol. 2020, 11, 1820. [Google Scholar] [CrossRef]
- Yang, K.; Park, H.J.; Han, S.; Lee, J.; Ko, E.; Kim, J.; Lee, J.S.; Yu, J.H.; Song, K.Y.; Cheong, E.; et al. Recapitulation of in vivo-like paracrine signals of human mesenchymal stem cells for functional neuronal differentiation of human neural stem cells in a 3D microfluidic system. Biomaterials 2015, 63, 177–188. [Google Scholar] [CrossRef]
- Hutmacher, D.W.; Loessner, D.; Rizzi, S.; Kaplan, D.L.; Mooney, D.J.; Clements, J.A. Can tissue engineering concepts advance tumor biology research? Trends Biotechnol. 2010, 28, 125–133. [Google Scholar] [CrossRef]
- Al-Lamki, R.S.; Bradley, J.R.; Pober, J.S. Human Organ Culture: Updating the Approach to Bridge the Gap from In Vitro to In Vivo in Inflammation, Cancer, and Stem Cell Biology. Front. Med. 2017, 4, 148. [Google Scholar] [CrossRef] [PubMed]
- Luca, A.C.; Mersch, S.; Deenen, R.; Schmidt, S.; Messner, I.; Schafer, K.L.; Baldus, S.E.; Huckenbeck, W.; Piekorz, R.P.; Knoefel, W.T.; et al. Impact of the 3D microenvironment on phenotype, gene expression, and EGFR inhibition of colorectal cancer cell lines. PLoS ONE 2013, 8, e59689. [Google Scholar] [CrossRef]
- Carletti, E.; Motta, A.; Migliaresi, C. Scaffolds for tissue engineering and 3D cell culture. Methods Mol. Biol. 2011, 695, 17–39. [Google Scholar] [CrossRef] [PubMed]
- Aboulkheyr Es, H.; Montazeri, L.; Aref, A.R.; Vosough, M.; Baharvand, H. Personalized Cancer Medicine: An Organoid Approach. Trends Biotechnol. 2018, 36, 358–371. [Google Scholar] [CrossRef] [PubMed]
- Al-Lamki, R.S.; Wang, J.; Yang, J.; Burrows, N.; Maxwell, P.H.; Eisen, T.; Warren, A.Y.; Vanharanta, S.; Pacey, S.; Vandenabeele, P.; et al. Tumor necrosis factor receptor 2-signaling in CD133-expressing cells in renal clear cell carcinoma. Oncotarget 2016, 7, 24111–24124. [Google Scholar] [CrossRef]
- Raghavan, S.; Mehta, P.; Ward, M.R.; Bregenzer, M.E.; Fleck, E.M.A.; Tan, L.; McLean, K.; Buckanovich, R.J.; Mehta, G. Personalized Medicine-Based Approach to Model Patterns of Chemoresistance and Tumor Recurrence Using Ovarian Cancer Stem Cell Spheroids. Clin. Cancer Res. 2017, 23, 6934–6945. [Google Scholar] [CrossRef]
- Raghavan, S.; Mehta, P.; Horst, E.N.; Ward, M.R.; Rowley, K.R.; Mehta, G. Comparative analysis of tumor spheroid generation techniques for differential in vitro drug toxicity. Oncotarget 2016, 7, 16948–16961. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.; Li, X.; Tan, S.; Zhou, H.J.; Ji, W.; Bellone, S.; Xu, X.; Zhang, H.; Santin, A.D.; Lou, G.; et al. Tumor-associated macrophages drive spheroid formation during early transcoelomic metastasis of ovarian cancer. J. Clin. Invest. 2016, 126, 4157–4173. [Google Scholar] [CrossRef] [PubMed]
- Raghavan, S.; Mehta, P.; Xie, Y.; Lei, Y.L.; Mehta, G. Ovarian cancer stem cells and macrophages reciprocally interact through the WNT pathway to promote pro-tumoral and malignant phenotypes in 3D engineered microenvironments. J. Immunother. Cancer 2019, 7, 190. [Google Scholar] [CrossRef]
- Faraci, M.; Vigeant, C.; Yale, J.F. Toxic effects of cyclosporine A and G in Wistar rats. Transpl. Proc. 1988, 20, 963–968. [Google Scholar]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef]
- Leahy, A.B.; Elgarten, C.W.; Grupp, S.A.; Maude, S.L.; Teachey, D.T. Tisagenlecleucel for the treatment of B-cell acute lymphoblastic leukemia. Expert Rev. Anticancer. Ther. 2018, 18, 959–971. [Google Scholar] [CrossRef]
- Fournier, C.; Rivera Vargas, T.; Martin, T.; Melis, A.; Apetoh, L. Immunotherapeutic properties of chemotherapy. Curr. Opin. Pharmacol. 2017, 35, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Wu, C.; Lu, B. Cytokine-induced killer cells promote antitumor immunity. J. Transl. Med. 2013, 11, 83. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Wu, Y.; Ma, W.; Zhang, S.; Zhang, Y.Q. Adoptive T-cell therapy of prostate cancer targeting the cancer stem cell antigen EpCAM. BMC Immunol. 2015, 16, 1. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, S.; Kochin, V.; Kanaseki, T.; Hongo, A.; Tokita, S.; Kikuchi, Y.; Takaya, A.; Hirohashi, Y.; Tsukahara, T.; Terui, T.; et al. The Antigen ASB4 on Cancer Stem Cells Serves as a Target for CTL Immunotherapy of Colorectal Cancer. Cancer Immunol. Res. 2018, 6, 358–369. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Prasad, S.; Gaedicke, S.; Hettich, M.; Firat, E.; Niedermann, G. Patient-derived glioblastoma stem cells are killed by CD133-specific CAR T cells but induce the T cell aging marker CD57. Oncotarget 2015, 6, 171–184. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, M.; Wu, Z.; Tong, C.; Dai, H.; Guo, Y.; Liu, Y.; Huang, J.; Lv, H.; Luo, C.; et al. CD133-directed CAR T cells for advanced metastasis malignancies: A phase I trial. Oncoimmunology 2018, 7, e1440169. [Google Scholar] [CrossRef]
- Morgan, R.A.; Johnson, L.A.; Davis, J.L.; Zheng, Z.; Woolard, K.D.; Reap, E.A.; Feldman, S.A.; Chinnasamy, N.; Kuan, C.T.; Song, H.; et al. Recognition of glioma stem cells by genetically modified T cells targeting EGFRvIII and development of adoptive cell therapy for glioma. Hum. Gene Ther. 2012, 23, 1043–1053. [Google Scholar] [CrossRef]
- Tettamanti, S.; Marin, V.; Pizzitola, I.; Magnani, C.F.; Giordano Attianese, G.M.; Cribioli, E.; Maltese, F.; Galimberti, S.; Lopez, A.F.; Biondi, A.; et al. Targeting of acute myeloid leukaemia by cytokine-induced killer cells redirected with a novel CD123-specific chimeric antigen receptor. Br. J. Haematol. 2013, 161, 389–401. [Google Scholar] [CrossRef] [PubMed]
- Feng, K.C.; Guo, Y.L.; Liu, Y.; Dai, H.R.; Wang, Y.; Lv, H.Y.; Huang, J.H.; Yang, Q.M.; Han, W.D. Cocktail treatment with EGFR-specific and CD133-specific chimeric antigen receptor-modified T cells in a patient with advanced cholangiocarcinoma. J. Hematol. Oncol. 2017, 10, 4. [Google Scholar] [CrossRef]
- Introna, M.; Correnti, F. Innovative Clinical Perspectives for CIK Cells in Cancer Patients. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.H.; Negrin, R.S. A novel population of expanded human CD3+CD56+ cells derived from T cells with potent in vivo antitumor activity in mice with severe combined immunodeficiency. J. Immunol. 1994, 153, 1687–1696. [Google Scholar] [PubMed]
- Franceschetti, M.; Pievani, A.; Borleri, G.; Vago, L.; Fleischhauer, K.; Golay, J.; Introna, M. Cytokine-induced killer cells are terminally differentiated activated CD8 cytotoxic T-EMRA lymphocytes. Exp. Hematol. 2009, 37, 616–628 e612. [Google Scholar] [CrossRef]
- Gammaitoni, L.; Giraudo, L.; Macagno, M.; Leuci, V.; Mesiano, G.; Rotolo, R.; Sassi, F.; Sanlorenzo, M.; Zaccagna, A.; Pisacane, A.; et al. Cytokine-Induced Killer Cells Kill Chemo-surviving Melanoma Cancer Stem Cells. Clin. Cancer Res. 2017, 23, 2277–2288. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Zhang, W.; Wang, L.; Xiao, C.; Wang, L.; Gong, Y.; Huang, D.; Guo, B.; Li, Q.; Xiang, Y.; et al. Co-culture of dendritic cells and cytokine-induced killer cells effectively suppresses liver cancer stem cell growth by inhibiting pathways in the immune system. BMC Cancer 2018, 18, 984. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, Y.; Wang, Y.; Wu, D.; Lau, A.H.Y.; Zhao, P.; Zou, C.; Dai, Y.; Chan, F.L. Targeting prostate cancer stem-like cells by an immunotherapeutic platform based on immunogenic peptide-sensitized dendritic cells-cytokine-induced killer cells. Stem Cell Res. Ther. 2020, 11, 123. [Google Scholar] [CrossRef] [PubMed]
- Rong, X.X.; Wei, F.; Lin, X.L.; Qin, Y.J.; Chen, L.; Wang, H.Y.; Shen, H.F.; Jia, L.T.; Xie, R.Y.; Lin, T.Y.; et al. Recognition and killing of cancer stem-like cell population in hepatocellular carcinoma cells by cytokine-induced killer cells via NKG2d-ligands recognition. Oncoimmunology 2016, 5, e1086060. [Google Scholar] [CrossRef] [PubMed]
- Kaur, K.; Cook, J.; Park, S.H.; Topchyan, P.; Kozlowska, A.; Ohanian, N.; Fang, C.; Nishimura, I.; Jewett, A. Novel Strategy to Expand Super-Charged NK Cells with Significant Potential to Lyse and Differentiate Cancer Stem Cells: Differences in NK Expansion and Function between Healthy and Cancer Patients. Front. Immunol 2017, 8, 297. [Google Scholar] [CrossRef] [PubMed]
- Mesiano, G.; Grignani, G.; Fiorino, E.; Leuci, V.; Rotolo, R.; D’Ambrosio, L.; Salfi, C.; Gammaitoni, L.; Giraudo, L.; Pisacane, A.; et al. Cytokine Induced Killer cells are effective against sarcoma cancer stem cells spared by chemotherapy and target therapy. Oncoimmunology 2018, 7, e1465161. [Google Scholar] [CrossRef] [PubMed]
- Landsberg, J.; Kohlmeyer, J.; Renn, M.; Bald, T.; Rogava, M.; Cron, M.; Fatho, M.; Lennerz, V.; Wolfel, T.; Holzel, M.; et al. Melanomas resist T-cell therapy through inflammation-induced reversible dedifferentiation. Nature 2012, 490, 412–416. [Google Scholar] [CrossRef]
- Chen, H.C.; Joalland, N.; Bridgeman, J.S.; Alchami, F.S.; Jarry, U.; Khan, M.W.A.; Piggott, L.; Shanneik, Y.; Li, J.; Herold, M.J.; et al. Synergistic targeting of breast cancer stem-like cells by human gammadelta T cells and CD8(+) T cells. Immunol. Cell Biol. 2017, 95, 620–629. [Google Scholar] [CrossRef] [PubMed]
- Visus, C.; Wang, Y.; Lozano-Leon, A.; Ferris, R.L.; Silver, S.; Szczepanski, M.J.; Brand, R.E.; Ferrone, C.R.; Whiteside, T.L.; Ferrone, S.; et al. Targeting ALDH(bright) human carcinoma-initiating cells with ALDH1A1-specific CD8(+) T cells. Clin. Cancer Res. 2011, 17, 6174–6184. [Google Scholar] [CrossRef]
- Lamers, C.H.; Sleijfer, S.; van Steenbergen, S.; van Elzakker, P.; van Krimpen, B.; Groot, C.; Vulto, A.; den Bakker, M.; Oosterwijk, E.; Debets, R.; et al. Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: Clinical evaluation and management of on-target toxicity. Mol. Ther. 2013, 21, 904–912. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Hao, H.; Yang, G.; Zhang, Y.; Fu, Y. Immunotherapy with CAR-Modified T Cells: Toxicities and Overcoming Strategies. J. Immunol. Res. 2018, 2018, 2386187. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.N.; Fry, T.J. Mechanisms of resistance to CAR T cell therapy. Nat. Rev. Clin. Oncol. 2019, 16, 372–385. [Google Scholar] [CrossRef]
- Rafiq, S.; Hackett, C.S.; Brentjens, R.J. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat. Rev. Clin. Oncol. 2020, 17, 147–167. [Google Scholar] [CrossRef] [PubMed]
- Bol, K.F.; Schreibelt, G.; Gerritsen, W.R.; de Vries, I.J.; Figdor, C.G. Dendritic Cell-Based Immunotherapy: State of the Art and Beyond. Clin. Cancer Res. 2016, 22, 1897–1906. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, L.H. Dendritic cells in cancer immunotherapy clinical trials: Are we making progress? Front. Immunol. 2013, 4, 454. [Google Scholar] [CrossRef] [PubMed]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef]
- Lopez, M.N.; Pereda, C.; Segal, G.; Munoz, L.; Aguilera, R.; Gonzalez, F.E.; Escobar, A.; Ginesta, A.; Reyes, D.; Gonzalez, R.; et al. Prolonged survival of dendritic cell-vaccinated melanoma patients correlates with tumor-specific delayed type IV hypersensitivity response and reduction of tumor growth factor beta-expressing T cells. J. Clin. Oncol. 2009, 27, 945–952. [Google Scholar] [CrossRef] [PubMed]
- Benteyn, D.; Van Nuffel, A.M.; Wilgenhof, S.; Corthals, J.; Heirman, C.; Neyns, B.; Thielemans, K.; Bonehill, A. Characterization of CD8+ T-cell responses in the peripheral blood and skin injection sites of melanoma patients treated with mRNA electroporated autologous dendritic cells (TriMixDC-MEL). Biomed. Res. Int. 2013, 2013, 976383. [Google Scholar] [CrossRef]
- Wefers, C.; Schreibelt, G.; Massuger, L.; de Vries, I.J.M.; Torensma, R. Immune Curbing of Cancer Stem Cells by CTLs Directed to NANOG. Front. Immunol. 2018, 9, 1412. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Lu, L.; Xia, Y.; Chen, X.; Chang, A.E.; Hollingsworth, R.E.; Hurt, E.; Owen, J.; Moyer, J.S.; Prince, M.E.; et al. Therapeutic Efficacy of Cancer Stem Cell Vaccines in the Adjuvant Setting. Cancer Res. 2016, 76, 4661–4672. [Google Scholar] [CrossRef] [PubMed]
- Bommareddy, P.K.; Shettigar, M.; Kaufman, H.L. Integrating oncolytic viruses in combination cancer immunotherapy. Nat. Rev. Immunol. 2018, 18, 498–513. [Google Scholar] [CrossRef]
- Nakao, S.; Arai, Y.; Tasaki, M.; Yamashita, M.; Murakami, R.; Kawase, T.; Amino, N.; Nakatake, M.; Kurosaki, H.; Mori, M.; et al. Intratumoral expression of IL-7 and IL-12 using an oncolytic virus increases systemic sensitivity to immune checkpoint blockade. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Andtbacka, R.H.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J. Clin. Oncol. 2015, 33, 2780–2788. [Google Scholar] [CrossRef]
- Zheng, F.; Dang, J.; Zhang, H.; Xu, F.; Ba, D.; Zhang, B.; Cheng, F.; Chang, A.E.; Wicha, M.S.; Li, Q. Cancer Stem Cell Vaccination With PD-L1 and CTLA-4 Blockades Enhances the Eradication of Melanoma Stem Cells in a Mouse Tumor Model. J. Immunother. 2018, 41, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Saha, D.; Martuza, R.L.; Rabkin, S.D. Macrophage Polarization Contributes to Glioblastoma Eradication by Combination Immunovirotherapy and Immune Checkpoint Blockade. Cancer Cell 2017, 32, 253–267 e255. [Google Scholar] [CrossRef]
- Zhu, S.; Lv, X.; Zhang, X.; Li, T.; Zang, G.; Yang, N.; Wang, X.; Wu, J.; Chen, W.; Liu, Y.J.; et al. An effective dendritic cell-based vaccine containing glioma stem-like cell lysate and CpG adjuvant for an orthotopic mouse model of glioma. Int. J. Cancer 2019, 144, 2867–2879. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Zhang, X.; Li, J.; Mo, L.; Zhao, H.; Zhu, Y.; Hu, Z.; Gao, J.; Tan, W. PD-1 blockade enhances the antitumor efficacy of GM-CSF surface-modified bladder cancer stem cells vaccine. Int. J. Cancer 2018, 142, 2106–2117. [Google Scholar] [CrossRef] [PubMed]
- Saha, D.; Wakimoto, H.; Peters, C.W.; Antoszczyk, S.J.; Rabkin, S.D.; Martuza, R.L. Combinatorial Effects of VEGFR Kinase Inhibitor Axitinib and Oncolytic Virotherapy in Mouse and Human Glioblastoma Stem-Like Cell Models. Clin. Cancer Res. 2018, 24, 3409–3422. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galassi, C.; Musella, M.; Manduca, N.; Maccafeo, E.; Sistigu, A. The Immune Privilege of Cancer Stem Cells: A Key to Understanding Tumor Immune Escape and Therapy Failure. Cells 2021, 10, 2361. https://doi.org/10.3390/cells10092361
Galassi C, Musella M, Manduca N, Maccafeo E, Sistigu A. The Immune Privilege of Cancer Stem Cells: A Key to Understanding Tumor Immune Escape and Therapy Failure. Cells. 2021; 10(9):2361. https://doi.org/10.3390/cells10092361
Chicago/Turabian StyleGalassi, Claudia, Martina Musella, Nicoletta Manduca, Ester Maccafeo, and Antonella Sistigu. 2021. "The Immune Privilege of Cancer Stem Cells: A Key to Understanding Tumor Immune Escape and Therapy Failure" Cells 10, no. 9: 2361. https://doi.org/10.3390/cells10092361
APA StyleGalassi, C., Musella, M., Manduca, N., Maccafeo, E., & Sistigu, A. (2021). The Immune Privilege of Cancer Stem Cells: A Key to Understanding Tumor Immune Escape and Therapy Failure. Cells, 10(9), 2361. https://doi.org/10.3390/cells10092361