
Perspectives on Mitochondria–ER and Mitochondria–Lipid Droplet Contact in Hepatocytes and Hepatic Lipid Metabolism



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
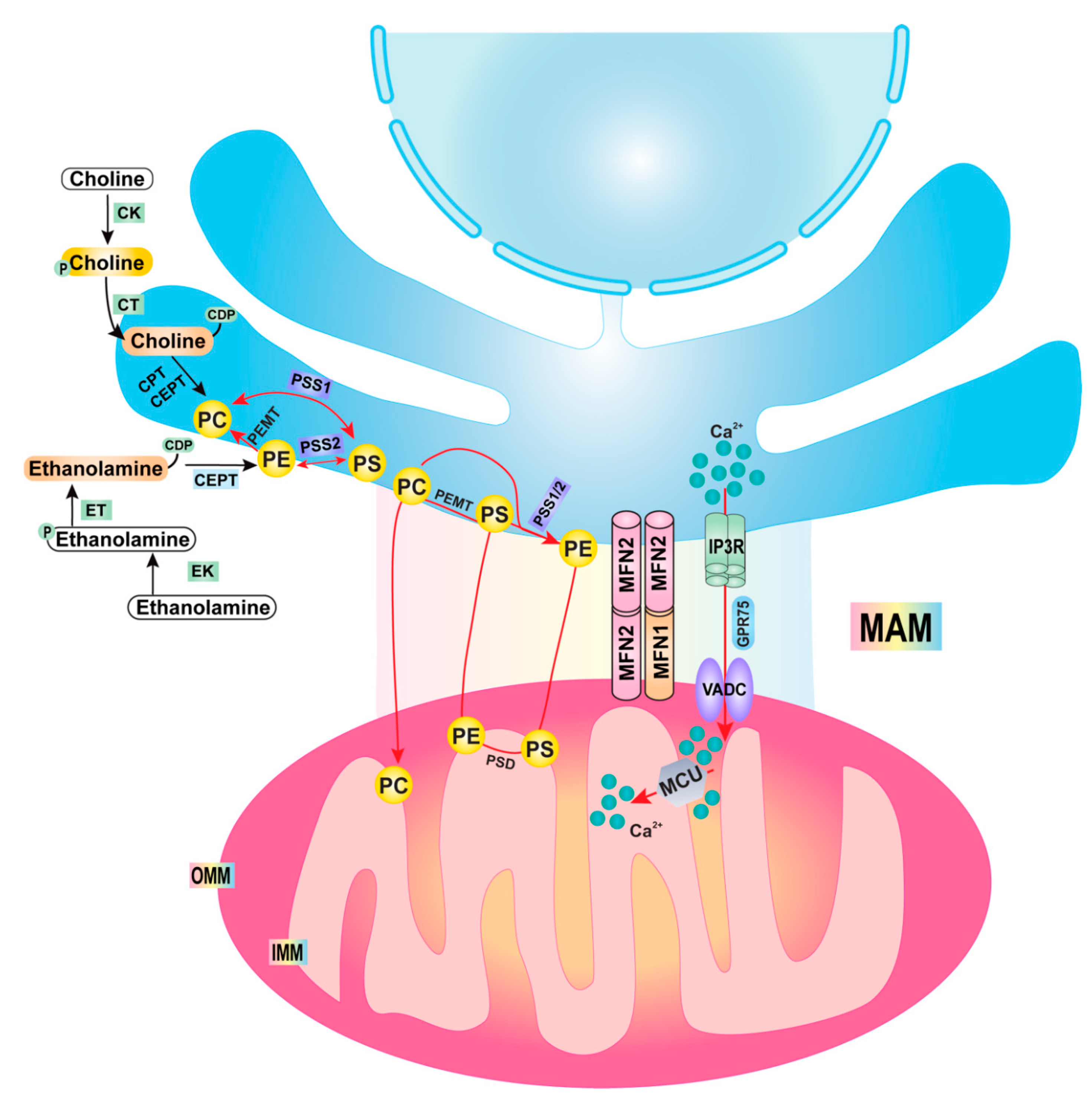
2. Mitochondria–ER Contact/Mitochondrial-Associated Membrane (MAM)
3. Lipid Droplets (LDs) and Autophagy
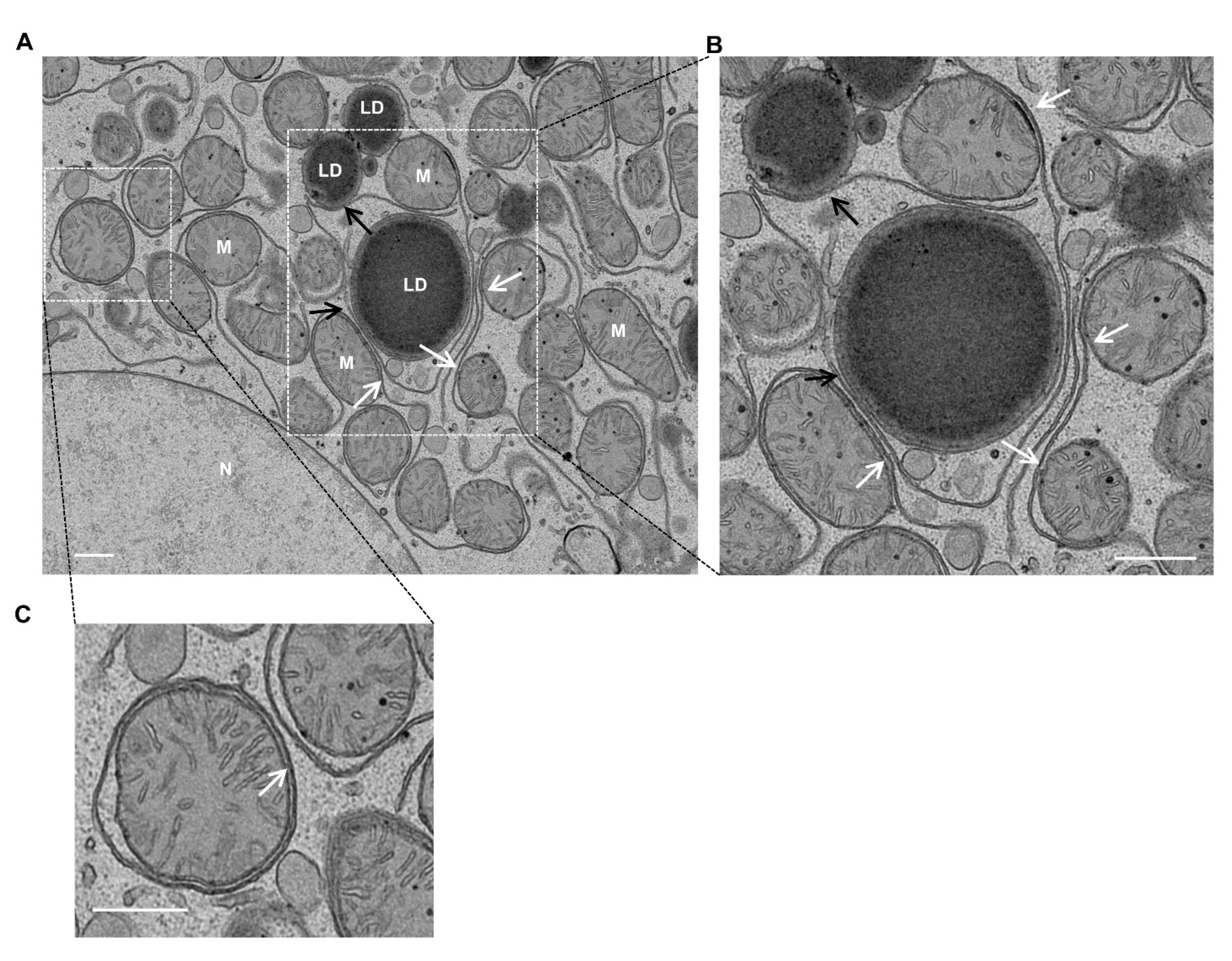
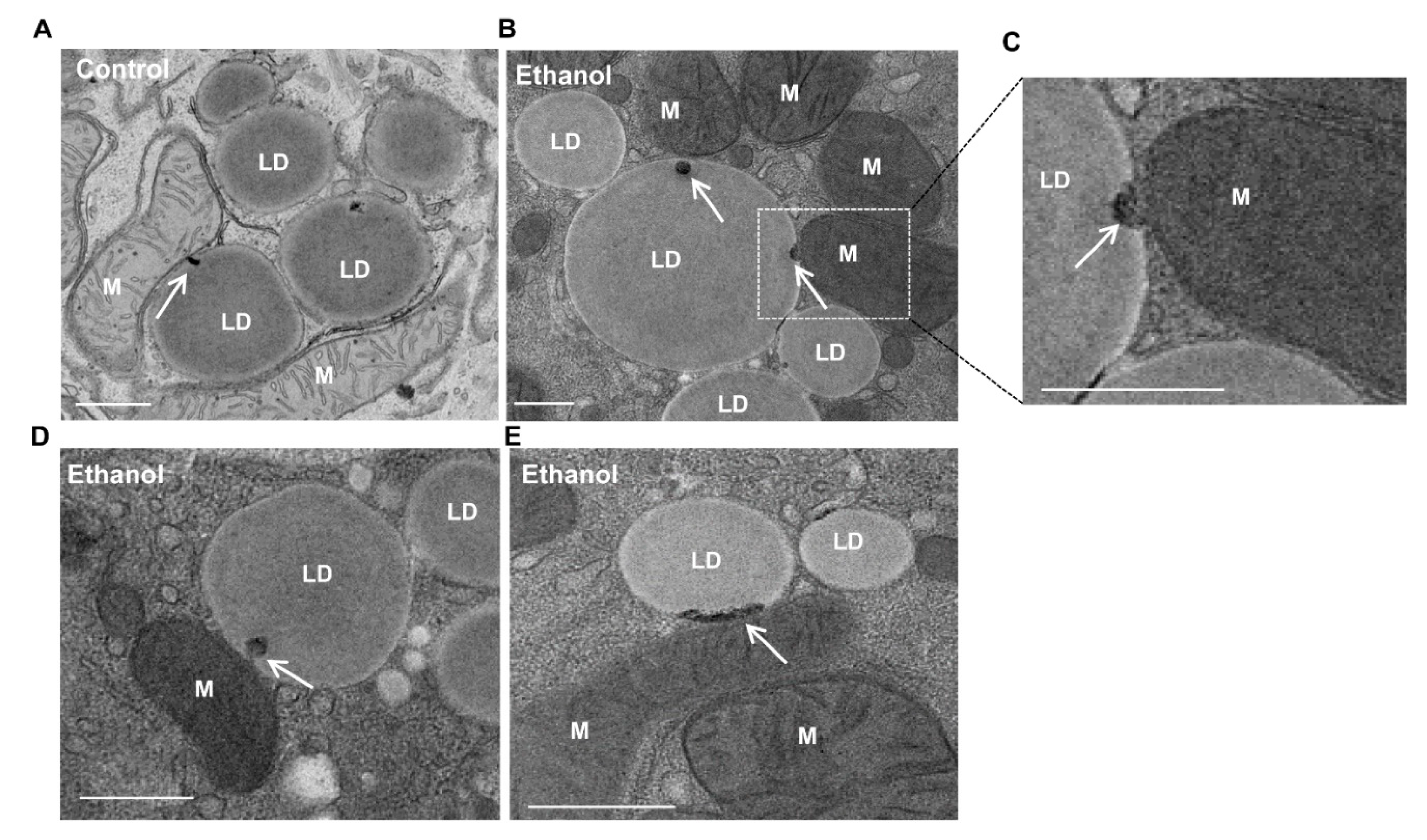
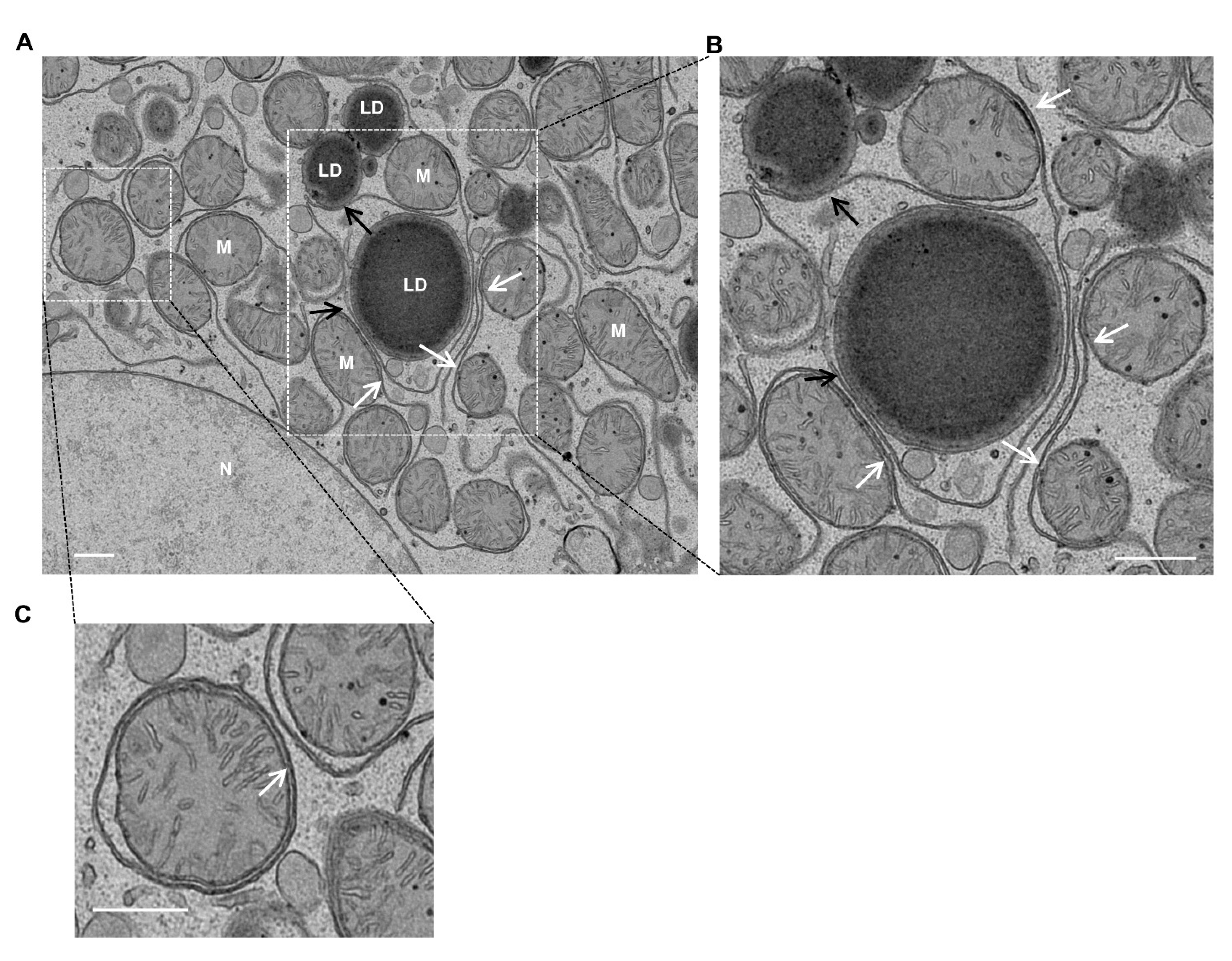
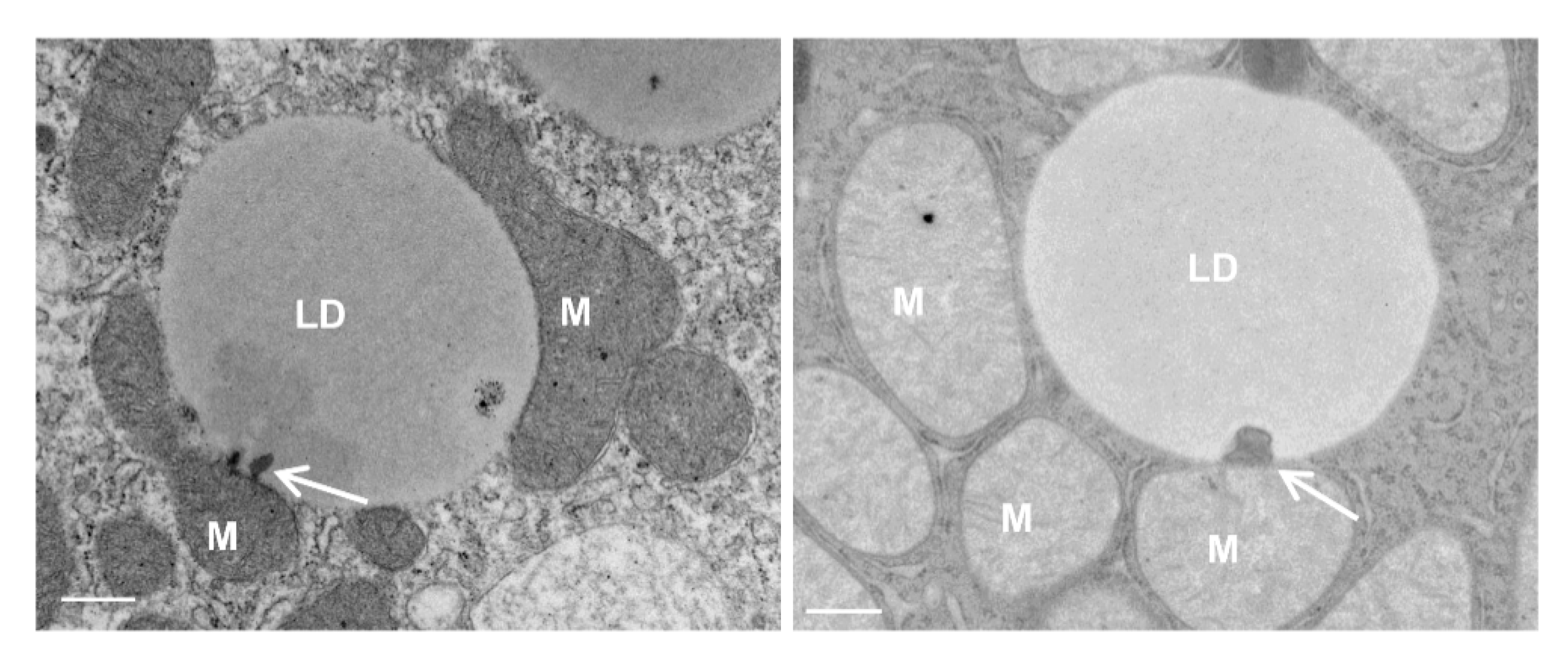
4. Mitochondria–LD Contact
5. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
List of Abbreviations
References
- Gao, B.; Bataller, R. Alcoholic liver disease: Pathogenesis and new therapeutic targets. Gastroenterology 2011, 141, 1572–1585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanyal, A.J. Past, present and future perspectives in nonalcoholic fatty liver disease. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 377–386. [Google Scholar] [CrossRef]
- Williams, J.A.; Manley, S.; Ding, W.X. New advances in molecular mechanisms and emerging therapeutic targets in alcoholic liver diseases. World J. Gastroenterol. 2014, 20, 12908–12933. [Google Scholar] [CrossRef]
- Loomba, R.; Friedman, S.L.; Shulman, G.I. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell 2021, 184, 2537–2564. [Google Scholar] [CrossRef] [PubMed]
- Valm, A.M.; Cohen, S.; Legant, W.R.; Melunis, J.; Hershberg, U.; Wait, E.; Cohen, A.R.; Davidson, M.W.; Betzig, E.; Lippincott-Schwartz, J. Applying systems-level spectral imaging and analysis to reveal the organelle interactome. Nature 2017, 546, 162–167. [Google Scholar] [CrossRef] [PubMed]
- Scorrano, L.; De Matteis, M.A.; Emr, S.; Giordano, F.; Hajnoczky, G.; Kornmann, B.; Lackner, L.L.; Levine, T.P.; Pellegrini, L.; Reinisch, K.; et al. Coming together to define membrane contact sites. Nat. Commun. 2019, 10, 1287. [Google Scholar] [CrossRef] [PubMed]
- Lackner, L.L. The Expanding and Unexpected Functions of Mitochondria Contact Sites. Trends Cell Biol. 2019, 29, 580–590. [Google Scholar] [CrossRef]
- Prinz, W.A.; Toulmay, A.; Balla, T. The functional universe of membrane contact sites. Nat. Rev. Mol. Cell Biol. 2020, 21, 7–24. [Google Scholar] [CrossRef]
- Vance, J.E. Inter-organelle membrane contact sites: Implications for lipid metabolism. Biol. Direct 2020, 15, 24. [Google Scholar] [CrossRef]
- Wang, J.; He, W.; Tsai, P.J.; Chen, P.H.; Ye, M.; Guo, J.; Su, Z. Mutual interaction between endoplasmic reticulum and mitochondria in nonalcoholic fatty liver disease. Lipids Health Dis. 2020, 19, 72. [Google Scholar] [CrossRef] [Green Version]
- Bernhard, W.; Rouiller, C. Close topographical relationship between mitochondria and ergastoplasm of liver cells in a definite phase of cellular activity. J. Cell Biol. 1956, 2, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Vance, J.E. Phospholipid synthesis in a membrane fraction associated with mitochondria. J. Biol. Chem. 1990, 265, 7248–7256. [Google Scholar] [CrossRef]
- Xu, L.; Wang, X.; Tong, C. Endoplasmic Reticulum-Mitochondria Contact Sites and Neurodegeneration. Front. Cell Dev. Biol. 2020, 8, 428. [Google Scholar] [CrossRef]
- Iwasawa, R.; Mahul-Mellier, A.L.; Datler, C.; Pazarentzos, E.; Grimm, S. Fis1 and Bap31 bridge the mitochondria-ER interface to establish a platform for apoptosis induction. EMBO J. 2011, 30, 556–568. [Google Scholar] [CrossRef] [Green Version]
- Stoica, R.; De Vos, K.J.; Paillusson, S.; Mueller, S.; Sancho, R.M.; Lau, K.F.; Vizcay-Barrena, G.; Lin, W.L.; Xu, Y.F.; Lewis, J.; et al. ER-mitochondria associations are regulated by the VAPB-PTPIP51 interaction and are disrupted by ALS/FTD-associated TDP-43. Nat. Commun. 2014, 5, 3996. [Google Scholar] [CrossRef] [Green Version]
- D’Eletto, M.; Rossin, F.; Occhigrossi, L.; Farrace, M.G.; Faccenda, D.; Desai, R.; Marchi, S.; Refolo, G.; Falasca, L.; Antonioli, M.; et al. Transglutaminase Type 2 Regulates ER-Mitochondria Contact Sites by Interacting with GRP75. Cell Rep. 2018, 25, 3573–3581. [Google Scholar] [CrossRef] [Green Version]
- Hoppe, U.C. Mitochondrial calcium channels. FEBS Lett. 2010, 584, 1975–1981. [Google Scholar] [CrossRef] [Green Version]
- Ali, E.S.; Petrovsky, N. Calcium Signaling As a Therapeutic Target for Liver Steatosis. Trends Endocrinol. Metab. 2019, 30, 270–281. [Google Scholar] [CrossRef]
- Feriod, C.N.; Oliveira, A.G.; Guerra, M.T.; Nguyen, L.; Richards, K.M.; Jurczak, M.J.; Ruan, H.B.; Camporez, J.P.; Yang, X.; Shulman, G.I.; et al. Hepatic Inositol 1,4,5 Trisphosphate Receptor Type 1 Mediates Fatty Liver. Hepatol. Commun. 2017, 1, 23–35. [Google Scholar] [CrossRef]
- Ni, H.M.; Williams, J.A.; Ding, W.X. Mitochondrial dynamics and mitochondrial quality control. Redox Biol. 2015, 4, 6–13. [Google Scholar] [CrossRef] [Green Version]
- Friedman, J.R.; Lackner, L.L.; West, M.; DiBenedetto, J.R.; Nunnari, J.; Voeltz, G.K. ER tubules mark sites of mitochondrial division. Science 2011, 334, 358–362. [Google Scholar] [CrossRef] [Green Version]
- Bononi, A.; Missiroli, S.; Poletti, F.; Suski, J.M.; Agnoletto, C.; Bonora, M.; De Marchi, E.; Giorgi, C.; Marchi, S.; Patergnani, S.; et al. Mitochondria-associated membranes (MAMs) as hotspot Ca2+ signaling units. Calcium Signal. 2012, 740, 411–437. [Google Scholar] [CrossRef]
- Szabadkai, G.; Bianchi, K.; Varnai, P.; De Stefani, D.; Wieckowski, M.R.; Cavagna, D.; Nagy, A.I.; Balla, T.; Rizzuto, R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 2006, 175, 901–911. [Google Scholar] [CrossRef] [Green Version]
- Anastasia, I.; Ilacqua, N.; Raimondi, A.; Lemieux, P.; Ghandehari-Alavijeh, R.; Faure, G.; Mekhedov, S.L.; Williams, K.J.; Caicci, F.; Valle, G.; et al. Mitochondria-rough-ER contacts in the liver regulate systemic lipid homeostasis. Cell Rep. 2021, 34, 108873. [Google Scholar] [CrossRef]
- Wang, B.; Tontonoz, P. Phospholipid Remodeling in Physiology and Disease. Annu. Rev. Physiol. 2019, 81, 165–188. [Google Scholar] [CrossRef]
- Spector, A.A.; Yorek, M.A. Membrane lipid composition and cellular function. J. Lipid Res. 1985, 26, 1015–1035. [Google Scholar] [CrossRef]
- Vance, J.E. Phospholipid synthesis and transport in mammalian cells. Traffic 2015, 16, 1–18. [Google Scholar] [CrossRef]
- Agren, J.J.; Kurvinen, J.P.; Kuksis, A. Isolation of very low density lipoprotein phospholipids enriched in ethanolamine phospholipids from rats injected with Triton WR 1339. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2005, 1734, 34–43. [Google Scholar] [CrossRef]
- Li, Z.; Agellon, L.B.; Allen, T.M.; Umeda, M.; Jewell, L.; Mason, A.; Vance, D.E. The ratio of phosphatidylcholine to phosphatidylethanolamine influences membrane integrity and steatohepatitis. Cell Metab. 2006, 3, 321–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Veen, J.N.; Kennelly, J.P.; Wan, S.; Vance, J.E.; Vance, D.E.; Jacobs, R.L. The critical role of phosphatidylcholine and phosphatidylethanolamine metabolism in health and disease. Biochim. Biophys. Acta (BBA)-Biomembr. 2017, 1859, 1558–1572. [Google Scholar] [CrossRef]
- Vance, D.E. Role of phosphatidylcholine biosynthesis in the regulation of lipoprotein homeostasis. Curr. Opin. Lipidol. 2008, 19, 229–234. [Google Scholar] [CrossRef]
- Fast, D.G.; Vance, D.E. Nascent VLDL phospholipid composition is altered when phosphatidylcholine biosynthesis is inhibited: Evidence for a novel mechanism that regulates VLDL secretion. Biochim. Biophys. Acta (BBA)-Lipids Lipid Metab. 1995, 1258, 159–168. [Google Scholar] [CrossRef]
- Verkade, H.J.; Fast, D.G.; Rusinol, A.E.; Scraba, D.G.; Vance, D.E. Impaired biosynthesis of phosphatidylcholine causes a decrease in the number of very low density lipoprotein particles in the Golgi but not in the endoplasmic reticulum of rat liver. J. Biol. Chem. 1993, 268, 24990–24996. [Google Scholar] [CrossRef]
- Jacobs, R.L.; Devlin, C.; Tabas, I.; Vance, D.E. Targeted deletion of hepatic CTP:phosphocholine cytidylyltransferase alpha in mice decreases plasma high density and very low density lipoproteins. J. Biol. Chem. 2004, 279, 47402–47410. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, R.L.; Lingrell, S.; Zhao, Y.; Francis, G.A.; Vance, D.E. Hepatic CTP:phosphocholine cytidylyltransferase-alpha is a critical predictor of plasma high density lipoprotein and very low density lipoprotein. J. Biol. Chem. 2008, 283, 2147–2155. [Google Scholar] [CrossRef] [Green Version]
- Leonardi, R.; Frank, M.W.; Jackson, P.D.; Rock, C.O.; Jackowski, S. Elimination of the CDP-ethanolamine pathway disrupts hepatic lipid homeostasis. J. Biol. Chem. 2009, 284, 27077–27089. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Su, B.; Jacobs, R.L.; Kennedy, B.; Francis, G.A.; Waddington, E.; Brosnan, J.T.; Vance, J.E.; Vance, D.E. Lack of phosphatidylethanolamine N-methyltransferase alters plasma VLDL phospholipids and attenuates atherosclerosis in mice. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1349–1355. [Google Scholar] [CrossRef] [Green Version]
- Lands, W.E. Metabolism of glycerolipides; a comparison of lecithin and triglyceride synthesis. J. Biol. Chem. 1958, 231, 883–888. [Google Scholar] [CrossRef]
- Harayama, T.; Riezman, H. Understanding the diversity of membrane lipid composition. Nat. Rev. Mol. Cell Biol. 2018, 19, 281–296. [Google Scholar] [CrossRef]
- Rong, X.; Wang, B.; Dunham, M.M.; Hedde, P.N.; Wong, J.S.; Gratton, E.; Young, S.G.; Ford, D.A.; Tontonoz, P. Lpcat3-dependent production of arachidonoyl phospholipids is a key determinant of triglyceride secretion. Elife 2015, 4, e06557. [Google Scholar] [CrossRef]
- Hashidate-Yoshida, T.; Harayama, T.; Hishikawa, D.; Morimoto, R.; Hamano, F.; Tokuoka, S.M.; Eto, M.; Tamura-Nakano, M.; Yanobu-Takanashi, R.; Mukumoto, Y.; et al. Fatty acid remodeling by LPCAT3 enriches arachidonate in phospholipid membranes and regulates triglyceride transport. Elife 2015, 4, e06328. [Google Scholar] [CrossRef]
- Lewin, T.M.; Van Horn, C.G.; Krisans, S.K.; Coleman, R.A. Rat liver acyl-CoA synthetase 4 is a peripheral-membrane protein located in two distinct subcellular organelles, peroxisomes, and mitochondrial-associated membrane. Arch. Biochem. Biophys. 2002, 404, 263–270. [Google Scholar] [CrossRef]
- Stone, S.J.; Levin, M.C.; Zhou, P.; Han, J.; Walther, T.C.; Farese, R.V., Jr. The endoplasmic reticulum enzyme DGAT2 is found in mitochondria-associated membranes and has a mitochondrial targeting signal that promotes its association with mitochondria. J. Biol. Chem. 2009, 284, 5352–5361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewin, T.M.; Kim, J.H.; Granger, D.A.; Vance, J.E.; Coleman, R.A. Acyl-CoA synthetase isoforms 1, 4, and 5 are present in different subcellular membranes in rat liver and can be inhibited independently. J. Biol. Chem. 2001, 276, 24674–24679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobs, R.L.; Zhao, Y.; Koonen, D.P.; Sletten, T.; Su, B.; Lingrell, S.; Cao, G.; Peake, D.A.; Kuo, M.S.; Proctor, S.D.; et al. Impaired de novo choline synthesis explains why phosphatidylethanolamine N-methyltransferase-deficient mice are protected from diet-induced obesity. J. Biol. Chem. 2010, 285, 22403–22413. [Google Scholar] [CrossRef] [Green Version]
- Noga, A.A.; Zhao, Y.; Vance, D.E. An unexpected requirement for phosphatidylethanolamine N-methyltransferase in the secretion of very low density lipoproteins. J. Biol. Chem. 2002, 277, 42358–42365. [Google Scholar] [CrossRef] [Green Version]
- van der Veen, J.N.; Lingrell, S.; da Silva, R.P.; Jacobs, R.L.; Vance, D.E. The concentration of phosphatidylethanolamine in mitochondria can modulate ATP production and glucose metabolism in mice. Diabetes 2014, 63, 2620–2630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamasaki, M.; Furuta, N.; Matsuda, A.; Nezu, A.; Yamamoto, A.; Fujita, N.; Oomori, H.; Noda, T.; Haraguchi, T.; Hiraoka, Y.; et al. Autophagosomes form at ER-mitochondria contact sites. Nature 2013, 495, 389–393. [Google Scholar] [CrossRef]
- Valverde, D.P.; Yu, S.; Boggavarapu, V.; Kumar, N.; Lees, J.A.; Walz, T.; Reinisch, K.M.; Melia, T.J. ATG2 transports lipids to promote autophagosome biogenesis. J. Cell Biol. 2019, 218, 1787–1798. [Google Scholar] [CrossRef] [Green Version]
- Bevers, E.M.; Williamson, P.L. Phospholipid scramblase: An update. FEBS Lett. 2010, 584, 2724–2730. [Google Scholar] [CrossRef] [Green Version]
- Morishita, H.; Zhao, Y.G.; Tamura, N.; Nishimura, T.; Kanda, Y.; Sakamaki, Y.; Okazaki, M.; Li, D.; Mizushima, N. A critical role of VMP1 in lipoprotein secretion. Elife 2019, 8, e48834. [Google Scholar] [CrossRef]
- Morita, K.; Hama, Y.; Mizushima, N. TMEM41B functions with VMP1 in autophagosome formation. Autophagy 2019, 15, 922–923. [Google Scholar] [CrossRef] [PubMed]
- Moretti, F.; Bergman, P.; Dodgson, S.; Marcellin, D.; Claerr, I.; Goodwin, J.M.; DeJesus, R.; Kang, Z.; Antczak, C.; Begue, D.; et al. TMEM41B is a novel regulator of autophagy and lipid mobilization. EMBO Rep. 2018, 19, e45889. [Google Scholar] [CrossRef]
- Huang, D.; Xu, B.; Liu, L.; Wu, L.; Zhu, Y.; Ghanbarpour, A.; Wang, Y.; Chen, F.J.; Lyu, J.; Hu, Y.; et al. TMEM41B acts as an ER scramblase required for lipoprotein biogenesis and lipid homeostasis. Cell Metab. 2021, 33, 1655–1670.e8. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.E.; Wang, Y.; Du, X.; Zhang, T.; Mak, H.Y.; Hancock, S.E.; McEwen, H.; Pandzic, E.; Whan, R.M.; Aw, Y.C.; et al. TMEM41B and VMP1 are scramblases and regulate the distribution of cholesterol and phosphatidylserine. J. Cell Biol. 2021, 220, e202103105. [Google Scholar] [CrossRef]
- Ghanbarpour, A.; Valverde, D.P.; Melia, T.J.; Reinisch, K.M. A model for a partnership of lipid transfer proteins and scramblases in membrane expansion and organelle biogenesis. Proc. Natl. Acad. Sci. USA 2021, 118, e2101562118. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.G.; Chen, Y.; Miao, G.; Zhao, H.; Qu, W.; Li, D.; Wang, Z.; Liu, N.; Li, L.; Chen, S.; et al. The ER-Localized Transmembrane Protein EPG-3/VMP1 Regulates SERCA Activity to Control ER-Isolation Membrane Contacts for Autophagosome Formation. Mol. Cell 2017, 67, 974–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, J.L.; Fortier, T.M.; Zhao, Y.G.; Wang, R.; Burmeister, M.; Baehrecke, E.H. Vmp1, Vps13D, and Marf/Mfn2 function in a conserved pathway to regulate mitochondria and ER contact in development and disease. Curr. Biol. 2021, 31, 3028–3039.e7. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Leonzino, M.; Hancock-Cerutti, W.; Horenkamp, F.A.; Li, P.; Lees, J.A.; Wheeler, H.; Reinisch, K.M.; De Camilli, P. VPS13A and VPS13C are lipid transport proteins differentially localized at ER contact sites. J. Cell Biol. 2018, 217, 3625–3639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seifert, W.; Kuhnisch, J.; Maritzen, T.; Horn, D.; Haucke, V.; Hennies, H.C. Cohen syndrome-associated protein, COH1, is a novel, giant Golgi matrix protein required for Golgi integrity. J. Biol. Chem. 2011, 286, 37665–37675. [Google Scholar] [CrossRef] [Green Version]
- Anding, A.L.; Wang, C.; Chang, T.K.; Sliter, D.A.; Powers, C.M.; Hofmann, K.; Youle, R.J.; Baehrecke, E.H. Vps13D Encodes a Ubiquitin-Binding Protein that Is Required for the Regulation of Mitochondrial Size and Clearance. Curr. Biol. 2018, 28, 287–295. [Google Scholar] [CrossRef] [Green Version]
- Guillen-Samander, A.; Leonzino, M.; Hanna, M.G.; Tang, N.; Shen, H.; De Camilli, P. VPS13D bridges the ER to mitochondria and peroxisomes via Miro. J. Cell Biol. 2021, 220, e202010004. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, H.A.; Wang, C.; Kanfer, G.; Shah, H.V.; Velayos-Baeza, A.; Dulovic-Mahlow, M.; Bruggemann, N.; Anding, A.; Baehrecke, E.H.; Maric, D.; et al. VPS13D promotes peroxisome biogenesis. J. Cell Biol. 2021, 220, e202001188. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Fang, N.; Xiong, J.; Du, Y.; Cao, Y.; Ji, W.K. An ESCRT-dependent step in fatty acid transfer from lipid droplets to mitochondria through VPS13D-TSG101 interactions. Nat. Commun. 2021, 12, 1252. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, J.; Meijer, I.A.; Lessel, D.; Mencacci, N.E.; Krainc, D.; Hempel, M.; Tsiakas, K.; Prokisch, H.; Rossignol, E.; Helm, M.H.; et al. Recessive mutations in VPS13D cause childhood onset movement disorders. Ann. Neurol. 2018, 83, 1089–1095. [Google Scholar] [CrossRef]
- Rampoldi, L.; Dobson-Stone, C.; Rubio, J.P.; Danek, A.; Chalmers, R.M.; Wood, N.W.; Verellen, C.; Ferrer, X.; Malandrini, A.; Fabrizi, G.M.; et al. A conserved sorting-associated protein is mutant in chorea-acanthocytosis. Nat. Genet. 2001, 28, 119–120. [Google Scholar] [CrossRef]
- Seong, E.; Insolera, R.; Dulovic, M.; Kamsteeg, E.J.; Trinh, J.; Bruggemann, N.; Sandford, E.; Li, S.; Ozel, A.B.; Li, J.Z.; et al. Mutations in VPS13D lead to a new recessive ataxia with spasticity and mitochondrial defects. Ann. Neurol. 2018, 83, 1075–1088. [Google Scholar] [CrossRef]
- Olzmann, J.A.; Carvalho, P. Dynamics and functions of lipid droplets. Nat. Rev. Mol. Cell Biol. 2019, 20, 137–155. [Google Scholar] [CrossRef]
- Wilfling, F.; Haas, J.T.; Walther, T.C.; Farese, R.V., Jr. Lipid droplet biogenesis. Curr. Opin. Cell Biol. 2014, 29, 39–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walther, T.C.; Chung, J.; Farese, R.V., Jr. Lipid Droplet Biogenesis. Annu. Rev. Cell Dev. Biol. 2017, 33, 491–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.; Hirose, H.; Ohneda, M.; Johnson, J.H.; McGarry, J.D.; Unger, R.H. Beta-cell lipotoxicity in the pathogenesis of non-insulin-dependent diabetes mellitus of obese rats: Impairment in adipocyte-beta-cell relationships. Proc. Natl. Acad. Sci. USA 1994, 91, 10878–10882. [Google Scholar] [CrossRef] [Green Version]
- Schaffer, J.E. Lipotoxicity: When tissues overeat. Curr. Opin. Lipidol. 2003, 14, 281–287. [Google Scholar] [CrossRef]
- Mei, S.; Ni, H.M.; Manley, S.; Bockus, A.; Kassel, K.M.; Luyendyk, J.P.; Copple, B.L.; Ding, W.X. Differential roles of unsaturated and saturated fatty acids on autophagy and apoptosis in hepatocytes. J. Pharmacol. Exp. Ther. 2011, 339, 487–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Zong, W.X.; Ding, W.X. Recycling the danger via lipid droplet biogenesis after autophagy. Autophagy 2017, 13, 1995–1997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.B.; Louie, S.M.; Daniele, J.R.; Tran, Q.; Dillin, A.; Zoncu, R.; Nomura, D.K.; Olzmann, J.A. DGAT1-Dependent Lipid Droplet Biogenesis Protects Mitochondrial Function during Starvation-Induced Autophagy. Dev. Cell 2017, 42, 9–21. [Google Scholar] [CrossRef] [Green Version]
- Zechner, R.; Strauss, J.G.; Haemmerle, G.; Lass, A.; Zimmermann, R. Lipolysis: Pathway under construction. Curr. Opin. Lipidol. 2005, 16, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chao, X.; Yang, L.; Lu, Q.; Li, T.; Ding, W.X.; Ni, H.M. Impaired Fasting-Induced Adaptive Lipid Droplet Biogenesis in Liver-Specific Atg5-Deficient Mouse Liver Is Mediated by Persistent Nuclear Factor-Like 2 Activation. Am. J. Pathol. 2018, 188, 1833–1846. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Cook, W.S.; Qi, C.; Yeldandi, A.V.; Reddy, J.K.; Rao, M.S. Defect in peroxisome proliferator-activated receptor alpha-inducible fatty acid oxidation determines the severity of hepatic steatosis in response to fasting. J. Biol. Chem. 2000, 275, 28918–28928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Ginneken, V.; Verhey, E.; Poelmann, R.; Ramakers, R.; van Dijk, K.W.; Ham, L.; Voshol, P.; Havekes, L.; Van Eck, M.; van der Greef, J. Metabolomics (liver and blood profiling) in a mouse model in response to fasting: A study of hepatic steatosis. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2007, 1771, 1263–1270. [Google Scholar] [CrossRef] [PubMed]
- Zechner, R.; Madeo, F.; Kratky, D. Cytosolic lipolysis and lipophagy: Two sides of the same coin. Nat. Rev. Mol. Cell Biol. 2017, 18, 671–684. [Google Scholar] [CrossRef]
- Badman, M.K.; Pissios, P.; Kennedy, A.R.; Koukos, G.; Flier, J.S.; Maratos-Flier, E. Hepatic fibroblast growth factor 21 is regulated by PPARalpha and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab. 2007, 5, 426–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inagaki, T.; Dutchak, P.; Zhao, G.; Ding, X.; Gautron, L.; Parameswara, V.; Li, Y.; Goetz, R.; Mohammadi, M.; Esser, V.; et al. Endocrine regulation of the fasting response by PPARalpha-mediated induction of fibroblast growth factor 21. Cell Metab. 2007, 5, 415–425. [Google Scholar] [CrossRef] [Green Version]
- Benador, I.Y.; Veliova, M.; Liesa, M.; Shirihai, O.S. Mitochondria Bound to Lipid Droplets: Where Mitochondrial Dynamics Regulate Lipid Storage and Utilization. Cell Metab. 2019, 29, 827–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, P.A.; Senkal, C.E.; Suchanek, A.L.; Grevengoed, T.J.; Lin, D.D.; Zhao, L.; Crunk, A.E.; Klett, E.L.; Fullekrug, J.; Obeid, L.M.; et al. Long-chain acyl-CoA synthetase 1 interacts with key proteins that activate and direct fatty acids into niche hepatic pathways. J. Biol. Chem. 2018, 293, 16724–16740. [Google Scholar] [CrossRef] [Green Version]
- Jagerstrom, S.; Polesie, S.; Wickstrom, Y.; Johansson, B.R.; Schroder, H.D.; Hojlund, K.; Bostrom, P. Lipid droplets interact with mitochondria using SNAP23. Cell Biol. Int. 2009, 33, 934–940. [Google Scholar] [CrossRef]
- Boutant, M.; Kulkarni, S.S.; Joffraud, M.; Ratajczak, J.; Valera-Alberni, M.; Combe, R.; Zorzano, A.; Canto, C. Mfn2 is critical for brown adipose tissue thermogenic function. EMBO J. 2017, 36, 1543–1558. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Sreenivasan, U.; Hu, H.; Saladino, A.; Polster, B.M.; Lund, L.M.; Gong, D.W.; Stanley, W.C.; Sztalryd, C. Perilipin 5, a lipid droplet-associated protein, provides physical and metabolic linkage to mitochondria. J. Lipid Res. 2011, 52, 2159–2168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freyre, C.A.C.; Rauher, P.C.; Ejsing, C.S.; Klemm, R.W. MIGA2 Links Mitochondria, the ER, and Lipid Droplets and Promotes De Novo Lipogenesis in Adipocytes. Mol. Cell 2019, 76, 811–825. [Google Scholar] [CrossRef] [PubMed]
- Czaja, M.J.; Ding, W.X.; Donohue, T.M., Jr.; Friedman, S.L.; Kim, J.S.; Komatsu, M.; Lemasters, J.J.; Lemoine, A.; Lin, J.D.; Ou, J.H.; et al. Functions of autophagy in normal and diseased liver. Autophagy 2013, 9, 1131–1158. [Google Scholar] [CrossRef] [Green Version]
- Holm, C. Molecular mechanisms regulating hormone-sensitive lipase and lipolysis. Biochem. Soc. Trans. 2003, 31, 1120–1124. [Google Scholar] [CrossRef]
- Krahmer, N.; Guo, Y.; Wilfling, F.; Hilger, M.; Lingrell, S.; Heger, K.; Newman, H.W.; Schmidt-Supprian, M.; Vance, D.E.; Mann, M.; et al. Phosphatidylcholine synthesis for lipid droplet expansion is mediated by localized activation of CTP:phosphocholine cytidylyltransferase. Cell Metab. 2011, 14, 504–515. [Google Scholar] [CrossRef] [Green Version]
- Onal, G.; Kutlu, O.; Gozuacik, D.; Dokmeci Emre, S. Lipid Droplets in Health and Disease. Lipids Health Dis. 2017, 16, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, W.X.; Yin, X.M. Mitophagy: Mechanisms, pathophysiological roles, and analysis. Biol. Chem. 2012, 393, 547–564. [Google Scholar] [CrossRef] [Green Version]
- Benador, I.Y.; Veliova, M.; Mahdaviani, K.; Petcherski, A.; Wikstrom, J.D.; Assali, E.A.; Acin-Perez, R.; Shum, M.; Oliveira, M.F.; Cinti, S.; et al. Mitochondria Bound to Lipid Droplets Have Unique Bioenergetics, Composition, and Dynamics that Support Lipid Droplet Expansion. Cell Metab. 2018, 27, 869–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, L.; Liu, P. Two Types of Contact Between Lipid Droplets and Mitochondria. Front. Cell Dev. Biol. 2020, 8, 618322. [Google Scholar] [CrossRef]
- Cui, L.; Mirza, A.H.; Zhang, S.; Liang, B.; Liu, P. Lipid droplets and mitochondria are anchored during brown adipocyte differentiation. Protein Cell 2019, 10, 921–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, X.; Qian, H.; Chen, A.; Ni, H.-M.; Ding, W.-X. Perspectives on Mitochondria–ER and Mitochondria–Lipid Droplet Contact in Hepatocytes and Hepatic Lipid Metabolism. Cells 2021, 10, 2273. https://doi.org/10.3390/cells10092273
Ma X, Qian H, Chen A, Ni H-M, Ding W-X. Perspectives on Mitochondria–ER and Mitochondria–Lipid Droplet Contact in Hepatocytes and Hepatic Lipid Metabolism. Cells. 2021; 10(9):2273. https://doi.org/10.3390/cells10092273
Chicago/Turabian StyleMa, Xiaowen, Hui Qian, Allen Chen, Hong-Min Ni, and Wen-Xing Ding. 2021. "Perspectives on Mitochondria–ER and Mitochondria–Lipid Droplet Contact in Hepatocytes and Hepatic Lipid Metabolism" Cells 10, no. 9: 2273. https://doi.org/10.3390/cells10092273
APA StyleMa, X., Qian, H., Chen, A., Ni, H.-M., & Ding, W.-X. (2021). Perspectives on Mitochondria–ER and Mitochondria–Lipid Droplet Contact in Hepatocytes and Hepatic Lipid Metabolism. Cells, 10(9), 2273. https://doi.org/10.3390/cells10092273