
The TGF-β/NADPH Oxidases Axis in the Regulation of Liver Cell Biology in Health and Disease



Abstract
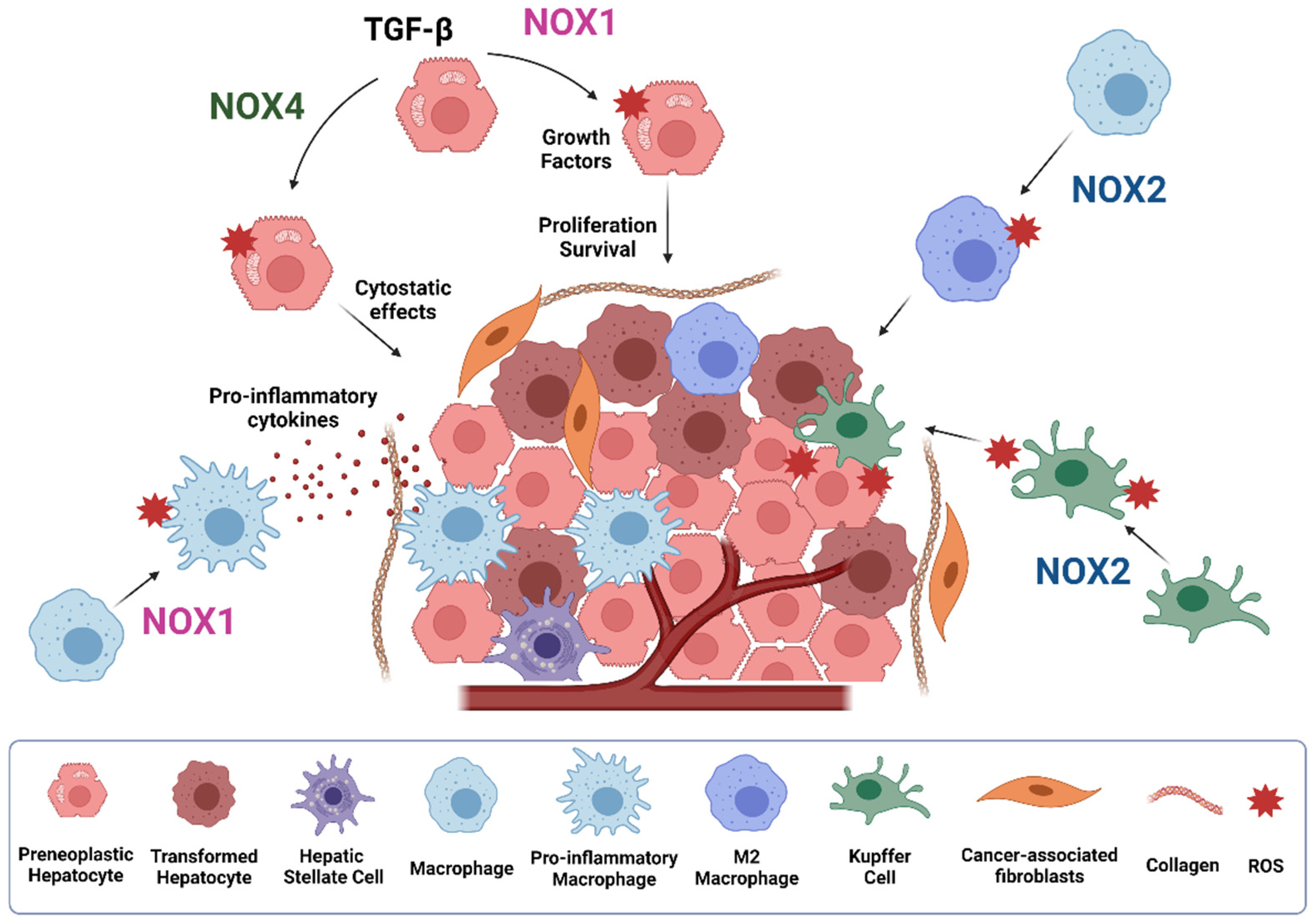
1. Introduction
2. NADPH Oxidase Family Members
2.1. Structural Properties
2.2. Subcellular Localization of NOXs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Type | Type of NOX | References |
|---|---|---|
| Hepatocyte | NOX1, NOX2, NOX4, DUOX1, DUOX2 | [33] |
| HSC * | NOX1, NOX2, NOX4 | [33,34] |
| EC * | NOX1, NOX2, NOX4, DUOX2 | [33,34,35,36] |
| KC * | NOX2 | [34,37] |
| Liver Infiltrating Leukocyte | NOX1, NOX2 | [34,38] |
2.2.1. NOX1 and NOX2
2.2.2. NOX4
3. TGF-β in Human Liver Physiology and Pathology
4. Role of the TGF-β/NOX Axis in Liver Regeneration
5. Role of TGF-β/NOX Axis in Liver Fibrosis
6. Role of NOXs in HCC
6.1. NOXs in HCC
6.2. TGF-β, NOXs and HCC
7. NOX Inhibitors as Therapeutic Tools in Liver Diseases
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fabregat, I.; Moreno-Càceres, J.; Sánchez, A.; Dooley, S.; Dewidar, B.; Giannelli, G.; ten Dijke, P.; the IT-LIVER Consortium. TGF-β Signalling and Liver Disease. FEBS J. 2016, 283, 2219–2232. [Google Scholar] [CrossRef]
- Katsuno, Y.; Derynck, R. Epithelial Plasticity, Epithelial-Mesenchymal Transition, and the TGF-β Family. Dev. Cell 2021, 56, 726–746. [Google Scholar] [CrossRef]
- Vermot, A.; Petit-Härtlein, I.; Smith, S.M.E.; Fieschi, F. NADPH Oxidases (NOX): An Overview from Discovery, Molecular Mechanisms to Physiology and Pathology. Antioxidants 2021, 10, 890. [Google Scholar] [CrossRef]
- Crosas-Molist, E.; Bertran, E.; Fabregat, I. Cross-Talk between TGF-β and NADPH Oxidases during Liver Fibrosis and Hepatocarcinogenesis. Curr. Pharm. Des. 2015, 21, 5964–5976. [Google Scholar] [CrossRef]
- Babior, B.; Lambeth, J.D.; Nauseef, W.M. The Neutrophil NADPH Oxidase. Arch. Biochem. Biophys. 2002, 397, 342–344. [Google Scholar] [CrossRef] [PubMed]
- Lambeth, J.D.; Neish, A.S. Nox Enzymes and New Thinking on Reactive Oxygen: A Double-Edged Sword Revisited. Annu. Rev. Pathol. Mech. Dis. 2014, 9, 119–145. [Google Scholar] [CrossRef] [PubMed]
- Waghela, B.N.; Vaidya, F.U.; Agrawal, Y.; Santra, M.K.; Mishra, V.; Pathak, C. Molecular Insights of NADPH Oxidases and Its Pathological Consequences. Cell Biochem. Funct. 2021, 39, 218–234. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Krause, K.-H. The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Buvelot, H.; Jaquet, V.; Krause, K.-H. Mammalian NADPH Oxidases. In NADPH Oxidases: Methods and Protocols; Methods in Molecular, Biology; Knaus, U., Leto, T., Eds.; Springer: New York, NY, USA, 2019; Volume 1982, pp. 17–36. [Google Scholar] [CrossRef]
- Nakano, Y.; Banfi, B.; Jesaitis, A.J.; Dinauer, M.C.; Allen, L.-A.H.; Nauseef, W.M. Critical Roles for P22phox in the Structural Maturation and Subcellular Targeting of Nox3. Biochem. J. 2007, 403, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Miyano, K.; Sumimoto, H. N-Linked Glycosylation of the Superoxide-Producing NADPH Oxidase Nox1. Biochem. Biophys. Res. Commun. 2014, 443, 1060–1065. [Google Scholar] [CrossRef] [PubMed]
- Dinauer, M.C.; Pierce, E.A.; Erickson, R.W.; Muhlebach, T.J.; Messner, H.; Orkin, S.H.; Seger, R.A.; Curnutte, J.T. Point Mutation in the Cytoplasmic Domain of the Neutrophil P22-Phox Cytochrome b Subunit Is Associated with a Nonfunctional NADPH Oxidase and Chronic Granulomatous Disease. Proc. Natl. Acad. Sci. USA 1991, 88, 11231–11235. [Google Scholar] [CrossRef] [PubMed]
- Knaus, U.; Heyworth, P.; Evans, T.; Curnutte, J.; Bokoch, G. Regulation of Phagocyte Oxygen Radical Production by the GTP-Binding Protein Rac 2. Science 1991, 254, 1512–1515. [Google Scholar] [CrossRef] [PubMed]
- Abo, A.; Pick, E.; Hall, A.; Totty, N.; Teahan, C.; Segal, A.W. Activation of the NADPH Oxidase Involves the Small GTP-Binding Protein P21rac1. Nature 1991, 353, 668–670. [Google Scholar] [CrossRef]
- Volpp, B.D.; Nauseef, W.M.; Clark, R.A. Two Cytosolic Neutrophil Oxidase Components Absent in Autosomal Chronic Granulomatous Disease. Sci. New Ser. 1988, 242, 1295–1297. [Google Scholar] [CrossRef] [PubMed]
- Wientjes, F.B.; Hsuan, J.; Totty, N.; Segal, A.W. P4OPhox, a Third Cytosolic Component of the Activation Complex of the NADPH Oxidase to Contain Src Homology 3 Domains. Biochem. J. 1993, 296, 5. [Google Scholar] [CrossRef]
- Bánfi, B.; Clark, R.A.; Steger, K.; Krause, K.-H. Two Novel Proteins Activate Superoxide Generation by the NADPH Oxidase NOX1. J. Biol. Chem. 2003, 278, 3510–3513. [Google Scholar] [CrossRef]
- Geiszt, M.; Lekstrom, K.; Witta, J.; Leto, T.L. Proteins Homologous to P47 and P67 Support Superoxide Production by NAD(P)H Oxidase 1 in Colon Epithelial Cells. J. Biol. Chem. 2003, 278, 20006–20012. [Google Scholar] [CrossRef]
- Takeya, R.; Ueno, N.; Kami, K.; Taura, M.; Kohjima, M.; Izaki, T.; Nunoi, H.; Sumimoto, H. Novel Human Homologues of P47 and P67 Participate in Activation of Superoxide-Producing NADPH Oxidases. J. Biol. Chem. 2003, 278, 25234–25246. [Google Scholar] [CrossRef]
- Suh, Y.-A.; Arnold, R.S.; Lassegue, B.; Shi, J.; Xu, X.; Sorescu, D.; Chung, A.B.; Griendling, K.K.; Lambeth, J.D. Cell Transformation by the Superoxide-Generating Oxidase Mox1. Nature 1999, 401, 79–82. [Google Scholar] [CrossRef]
- Babior, G.L.; Rosin, R.E.; McMurrich, B.J.; Peters, W.A.; Babior, B.M. Arrangement of the Respiratory Burst Oxidase in the Plasma Membrane of the Neutrophil. J. Clin. Investig. 1981, 67, 1724–1728. [Google Scholar] [CrossRef]
- Bánfi, B.; Malgrange, B.; Knisz, J.; Steger, K.; Dubois-Dauphin, M.; Krause, K.-H. NOX3, a Superoxide-Generating NADPH Oxidase of the Inner Ear. J. Biol. Chem. 2004, 279, 46065–46072. [Google Scholar] [CrossRef] [PubMed]
- Bánfi, B.; Molnár, G.; Maturana, A.; Steger, K.; Hegedûs, B.; Demaurex, N.; Krause, K.-H. A Ca2+-Activated NADPH Oxidase in Testis, Spleen, and Lymph Nodes. J. Biol. Chem. 2001, 276, 37594–37601. [Google Scholar] [CrossRef] [PubMed]
- Bánfi, B.; Tirone, F.; Durussel, I.; Knisz, J.; Moskwa, P.; Molnár, G.Z.; Krause, K.-H.; Cox, J.A. Mechanism of Ca2+ Activation of the NADPH Oxidase 5 (NOX5). J. Biol. Chem. 2004, 279, 18583–18591. [Google Scholar] [CrossRef] [PubMed]
- Dupuy, C.; Ohayon, R.; Valent, A.; Noël-Hudson, M.-S.; Dème, D.; Virion, A. Purification of a Novel Flavoprotein Involved in the Thyroid NADPH Oxidase. J. Biol. Chem. 1999, 274, 37265–37269. [Google Scholar] [CrossRef] [PubMed]
- Ueyama, T.; Sakuma, M.; Ninoyu, Y.; Hamada, T.; Dupuy, C.; Geiszt, M.; Leto, T.L.; Saito, N. The Extracellular A-Loop of Dual Oxidases Affects the Specificity of Reactive Oxygen Species Release. J. Biol. Chem. 2015, 290, 6495–6506. [Google Scholar] [CrossRef]
- Martyn, K.D.; Frederick, L.M.; von Loehneysen, K.; Dinauer, M.C.; Knaus, U.G. Functional Analysis of Nox4 Reveals Unique Characteristics Compared to Other NADPH Oxidases. Cell. Signal. 2006, 18, 69–82. [Google Scholar] [CrossRef]
- Lyle, A.N.; Deshpande, N.N.; Taniyama, Y.; Seidel-Rogol, B.; Pounkova, L.; Du, P.; Papaharalambus, C.; Lassègue, B.; Griendling, K.K. Poldip2, a Novel Regulator of Nox4 and Cytoskeletal Integrity in Vascular Smooth Muscle Cells. Circ. Res. 2009, 105, 249–259. [Google Scholar] [CrossRef]
- Datla, S.R.; McGrail, D.J.; Vukelic, S.; Huff, L.P.; Lyle, A.N.; Pounkova, L.; Lee, M.; Seidel-Rogol, B.; Khalil, M.K.; Hilenski, L.L.; et al. Poldip2 Controls Vascular Smooth Muscle Cell Migration by Regulating Focal Adhesion Turnover and Force Polarization. Am. J. Physiol.-Heart Circ. Physiol. 2014, 307, H945–H957. [Google Scholar] [CrossRef] [PubMed]
- Takac, I.; Schröder, K.; Zhang, L.; Lardy, B.; Anilkumar, N.; Lambeth, J.D.; Shah, A.M.; Morel, F.; Brandes, R.P. The E-Loop Is Involved in Hydrogen Peroxide Formation by the NADPH Oxidase Nox4. J. Biol. Chem. 2011, 286, 13304–13313. [Google Scholar] [CrossRef] [PubMed]
- Anilkumar, N.; Jose, G.S.; Sawyer, I.; Santos, C.X.C.; Sand, C.; Brewer, A.C.; Warren, D.; Shah, A.M. A 28-KDa Splice Variant of NADPH Oxidase-4 Is Nuclear-Localized and Involved in Redox Signaling in Vascular Cells. Arterioscler. Thromb. Vasc. Biol. 2013, 33, e104–e112. [Google Scholar] [CrossRef]
- Diebold, B.A.; Wilder, S.G.; De Deken, X.; Meitzler, J.L.; Doroshow, J.H.; McCoy, J.W.; Zhu, Y.; Lambeth, J.D. Guidelines for the Detection of NADPH Oxidases by Immunoblot and RT-qPCR. In NADPH Oxidases: Methods and Protocols; Methods in Molecular Biology; Knaus, U., Leto, T., Eds.; Springer: New York, NY, USA, 2019; Volume 1982, pp. 191–229. [Google Scholar] [CrossRef]
- Liang, S.; Kisseleva, T.; Brenner, D.A. The Role of NADPH Oxidases (NOXs) in Liver Fibrosis and the Activation of Myofibroblasts. Front. Physiol. 2016, 7, 17. [Google Scholar] [CrossRef]
- Paik, Y.-H.; Iwaisako, K.; Seki, E.; Inokuchi, S.; Schnabl, B.; Österreicher, C.H.; Kisseleva, T.; Brenner, D.A. The Nicotinamide Adenine Dinucleotide Phosphate Oxidase (NOX) Homologues NOX1 and NOX2/Gp91phox Mediate Hepatic Fibrosis in Mice. Hepatology 2011, 53, 1730–1741. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Ye, T.; Sun, Y.; Ji, G.; Shido, K.; Chen, Y.; Luo, L.; Na, F.; Li, X.; Huang, Z.; et al. Targeting the Vascular and Perivascular Niches as a Regenerative Therapy for Lung and Liver Fibrosis. Sci. Transl. Med. 2017, 9, eaai8710. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; O’Donnell, V.; Wood, J.; Broughton, J.; Hughes, E.; Jones, O. Expression of Phagocyte NADPH Oxidase Components in Human Endothelial Cells. Am. J. Physiol. 1996, 271 Pt 2, H1626–H1634. [Google Scholar] [CrossRef]
- De Minicis, S.; Brenner, D.A. NOX in Liver Fibrosis. Arch. Biochem. Biophys. 2007, 462, 266–272. [Google Scholar] [CrossRef]
- Liang, S.; Ma, H.-Y.; Zhong, Z.; Dhar, D.; Liu, X.; Xu, J.; Koyama, Y.; Nishio, T.; Karin, D.; Karin, G.; et al. NADPH Oxidase 1 in Liver Macrophages Promotes Inflammation and Tumor Development in Mice. Gastroenterology 2019, 156, 1156–1172.e6. [Google Scholar] [CrossRef] [PubMed]
- Hilenski, L.L.; Clempus, R.E.; Quinn, M.T.; Lambeth, J.D.; Griendling, K.K. Distinct Subcellular Localizations of Nox1 and Nox4 in Vascular Smooth Muscle Cells. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Helmcke, I.; Heumüller, S.; Tikkanen, R.; Schröder, K.; Brandes, R.P. Identification of Structural Elements in Nox1 and Nox4 Controlling Localization and Activity. Antioxid. Redox Signal. 2009, 11, 1279–1287. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Càceres, J.; Mainez, J.; Mayoral, R.; Martín-Sanz, P.; Egea, G.; Fabregat, I. Caveolin-1-Dependent Activation of the Metalloprotease TACE/ADAM17 by TGF-β in Hepatocytes Requires Activation of Src and the NADPH Oxidase NOX1. FEBS J. 2016, 283, 1300–1310. [Google Scholar] [CrossRef]
- Li, Q.; Zhang, Y.; Marden, J.J.; Banfi, B.; Engelhardt, J.F. Endosomal NADPH Oxidase Regulates C-Src Activation Following Hypoxia/Reoxygenation Injury. Biochem. J. 2008, 411, 531–541. [Google Scholar] [CrossRef]
- Ikeda, S.; Yamaoka-Tojo, M.; Hilenski, L.; Patrushev, N.A.; Anwar, G.M.; Quinn, M.T.; Ushio-Fukai, M. IQGAP1 Regulates Reactive Oxygen Species–Dependent Endothelial Cell Migration Through Interacting with Nox2. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2295–2300. [Google Scholar] [CrossRef]
- Wu, R.F.; Xu, Y.C.; Ma, Z.; Nwariaku, F.E.; Sarosi, G.A.; Terada, L.S. Subcellular Targeting of Oxidants during Endothelial Cell Migration. J. Cell Biol. 2005, 171, 893–904. [Google Scholar] [CrossRef] [PubMed]
- Chamulitrat, W.; Schmidt, R.; Tomakidi, P.; Stremmel, W.; Chunglok, W.; Kawahara, T.; Rokutan, K. Association of Gp91phox Homolog Nox1 with Anchorage-Independent Growth and MAP Kinase-Activation of Transformed Human Keratinocytes. Oncogene 2003, 22, 6045–6053. [Google Scholar] [CrossRef] [PubMed]
- Buul, J.D.V.; Fernandez-Borja, M.; Anthony, E.C.; Hordijk, P.L. Expression and Localization of NOX2 and NOX4 in Primary Human Endothelial Cells. Antioxid. Redox Signal. 2005, 7, 308–317. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Kirber, M.T.; Xiao, H.; Yang, Y.; Keaney, J.F. Regulation of ROS Signal Transduction by NADPH Oxidase 4 Localization. J. Cell Biol. 2008, 181, 1129–1139. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.-F.; Ma, Z.; Liu, Z.; Terada, L.S. Nox4-Derived H2O2 Mediates Endoplasmic Reticulum Signaling through Local Ras Activation. Mol. Cell. Biol. 2010, 30, 3553–3568. [Google Scholar] [CrossRef]
- Prior, K.-K.; Wittig, I.; Leisegang, M.S.; Groenendyk, J.; Weissmann, N.; Michalak, M.; Jansen-Dürr, P.; Shah, A.M.; Brandes, R.P. The Endoplasmic Reticulum Chaperone Calnexin Is a NADPH Oxidase NOX4 Interacting Protein. J. Biol. Chem. 2016, 291, 7045–7059. [Google Scholar] [CrossRef] [PubMed]
- Auer, S.; Rinnerthaler, M.; Bischof, J.; Streubel, M.K.; Breitenbach-Koller, H.; Geisberger, R.; Aigner, E.; Cadamuro, J.; Richter, K.; Sopjani, M.; et al. The Human NADPH Oxidase, Nox4, Regulates Cytoskeletal Organization in Two Cancer Cell Lines, HepG2 and SH-SY5Y. Front. Oncol. 2017, 7, 111. [Google Scholar] [CrossRef]
- Clempus, R.E.; Sorescu, D.; Dikalova, A.E.; Pounkova, L.; Jo, P.; Sorescu, G.P.; Lassègue, B.; Griendling, K.K. Nox4 Is Required for Maintenance of the Differentiated Vascular Smooth Muscle Cell Phenotype. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.R.; Berton, G. Regulation of Src Family Tyrosine Kinase Activities in Adherent Human Neutrophils. J. Biol. Chem. 1996, 271, 23464–23471. [Google Scholar] [CrossRef][Green Version]
- Vukelic, S.; Xu, Q.; Seidel-Rogol, B.; Faidley, E.A.; Dikalova, A.E.; Hilenski, L.L.; Jorde, U.; Poole, L.B.; Lassègue, B.; Zhang, G.; et al. NOX4 (NADPH Oxidase 4) and Poldip2 (Polymerase δ-Interacting Protein 2) Induce Filamentous Actin Oxidation and Promote Its Interaction with Vinculin during Integrin-Mediated Cell Adhesion. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 2423–2434. [Google Scholar] [CrossRef]
- Block, K.; Gorin, Y.; Abboud, H.E. Subcellular Localization of Nox4 and Regulation in Diabetes. Proc. Natl. Acad. Sci. USA 2009, 106, 14385–14390. [Google Scholar] [CrossRef]
- Shanmugasundaram, K.; Nayak, B.K.; Friedrichs, W.E.; Kaushik, D.; Rodriguez, R.; Block, K. NOX4 Functions as a Mitochondrial Energetic Sensor Coupling Cancer Metabolic Reprogramming to Drug Resistance. Nat. Commun. 2017, 8, 997. [Google Scholar] [CrossRef]
- Beretta, M.; Santos, C.X.; Molenaar, C.; Hafstad, A.D.; Miller, C.C.; Revazian, A.; Betteridge, K.; Schröder, K.; Streckfuß-Bömeke, K.; Doroshow, J.H.; et al. Nox4 Regulates InsP 3 Receptor-dependent Ca 2+ Release into Mitochondria to Promote Cell Survival. EMBO J. 2020, 39, e103530. [Google Scholar] [CrossRef] [PubMed]
- Spencer, N.; Yan, Z.; Boudreau, R.; Zhang, Y.; Luo, M.; Li, Q.; Tian, X.; Shah, A.; Davisson, R.; Davidson, B.; et al. Control of Hepatic Nuclear Superoxide Production by Glucose 6-Phosphate Dehydrogenase and NADPH Oxidase-4. Biol. Chem. 2011, 286, 8977–8987. [Google Scholar] [CrossRef]
- Eun, H.S.; Chun, K.; Song, I.-S.; Oh, C.-H.; Seong, I.-O.; Yeo, M.-K.; Kim, K.-H. High Nuclear NADPH Oxidase 4 Expression Levels Are Correlated with Cancer Development and Poor Prognosis in Hepatocellular Carcinoma. Pathology 2019, 51, 579–585. [Google Scholar] [CrossRef] [PubMed]
- Dröge, W. Free Radicals in the Physiological Control of Cell Function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.-M.; Choi, J.; Wu, J.-H.; Gaston Pravia, K.A.; Lewis, K.M.; Brand, J.D.; Mochel, N.S.R.; Krzywanski, D.M.; Lambeth, J.D.; Hagood, J.S.; et al. Oxidative Modification of Nuclear Mitogen-Activated Protein Kinase Phosphatase 1 Is Involved in Transforming Growth Factor Β1-Induced Expression of Plasminogen Activator Inhibitor 1 in Fibroblasts. J. Biol. Chem. 2010, 285, 16239–16247. [Google Scholar] [CrossRef]
- von Löhneysen, K.; Noack, D.; Jesaitis, A.J.; Dinauer, M.C.; Knaus, U.G. Mutational Analysis Reveals Distinct Features of the Nox4-P22 Complex. J. Biol. Chem. 2008, 283, 35273–35282. [Google Scholar] [CrossRef] [PubMed]
- Batlle, E.; Massagué, J. Transforming Growth Factor-β Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef]
- Santibanez, J.F.; Obradović, H.; Kukolj, T.; Krstić, J. Transforming Growth Factor-β, Matrix Metalloproteinases, and Urokinase-Type Plasminogen Activator Interaction in the Cancer Epithelial to Mesenchymal Transition: TGF-β, MMPs, and UPA Interplay in Cancer EMT. Dev. Dyn. 2018, 247, 382–395. [Google Scholar] [CrossRef] [PubMed]
- Tzavlaki, K.; Moustakas, A. TGF-β Signaling. Biomolecules 2020, 10, 487. [Google Scholar] [CrossRef] [PubMed]
- Seoane, J. Escaping from the TGF-β Anti-Proliferative Control. Carcinogenesis 2006, 27, 2148–2156. [Google Scholar] [CrossRef] [PubMed]
- Fabregat, I.; Caballero-Díaz, D. Transforming Growth Factor-β-Induced Cell Plasticity in Liver Fibrosis and Hepatocarcinogenesis. Front. Oncol. 2018, 8, 357. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Chen, P. Proliferation-Inhibiting Pathways in Liver Regeneration. Mol. Med. Rep. 2017, 16, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Datto, M.B.; Li, Y.; Panus, J.; Howe, D.; Xiong, Y.; Wang, X.-F. Transforming Growth Factor Beta Induces the Cyclin-Dependent Kinase Inhibitor P21 through a P53-Independent Mechanism. Proc. Natl. Acad. Sci. USA 1995, 92, 5545–5549. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-M.; Nichols, M.A.; Chandrasekharan, S.; Xiong, Y.; Wang, X.-F. Transforming Growth Factor β Activates the Promoter of Cyclin-Dependent Kinase Inhibitor P15 through an Sp1 Consensus Site. J. Biol. Chem. 1995, 270, 26750–26753. [Google Scholar] [CrossRef]
- Hannon, G.; Beach, D. P15INK4B Is a Potential Effector of TGF-Beta-Induced Cell Cycle Arrest. Br. J. Cancer 1994, 371, 257–261. [Google Scholar] [CrossRef]
- Sánchez, A.; Álvarez, A.M.; Benito, M.; Fabregat, I. Apoptosis Induced by Transforming Growth Factor-Beta in Fetal Hepatocyte Primary Cultures: Involvement of Reactive Oxygen Intermediates. J. Biol. Chem. 1996, 271, 7416–7422. [Google Scholar] [CrossRef]
- Carr, B.I.; Hayashi, I.; Branum, E.L.; Moses, H.L. Inhibition of DNA Synthesis in Rat Hepatocytes by Platelet-Derived Type Beta Transforming Growth Factor. Cancer Res. 1986, 46, 6. [Google Scholar]
- De Juan, C.; Sanchez, A.; Nakamura, T.; Fabregat, I.; Benito, M. Hepatocyte Growth Factor Up-Regulates Met Expression in Rat Fetal Hepatocytes in Primary Culture. Biochem. Biophys. Res. Commun. 1994, 204, 1364–1370. [Google Scholar] [CrossRef]
- Sugiyama, A.; Nagaki, M.; Shidoji, Y.; Moriwaki, H.; Muto, Y. Regulation of Cell Cycle-Related Genes in Rat Hepatocytes by Transforming Growth Factor Beta. Biochem. Biophys. Res. Commun. 1997, 238, 5. [Google Scholar] [CrossRef]
- Moustakas, A.; Kardassis, D. Regulation of the Human P21/WAF1/Cip1 Promoter in Hepatic Cells by Functional Interactions between Sp1 and Smad Family Members. Proc. Natl. Acad. Sci. USA 1998, 95, 6733–6738. [Google Scholar] [CrossRef]
- Coffey Jr, R.; Bascom, C.; Sipes, N.J.; Graves-Deal, R.; Weissmann, B.; Moses’, H.L. Selective Inhibition of Growth-Related Gene Expression in Murine Keratinocytes by Transforming Growth Factor Beta. Mol. Cell. Biol. 1988, 8, 6. [Google Scholar] [CrossRef]
- Pietenpol, J.A.; Holt, J.T.; Stein, R.W.; Moses, H.L. Transforming Growth Factor Beta 1 Suppression of C-Myc Gene Transcription: Role in Inhibition of Keratinocyte Proliferation. Proc. Natl. Acad. Sci. USA 1990, 87, 5. [Google Scholar] [CrossRef] [PubMed]
- Ling, M.T.; Wang, X.; Tsao, S.W.; Wong, Y.C. Down-Regulation of Id-1 Expression Is Associated with TGFbeta1-Induced Growth Arrest in Prostate Epithelial Cells. Biochim. Biophys. Acta 2002, 1570, 8. [Google Scholar] [CrossRef]
- Sanchez, A.; Alvarez, A.M.; Benito, M.; Fabregat, I. Transforming Growth Factor Beta Modulates Growth and Differentiation of Fetal Hepatocytes in Primary Culture. J. Cell. Physiol. 1995, 165, 398–405. [Google Scholar] [CrossRef]
- Senturk, S.; Mumcuoglu, M.; Gursoy-Yuzugullu, O.; Cingoz, B.; Akcali, K.C.; Ozturk, M. Transforming Growth Factor-Beta Induces Senescence in Hepatocellular Carcinoma Cells and Inhibits Tumor Growth. Hepatology 2010, 52, 966–974. [Google Scholar] [CrossRef] [PubMed]
- Dzieran, J.; Fabian, J.; Feng, T.; Coulouarn, C.; Ilkavets, I.; Kyselova, A.; Breuhahn, K.; Dooley, S.; Meindl-Beinker, N.M. Comparative Analysis of TGF-β/Smad Signaling Dependent Cytostasis in Human Hepatocellular Carcinoma Cell Lines. PLoS ONE 2013, 8, e72252. [Google Scholar] [CrossRef] [PubMed]
- Franklin, C.C.; Rosenfeld-Franklin, M.E.; White, C.; Kavanagh, T.J.; Fausto, N. TGFβ1-induced Suppression of Glutathione Antioxidant Defenses in Hepatocytes: Caspase-dependent Posttranslational and Caspase-independent Transcriptional Regulatory Mechanisms. FASEB J. 2003, 17, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Herrera, B.; Fernández, M.; Álvarez, A.M.; Roncero, C.; Benito, M.; Gil, J.; Fabregat, I. Activation of Caspases Occurs Downstream from Radical Oxygen Species Production, Bcl-XL down-Regulation, and Early Cytochrome C Release in Apoptosis Induced by Transforming Growth Factor β in Rat Fetal Hepatocytes. Hepatology 2001, 34, 548–556. [Google Scholar] [CrossRef]
- Kanamaru, C. Involvement of Smad Proteins in TGF-β and Activin A-Induced Apoptosis and Growth Inhibition of Liver Cells. Hepatol. Res. 2002, 23, 211–219. [Google Scholar] [CrossRef]
- Teramoto, T.; Kiss, A.; Thorgeirsson, S.S. Induction of P53 and Bax during TGF-Beta1 Initiated Apoptosis in Rat Liver Epithelial Cells. Biochem. Biophys. Res. Commun. 1998, 251, 5. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Zhang, L.; Chen, A.; Xiang, G.; Wang, Y.; Wu, J.; Mitchelson, K.; Cheng, J.; Zhou, Y. Identification of the Gene Transcription and Apoptosis Mediated by TGF-β-Smad2/3-Smad4 Signaling. J. Cell. Physiol. 2008, 215, 422–433. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, S.; Qi, X.; Wildey, G.M.; Robinson, J.; Molkentin, J.; Letterio, J.; Howe, P.H. TGFβ-mediated BIM Expression and Apoptosis Are Regulated through SMAD3-dependent Expression of the MAPK Phosphatase MKP2. EMBO Rep. 2008, 9, 990–997. [Google Scholar] [CrossRef]
- Albright, C.D.; Salganik, R.I.; Craciunescu, C.N.; Mar, M.-H.; Zeisel, S.H. Mitochondrial and Microsomal Derived Reactive Oxygen Species Mediate Apoptosis Induced by Transforming Growth Factor-Beta 1 in Immortalized Rat Hepatocytes. J. Cell. Biochem. 2003, 89, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Carmona-Cuenca, I.; Roncero, C.; Sancho, P.; Caja, L.; Fausto, N.; Fernández, M.; Fabregat, I. Upregulation of the NADPH Oxidase NOX4 by TGF-Beta in Hepatocytes Is Required for Its pro-Apoptotic Activity. J. Hepatol. 2008, 49, 965–976. [Google Scholar] [CrossRef]
- Herrera, B.; Murillo, M.M.; Álvarez-Barrientos, A.; Beltrán, J.; Fernández, M.; Fabregat, I. Source of Early Reactive Oxygen Species in the Apoptosis Induced by Transforming Growth Factor-β in Fetal Rat Hepatocytes. Free Radic. Biol. Med. 2004, 36, 16–26. [Google Scholar] [CrossRef]
- Carmona-Cuenca, I.; Herrera, B.; Ventura, J.-J.; Roncero, C.; Fernández, M.; Fabregat, I. EGF Blocks NADPH Oxidase Activation by TGF-β in Fetal Rat Hepatocytes, Impairing Oxidative Stress, and Cell Death. J. Cell. Physiol. 2006, 207, 322–330. [Google Scholar] [CrossRef]
- Fabregat, I.; Herrera, B.; Fernández, M.; Álvarez, A.M.; Sánchez, A.; Roncero, C.; Ventura, J.-J.; Valverde, Á.M.; Benito, M. Epidermal Growth Factor Impairs the Cytochrome C/Caspase-3 Apoptotic Pathway Induced by Transforming Growth Factorβ in Rat Fetal Hepatocytes Via a Phosphoinositide 3-Kinase–Dependent Pathway. Hepatology 2000, 32, 528–535. [Google Scholar] [CrossRef]
- Murillo, M.M.; del Castillo, G.; Sánchez, A.; Fernández, M.; Fabregat, I. Involvement of EGF Receptor and C-Src in the Survival Signals Induced by TGF-Β1 in Hepatocytes. Oncogene 2005, 24, 4580–4587. [Google Scholar] [CrossRef] [PubMed]
- Caja, L.; Ortiz, C.; Bertran, E.; Murillo, M.M.; Miró-Obradors, M.J.; Palacios, E.; Fabregat, I. Differential Intracellular Signalling Induced by TGF-β in Rat Adult Hepatocytes and Hepatoma Cells: Implications in Liver Carcinogenesis. Cell. Signal. 2007, 19, 683–694. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Càceres, J.; Caja, L.; Mainez, J.; Mayoral, R.; Martín-Sanz, P.; Moreno-Vicente, R.; del Pozo, M.Á.; Dooley, S.; Egea, G.; Fabregat, I. Caveolin-1 Is Required for TGF-β-Induced Transactivation of the EGF Receptor Pathway in Hepatocytes through the Activation of the Metalloprotease TACE/ADAM17. Cell Death Dis. 2014, 5, e1326. [Google Scholar] [CrossRef] [PubMed]
- Murillo, M.M.; Carmona-Cuenca, I.; del Castillo, G.; Ortiz, C.; Roncero, C.; Sánchez, A.; Fernández, M.; Fabregat, I. Activation of NADPH Oxidase by Transforming Growth Factor-β in Hepatocytes Mediates up-Regulation of Epidermal Growth Factor Receptor Ligands through a Nuclear Factor-ΚB-Dependent Mechanism. Biochem. J. 2007, 405, 251–259. [Google Scholar] [CrossRef]
- Sancho, P.; Fabregat, I. NADPH Oxidase NOX1 Controls Autocrine Growth of Liver Tumor Cells through Up-Regulation of the Epidermal Growth Factor Receptor Pathway. J. Biol. Chem. 2010, 285, 24815–24824. [Google Scholar] [CrossRef]
- López-Luque, J.; Fabregat, I. Revisiting the Liver: From Development to Regeneration—What We Ought to Know! Int. J. Dev. Biol. 2018, 62, 441–451. [Google Scholar] [CrossRef]
- Fausto, N.; Campbell, J.S.; Riehle, K.J. Liver Regeneration. Hepatology 2006, 43 (Suppl. S1), S45–S53. [Google Scholar] [CrossRef]
- Michalopoulos, G.K. Liver Regeneration. In The Liver; Arias, I.M., Alter, H.J., Boyer, J.L., Cohen, D.E., Shafritz, D.A., Thorgeirsson, S.S., Wolkoff, A.W., Eds.; Wiley: Hoboken, NJ, USA, 2020; pp. 566–584. [Google Scholar] [CrossRef]
- Ozaki, M. Cellular and Molecular Mechanisms of Liver Regeneration: Proliferation, Growth, Death and Protection of Hepatocytes. Semin. Cell Dev. Biol. 2020, 100, 62–73. [Google Scholar] [CrossRef]
- Chart, R.S.; Price, D.T.; Sue, S.R.; Meyers, W.C.; Jirtle, R.L. Down-Regulation of Transforming Growth Factor Beta Receptor Type I, II, and III during Liver Regeneration. Am. J. Surg. 1995, 169, 126–132. [Google Scholar] [CrossRef]
- Herrera, B.; Álvarez, A.M.; Beltrán, J.; Valdés, F.; Fabregat, I.; Fernández, M. Resistance to TGF-β-Induced Apoptosis in Regenerating Hepatocytes: TGF-β-Apoptosis in Regenerating Hepatocytes. J. Cell. Physiol. 2004, 201, 385–392. [Google Scholar] [CrossRef]
- Russell, W.E. Type Beta Transforming Growth Factor Reversibly Inhibits the Early Proliferative Response to Partial Hepatectomy in the Rat. Cell Biol. 1988, 85, 5. [Google Scholar] [CrossRef]
- Oe, S.; Lemmer, E.R.; Conner, E.A.; Factor, V.M.; Levéen, P.; Larsson, J.; Karlsson, S.; Thorgeirsson, S.S. Intact Signaling by Transforming Growth Factor β Is Not Required for Termination of Liver Regeneration in Mice. Hepatology 2004, 40, 1098–1105. [Google Scholar] [CrossRef]
- Romero-Gallo, J.; Sozmen, E.G.; Chytil, A.; Russell, W.E.; Whitehead, R.; Parks, W.T.; Holdren, M.S.; Her, M.F.; Gautam, S.; Magnuson, M.; et al. Inactivation of TGF-β Signaling in Hepatocytes Results in an Increased Proliferative Response after Partial Hepatectomy. Oncogene 2005, 24, 3028–3041. [Google Scholar] [CrossRef]
- Nakamura, T.; Sakata, R.; Ueno, T.; Sata, M.; Ueno, H. Inhibition of Transforming Growth Factorβ Prevents Progression of Liver Fibrosis and Enhances Hepatocyte Regeneration in Dimethylnitrosamine-Treated Rats. Hepatology 2000, 32, 247–255. [Google Scholar] [CrossRef]
- Karkampouna, S.; Goumans, M.-J.; ten Dijke, P.; Dooley, S.; Kruithof-de Julio, M. Inhibition of TGFβ Type I Receptor Activity Facilitates Liver Regeneration upon Acute CCl4 Intoxication in Mice. Arch. Toxicol. 2016, 90, 347–357. [Google Scholar] [CrossRef]
- Masuda, A.; Nakamura, T.; Abe, M.; Iwamoto, H.; Sakaue, T.; Tanaka, T.; Suzuki, H.; Koga, H.; Torimura, T. Promotion of Liver Regeneration and Anti-fibrotic Effects of the TGF-β Receptor Kinase Inhibitor Galunisertib in CCl4-treated Mice. Int. J. Mol. Med. 2020, 46, 427–438. [Google Scholar] [CrossRef]
- Zeng, W.; Xiao, J.; Zheng, G.; Xing, F.; Tipoe, G.L.; Wang, X.; He, C.; Chen, Z.-Y.; Liu, Y. Antioxidant Treatment Enhances Human Mesenchymal Stem Cell Anti-Stress Ability and Therapeutic Efficacy in an Acute Liver Failure Model. Sci. Rep. 2015, 5, 11100. [Google Scholar] [CrossRef] [PubMed]
- Córdoba-Jover, B.; Arce-Cerezo, A.; Ribera, J.; Pauta, M.; Oró, D.; Casals, G.; Fernández-Varo, G.; Casals, E.; Puntes, V.; Jiménez, W.; et al. Cerium Oxide Nanoparticles Improve Liver Regeneration after Acetaminophen-Induced Liver Injury and Partial Hepatectomy in Rats. J. Nanobiotechnol. 2019, 17, 112. [Google Scholar] [CrossRef] [PubMed]
- Beyer, T.A.; Xu, W.; Teupser, D.; auf dem Keller, U.; Bugnon, P.; Hildt, E.; Thiery, J.; Kan, Y.W.; Werner, S. Impaired Liver Regeneration in Nrf2 Knockout Mice: Role of ROS-Mediated Insulin/IGF-1 Resistance. EMBO J. 2008, 27, 212–223. [Google Scholar] [CrossRef] [PubMed]
- Bai, H.; Zhang, W.; Qin, X.-J.; Zhang, T.; Wu, H.; Liu, J.-Z.; Hai, C.-X. Hydrogen Peroxide Modulates the Proliferation/Quiescence Switch in the Liver during Embryonic Development and Posthepatectomy Regeneration. Antioxid. Redox Signal. 2015, 22, 921–937. [Google Scholar] [CrossRef] [PubMed]
- Ueno, S.; Campbell, J.; Fausto, N. Reactive Oxygen Species Derived from NADPH Oxidase System Is Not Essential for Liver Regeneration After Partial Hepatectomy. J. Surg. Res. 2006, 136, 260–265. [Google Scholar] [CrossRef]
- Crosas-Molist, E.; Bertran, E.; Sancho, P.; López-Luque, J.; Fernando, J.; Sánchez, A.; Fernández, M.; Navarro, E.; Fabregat, I. The NADPH Oxidase NOX4 Inhibits Hepatocyte Proliferation and Liver Cancer Progression. Free Radic. Biol. Med. 2014, 69, 338–347. [Google Scholar] [CrossRef] [PubMed]
- Herranz-Itúrbide, M.; López-Luque, J.; Gonzalez-Sanchez, E.; Caballero-Díaz, D.; Crosas-Molist, E.; Martín-Mur, B.; Gut, M.; Esteve-Codina, A.; Jaquet, V.; Jiang, J.X.; et al. NADPH Oxidase 4 (Nox4) Deletion Accelerates Liver Regeneration in Mice. Redox Biol. 2021, 40, 101841. [Google Scholar] [CrossRef]
- Crosas-Molist, E.; Fabregat, I. Role of NADPH Oxidases in the Redox Biology of Liver Fibrosis. Redox Biol. 2015, 6, 106–111. [Google Scholar] [CrossRef]
- Dunning, S.; ur Rehman, A.; Tiebosch, M.H.; Hannivoort, R.A.; Haijer, F.W.; Woudenberg, J.; van den Heuvel, F.A.J.; Buist-Homan, M.; Faber, K.N.; Moshage, H. Glutathione and Antioxidant Enzymes Serve Complementary Roles in Protecting Activated Hepatic Stellate Cells against Hydrogen Peroxide-Induced Cell Death. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2013, 1832, 2027–2034. [Google Scholar] [CrossRef] [PubMed]
- Foo, N.-P.; Lin, S.-H.; Lee, Y.-H.; Wu, M.-J.; Wang, Y.-J. α-Lipoic Acid Inhibits Liver Fibrosis through the Attenuation of ROS-Triggered Signaling in Hepatic Stellate Cells Activated by PDGF and TGF-β. Toxicology 2011, 282, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Abhilash, P.A.; Harikrishnan, R.; Indira, M. Ascorbic Acid Supplementation Down-Regulates the Alcohol Induced Oxidative Stress, Hepatic Stellate Cell Activation, Cytotoxicity and MRNA Levels of Selected Fibrotic Genes in Guinea Pigs. Free Radic. Res. 2012, 46, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Sancho, P.; Mainez, J.; Crosas-Molist, E.; Roncero, C.; Fernández-Rodriguez, C.M.; Pinedo, F.; Huber, H.; Eferl, R.; Mikulits, W.; Fabregat, I. NADPH Oxidase NOX4 Mediates Stellate Cell Activation and Hepatocyte Cell Death during Liver Fibrosis Development. PLoS ONE 2012, 7, e45285. [Google Scholar] [CrossRef]
- Bettaieb, A.; Jiang, J.X.; Sasaki, Y.; Chao, T.-I.; Kiss, Z.; Chen, X.; Tian, J.; Katsuyama, M.; Yabe-Nishimura, C.; Xi, Y.; et al. Hepatocyte Nicotinamide Adenine Dinucleotide Phosphate Reduced Oxidase 4 Regulates Stress Signaling, Fibrosis, and Insulin Sensitivity during Development of Steatohepatitis in Mice. Gastroenterology 2015, 149, 468–480.e10. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Matsuno, K.; Iwata, K.; Ibi, M.; Matsumoto, M.; Zhang, J.; Zhu, K.; Katsuyama, M.; Torok, N.J.; Yabe-Nishimura, C. NOX1/Nicotinamide Adenine Dinucleotide Phosphate, Reduced Form (NADPH) Oxidase Promotes Proliferation of Stellate Cells and Aggravates Liver Fibrosis Induced by Bile Duct Ligation. Hepatology 2011, 54, 949–958. [Google Scholar] [CrossRef] [PubMed]
- Lan, T.; Kisseleva, T.; Brenner, D.A. Deficiency of NOX1 or NOX4 Prevents Liver Inflammation and Fibrosis in Mice through Inhibition of Hepatic Stellate Cell Activation. PLoS ONE 2015, 10, e0129743. [Google Scholar] [CrossRef]
- Jiang, J.X.; Fish, S.R.; Tomilov, A.; Li, Y.; Fan, W.; Dehnad, A.; Gae, D.; Das, S.; Mozes, G.; Charville, G.W.; et al. Nonphagocytic Activation of NOX2 Is Implicated in Progressive Nonalcoholic Steatohepatitis during Aging. Hepatology 2020, 72, 1204–1218. [Google Scholar] [CrossRef] [PubMed]
- Andueza, A.; Garde, N.; García-Garzón, A.; Ansorena, E.; López-Zabalza, M.J.; Iraburu, M.J.; Zalba, G.; Martínez-Irujo, J.J. NADPH Oxidase 5 Promotes Proliferation and Fibrosis in Human Hepatic Stellate Cells. Free Radic. Biol. Med. 2018, 126, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.X.; Mikami, K.; Venugopal, S.; Li, Y.; Török, N.J. Apoptotic Body Engulfment by Hepatic Stellate Cells Promotes Their Survival by the JAK/STAT and Akt/NF-ΚB-Dependent Pathways. J. Hepatol. 2009, 51, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Palacián, A.; del Castillo, G.; Suárez-Causado, A.; García-Álvaro, M.; de la Morena-Frutos, D.; Fernández, M.; Roncero, C.; Fabregat, I.; Herrera, B.; Sánchez, A. Mouse Hepatic Oval Cells Require Met-Dependent PI3K to Impair TGF-β-Induced Oxidative Stress and Apoptosis. PLoS ONE 2013, 8, e53108. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.X.; Chen, X.; Serizawa, N.; Szyndralewiez, C.; Page, P.; Schröder, K.; Brandes, R.P.; Devaraj, S.; Török, N.J. Liver Fibrosis and Hepatocyte Apoptosis Are Attenuated by GKT137831, a Novel NOX4/NOX1 Inhibitor in Vivo. Free Radic. Biol. Med. 2012, 53, 289–296. [Google Scholar] [CrossRef]
- Yu, J.H.; Zhu, B.-M.; Riedlinger, G.; Kang, K.; Hennighausen, L. The Liver-Specific Tumor Suppressor STAT5 Controls Expression of the Reactive Oxygen Species-Generating Enzyme NOX4 and the Proapoptotic Proteins PUMA and BIM in Mice. Hepatology 2012, 56, 2375–2386. [Google Scholar] [CrossRef]
- Jiang, J.X.; Venugopal, S.; Serizawa, N.; Chen, X.; Scott, F.; Li, Y.; Adamson, R.; Devaraj, S.; Shah, V.; Gershwin, M.E.; et al. Reduced Nicotinamide Adenine Dinucleotide Phosphate Oxidase 2 Plays a Key Role in Stellate Cell Activation and Liver Fibrogenesis In Vivo. Gastroenterology 2010, 139, 1375–1384.e4. [Google Scholar] [CrossRef]
- Brenner, C.; Galluzzi, L.; Kepp, O.; Kroemer, G. Decoding cell death signals in liver inflammation. J. Hepatol. 2013, 59, 583–594. [Google Scholar] [CrossRef]
- Zeisberg, M.; Yang, C.; Martino, M.; Duncan, M.B.; Rieder, F.; Tanjore, H.; Kalluri, R. Fibroblasts derive from hepatocytes in liver fibrosis via epithelial to mesenchymal transition. J. Biol. Chem. 2007, 282, 23337–23347. [Google Scholar] [CrossRef]
- Alyaseer, A.; de Lima, M.; Braga, T.T. The Role of NLRP3 Inflammasome Activation in the Epithelial to Mesenchymal Transition Process during the Fibrosis. Front. Immunol. 2020, 11, 883. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Tan, L.; Hu, M. The role of autophagy in hepatic fibrosis. Am. J. Transl. Res. 2021, 13, 5747–5757. [Google Scholar]
- Zhang, Y.; Tang, H.M.; Liu, C.F.; Yuan, X.F.; Wang, X.Y.; Ma, N.; Xu, G.F.; Wang, S.P.; Deng, J.; Wang, X. TGF-β3 Induces Autophagic Activity by Increasing ROS Generation in a NOX4-Dependent Pathway. Mediat. Inflamm. 2019, 2019, 3153240. [Google Scholar] [CrossRef]
- Zhang, J.; Jiang, N.; Ping, J.; Xu, L. TGF-β1-induced autophagy activates hepatic stellate cells via the ERK and JNK signaling pathways. Int. J. Mol. Med. 2021, 47, 256–266. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.Y.; Li, Y.; Jiang, W.Q.; Zhou, L.F. MAPK/JNK signalling: A potential autophagy regulation pathway. Biosci. Rep. 2015, 35, e00199. [Google Scholar] [CrossRef] [PubMed]
- Lucantoni, F.; Martínez-Cerezuela, A.; Gruevska, A.; Moragrega, Á.B.; Víctor, V.M.; Esplugues, J.V.; Blas-García, A.; Apostolova, N. Understanding the implication of autophagy in the activation of hepatic stellate cells in liver fibrosis: Are we there yet? J. Pathol. 2021, 254, 216–228. [Google Scholar] [CrossRef]
- Huang, Y.; Li, Y.; Lou, A.; Wang, G.Z.; Hu, Y.; Zhang, Y.; Huang, W.; Wang, J.; Li, Y.; Zhu, X.; et al. Alamandine attenuates hepatic fibrosis by regulating autophagy induced by NOX4-dependent ROS. Clin. Sci. 2020, 134, 853–869. [Google Scholar] [CrossRef]
- Ayuso, C.; Rimola, J.; Vilana, R.; Burrel, M.; Darnell, A.; García-Criado, Á.; Bianchi, L.; Belmonte, E.; Caparroz, C.; Barrufet, M.; et al. Diagnosis and Staging of Hepatocellular Carcinoma (HCC): Current Guidelines. Eur. J. Radiol. 2018, 101, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Bruix, J.; da Fonseca, L.G.; Reig, M. Insights into the Success and Failure of Systemic Therapy for Hepatocellular Carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 617–630. [Google Scholar] [CrossRef]
- Müller, M.; Bird, T.G.; Nault, J.-C. The Landscape of Gene Mutations in Cirrhosis and Hepatocellular Carcinoma. J. Hepatol. 2020, 72, 990–1002. [Google Scholar] [CrossRef]
- Caja, L.; Dituri, F.; Mancarella, S.; Caballero-Diaz, D.; Moustakas, A.; Giannelli, G.; Fabregat, I. TGF-β and the Tissue Microenvironment: Relevance in Fibrosis and Cancer. Int. J. Mol. Sci. 2018, 19, 1294. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Corder, N.L.B.; Koduru, B.; Wang, Y. Oxidative Stress and Hepatic Nox Proteins in Chronic Hepatitis C and Hepatocellular Carcinoma. Free Radic. Biol. Med. 2014, 72, 267–284. [Google Scholar] [CrossRef] [PubMed]
- Vandierendonck, A.; Degroote, H.; Vanderborght, B.; Verhelst, X.; Geerts, A.; Devisscher, L.; Van Vlierberghe, H. NOX1 Inhibition Attenuates the Development of a Pro-tumorigenic Environment in Experimental Hepatocellular Carcinoma. J. Exp. Clin. Cancer Res. 2021, 40, 40. [Google Scholar] [CrossRef] [PubMed]
- Shiau, D.-J.; Kuo, W.-T.; Davuluri, G.V.N.; Shieh, C.-C.; Tsai, P.-J.; Chen, C.-C.; Lin, Y.-S.; Wu, Y.-Z.; Hsiao, Y.-P.; Chang, C.-P. Hepatocellular Carcinoma-Derived High Mobility Group Box 1 Triggers M2 Macrophage Polarization via a TLR2/NOX2/Autophagy Axis. Sci. Rep. 2020, 10, 13582. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.-X.; Yan, H.-X.; Liu, Q.; Yang, W.; Wu, H.-P.; Dong, W.; Tang, L.; Lin, Y.; He, Y.-Q.; Zou, S.-S.; et al. Endotoxin Accumulation Prevents Carcinogen-Induced Apoptosis and Promotes Liver Tumorigenesis in Rodents. Hepatology 2010, 52, 1322–1333. [Google Scholar] [CrossRef] [PubMed]
- Dapito, D.H.; Mencin, A.; Gwak, G.-Y.; Pradere, J.-P.; Jang, M.-K.; Mederacke, I.; Caviglia, J.M.; Khiabanian, H.; Adeyemi, A.; Bataller, R.; et al. Promotion of Hepatocellular Carcinoma by the Intestinal Microbiota and TLR4. Cancer Cell 2012, 21, 504–516. [Google Scholar] [CrossRef]
- DeLeo, F.R.; Renee, J.; McCormick, S.; Nakamura, M.; Apicella, M.; Weiss, J.P.; Nauseef, W.M. Neutrophils Exposed to Bacterial Lipopolysaccharide Upregulate NADPH Oxidase Assembly. J. Clin. Investig. 1998, 101, 455–463. [Google Scholar] [CrossRef]
- Kono, H.; Rusyn, I.; Yin, M.; Gäbele, E.; Yamashina, S.; Dikalova, A.; Kadiiska, M.B.; Connor, H.D.; Mason, R.P.; Segal, B.H.; et al. NADPH Oxidase–Derived Free Radicals Are Key Oxidants in Alcohol-Induced Liver Disease. J. Clin. Investig. 2000, 106, 867–872. [Google Scholar] [CrossRef]
- Teufelhofer, O. Superoxide Generation from Kupffer Cells Contributes to Hepatocarcinogenesis: Studies on NADPH Oxidase Knockout Mice. Carcinogenesis 2004, 26, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Parzefall, W.; Freiler, C.; Lorenz, O.; Koudelka, H.; Riegler, T.; Nejabat, M.; Kainzbauer, E.; Grasl-Kraupp, B.; Schulte-Hermann, R. Superoxide Deficiency Attenuates Promotion of Hepatocarcinogenesis by Cytotoxicity in NADPH Oxidase Knockout Mice. Arch. Toxicol. 2015, 89, 1383–1393. [Google Scholar] [CrossRef]
- Bertram, K.; Valcu, C.-M.; Weitnauer, M.; Linne, U.; Görlach, A. NOX1 Supports the Metabolic Remodeling of HepG2 Cells. PLoS ONE 2015, 10, e0122002. [Google Scholar] [CrossRef] [PubMed]
- Eun, H.S.; Cho, S.Y.; Joo, J.S.; Kang, S.H.; Moon, H.S.; Lee, E.S.; Kim, S.H.; Lee, B.S. Gene Expression of NOX Family Members and Their Clinical Significance in Hepatocellular Carcinoma. Sci. Rep. 2017, 7, 11060. [Google Scholar] [CrossRef] [PubMed]
- Dou, C.; Xu, Q.; Liu, J.; Wang, Y.; Zhou, Z.; Yao, W.; Jiang, K.; Cheng, J.; Zhang, C.; Tu, K. SHMT1 Inhibits the Metastasis of HCC by Repressing NOX1-Mediated ROS Production. J. Exp. Clin. Cancer Res. 2019, 38, 70. [Google Scholar] [CrossRef] [PubMed]
- Ha, S.Y.; Paik, Y.-H.; Yang, J.W.; Lee, M.J.; Bae, H.; Park, C.-K. NADPH Oxidase 1 and NADPH Oxidase 4 Have Opposite Prognostic Effects for Patients with Hepatocellular Carcinoma after Hepatectomy. Gut Liver 2016, 10, 826–835. [Google Scholar] [CrossRef] [PubMed]
- De Mochel, N.S.R.; Seronello, S.; Wang, S.H.; Ito, C.; Zheng, J.X.; Liang, T.J.; Lambeth, J.D.; Choi, J. Hepatocyte NAD(P)H Oxidases as an Endogenous Source of Reactive Oxygen Species during Hepatitis C Virus Infection. Hepatology 2010, 52, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Crosas-Molist, E.; Bertran, E.; Rodriguez-Hernandez, I.; Herraiz, C.; Cantelli, G.; Fabra, À.; Sanz-Moreno, V.; Fabregat, I. The NADPH Oxidase NOX4 Represses Epithelial to Amoeboid Transition and Efficient Tumour Dissemination. Oncogene 2017, 36, 3002–3014. [Google Scholar] [CrossRef]
- Caja, L.; Sancho, P.; Bertran, E.; Iglesias-Serret, D.; Gil, J.; Fabregat, I. Overactivation of the MEK/ERK Pathway in Liver Tumor Cells Confers Resistance to TGF-β–Induced Cell Death through Impairing Up-Regulation of the NADPH Oxidase NOX4. Cancer Res. 2009, 69, 7595–7602. [Google Scholar] [CrossRef]
- Sancho, P.; Fabregat, I. The NADPH Oxidase Inhibitor VAS2870 Impairs Cell Growth and Enhances TGF-β-Induced Apoptosis of Liver Tumor Cells. Biochem. Pharmacol. 2011, 81, 917–924. [Google Scholar] [CrossRef]
- Sancho, P.; Bertran, E.; Caja, L.; Carmona-Cuenca, I.; Murillo, M.M.; Fabregat, I. The Inhibition of the Epidermal Growth Factor (EGF) Pathway Enhances TGF-β-Induced Apoptosis in Rat Hepatoma Cells through Inducing Oxidative Stress Coincident with a Change in the Expression Pattern of the NADPH Oxidases (NOX) Isoforms. Biochim. Biophys. Acta BBA Mol. Cell Res. 2009, 1793, 253–263. [Google Scholar] [CrossRef]
- Caja, L.; Sancho, P.; Bertran, E.; Fabregat, I. Dissecting the Effect of Targeting the Epidermal Growth Factor Receptor on TGF-β-Induced-Apoptosis in Human Hepatocellular Carcinoma Cells. J. Hepatol. 2011, 55, 351–358. [Google Scholar] [CrossRef]
- del Castillo, G.; Murillo, M.M.; Álvarez-Barrientos, A.; Bertran, E.; Fernández, M.; Sánchez, A.; Fabregat, I. Autocrine Production of TGF-β Confers Resistance to Apoptosis after an Epithelial–Mesenchymal Transition Process in Hepatocytes: Role of EGF Receptor Ligands. Exp. Cell Res. 2006, 312, 2860–2871. [Google Scholar] [CrossRef] [PubMed]
- Caballero-Díaz, D.; Bertran, E.; Peñuelas-Haro, I.; Moreno-Càceres, J.; Malfettone, A.; López-Luque, J.; Addante, A.; Herrera, B.; Sánchez, A.; Alay, A.; et al. Clathrin Switches Transforming Growth Factor-β Role to pro-Tumorigenic in Liver Cancer. J. Hepatol. 2020, 72, 125–134. [Google Scholar] [CrossRef]
- Ortiz, C.; Caja, L.; Bertran, E.; Gonzalez-Rodriguez, Á.; Valverde, Á.M.; Fabregat, I.; Sancho, P. Protein-Tyrosine Phosphatase 1B (PTP1B) Deficiency Confers Resistance to Transforming Growth Factor-β (TGF-β)-Induced Suppressor Effects in Hepatocytes. J. Biol. Chem. 2012, 287, 15263–15274. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Sanchez, E.; Vaquero, J.; Férnandez-Barrena, M.G.; Lasarte, J.J.; Avila, M.A.; Sarobe, P.; Reig, M.; Calvo, M.; Fabregat, I. The TGF-β Pathway: A Pharmacological Target in Hepatocellular Carcinoma? Cancers 2021, 13, 3248. [Google Scholar] [CrossRef] [PubMed]
- Giannelli, G.; Mikulits, W.; Dooley, S.; Fabregat, I.; Moustakas, A.; ten Dijke, P.; Portincasa, P.; Winter, P.; Janssen, R.; Leporatti, S.; et al. The Rationale for Targeting TGF-β in Chronic Liver Diseases. Eur. J. Clin. Investig. 2016, 46, 349–361. [Google Scholar] [CrossRef]
- Bernard, K.; Thannickal, V.J. NADPH Oxidase Inhibition in Fibrotic Pathologies. Antioxid. Redox Signal. 2020, 33, 455–479. [Google Scholar] [CrossRef]
- Augsburger, F.; Filippova, A.; Rasti, D.; Seredenina, T.; Lam, M.; Maghzal, G.; Mahiout, Z.; Jansen-Dürr, P.; Knaus, U.G.; Doroshow, J.; et al. Pharmacological Characterization of the Seven Human NOX Isoforms and Their Inhibitors. Redox Biol. 2019, 26, 101272. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, T.; Paik, Y.-H.; Watanabe, S.; Laleu, B.; Gaggini, F.; Fioraso-Cartier, L.; Molango, S.; Heitz, F.; Merlot, C.; Szyndralewiez, C.; et al. Nicotinamide Adenine Dinucleotide Phosphate Oxidase in Experimental Liver Fibrosis: GKT137831 as a Novel Potential Therapeutic Agent. Hepatology 2012, 56, 2316–2327. [Google Scholar] [CrossRef] [PubMed]
- Nishio, T.; Hu, R.; Koyama, Y.; Liang, S.; Rosenthal, S.B.; Yamamoto, G.; Karin, D.; Baglieri, J.; Ma, H.-Y.; Xu, J.; et al. Activated Hepatic Stellate Cells and Portal Fibroblasts Contribute to Cholestatic Liver Fibrosis in MDR2 Knockout Mice. J. Hepatol. 2019, 71, 573–585. [Google Scholar] [CrossRef]
- Widjaja, A.A.; Dong, J.; Adami, E.; Viswanathan, S.; Ng, B.; Pakkiri, L.S.; Chothani, S.P.; Singh, B.K.; Lim, W.W.; Zhou, J.; et al. Redefining IL11 as a Regeneration-Limiting Hepatotoxin and Therapeutic Target in Acetaminophen-Induced Liver Injury. Sci. Transl. Med. 2021, 13, eaba8146. [Google Scholar] [CrossRef]
- Gianni, D.; Taulet, N.; Zhang, H.; DerMardirossian, C.; Kister, J.; Martinez, L.; Roush, W.R.; Brown, S.J.; Bokoch, G.M.; Rosen, H. A Novel and Specific NADPH Oxidase-1 (Nox1) Small-Molecule Inhibitor Blocks the Formation of Functional Invadopodia in Human Colon Cancer Cells. ACS Chem. Biol. 2010, 5, 981–993. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Herranz-Itúrbide, M.; Peñuelas-Haro, I.; Espinosa-Sotelo, R.; Bertran, E.; Fabregat, I. The TGF-β/NADPH Oxidases Axis in the Regulation of Liver Cell Biology in Health and Disease. Cells 2021, 10, 2312. https://doi.org/10.3390/cells10092312
Herranz-Itúrbide M, Peñuelas-Haro I, Espinosa-Sotelo R, Bertran E, Fabregat I. The TGF-β/NADPH Oxidases Axis in the Regulation of Liver Cell Biology in Health and Disease. Cells. 2021; 10(9):2312. https://doi.org/10.3390/cells10092312
Chicago/Turabian StyleHerranz-Itúrbide, Macarena, Irene Peñuelas-Haro, Rut Espinosa-Sotelo, Esther Bertran, and Isabel Fabregat. 2021. "The TGF-β/NADPH Oxidases Axis in the Regulation of Liver Cell Biology in Health and Disease" Cells 10, no. 9: 2312. https://doi.org/10.3390/cells10092312
APA StyleHerranz-Itúrbide, M., Peñuelas-Haro, I., Espinosa-Sotelo, R., Bertran, E., & Fabregat, I. (2021). The TGF-β/NADPH Oxidases Axis in the Regulation of Liver Cell Biology in Health and Disease. Cells, 10(9), 2312. https://doi.org/10.3390/cells10092312