
The Candidate IBD Risk Gene CCNY Is Dispensable for Intestinal Epithelial Homeostasis


Abstract
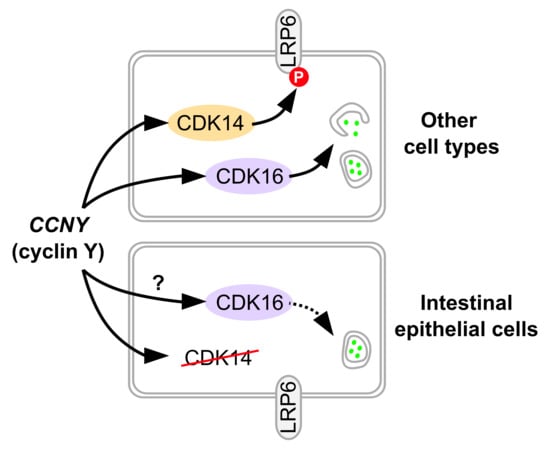
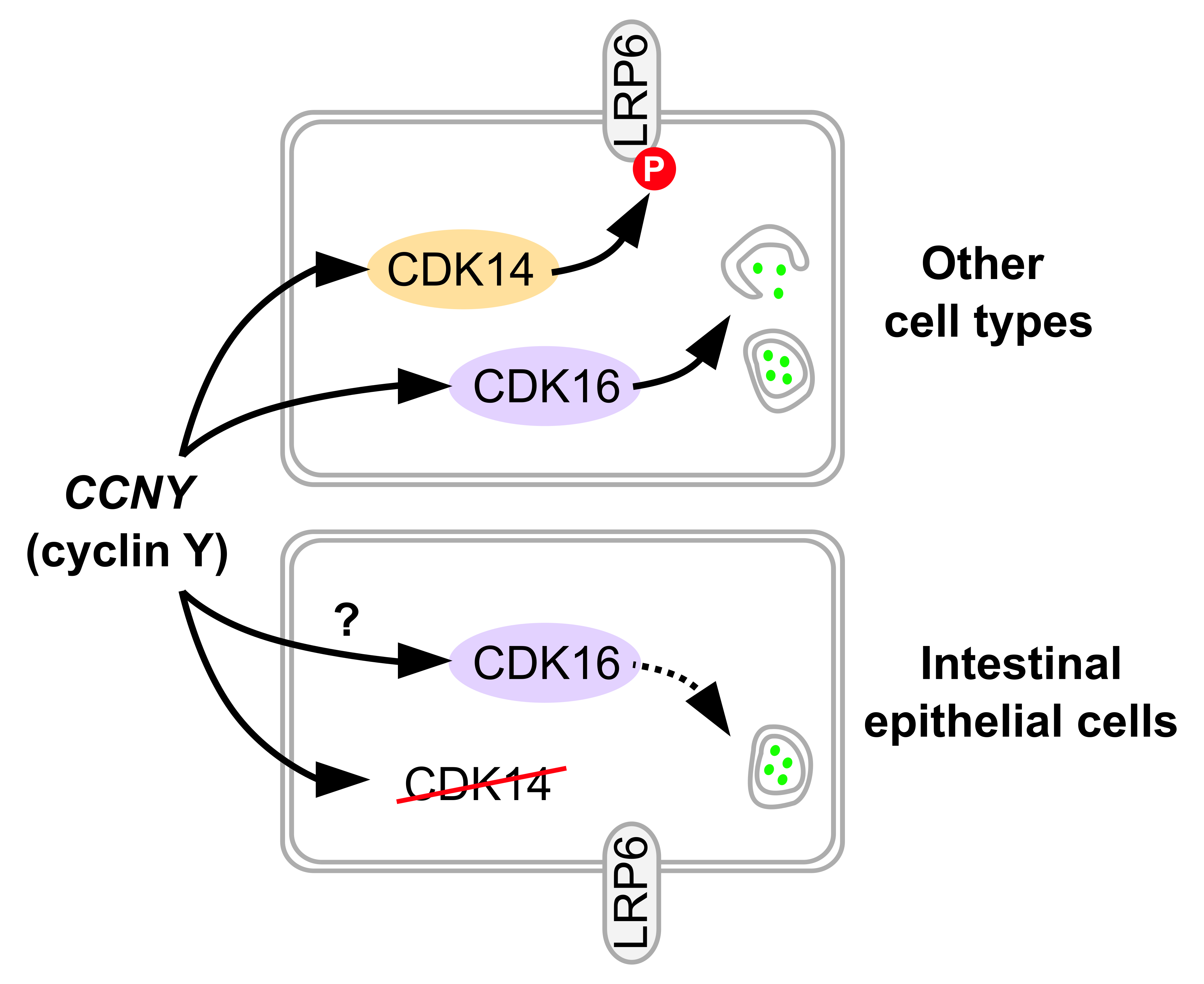
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Dextran Sulfate Sodium (DSS) Colitis
2.3. Cell Culture and siRNA Transfection
2.4. Immunoblotting
2.5. Immunostaining
2.6. MTT Assay
2.7. Luciferase Reporter Assay
2.8. Analysis of Public Datasets
2.9. Statistics and Data Analysis
3. Results
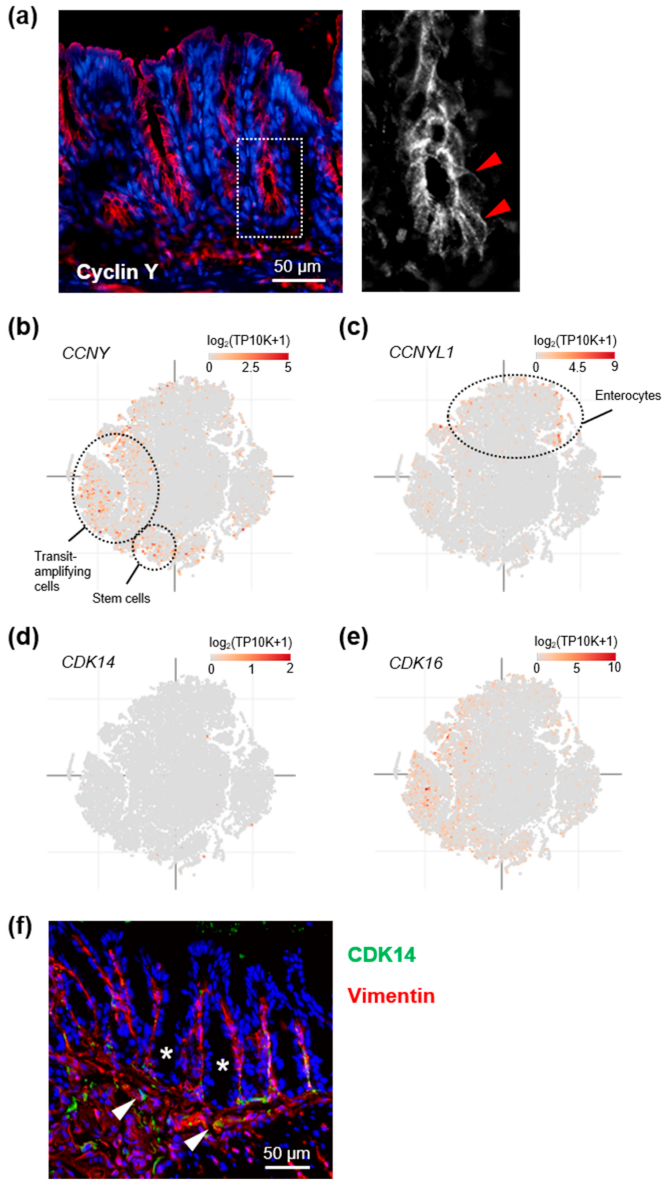
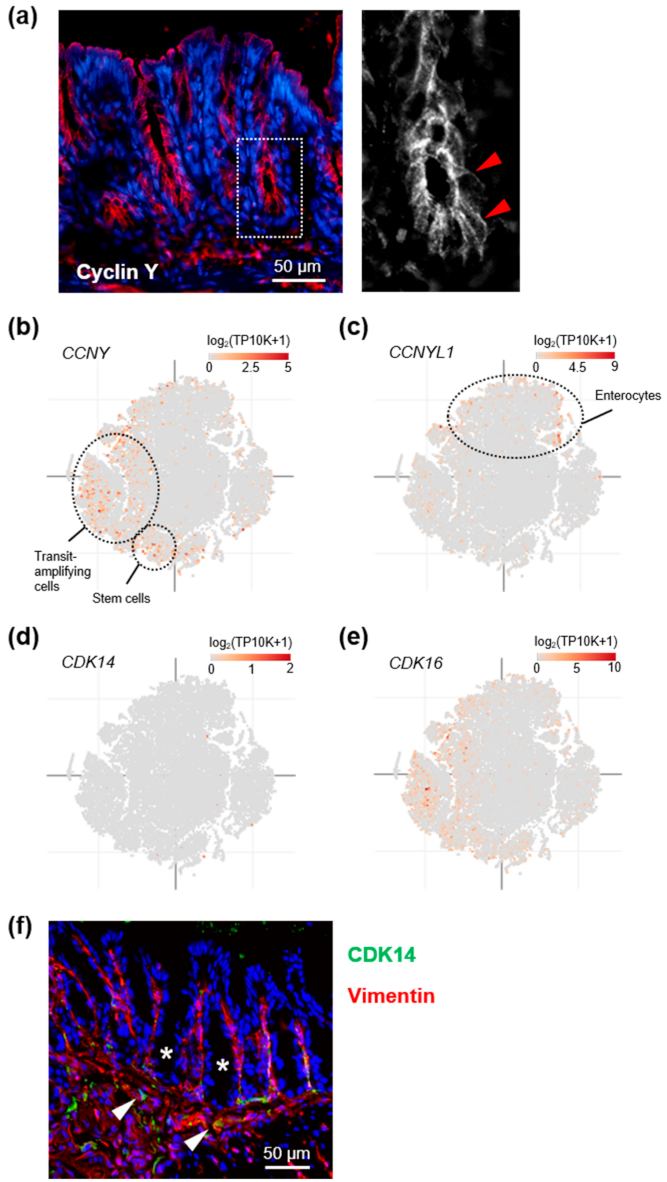
3.1. Cyclin Y Is Expressed in Crypt Base Intestinal Epithelial Cells
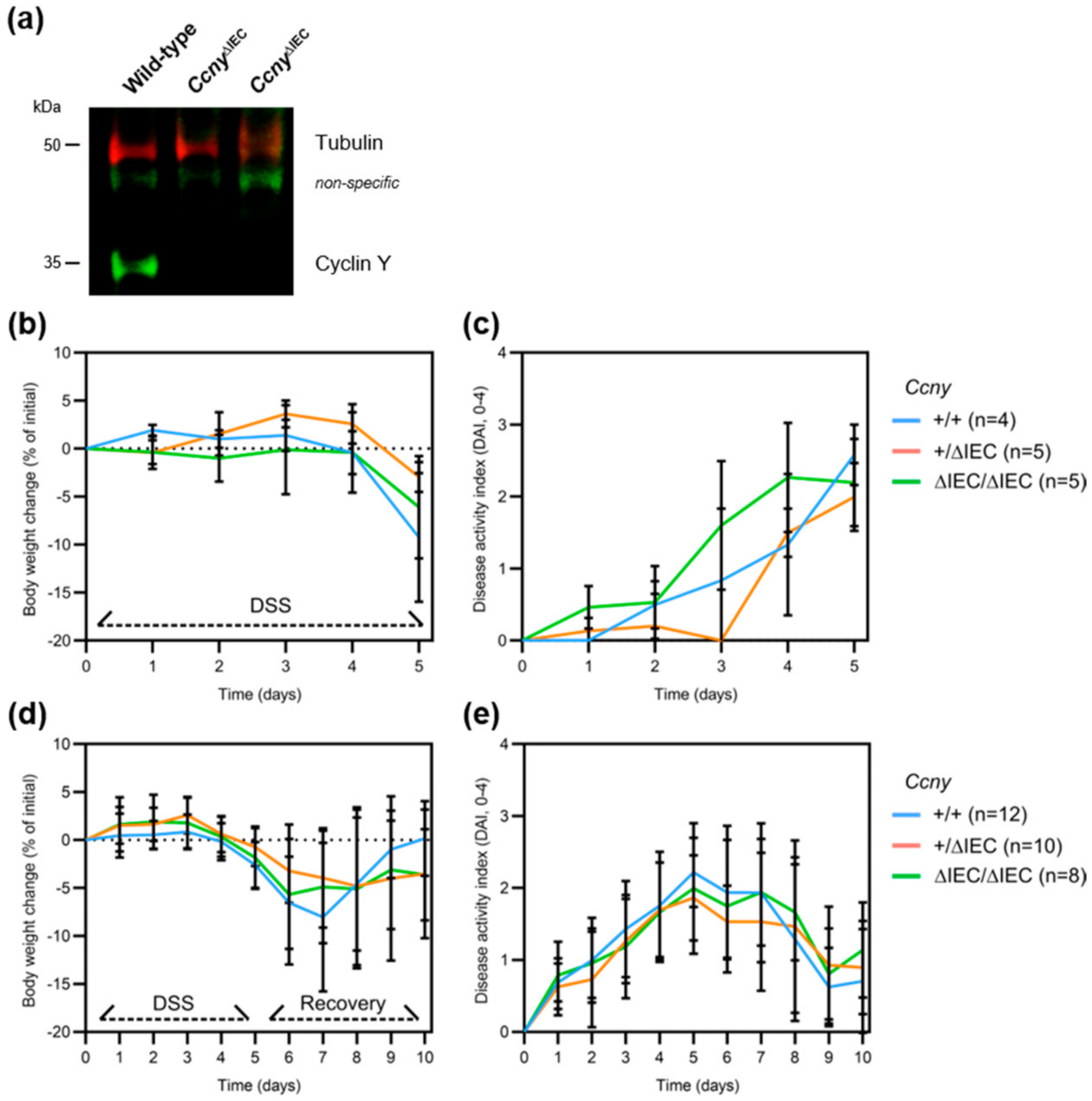
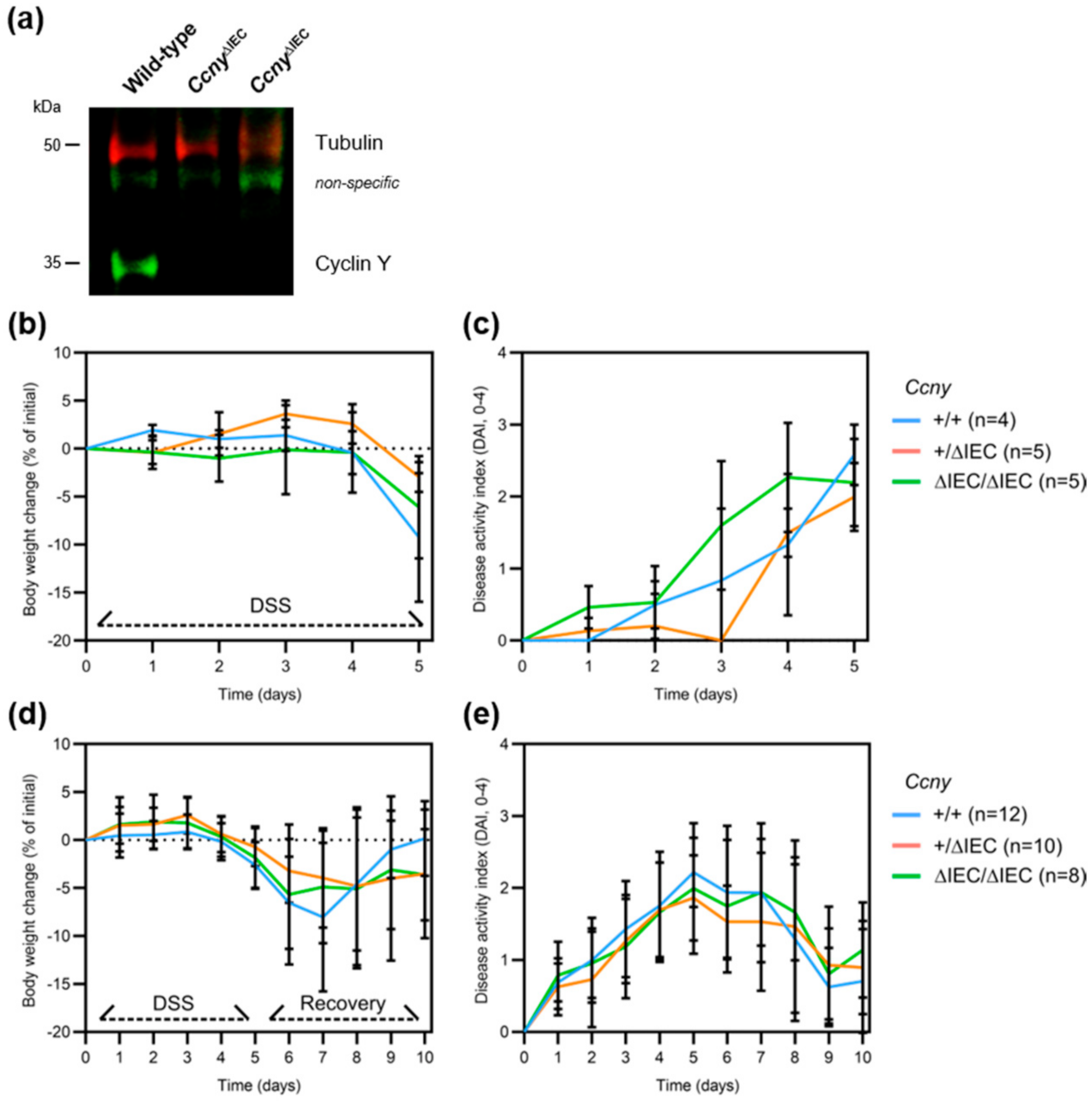
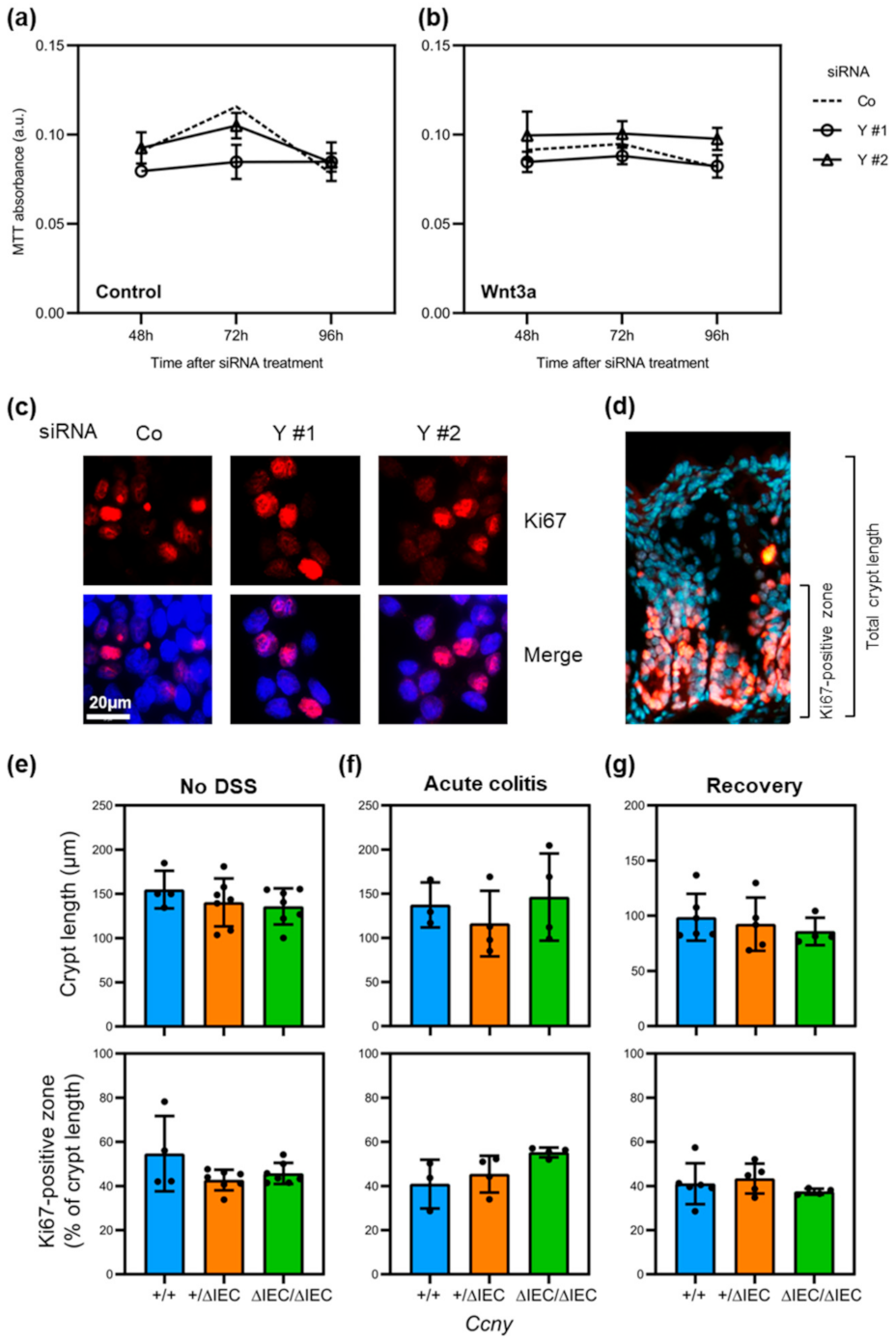
3.2. Loss of Ccny Does Not Exacerbate Experimental Colitis or Impair Injury Repair in Mice
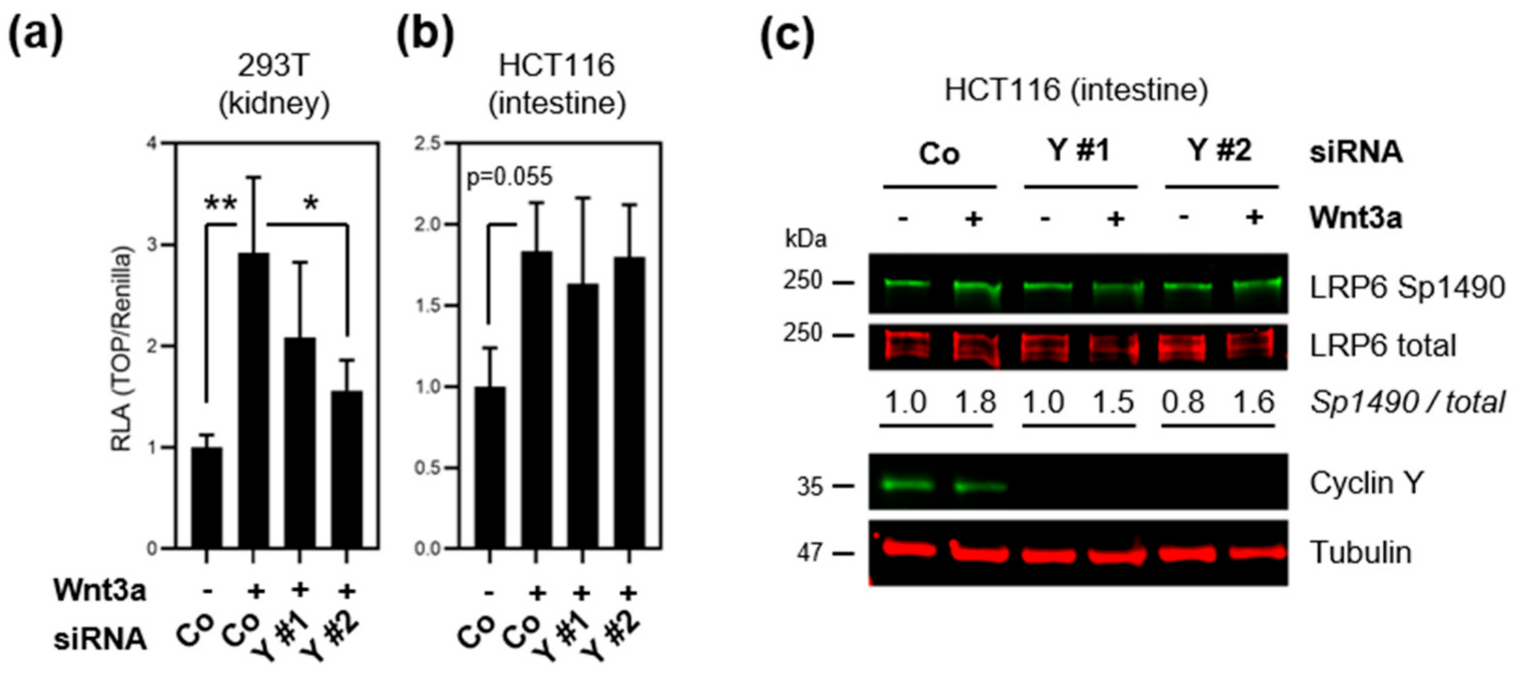
3.3. Loss of Cyclin Y Does Not Affect Wnt Signaling in Colorectal Cancer Cells
3.4. Loss of Cyclin Y Does Not Affect Cell Proliferation in Intestinal Epithelial Cells
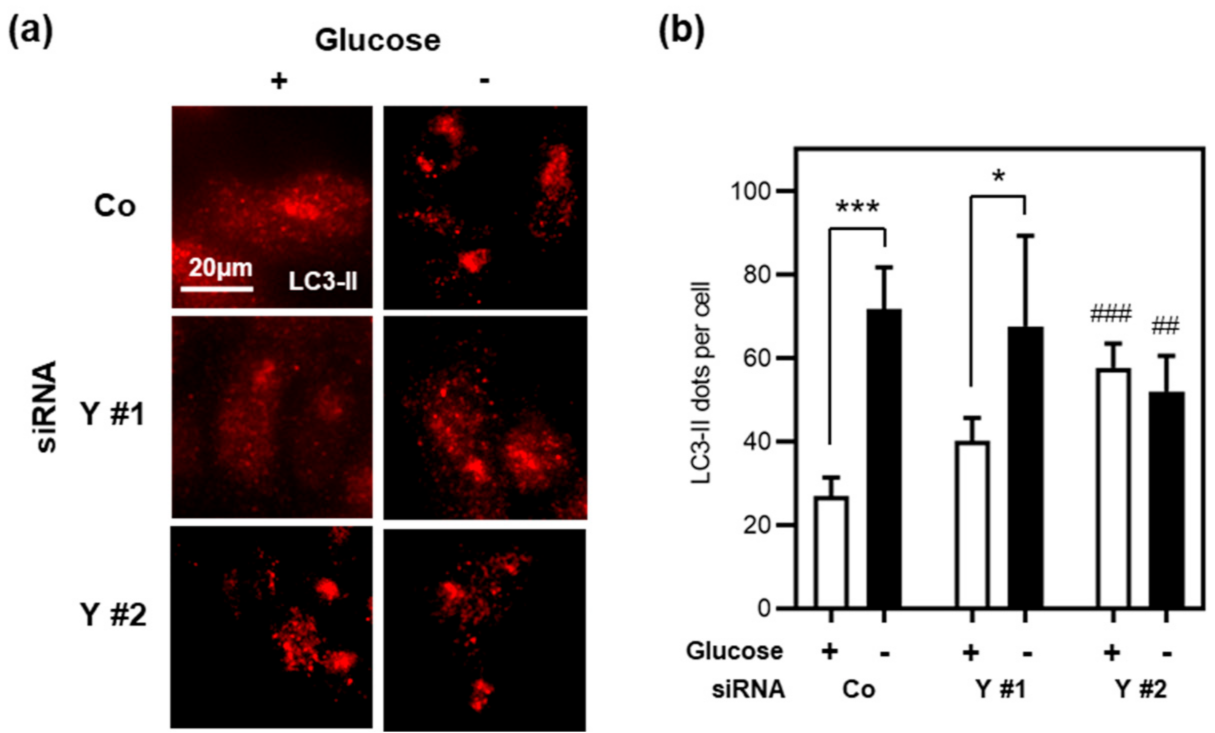
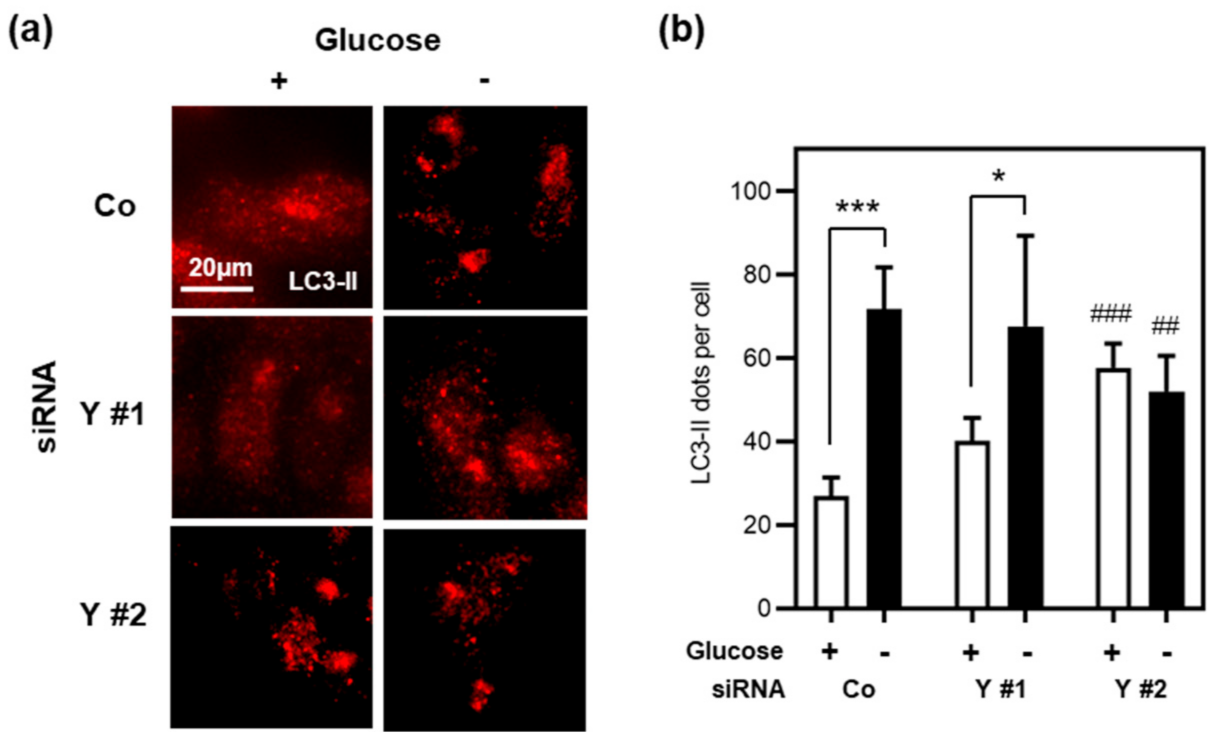
3.5. Cyclin Y Is Not Required for Autophagy in Stressed Intestinal Epithelial Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ng, S.C.; Shi, H.Y.; Hamidi, N.; Underwood, F.E.; Tang, W.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Wu, J.C.Y.; Chan, F.K.L.; et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: A systematic review of population-based studies. Lancet 2018, 390, 2769–2778. [Google Scholar] [CrossRef]
- Huang, H.; Fang, M.; Jostins, L.; Umićević Mirkov, M.; Boucher, G.; Anderson, C.A.; Andersen, V.; Cleynen, I.; Cortes, A.; Crins, F.; et al. Fine-mapping inflammatory bowel disease loci to single-variant resolution. Nature 2017, 547, 173–178. [Google Scholar] [CrossRef] [Green Version]
- Franke, A.; Balschun, T.; Karlsen, T.H.; Hedderich, J.; May, S.; Lu, T.; Schuldt, D.; Nikolaus, S.; Rosenstiel, P.; Krawczak, M.; et al. Replication of signals from recent studies of Crohn’s disease identifies previously unknown disease loci for ulcerative colitis. Nat. Genet. 2008, 40, 713–715. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.A.; Massey, D.C.; Barrett, J.C.; Prescott, N.J.; Tremelling, M.; Fisher, S.A.; Gwilliam, R.; Jacob, J.; Nimmo, E.R.; Drummond, H.; et al. Investigation of Crohn’s disease risk loci in ulcerative colitis further defines their molecular relationship. Gastroenterology 2009, 136, 523–529. [Google Scholar] [CrossRef] [Green Version]
- Weersma, R.K.; Stokkers, P.C.; Cleynen, I.; Wolfkamp, S.C.; Henckaerts, L.; Schreiber, S.; Dijkstra, G.; Franke, A.; Nolte, I.M.; Rutgeerts, P.; et al. Confirmation of multiple Crohn’s disease susceptibility loci in a large Dutch-Belgian cohort. Am. J. Gastroenterol. 2009, 104, 630–638. [Google Scholar] [CrossRef]
- Anderson, C.A.; Boucher, G.; Lees, C.W.; Franke, A.; D’Amato, M.; Taylor, K.D.; Lee, J.C.; Goyette, P.; Imielinski, M.; Latiano, A. Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat. Genet. 2011, 43, 246–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waterman, M.; Xu, W.; Stempak, J.M.; Milgrom, R.; Bernstein, C.N.; Griffiths, A.M.; Greenberg, G.R.; Steinhart, H.A.; Silverberg, M.S. Distinct and overlapping genetic loci in Crohn’s disease and ulcerative colitis: Correlations with pathogenesis. Inflamm. Bowel Dis. 2011, 17, 1936–1942. [Google Scholar] [CrossRef] [Green Version]
- An, W.; Zhang, Z.; Zeng, L.; Yang, Y.; Zhu, X.; Wu, J. Cyclin Y is involved in the regulation of adipogenesis and lipid production. PLoS ONE 2015, 10, e0132721. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Cai, C.; Li, S.; Wang, W.; Li, Y.; Chen, J.; Zhu, X.; Zeng, Y.A. Essential roles of cyclin Y-like 1 and cyclin Y in dividing Wnt-responsive mammary stem/progenitor cells. PLoS Genet. 2016, 12, e1006055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Silva, F.; Zhang, K.; Pinson, A.; Fatti, E.; Wilsch-Bräuninger, M.; Herbst, J.; Vidal, V.; Schedl, A.; Huttner, W.B.; Niehrs, C. Mitotic WNT signalling orchestrates neurogenesis in the developing neocortex. EMBO J. 2021, e108041. [Google Scholar] [CrossRef]
- Davidson, G.; Shen, J.; Huang, Y.L.; Su, Y.; Karaulanov, E.; Bartscherer, K.; Hassler, C.; Stannek, P.; Boutros, M.; Niehrs, C. Cell cycle control of wnt receptor activation. Dev. Cell 2009, 17, 788–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch, S.; Acebron, S.P.; Herbst, J.; Hatiboglu, G.; Niehrs, C. Post-transcriptional Wnt signaling governs epididymal sperm maturation. Cell 2015, 163, 1225–1236. [Google Scholar] [CrossRef] [Green Version]
- Moparthi, L.; Koch, S. Wnt signaling in intestinal inflammation. Differentiation 2019, 108, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Clevers, H. Reparative inflammation takes charge of tissue regeneration. Nature 2016, 529, 307–315. [Google Scholar] [CrossRef]
- Raisch, J.; Cote-Biron, A.; Langlois, M.J.; Leblanc, C.; Rivard, N. Unveiling the roles of low-density lipoprotein receptor-related protein 6 in intestinal homeostasis, regeneration and oncogenesis. Cells 2021, 10, 1792. [Google Scholar] [CrossRef] [PubMed]
- Dohmen, M.; Krieg, S.; Agalaridis, G.; Zhu, X.; Shehata, S.N.; Pfeiffenberger, E.; Amelang, J.; Butepage, M.; Buerova, E.; Pfaff, C.M.; et al. AMPK-dependent activation of the Cyclin Y/CDK16 complex controls autophagy. Nat. Commun. 2020, 11, 1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in immunity and inflammation. Nature 2011, 469, 323–335. [Google Scholar] [CrossRef]
- Nava, P.; Koch, S.; Laukoetter, M.G.; Lee, W.Y.; Kolegraff, K.; Capaldo, C.T.; Beeman, N.; Addis, C.; Gerner-Smidt, K.; Neumaier, I.; et al. Interferon-gamma regulates intestinal epithelial homeostasis through converging beta-catenin signaling pathways. Immunity 2010, 32, 392–402. [Google Scholar] [CrossRef] [Green Version]
- Cooper, H.S.; Murthy, S.; Shah, R.; Sedergran, D. Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Lab. Investig. 1993, 69, 238–249. [Google Scholar]
- Garcia-Hernandez, V.; Neumann, P.A.; Koch, S.; Lyons, R.; Nusrat, A.; Parkos, C.A. Systematic scoring analysis for intestinal inflammation in a murine dextran sodium sulfate-induced colitis model. J. Vis. Exp. 2021. [Google Scholar] [CrossRef]
- Moparthi, L.; Koch, S. A uniform expression library for the exploration of FOX transcription factor biology. Differentiation 2020, 115, 30–36. [Google Scholar] [CrossRef]
- Smillie, C.S.; Biton, M.; Ordovas-Montanes, J.; Sullivan, K.M.; Burgin, G.; Graham, D.B.; Herbst, R.H.; Rogel, N.; Slyper, M.; Waldman, J.; et al. Intra- and inter-cellular rewiring of the human colon during ulcerative colitis. Cell 2019, 178, 714–730. [Google Scholar] [CrossRef]
- Haberman, Y.; Karns, R.; Dexheimer, P.J.; Schirmer, M.; Somekh, J.; Jurickova, I.; Braun, T.; Novak, E.; Bauman, L.; Collins, M.H.; et al. Ulcerative colitis mucosal transcriptomes reveal mitochondriopathy and personalized mechanisms underlying disease severity and treatment response. Nat. Commun. 2019, 10, 38. [Google Scholar] [CrossRef] [Green Version]
- Klijn, C.; Durinck, S.; Stawiski, E.W.; Haverty, P.M.; Jiang, Z.; Liu, H.; Degenhardt, J.; Mayba, O.; Gnad, F.; Liu, J.; et al. A comprehensive transcriptional portrait of human cancer cell lines. Nat. Biotechnol. 2015, 33, 306–312. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Zi, Z.; Zhang, Z.; Li, Q.; An, W.; Zeng, L.; Gao, D.; Yang, Y.; Zhu, X.; Zeng, R.; Shum, W.W.; et al. CCNYL1, but not CCNY, cooperates with CDK16 to regulate spermatogenesis in mouse. PLoS Genet. 2015, 11, e1005485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khounlotham, M.; Kim, W.; Peatman, E.; Nava, P.; Medina-Contreras, O.; Addis, C.; Koch, S.; Fournier, B.; Nusrat, A.; Denning, T.L.; et al. Compromised intestinal epithelial barrier induces adaptive immune compensation that protects from colitis. Immunity 2012, 37, 563–573. [Google Scholar] [CrossRef] [Green Version]
- Koch, S.; Nava, P.; Addis, C.; Kim, W.; Denning, T.L.; Li, L.; Parkos, C.A.; Nusrat, A. The Wnt antagonist Dkk1 regulates intestinal epithelial homeostasis and wound repair. Gastroenterology 2011, 141, 259–268. [Google Scholar] [CrossRef] [Green Version]
- Miyoshi, H.; Ajima, R.; Luo, C.T.; Yamaguchi, T.P.; Stappenbeck, T.S. Wnt5a potentiates TGF-β signaling to promote colonic crypt regeneration after tissue injury. Science 2012, 338, 108–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinto, D.; Clevers, H. Wnt, stem cells and cancer in the intestine. Biol. Cell 2005, 97, 185–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voloshanenko, O.; Erdmann, G.; Dubash, T.D.; Augustin, I.; Metzig, M.; Moffa, G.; Hundsrucker, C.; Kerr, G.; Sandmann, T.; Anchang, B.; et al. Wnt secretion is required to maintain high levels of Wnt activity in colon cancer cells. Nat. Commun. 2013, 4, 2610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Shi, H.; Fan, Q.; Sun, X. Cyclin Y regulates the proliferation, migration, and invasion of ovarian cancer cells via Wnt signaling pathway. Tumor Biol. 2016, 37, 10161–10175. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Co, N.N.; Wong, N. PFTK1 interacts with cyclin Y to activate non-canonical Wnt signaling in hepatocellular carcinoma. Biochem. Biophys. Res. Commun. 2014, 449, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Stanyon, C.A.; Liu, G.; Mangiola, B.A.; Patel, N.; Giot, L.; Kuang, B.; Zhang, H.; Zhong, J.; Finley, R.L. A Drosophila protein-interaction map centered on cell-cycle regulators. Genome Biol. 2004, 5, R96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, X.; Wang, Y.; Wang, H.; Ni, Q.; Zhang, C.; Zhu, J.; Huang, W.; Xu, P.; Mao, G.; Yang, S. Upregulated PFTK1 promotes tumor cell proliferation, migration, and invasion in breast cancer. Med. Oncol. 2015, 32, 195. [Google Scholar] [CrossRef]
- Shu, F.; Lv, S.; Qin, Y.; Ma, X.; Wang, X.; Peng, X.; Luo, Y.; Xu, B.E.; Sun, X.; Wu, J. Functional characterization of human PFTK1 as a cyclin-dependent kinase. Proc. Natl. Acad. Sci. USA 2007, 104, 9248–9253. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Wang, Y.; Jiang, W.; Ni, R.; Wang, Y.; Ni, S. CDK14 involvement in proliferation migration and invasion of esophageal cancer. Ann. Transl. Med. 2019, 7, 681. [Google Scholar] [CrossRef] [PubMed]
- Mikolcevic, P.; Sigl, R.; Rauch, V.; Hess, M.W.; Pfaller, K.; Barisic, M.; Pelliniemi, L.J.; Boesl, M.; Geley, S. Cyclin-dependent kinase 16/PCTAIRE kinase 1 is activated by cyclin Y and is essential for spermatogenesis. Mol. Cell. Biol. 2012, 32, 868–879. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Jia, Y.; Fei, C.; Song, X.; Li, L. Activation/proliferation-associated protein 2 (caprin-2) positively regulates CDK14/cyclin Y-mediated lipoprotein receptor-related protein 5 and 6 (LRP5/6) constitutive phosphorylation. J. Biol. Chem. 2016, 291, 26427–26434. [Google Scholar] [CrossRef] [Green Version]
- Lauzier, A.; Normandeau-Guimond, J.; Vaillancourt-Lavigueur, V.; Boivin, V.; Charbonneau, M.; Rivard, N.; Scott, M.S.; Dubois, C.M.; Jean, S. Colorectal cancer cells respond differentially to autophagy inhibition in vivo. Sci. Rep. 2019, 9, 11316. [Google Scholar] [CrossRef] [Green Version]
- Cadwell, K.; Liu, J.Y.; Brown, S.L.; Miyoshi, H.; Loh, J.; Lennerz, J.K.; Kishi, C.; Kc, W.; Carrero, J.A.; Hunt, S.; et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 2008, 456, 259–263. [Google Scholar] [CrossRef] [Green Version]
- Loddo, I.; Romano, C. Inflammatory bowel disease: Genetics, epigenetics, and pathogenesis. Front. Immunol. 2015, 6, 551. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Molinas, A.; Heil, S.; Koch, S. The Candidate IBD Risk Gene CCNY Is Dispensable for Intestinal Epithelial Homeostasis. Cells 2021, 10, 2330. https://doi.org/10.3390/cells10092330
Molinas A, Heil S, Koch S. The Candidate IBD Risk Gene CCNY Is Dispensable for Intestinal Epithelial Homeostasis. Cells. 2021; 10(9):2330. https://doi.org/10.3390/cells10092330
Chicago/Turabian StyleMolinas, Andrea, Stéphanie Heil, and Stefan Koch. 2021. "The Candidate IBD Risk Gene CCNY Is Dispensable for Intestinal Epithelial Homeostasis" Cells 10, no. 9: 2330. https://doi.org/10.3390/cells10092330
APA StyleMolinas, A., Heil, S., & Koch, S. (2021). The Candidate IBD Risk Gene CCNY Is Dispensable for Intestinal Epithelial Homeostasis. Cells, 10(9), 2330. https://doi.org/10.3390/cells10092330