
Neutropenia and Large Granular Lymphocyte Leukemia: From Pathogenesis to Therapeutic Options

 , , ,
, , , 

Abstract
1. Introduction
2. Immunological Deregulations in LGLL Patients
3. Neutrophils Lifespan and Neutropenia
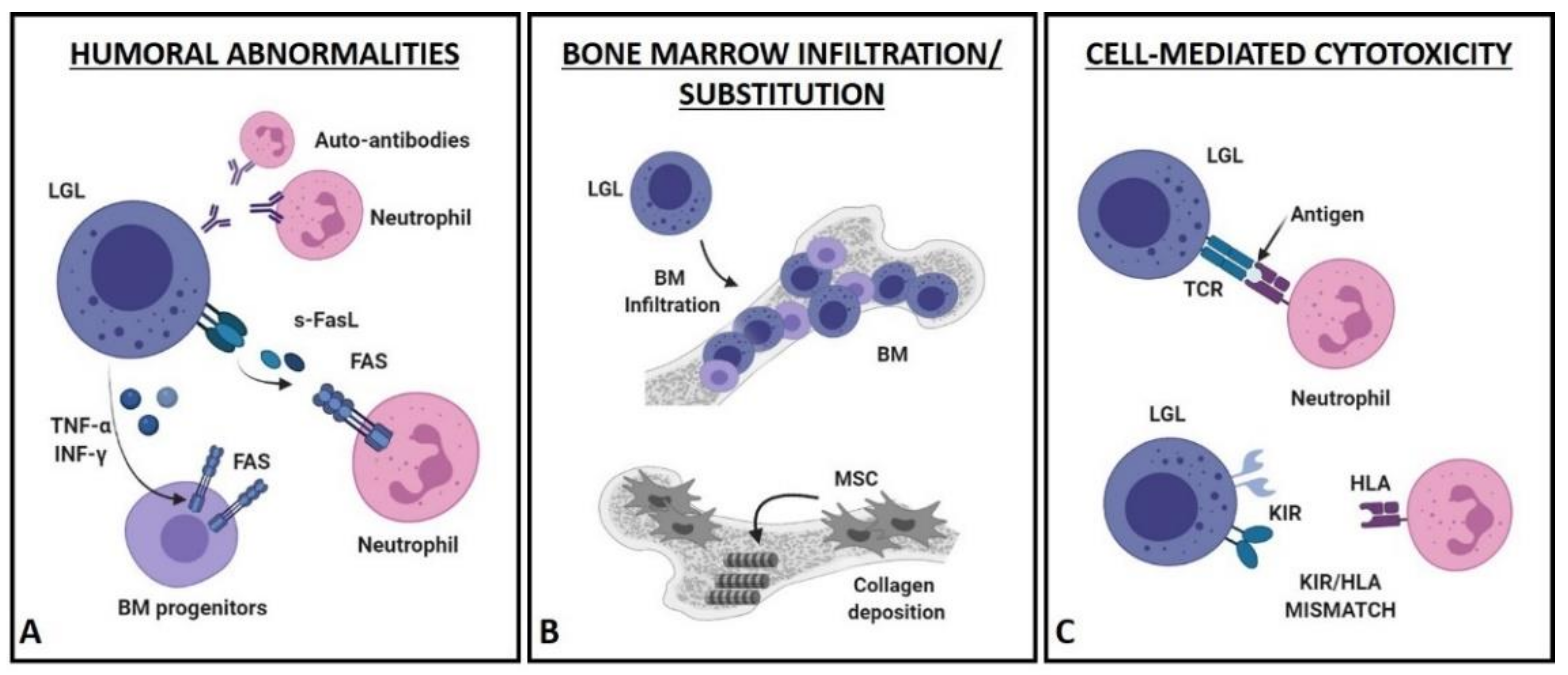
4. Pathogenetic Hypotheses of LGLL-Related Neutropenia
4.1. Humoral Mechanisms (Figure 2A)
4.2. Bone Marrow Infiltration/Substitution (Figure 2B)
4.3. Cell-Mediated Cytotoxic Mechanisms (Figure 2C)
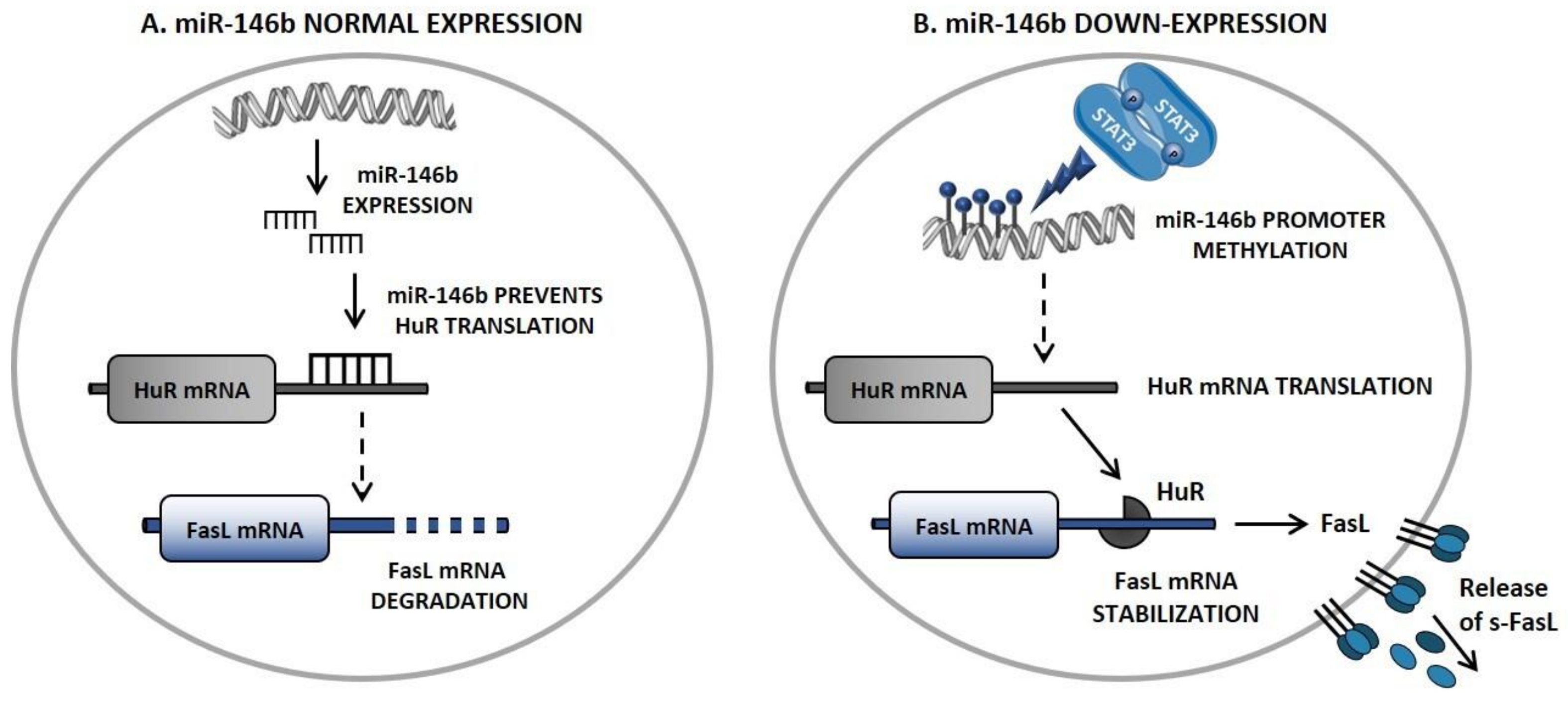
5. Novel Insights in FasL Regulation: The STAT3/miR-146b/FasL Axis
6. The Prognostic Value of Immunophenotype to Identify Neutropenic LGLL Patients
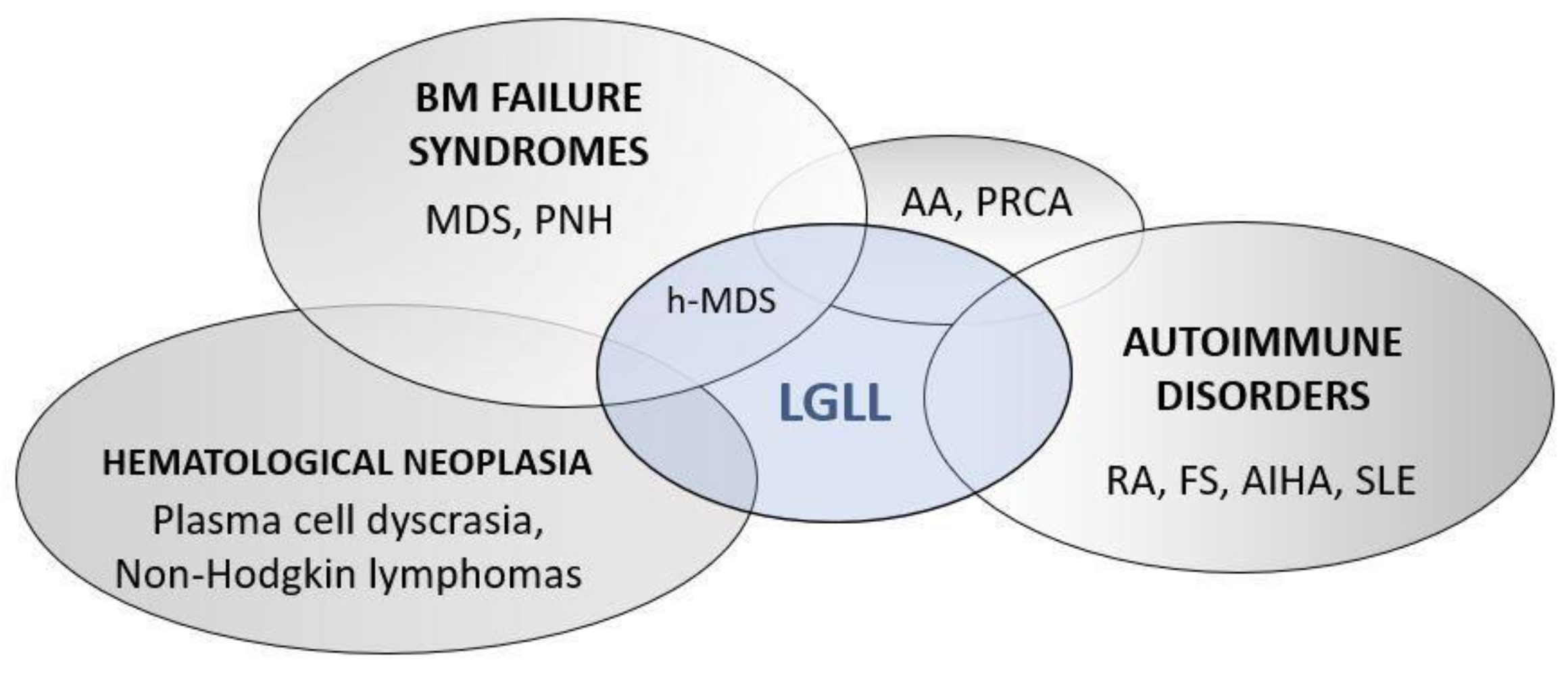
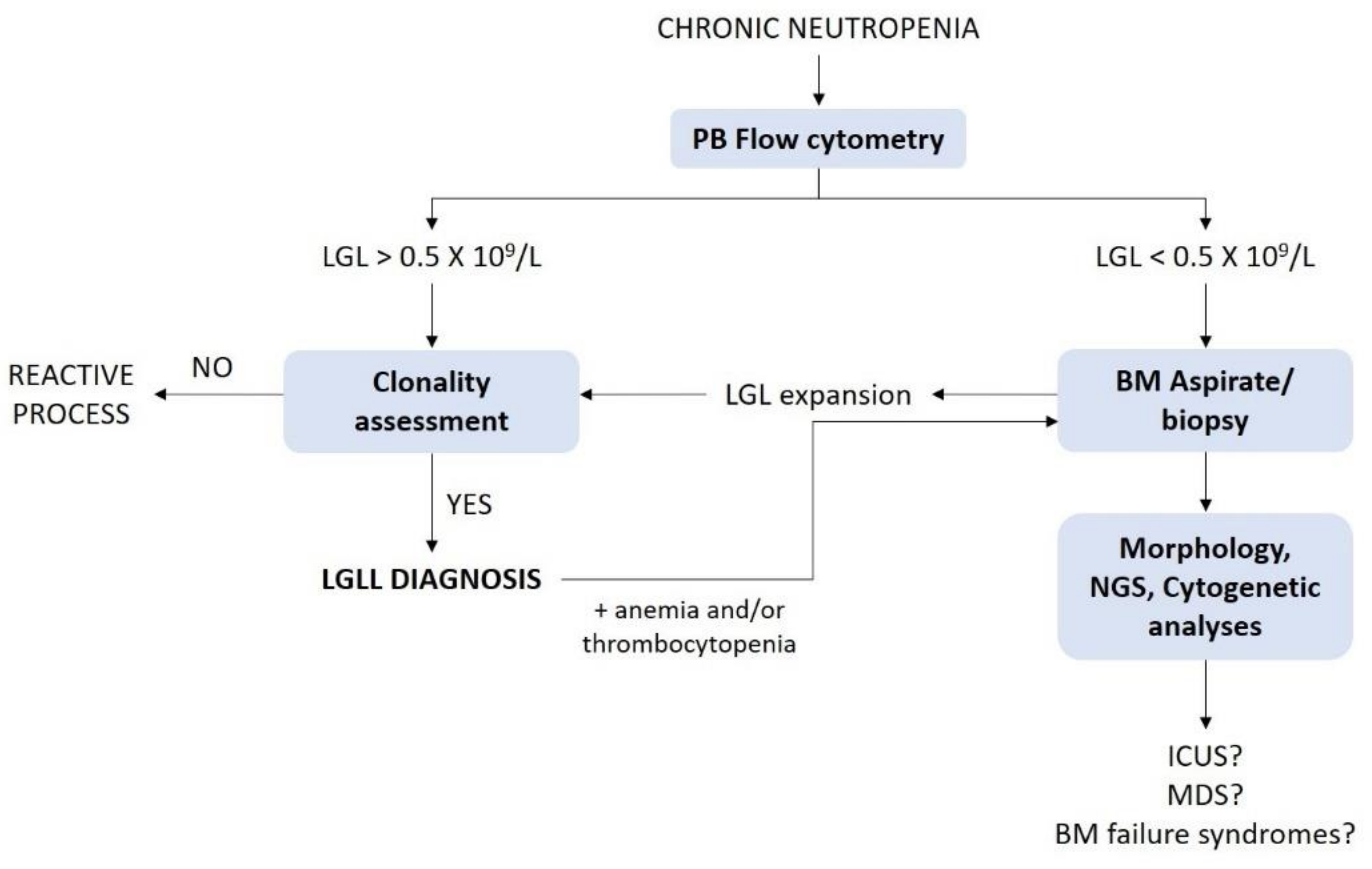
7. Differential Diagnosis of LGLL-Related Neutropenia
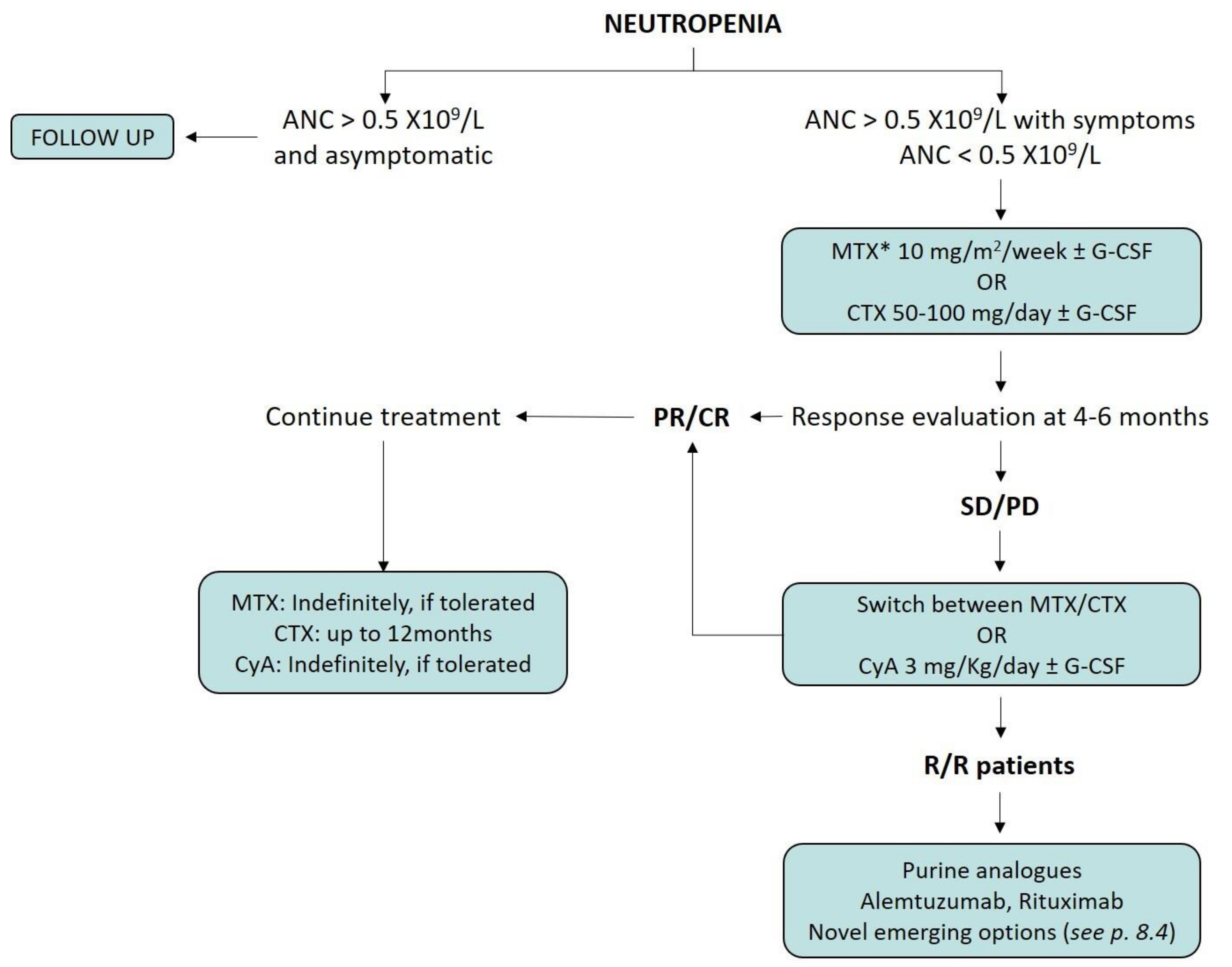
8. Treatment Indications for Neutropenic LGLL Patients
8.1. Immunosuppressive Therapy
8.2. Splenectomy and Supportive Therapy
8.3. Salvage Therapies
8.4. Emerging Therapeutic Options for LGLL Patients
9. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Loughran, T.P., Jr. Clonal Diseases of Large Granular Lymphocytes. Blood 1993, 82, 1–14. [Google Scholar] [CrossRef]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 Revision of the World Health Organization Classification of Lymphoid Neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef]
- Shah, M.V.; Hook, C.C.; Call, T.G.; Go, R.S. A Population-Based Study of Large Granular Lymphocyte Leukemia. Blood Cancer J. 2016, 6, e455. [Google Scholar] [CrossRef]
- Dinmohamed, A.G.; Brink, M.; Visser, O.; Jongen-Lavrencic, M. Population-Based Analyses among 184 Patients Diagnosed with Large Granular Lymphocyte Leukemia in the Netherlands between 2001 and 2013. Leukemia 2016, 30, 1449–1451. [Google Scholar] [CrossRef]
- Matutes, E. The 2017 WHO Update on Mature T- and Natural Killer (NK) Cell Neoplasms. Int. J. Lab. Hematol. 2018, 40 (Suppl. 1), 97–103. [Google Scholar] [CrossRef]
- Jerez, A.; Clemente, M.J.; Makishima, H.; Koskela, H.; Leblanc, F.; Peng, N.K.; Olson, T.; Przychodzen, B.; Afable, M.; Gomez-Segui, I.; et al. STAT3 Mutations Unify the Pathogenesis of Chronic Lymphoproliferative Disorders of NK Cells and T-Cell Large Granular Lymphocyte Leukemia. Blood 2012, 120, 3048–3057. [Google Scholar] [CrossRef]
- Barilà, G.; Calabretto, G.; Teramo, A.; Vicenzetto, C.; Gasparini, V.R.; Semenzato, G.; Zambello, R. T Cell Large Granular Lymphocyte Leukemia and Chronic NK Lymphocytosis. Best Pract. Res. Clin. Haematol. 2019, 32, 207–216. [Google Scholar] [CrossRef]
- Bareau, B.; Rey, J.; Hamidou, M.; Donadieu, J.; Morcet, J.; Reman, O.; Schleinitz, N.; Tournilhac, O.; Roussel, M.; Fest, T.; et al. Analysis of a French Cohort of Patients with Large Granular Lymphocyte Leukemia: A Report on 229 Cases. Haematologica 2010, 95, 1534–1541. [Google Scholar] [CrossRef]
- Sanikommu, S.R.; Clemente, M.J.; Chomczynski, P.; Afable, M.G., 2nd; Jerez, A.; Thota, S.; Patel, B.; Hirsch, C.; Nazha, A.; Desamito, J.; et al. Clinical Features and Treatment Outcomes in Large Granular Lymphocytic Leukemia (LGLL). Leuk. Lymphoma 2018, 59, 416–422. [Google Scholar] [CrossRef]
- Barilà, G.; Teramo, A.; Calabretto, G.; Vicenzetto, C.; Gasparini, V.R.; Pavan, L.; Leoncin, M.; Vedovato, S.; Frigo, A.C.; Facco, M.; et al. Stat3 Mutations Impact on Overall Survival in Large Granular Lymphocyte Leukemia: A Single-Center Experience of 205 Patients. Leukemia 2020, 34, 1116–1124. [Google Scholar] [CrossRef]
- Dong, N.; Tokumori, F.C.; Isenalumhe, L.; Zhang, Y.; Tandon, A.; Knepper, T.C.; Mo, Q.; Shao, H.; Zhang, L.; Sokol, L. Large Granular Lymphocytic Leukemia—A Retrospective Study of 319 Cases. Am. J. Hematol. 2021, 96, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Mariotti, B.; Calabretto, G.; Rossato, M.; Teramo, A.; Castellucci, M.; Barilà, G.; Leoncin, M.; Vicenzetto, C.; Facco, M.; Semenzato, G.; et al. Identification of a miR-146b-Fas Ligand Axis in the Development of Neutropenia in T Large Granular Lymphocyte Leukemia. Haematologica 2020, 105, 1351–1360. [Google Scholar] [CrossRef]
- Lamy, T.; Moignet, A.; Loughran, T.P. LGL Leukemia: From Pathogenesis to Treatment. Blood 2017, 129, 1082–1094. [Google Scholar] [CrossRef]
- Zambello, R.; Berno, T.; Cannas, G.; Baesso, I.; Binotto, G.; Bonoldi, E.; Bevilacqua, P.; Miorin, M.; Facco, M.; Trentin, L.; et al. Phenotypic and Functional Analyses of Dendritic Cells in Patients with Lymphoproliferative Disease of Granular Lymphocytes (LDGL). Blood 2005, 106, 3926–3931. [Google Scholar] [CrossRef][Green Version]
- Yoon, H.J.; Sugamura, K.; Loughran, T.P., Jr. Activation of Leukemic Large Granular Lymphocytes by Interleukin-2 via the p75 Interleukin-2 Receptor. Leukemia 1990, 4, 848–850. [Google Scholar]
- Teramo, A.; Gattazzo, C.; Passeri, F.; Lico, A.; Tasca, G.; Cabrelle, A.; Martini, V.; Frezzato, F.; Trimarco, V.; Ave, E.; et al. Intrinsic and Extrinsic Mechanisms Contribute to Maintain the JAK/STAT Pathway Aberrantly Activated in T-Type Large Granular Lymphocyte Leukemia. Blood 2013, 121, 3843–3854. [Google Scholar] [CrossRef]
- Zambello, R.; Facco, M.; Trentin, L.; Sancetta, R.; Tassinari, C.; Perin, A.; Milani, A.; Pizzolo, G.; Rodeghiero, F.; Agostini, C.; et al. Interleukin-15 Triggers the Proliferation and Cytotoxicity of Granular Lymphocytes in Patients with Lymphoproliferative Disease of Granular Lymphocytes. Blood 1997, 89, 201–211. [Google Scholar] [CrossRef]
- Mishra, A.; Liu, S.; Sams, G.H.; Curphey, D.P.; Santhanam, R.; Rush, L.J.; Schaefer, D.; Falkenberg, L.G.; Sullivan, L.; Jaroncyk, L.; et al. Aberrant Overexpression of IL-15 Initiates Large Granular Lymphocyte Leukemia through Chromosomal Instability and DNA Hypermethylation. Cancer Cell 2012, 22, 645–655. [Google Scholar] [CrossRef]
- Kothapalli, R.; Nyland, S.B.; Kusmartseva, I.; Bailey, R.D.; McKeown, T.M.; Loughran, T.P., Jr. Constitutive Production of Proinflammatory Cytokines RANTES, MIP-1beta and IL-18 Characterizes LGL Leukemia. Int. J. Oncol. 2005, 26, 529–535. [Google Scholar]
- Yang, J.; Liu, X.; Nyland, S.B.; Zhang, R.; Ryland, L.K.; Broeg, K.; Baab, K.T.; Jarbadan, N.R.; Irby, R.; Loughran, T.P., Jr. Platelet-Derived Growth Factor Mediates Survival of Leukemic Large Granular Lymphocytes via an Autocrine Regulatory Pathway. Blood 2010, 115, 51–60. [Google Scholar] [CrossRef]
- Sun, H.; Wei, S.; Yang, L. Dysfunction of Immune System in the Development of Large Granular Lymphocyte Leukemia. Hematology 2019, 24, 139–147. [Google Scholar] [CrossRef]
- Moignet, A.; Lamy, T. Latest Advances in the Diagnosis and Treatment of Large Granular Lymphocytic Leukemia. Am. Soc. Clin. Oncol. Educ. Book 2018, 38, 616–625. [Google Scholar] [CrossRef]
- Nauseef, W.M. Neutrophils, from Cradle to Grave and beyond. Immunol. Rev. 2016, 273, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Yvan-Charvet, L.; Ng, L.G. Granulopoiesis and Neutrophil Homeostasis: A Metabolic, Daily Balancing Act. Trends Immunol. 2019, 40, 598–612. [Google Scholar] [CrossRef] [PubMed]
- McCracken, J.M.; Allen, L.-A.H. Regulation of Human Neutrophil Apoptosis and Lifespan in Health and Disease. J. Cell Death 2014, 7, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, A.; Chilvers, E.R.; Summers, C.; Koenderman, L. The Neutrophil Life Cycle. Trends Immunol. 2019, 40, 584–597. [Google Scholar] [CrossRef]
- Boxer, L.A. How to Approach Neutropenia. Hematol. Am. Soc. Hematol. Educ. Program 2012, 2012, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Autrel-Moignet, A.; Lamy, T. Autoimmune Neutropenia. Presse Med. 2014, 43, e105–e118. [Google Scholar] [CrossRef] [PubMed]
- Gazitt, T.; Loughran, T.P., Jr. Chronic Neutropenia in LGL Leukemia and Rheumatoid Arthritis. Hematol. Am. Soc. Hematol. Educ. Program 2017, 2017, 181–186. [Google Scholar] [CrossRef]
- Burks, E.J.; Loughran, T.P., Jr. Pathogenesis of Neutropenia in Large Granular Lymphocyte Leukemia and Felty Syndrome. Blood Rev. 2006, 20, 245–266. [Google Scholar] [CrossRef]
- Pontikoglou, C.; Kalpadakis, C.; Papadaki, H.A. Pathophysiologic Mechanisms and Management of Neutropenia Associated with Large Granular Lymphocytic Leukemia. Expert Rev. Hematol. 2011, 4, 317–328. [Google Scholar] [CrossRef]
- Loughran, T.P., Jr.; Kadin, M.E.; Starkebaum, G.; Abkowitz, J.L.; Clark, E.A.; Disteche, C.; Lum, L.G.; Slichter, S.J. Leukemia of Large Granular Lymphocytes: Association with Clonal Chromosomal Abnormalities and Autoimmune Neutropenia, Thrombocytopenia, and Hemolytic Anemia. Ann. Intern. Med. 1985, 102, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Bassan, R.; Pronesti, M.; Buzzetti, M.; Allavena, P.; Rambaldi, A.; Mantovani, A.; Barbui, T. Autoimmunity and B-Cell Dysfunction in Chronic Proliferative Disorders of Large Granular Lymphocytes/natural Killer Cells. Cancer 1989, 63, 90–95. [Google Scholar] [CrossRef]
- Sivakumaran, M.; Richards, S. Immunological Abnormalities of Chronic Large Granular Lymphocytosis. Clin. Lab. Haematol. 1997, 19, 57–60. [Google Scholar] [CrossRef] [PubMed]
- Gentile, T.C.; Wener, M.H.; Starkebaum, G.; Loughran, T.P., Jr. Humoral Immune Abnormalities in T-Cell Large Granular Lymphocyte Leukemia. Leuk. Lymphoma 1996, 23, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Rustagi, P.K.; Han, T.; Ziolkowski, L.; Farolino, D.L.; Currie, M.S.; Logue, G.L. Granulocyte Antibodies in Leukaemic Chronic Lymphoproliferative Disorders. Br. J. Haematol. 1987, 66, 461–465. [Google Scholar] [CrossRef] [PubMed]
- Starkebaum, G.; Martin, P.J.; Singer, J.W.; Lum, L.G.; Price, T.H.; Kadin, M.E.; Raskind, W.H.; Fialkow, P.J. Chronic Lymphocytosis with Neutropenia: Evidence for a Novel, Abnormal T-Cell Population Associated with Antibody-Mediated Neutrophil Destruction. Clin. Immunol. Immunopathol. 1983, 27, 110–123. [Google Scholar] [CrossRef]
- Van der Veen, J.P.; Goldschmeding, R.; Miedema, F.; Smit, J.W.; Melief, C.J.; von dem Borne, A.E. K-Cell Lymphocytosis/neutropenia Syndrome: The Neutropenia Is Not Caused by Autoimmunity. Br. J. Haematol. 1986, 64, 777–787. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Suda, T.; Haze, K.; Nakamura, N.; Sato, K.; Kimura, F.; Motoyoshi, K.; Mizuki, M.; Tagawa, S.; Ohga, S.; et al. Fas Ligand in Human Serum. Nat. Med. 1996, 2, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.H.; Wei, S.; Lamy, T.; Epling-Burnette, P.K.; Starkebaum, G.; Djeu, J.Y.; Loughran, T.P. Chronic Neutropenia Mediated by Fas Ligand. Blood 2000, 95, 3219–3222. [Google Scholar] [CrossRef]
- Teramo, A.; Barilà, G.; Calabretto, G.; Ercolin, C.; Lamy, T.; Moignet, A.; Roussel, M.; Pastoret, C.; Leoncin, M.; Gattazzo, C.; et al. STAT3 Mutation Impacts Biological and Clinical Features of T-LGL Leukemia. Oncotarget 2017, 8, 61876–61889. [Google Scholar] [CrossRef] [PubMed]
- Perzova, R.; Loughran, T.P. Constitutive Expression of Fas Ligand in Large Granular Lymphocyte Leukaemia. Br. J. Haematol. 1997, 97, 123–126. [Google Scholar] [CrossRef]
- Lamy, T.; Liu, J.H.; Landowski, T.H.; Dalton, W.S.; Loughran, T.P. Dysregulation of CD95/CD95 Ligand-Apoptotic Pathway in CD3 Large Granular Lymphocyte Leukemia. Blood 1998, 92, 4771–4777. [Google Scholar] [CrossRef]
- Saitoh, T.; Karasawa, M.; Sakuraya, M.; Norio, N.; Junko, T.; Shirakawa, K.; Matsushima, T.; Tsukamoto, N.; Nojima, Y.; Murakami, H. Improvement of Extrathymic T Cell Type of Large Granular Lymphocyte (LGL) Leukemia by Cyclosporin A: The Serum Level of Fas Ligand Is a Marker of LGL Leukemia Activity. Eur. J. Haematol. 2000, 65, 272–275. [Google Scholar] [CrossRef]
- Liles, W.C.; Kiener, P.A.; Ledbetter, J.A.; Aruffo, A.; Klebanoff, S.J. Differential Expression of Fas (CD95) and Fas Ligand on Normal Human Phagocytes: Implications for the Regulation of Apoptosis in Neutrophils. J. Exp. Med. 1996, 184, 429–440. [Google Scholar] [CrossRef]
- Maciejewski, J.; Selleri, C.; Anderson, S.; Young, N.S. Fas Antigen Expression on CD34+ Human Marrow Cells Is Induced by Interferon Gamma and Tumor Necrosis Factor Alpha and Potentiates Cytokine-Mediated Hematopoietic Suppression in Vitro. Blood 1995, 85, 3183–3190. [Google Scholar] [CrossRef]
- Maciejewski, J.P.; Selleri, C.; Sato, T.; Anderson, S.; Young, N.S. Increased Expression of Fas Antigen on Bone Marrow CD34+ Cells of Patients with Aplastic Anaemia. Br. J. Haematol. 1995, 91, 245–252. [Google Scholar] [CrossRef]
- Hooks, J.J.; Haynes, B.F.; Detrick-Hooks, B.; Diehl, L.F.; Gerrard, T.L.; Fauci, A.S. Gamma (immune) Interferon Production by Leukocytes from a Patient with a TG Cell Proliferative Disease. Blood 1982, 59, 198–201. [Google Scholar] [CrossRef]
- Bank, I.; Cohen, L.; Kneller, A.; De Rosbo, N.K.; Book, M.; Ben-Nun, A. Aberrant T-Cell Receptor Signalling of Interferon-Gamma- and Tumour Necrosis Factor-Alpha-Producing Cytotoxic CD8+ Vdelta1/Vbeta16 T Cells in a Patient with Chronic Neutropenia. Scand. J. Immunol. 2003, 58, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Zambello, R.; Semenzato, G. Large Granular Lymphocyte Disorders: New Etiopathogenetic Clues as a Rationale for Innovative Therapeutic Approaches. Haematologica 2009, 94, 1341–1345. [Google Scholar] [CrossRef] [PubMed]
- Picker, L.J.; Furst, A.; Robinson, S.H.; Kadin, M.E. Immunoarchitecture of the Bone Marrow in Neutropenia: Increased HNK-1 + Cells Define a Subset of Neutropenic Patients. Am. J. Hematol. 1987, 25, 29–41. [Google Scholar] [CrossRef]
- Evans, H.L.; Burks, E.; Viswanatha, D.; Larson, R.S. Utility of Immunohistochemistry in Bone Marrow Evaluation of T-Lineage Large Granular Lymphocyte Leukemia. Hum. Pathol. 2000, 31, 1266–1273. [Google Scholar] [CrossRef]
- Osuji, N.; Beiske, K.; Randen, U.; Matutes, E.; Tjonnfjord, G.; Catovsky, D.; Wotherspoon, A. Characteristic Appearances of the Bone Marrow in T-Cell Large Granular Lymphocyte Leukaemia. Histopathology 2007, 50, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Morice, W.G.; Kurtin, P.J.; Tefferi, A.; Hanson, C.A. Distinct Bone Marrow Findings in T-Cell Granular Lymphocytic Leukemia Revealed by Paraffin Section Immunoperoxidase Stains for CD8, TIA-1, and Granzyme B. Blood 2002, 99, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Coakley, G.; Iqbal, M.; Brooks, D.; Panayi, G.S.; Lanchbury, J.S. CD8+, CD57+ T Cells from Healthy Elderly Subjects Suppress Neutrophil Development in Vitro: Implications for the Neutropenia of Felty’s and Large Granular Lymphocyte Syndromes. Arthritis Rheum. 2000, 43, 834–843. [Google Scholar] [CrossRef]
- Mailloux, A.W.; Zhang, L.; Moscinski, L.; Bennett, J.M.; Yang, L.; Yoder, S.J.; Bloom, G.; Wei, C.; Wei, S.; Sokol, L.; et al. Fibrosis and Subsequent Cytopenias Are Associated with Basic Fibroblast Growth Factor-Deficient Pluripotent Mesenchymal Stromal Cells in Large Granular Lymphocyte Leukemia. J. Immunol. 2013, 191, 3578–3593. [Google Scholar] [CrossRef] [PubMed]
- Zambello, R.; Semenzato, G. Natural Killer Receptors in Patients with Lymphoproliferative Diseases of Granular Lymphocytes. Semin. Hematol. 2003, 40, 201–212. [Google Scholar] [CrossRef]
- Zambello, R.; Falco, M.; Della Chiesa, M.; Trentin, L.; Carollo, D.; Castriconi, R.; Cannas, G.; Carlomagno, S.; Cabrelle, A.; Lamy, T.; et al. Expression and Function of KIR and Natural Cytotoxicity Receptors in NK-Type Lymphoproliferative Diseases of Granular Lymphocytes. Blood 2003, 102, 1797–1805. [Google Scholar] [CrossRef]
- Scquizzato, E.; Teramo, A.; Miorin, M.; Facco, M.; Piazza, F.; Noventa, F.; Trentin, L.; Agostini, C.; Zambello, R.; Semenzato, G. Genotypic Evaluation of Killer Immunoglobulin-like Receptors in NK-Type Lymphoproliferative Disease of Granular Lymphocytes. Leukemia 2007, 21, 1060–1069. [Google Scholar] [CrossRef][Green Version]
- Howe, E.C.; Wlodarski, M.; Ball, E.J.; Rybicki, L.; Maciejewski, J.P. Killer Immunoglobulin-like Receptor Genotype in Immune-Mediated Bone Marrow Failure Syndromes. Exp. Hematol. 2005, 33, 1357–1362. [Google Scholar] [CrossRef]
- Handgretinger, R.; Geiselhart, A.; Moris, A.; Grau, R.; Teuffel, O.; Bethge, W.; Kanz, L.; Fisch, P. Pure Red-Cell Aplasia Associated with Clonal Expansion of Granular Lymphocytes Expressing Killer-Cell Inhibitory Receptors. N. Engl. J. Med. 1999, 340, 278–284. [Google Scholar] [CrossRef]
- Nearman, Z.P.; Wlodarski, M.; Jankowska, A.M.; Howe, E.; Narvaez, Y.; Ball, E.; Maciejewski, J.P. Immunogenetic Factors Determining the Evolution of T-Cell Large Granular Lymphocyte Leukaemia and Associated Cytopenias. Br. J. Haematol. 2007, 136, 237–248. [Google Scholar] [CrossRef]
- Wlodarski, M.W.; O’Keefe, C.; Howe, E.C.; Risitano, A.M.; Rodriguez, A.; Warshawsky, I.; Loughran, T.P., Jr.; Maciejewski, J.P. Pathologic Clonal Cytotoxic T-Cell Responses: Nonrandom Nature of the T-Cell-Receptor Restriction in Large Granular Lymphocyte Leukemia. Blood 2005, 106, 2769–2780. [Google Scholar] [CrossRef]
- Zhang, D.; Loughran, T.P., Jr. Large Granular Lymphocytic Leukemia: Molecular Pathogenesis, Clinical Manifestations, and Treatment. Hematology Am. Soc. Hematol. Educ. Program 2012, 2012, 652–659. [Google Scholar] [CrossRef] [PubMed]
- Teramo, A.; Barilà, G.; Calabretto, G.; Vicenzetto, C.; Gasparini, V.R.; Semenzato, G.; Zambello, R. Insights Into Genetic Landscape of Large Granular Lymphocyte Leukemia. Front. Oncol. 2020, 10, 152. [Google Scholar] [CrossRef] [PubMed]
- Loughran, T.P., Jr.; Zickl, L.; Olson, T.L.; Wang, V.; Zhang, D.; Rajala, H.L.M.; Hasanali, Z.; Bennett, J.M.; Lazarus, H.M.; Litzow, M.R.; et al. Immunosuppressive Therapy of LGL Leukemia: Prospective Multicenter Phase II Study by the Eastern Cooperative Oncology Group (E5998). Leukemia 2015, 29, 886–894. [Google Scholar] [CrossRef] [PubMed]
- Gabrielli, S.; Ortolani, C.; Del Zotto, G.; Luchetti, F.; Canonico, B.; Buccella, F.; Artico, M.; Papa, S.; Zamai, L. The Memories of NK Cells: Innate-Adaptive Immune Intrinsic Crosstalk. J. Immunol. Res. 2016, 2016, 1376595. [Google Scholar] [CrossRef]
- Barilà, G.; Teramo, A.; Calabretto, G.; Ercolin, C.; Boscaro, E.; Trimarco, V.; Carraro, S.; Leoncin, M.; Vicenzetto, C.; Cabrelle, A.; et al. Dominant Cytotoxic NK Cell Subset within CLPD-NK Patients Identifies a More Aggressive NK Cell Proliferation. Blood Cancer J. 2018, 8, 51. [Google Scholar] [CrossRef] [PubMed]
- Mohan, S.R.; Maciejewski, J.P. Diagnosis and Therapy of Neutropenia in Large Granular Lymphocyte Leukemia. Curr. Opin. Hematol. 2009, 16, 27–34. [Google Scholar] [CrossRef]
- Savola, P.; Brück, O.; Olson, T.; Kelkka, T.; Kauppi, M.J.; Kovanen, P.E.; Kytölä, S.; Sokka-Isler, T.; Loughran, T.P.; Leirisalo-Repo, M.; et al. Somatic STAT3 Mutations in Felty Syndrome: An Implication for a Common Pathogenesis with Large Granular Lymphocyte Leukemia. Haematologica 2018, 103, 304–312. [Google Scholar] [CrossRef]
- Komrokji, R.S.; Ali, N.A.; Sallman, D.; Padron, E.; Lancet, J.; Sokol, L.; Varnadoe, C.; Burnette, P.K.; List, A. Characterization of Myelodysplastic Syndromes (MDS) with T-Cell Large Granular Lymphocyte Proliferations (LGL). Leukemia 2020, 34, 3097–3099. [Google Scholar] [CrossRef]
- Durrani, J.; Awada, H.; Kishtagari, A.; Visconte, V.; Kerr, C.; Adema, V.; Nagata, Y.; Kuzmanovic, T.; Hong, S.; Patel, B.; et al. Large Granular Lymphocytic Leukemia Coexists with Myeloid Clones and Myelodysplastic Syndrome. Leukemia 2020, 34, 957–962. [Google Scholar] [CrossRef]
- Jerez, A.; Clemente, M.J.; Makishima, H.; Rajala, H.; Gómez-Seguí, I.; Olson, T.; McGraw, K.; Przychodzen, B.; Kulasekararaj, A.; Afable, M.; et al. STAT3 Mutations Indicate the Presence of Subclinical T-Cell Clones in a Subset of Aplastic Anemia and Myelodysplastic Syndrome Patients. Blood 2013, 122, 2453–2459. [Google Scholar] [CrossRef] [PubMed]
- Cheon, H.; Dziewulska, K.H.; Moosic, K.B.; Olson, K.C.; Gru, A.A.; Feith, D.J.; Loughran, T.P., Jr. Advances in the Diagnosis and Treatment of Large Granular Lymphocytic Leukemia. Curr. Hematol. Malig. Rep. 2020, 15, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Wolfrom, C.M.; Lévy, V.; Deschatrette, J. Neutropenia Dynamics in a Case of T-LGL Lymphoproliferation Illustrate Rapid Turnover of Granulocyte Progenitors. Cell Prolif. 2010, 43, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Lamy, T.; Loughran, T.P. How I Treat LGL Leukemia. Blood 2011, 117, 2764–2774. [Google Scholar] [CrossRef]
- Lamy, T.; Pastoret, C.; Houot, R.; Ysebaert, L.; Hunault, M.; Damaj, G.; Banos, A.; Tournilhac, O.; Choufi, B.; Marolleau, J.-P.; et al. Prospective, Multicentric Phase II Randomized Trial Comparing the Efficacy of Methotrexate or Cyclophosphamide in Large Granular Lymphocytic Leukemia: A French National Study. Report on the Interim Analysis. Blood 2019, 134, 1545. [Google Scholar] [CrossRef]
- Moignet, A.; Hasanali, Z.; Zambello, R.; Pavan, L.; Bareau, B.; Tournilhac, O.; Roussel, M.; Fest, T.; Awwad, A.; Baab, K.; et al. Cyclophosphamide as a First-Line Therapy in LGL Leukemia. Leukemia 2014, 28, 1134–1136. [Google Scholar] [CrossRef]
- Osuji, N.; Matutes, E.; Catovsky, D.; Lampert, I.; Wotherspoon, A. Histopathology of the Spleen in T-Cell Large Granular Lymphocyte Leukemia and T-Cell Prolymphocytic Leukemia: A Comparative Review. Am. J. Surg. Pathol. 2005, 29, 935–941. [Google Scholar] [CrossRef]
- Subbiah, V.; Viny, A.D.; Rosenblatt, S.; Pohlman, B.; Lichtin, A.; Maciejewski, J.P. Outcomes of Splenectomy in T-Cell Large Granular Lymphocyte Leukemia with Splenomegaly and Cytopenia. Exp. Hematol. 2008, 36, 1078–1083. [Google Scholar] [CrossRef]
- Loughran, T.P., Jr.; Starkebaum, G.; Clark, E.; Wallace, P.; Kadin, M.E. Evaluation of Splenectomy in Large Granular Lymphocyte Leukaemia. Br. J. Haematol. 1987, 67, 135–140. [Google Scholar] [CrossRef]
- Dumitriu, B.; Ito, S.; Feng, X.; Stephens, N.; Yunce, M.; Kajigaya, S.; Melenhorst, J.J.; Rios, O.; Scheinberg, P.; Chinian, F.; et al. Alemtuzumab in T-Cell Large Granular Lymphocytic Leukaemia: Interim Results from a Single-Arm, Open-Label, Phase 2 Study. Lancet Haematol 2016, 3, e22–e29. [Google Scholar] [CrossRef]
- Cornec, D.; Devauchelle-Pensec, V.; Jousse-Joulin, S.; Marhadour, T.; Ugo, V.; Berthou, C.; Douet-Guilbert, N.; Saraux, A. Long-Term Remission of T-Cell Large Granular Lymphocyte Leukemia Associated with Rheumatoid Arthritis after Rituximab Therapy. Blood 2013, 122, 1583–1586. [Google Scholar] [CrossRef]
- Bilori, B.; Thota, S.; Clemente, M.J.; Patel, B.; Jerez, A.; Afable, M., II; Maciejewski, J.P. Tofacitinib as a Novel Salvage Therapy for Refractory T-Cell Large Granular Lymphocytic Leukemia. Leukemia 2015, 29, 2427–2429. [Google Scholar] [CrossRef]
- Poh, C.; Arora, M.; Ghuman, S.; Tuscano, J. Belinostat in Relapsed/Refractory T-Cell Large Granular Lymphocyte Leukemia. Acta Haematol. 2021, 144, 95–99. [Google Scholar] [CrossRef]
- Hodge, D.L.; Yang, J.; Buschman, M.D.; Schaughency, P.M.; Dang, H.; Bere, W.; Yang, Y.; Savan, R.; Subleski, J.J.; Yin, X.-M.; et al. Interleukin-15 Enhances Proteasomal Degradation of Bid in Normal Lymphocytes: Implications for Large Granular Lymphocyte Leukemias. Cancer Res. 2009, 69, 3986–3994. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; LeBlanc, F.R.; Dighe, S.A.; Hamele, C.E.; Olson, T.L.; Feith, D.J.; Loughran, T.P., Jr. TRAIL Mediates and Sustains Constitutive NF-κB Activation in LGL Leukemia. Blood 2018, 131, 2803–2815. [Google Scholar] [CrossRef]
- Pelliccia, S.; Di Napoli, A.; Naso, V.; Alma, E.; Rebecchini, C.; Cox, M.C. Very Long-Lasting Remission of Refractory T-Large Granular Lymphocytes Leukemia and Myeloma by Lenalidomide Treatment. Eur. J. Haematol. 2013, 91, 183–186. [Google Scholar] [CrossRef] [PubMed]
- Frohna, P.; Tagaya, Y.; Ratnayake, A.; Doerr, N.; Basheer, A.; Al-Mawsawi, L.; Kim, W.J.; Zapata, J.; Wu, X.; Azimi, N. Results from a First-in-Human Study with Bnz-1: A Novel Peptide Inhibitor of IL-2, IL-9 and IL-15 for the Treatment of T-Cell Malignancies That Safely and Selectively Decreases Regulatory T-Cells, Natural Killer Cells, and CD8 Central Memory T-Cells. Blood 2017, 130, 695. [Google Scholar] [CrossRef]
- Bajan, S.; Hutvagner, G. RNA-Based Therapeutics: From Antisense Oligonucleotides to miRNAs. Cells 2020, 9, 137. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.-Y.; Qin, Z.; Zhu, Y.-H.; He, Z.-Y.; Xu, T. Current RNA-Based Therapeutics in Clinical Trials. Curr. Gene Ther. 2019, 19, 172–196. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CD3 | CD8 | CD4 | CD16 | CD56 | CD57 | Neutropenia | |
|---|---|---|---|---|---|---|---|
| T-LGLL | + | + | - | + | - | ± | Frequent |
| + | + | - | - | - | + | Rare | |
| + | + | - | - | + | + | Rare | |
| + | + | - | + | + | + | Rare | |
| + | + | - | - | + | - | Rare | |
| + | -/Dim | + | ± | + | + | Rare | |
| CLPD-NK | - | ± | - | Bright | -/Dim | - | Frequent |
| - | ± | - | Bright | -/Dim | + | Rare | |
| - | ± | - | Dim | Dim | ± | Rare |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Calabretto, G.; Teramo, A.; Barilà, G.; Vicenzetto, C.; Gasparini, V.R.; Semenzato, G.; Zambello, R. Neutropenia and Large Granular Lymphocyte Leukemia: From Pathogenesis to Therapeutic Options. Cells 2021, 10, 2800. https://doi.org/10.3390/cells10102800
Calabretto G, Teramo A, Barilà G, Vicenzetto C, Gasparini VR, Semenzato G, Zambello R. Neutropenia and Large Granular Lymphocyte Leukemia: From Pathogenesis to Therapeutic Options. Cells. 2021; 10(10):2800. https://doi.org/10.3390/cells10102800
Chicago/Turabian StyleCalabretto, Giulia, Antonella Teramo, Gregorio Barilà, Cristina Vicenzetto, Vanessa Rebecca Gasparini, Gianpietro Semenzato, and Renato Zambello. 2021. "Neutropenia and Large Granular Lymphocyte Leukemia: From Pathogenesis to Therapeutic Options" Cells 10, no. 10: 2800. https://doi.org/10.3390/cells10102800
APA StyleCalabretto, G., Teramo, A., Barilà, G., Vicenzetto, C., Gasparini, V. R., Semenzato, G., & Zambello, R. (2021). Neutropenia and Large Granular Lymphocyte Leukemia: From Pathogenesis to Therapeutic Options. Cells, 10(10), 2800. https://doi.org/10.3390/cells10102800