
DYRK1A Kinase Inhibitors Promote β-Cell Survival and Insulin Homeostasis

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Protein Expression and Purification
2.3. MST Measurements
2.4. Cook Assay
2.5. NFAT Luciferase Reporter Assay
2.6. Insulinomas Cell Culture
2.7. Cytotoxicity and Cell Survival Assay
2.8. Glucose-Stimulated Insulin Secretion (GSIS)
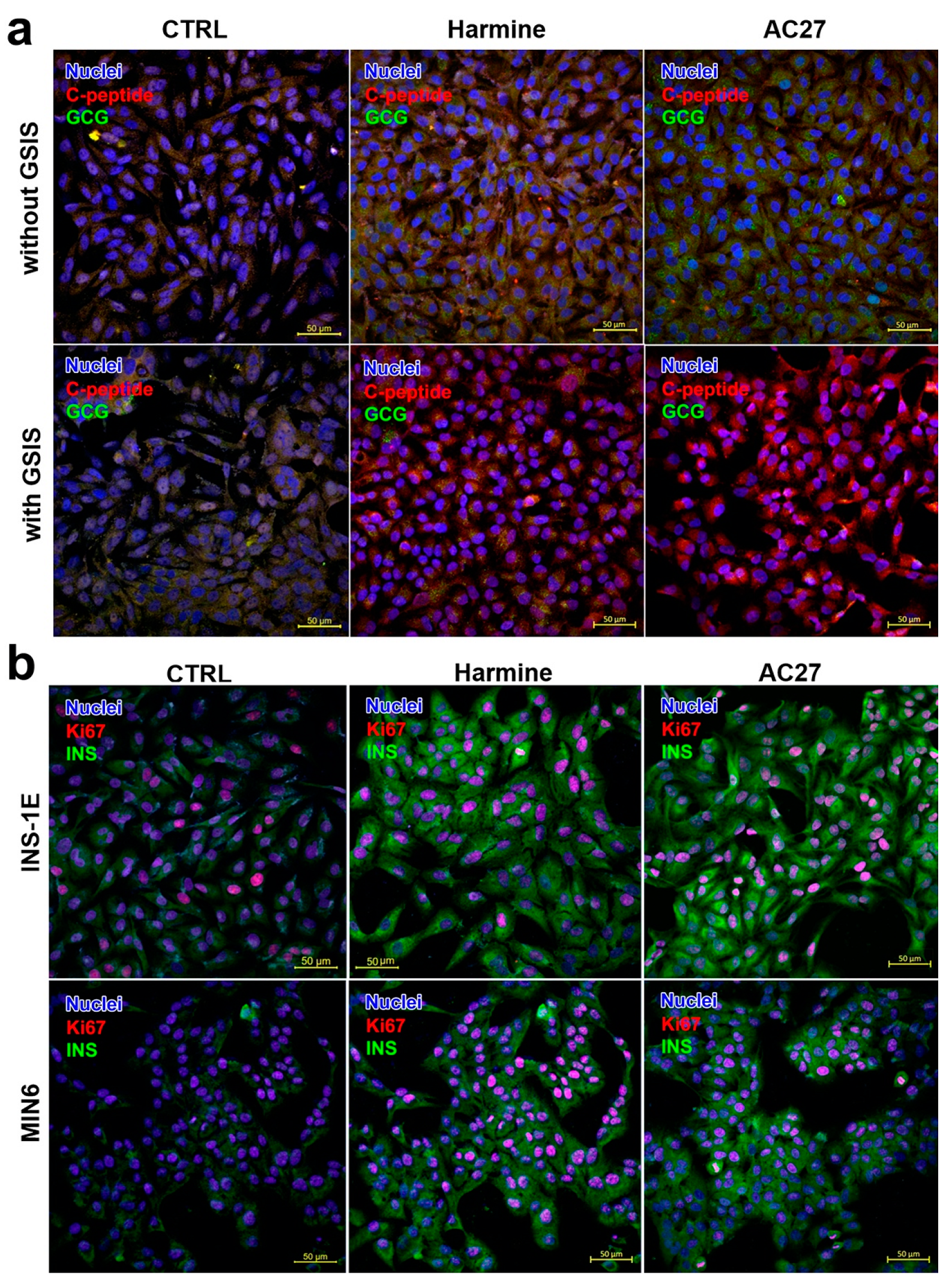
2.9. Staining for Insulin, Ki67, Glucagon, C-peptide and DYRK1A
2.10. Flow Cytometry Analysis
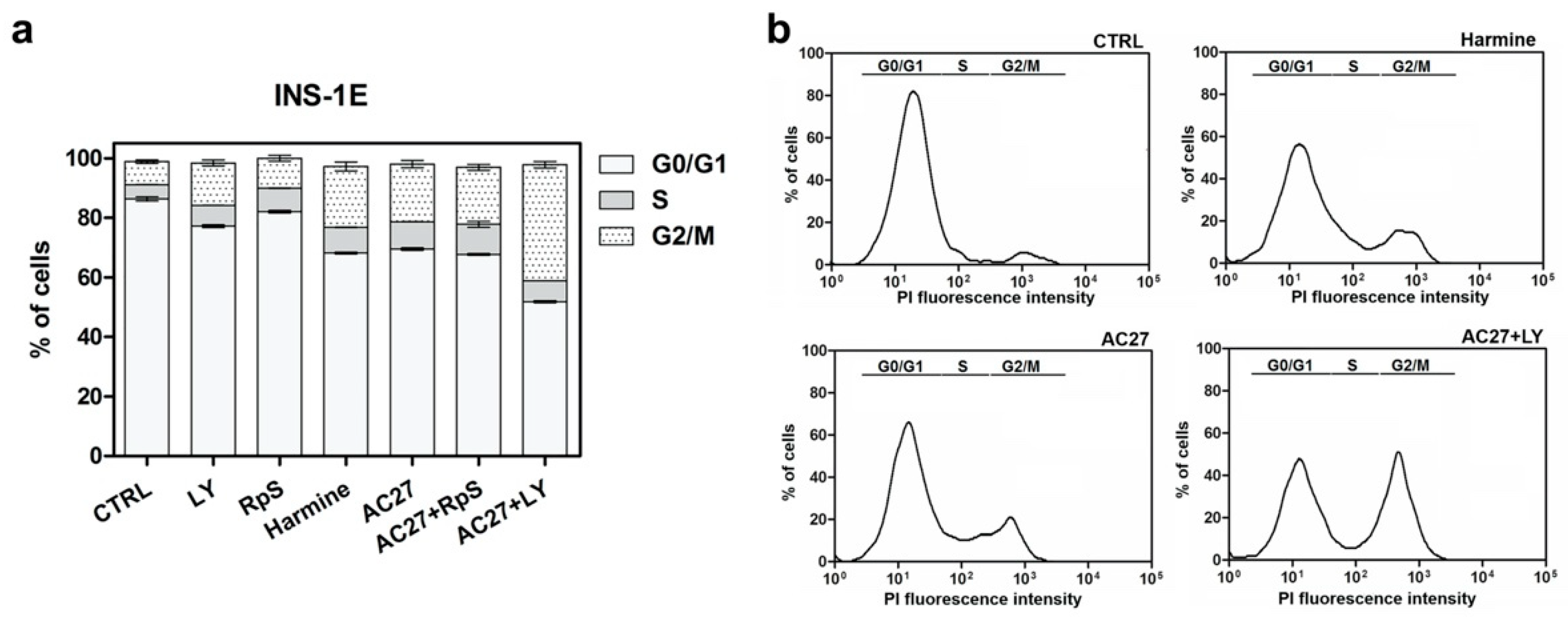
2.11. Cell Cycle Analysis
2.12. Differentiation hiPSC into Pancreatic Islets Cells
2.13. IPSC Staining Protocol—3D Cell Culture—Organoids
2.14. ELISA Assay
2.15. Mouse Islet Isolation
2.16. Mouse Islet Staining Protocol
2.17. Statistical Analysis
3. Results
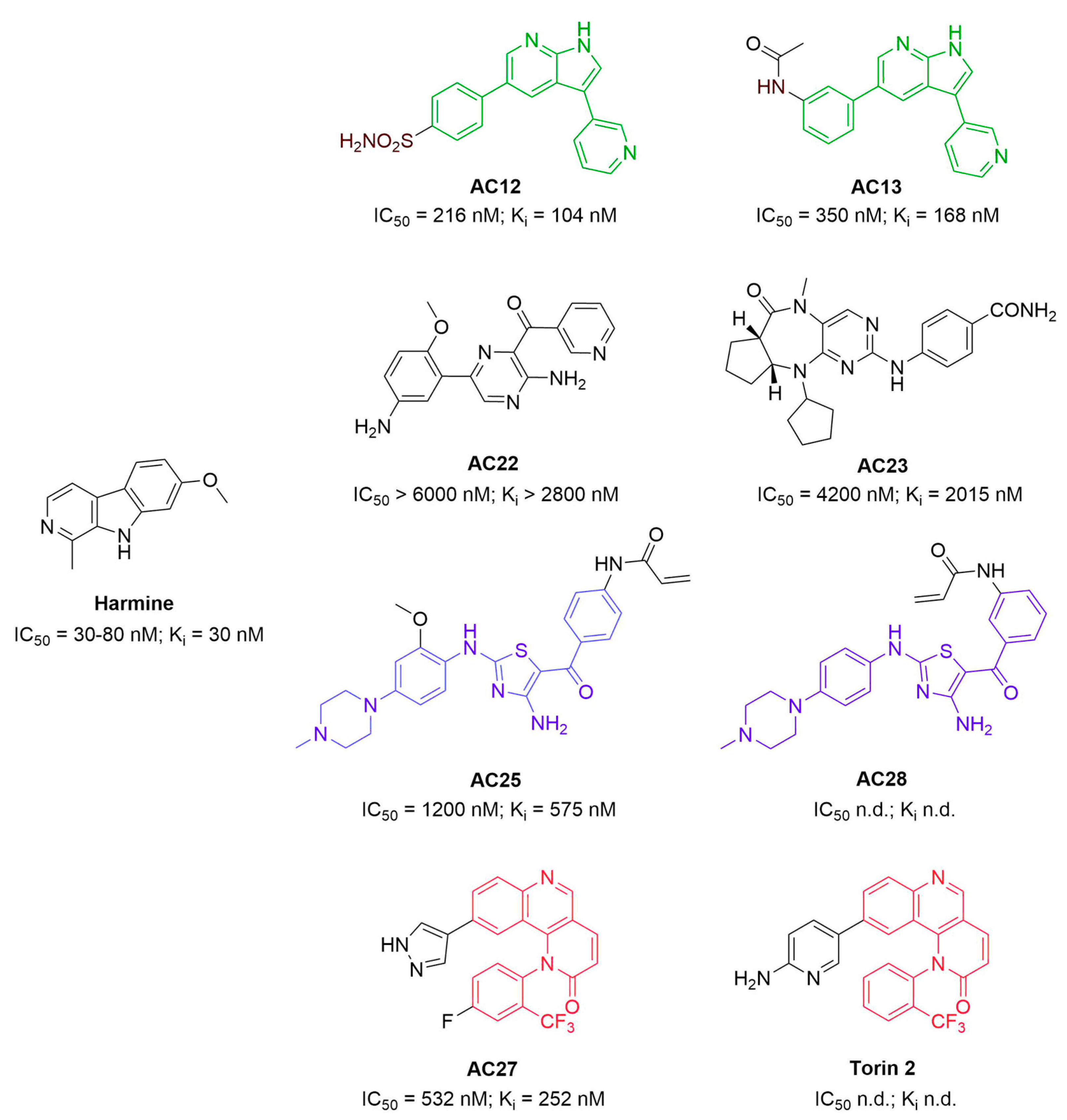
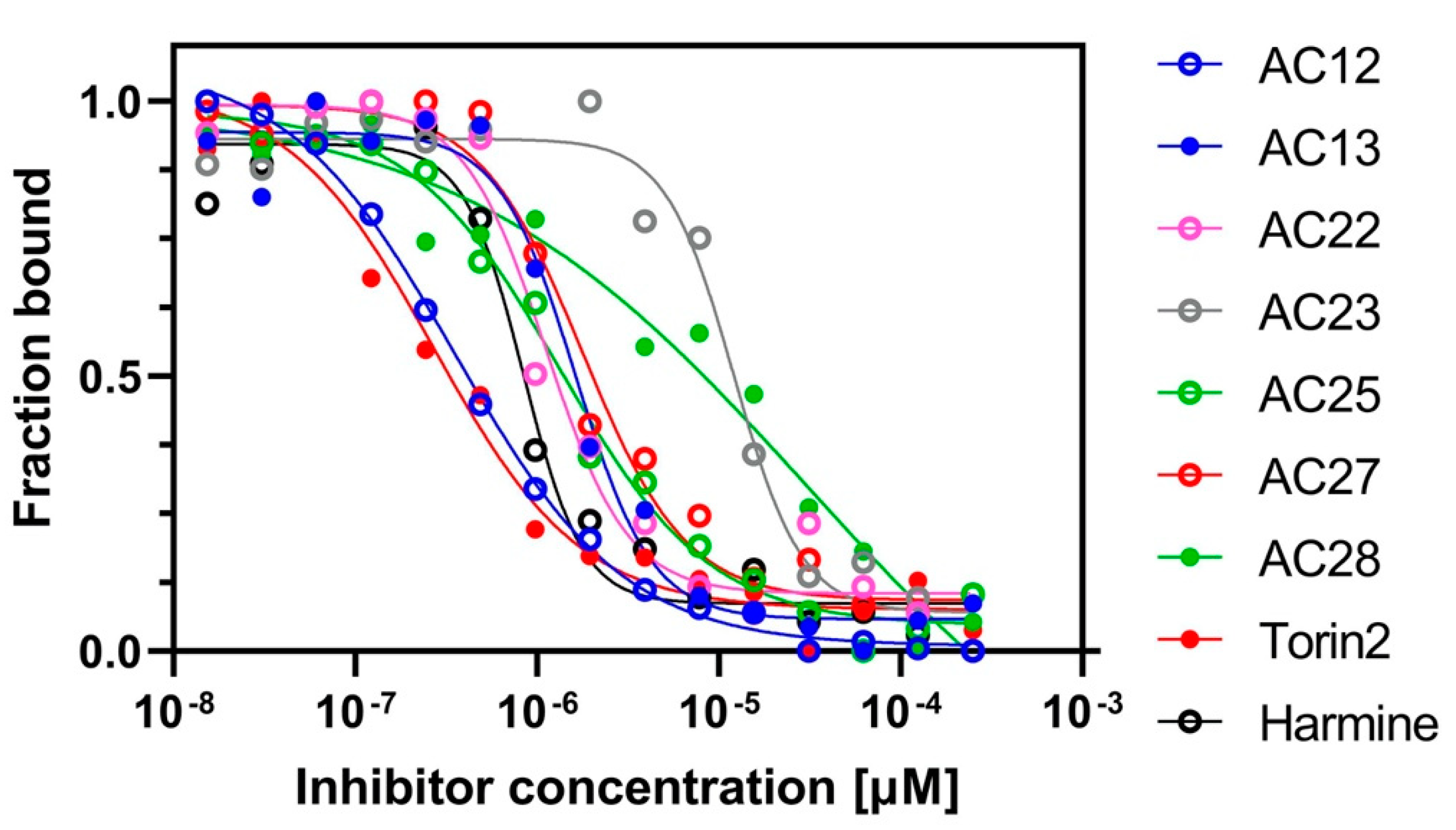
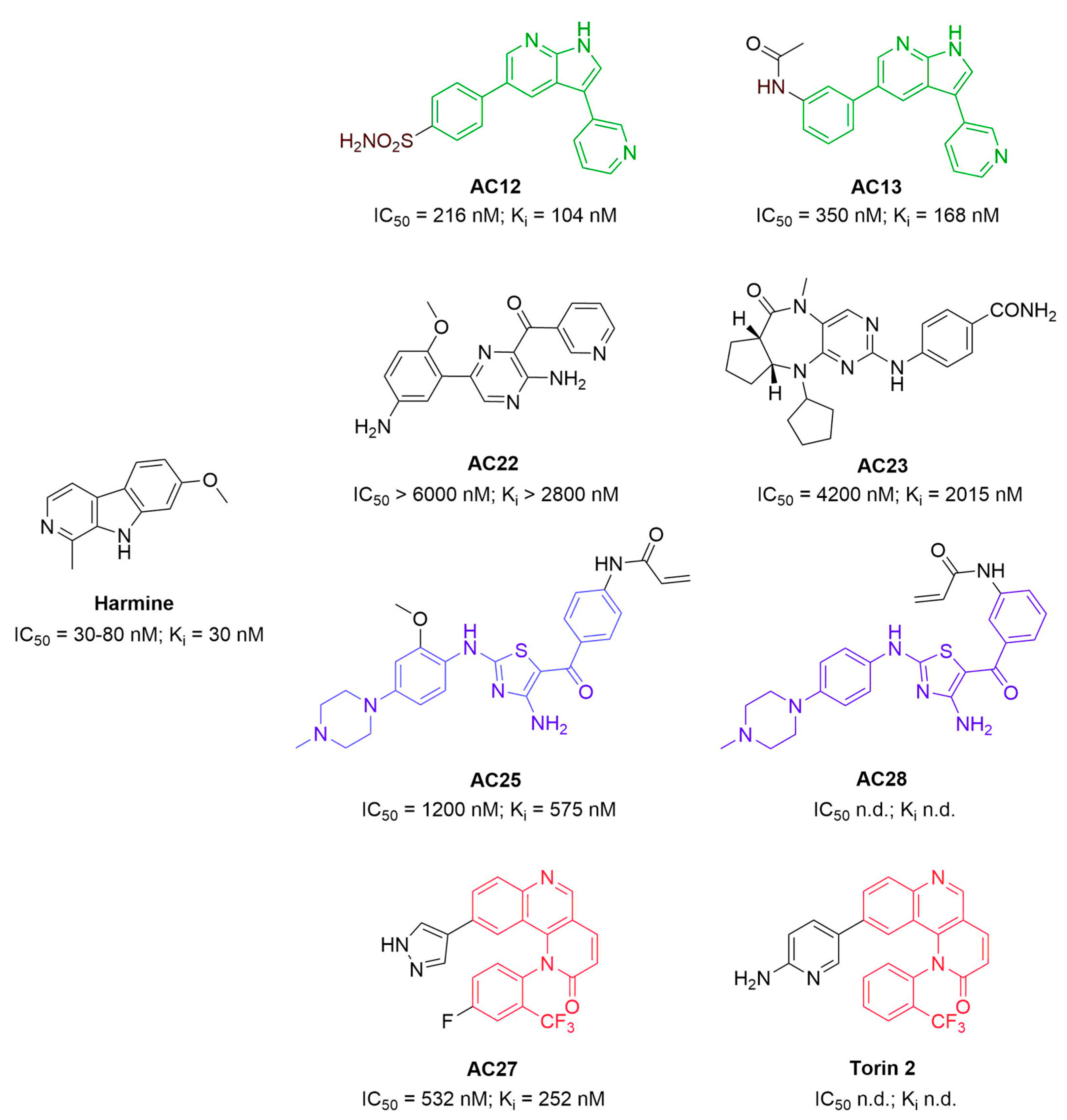
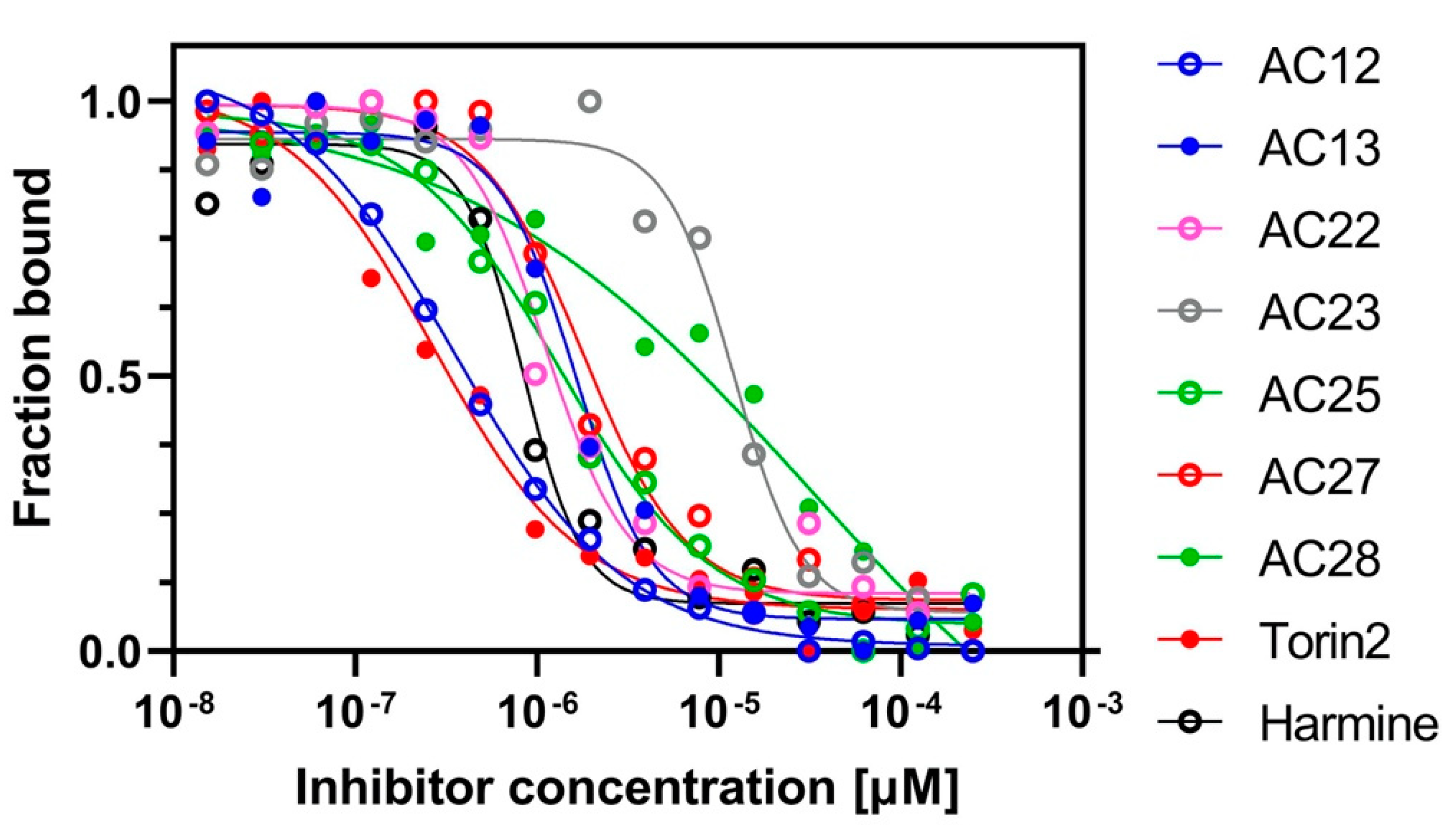
3.1. Binding Affinity and Inhibitory Potential of Selected DYRK1A Inhibitors
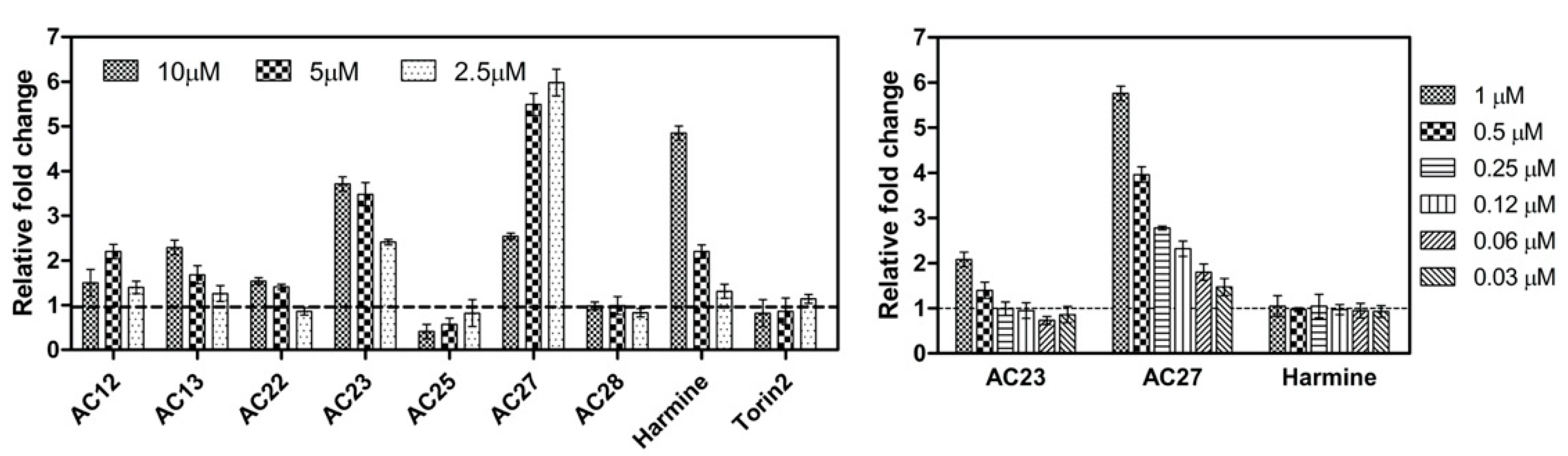
3.2. NFAT Luc Reporter Assay
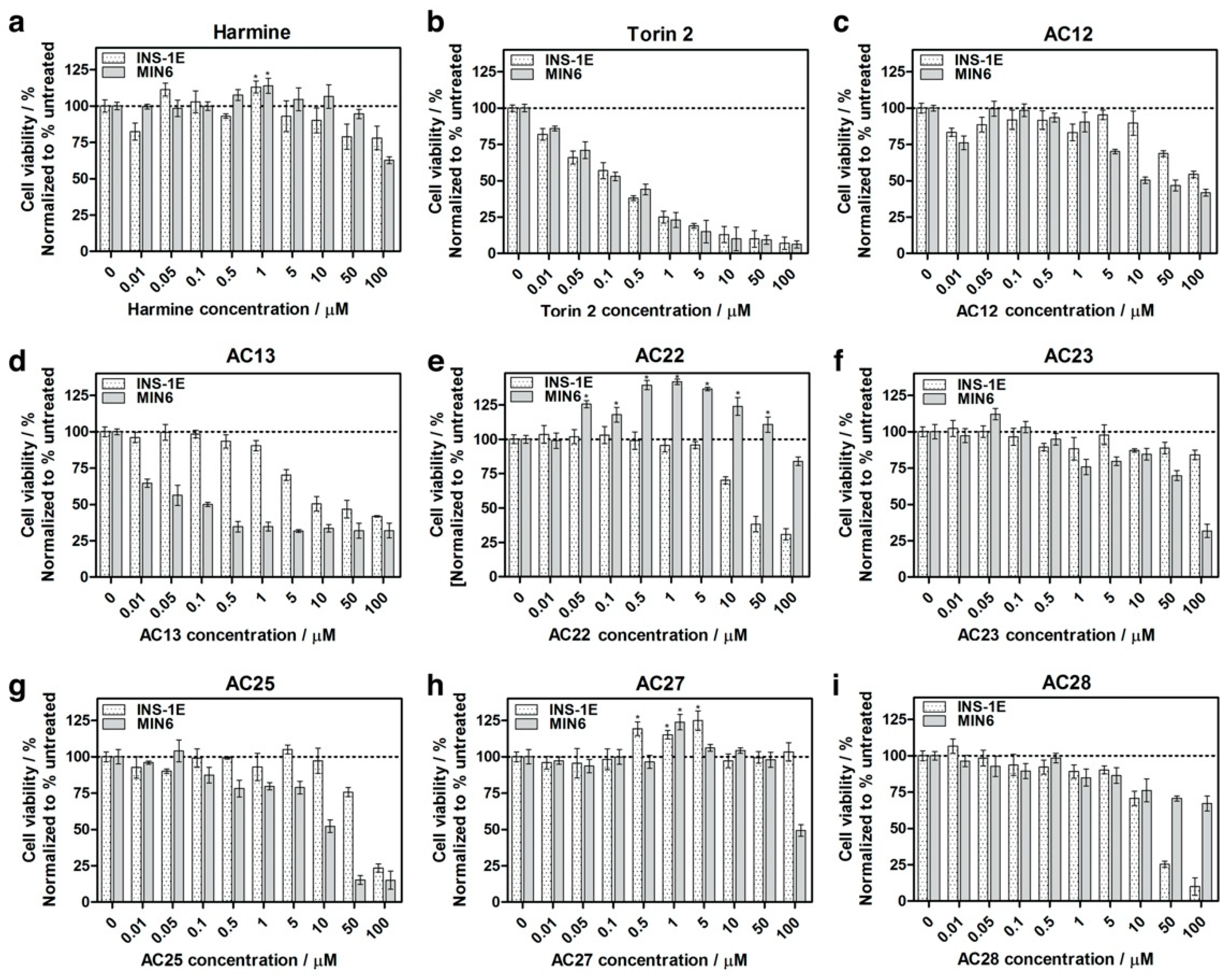
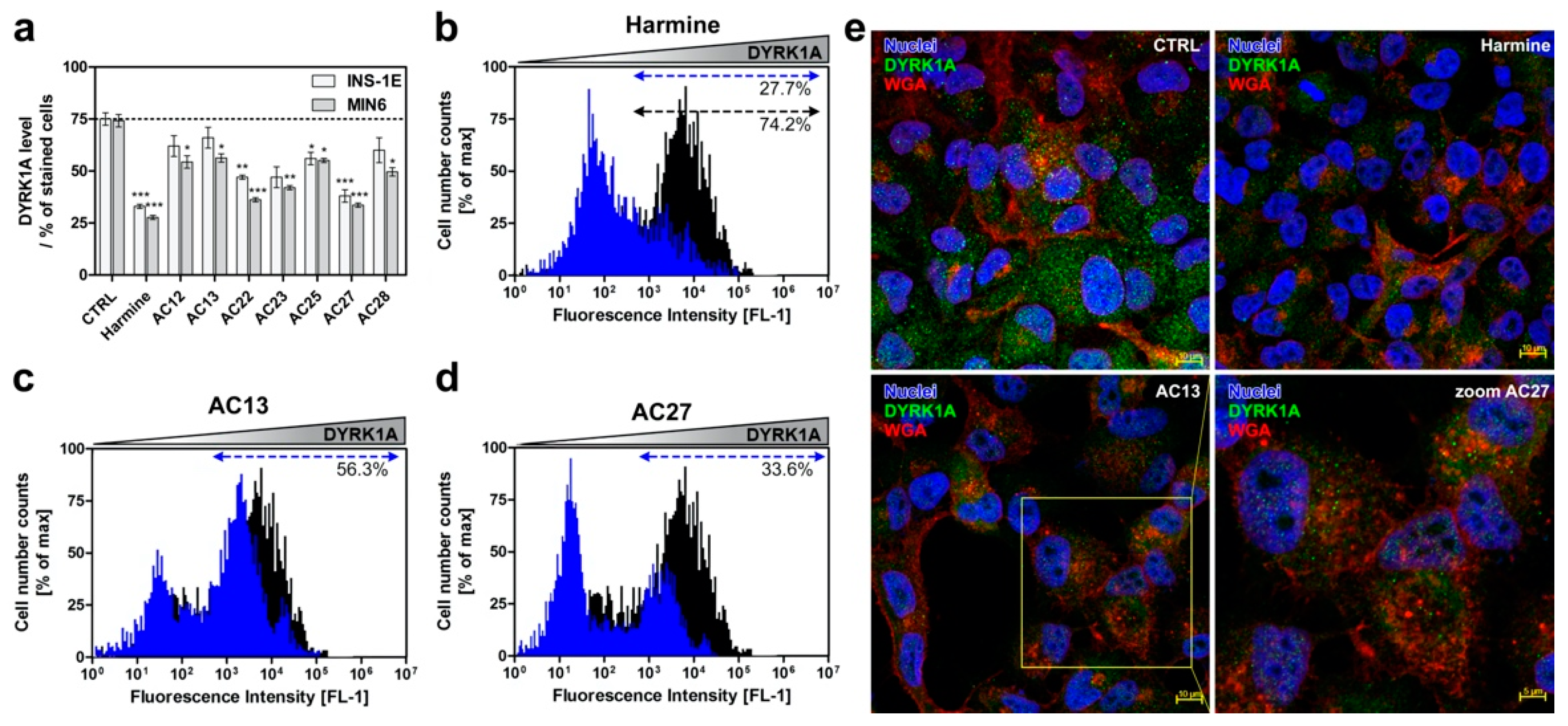
3.3. Inhibition of DYRK1A Activity Stimulates Growth of INS-1E and MIN6 β-Cells
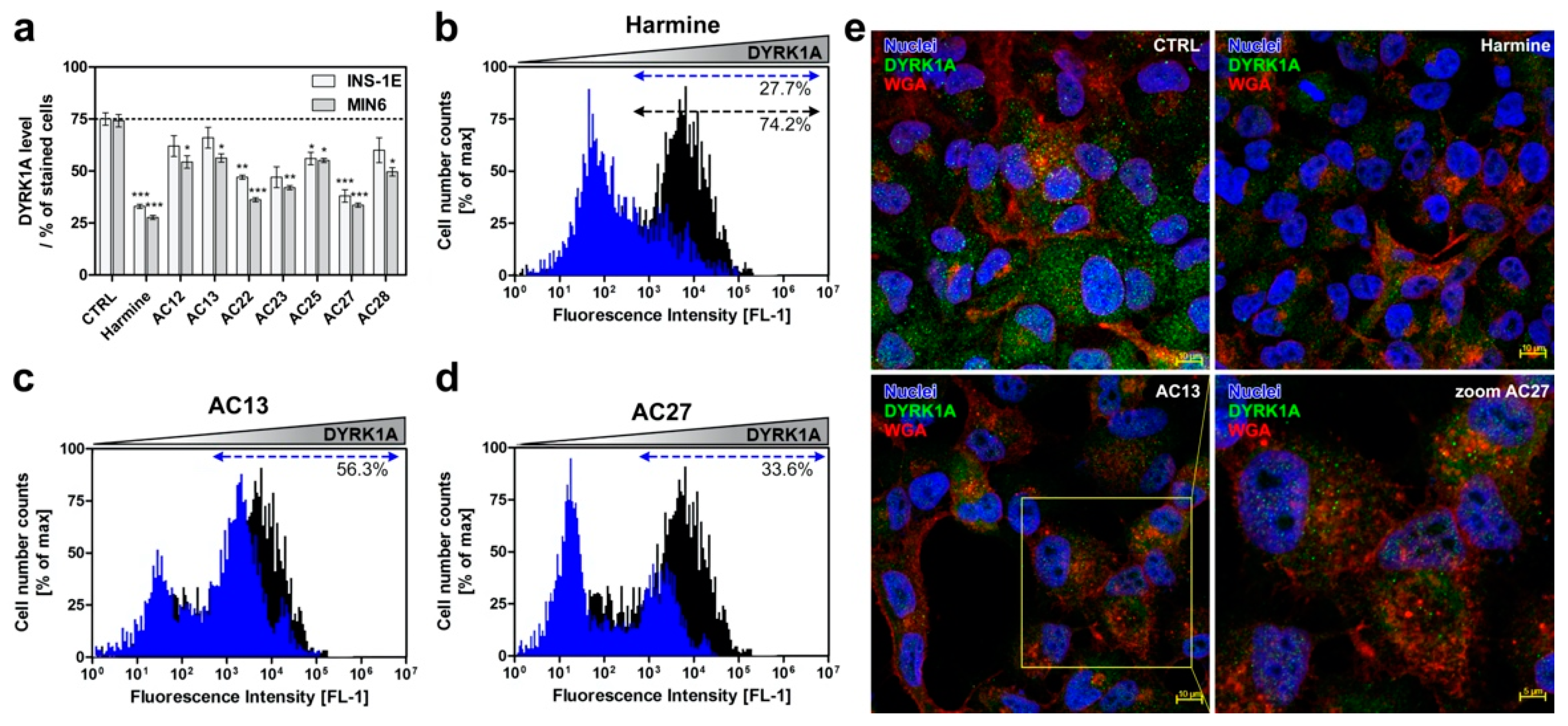
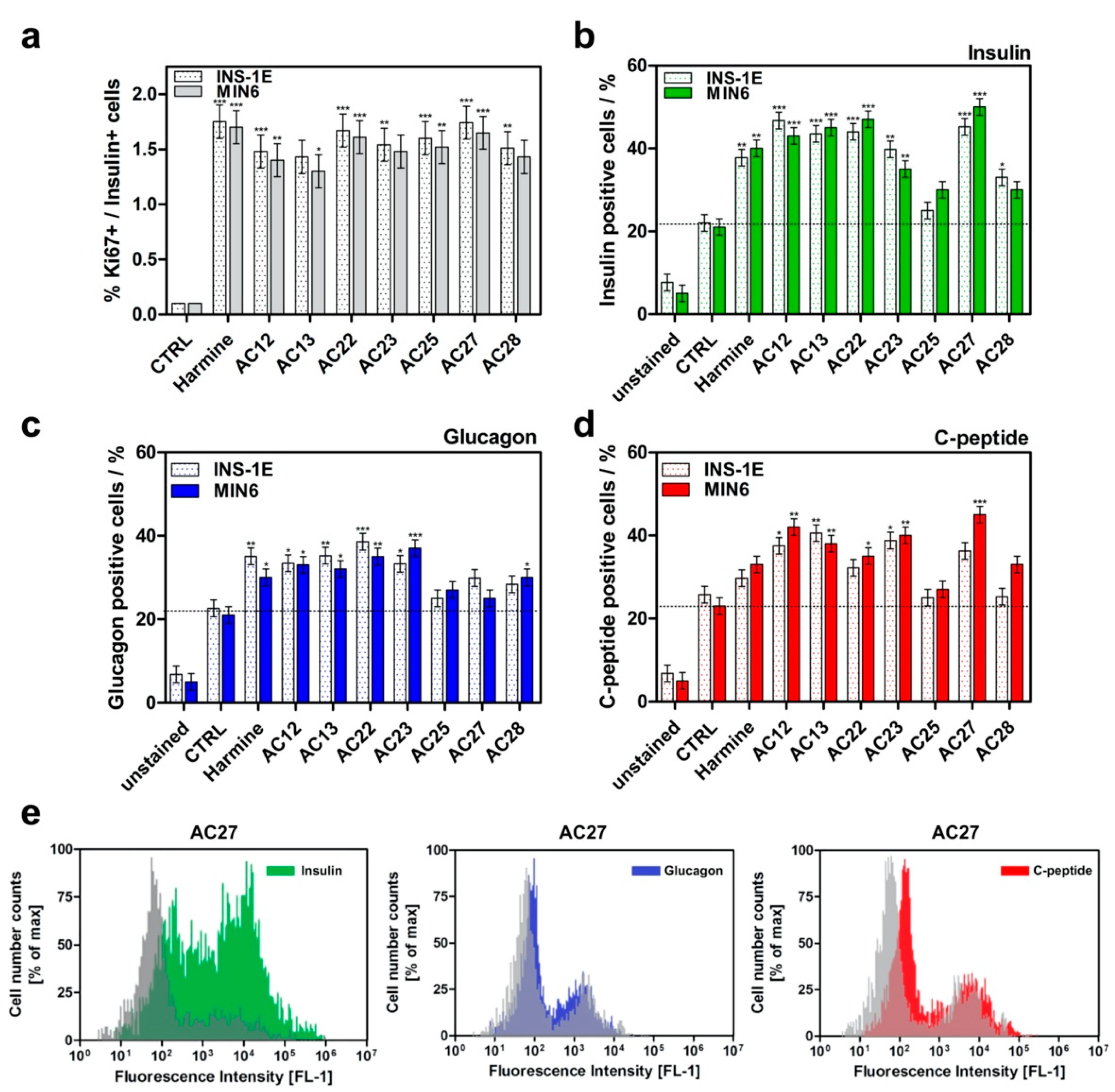
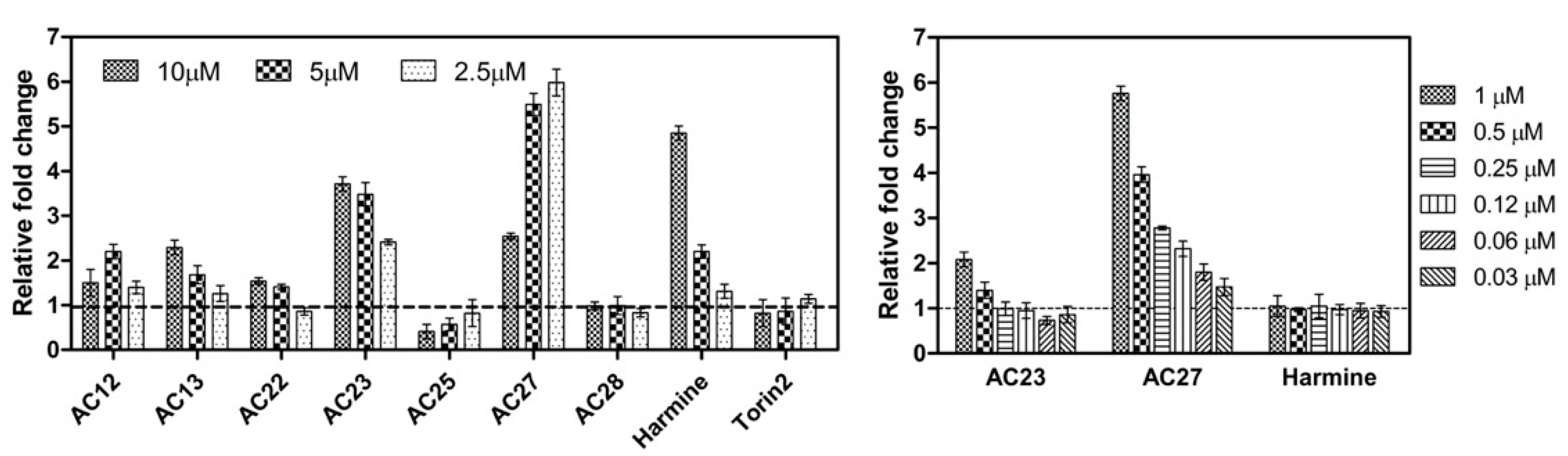
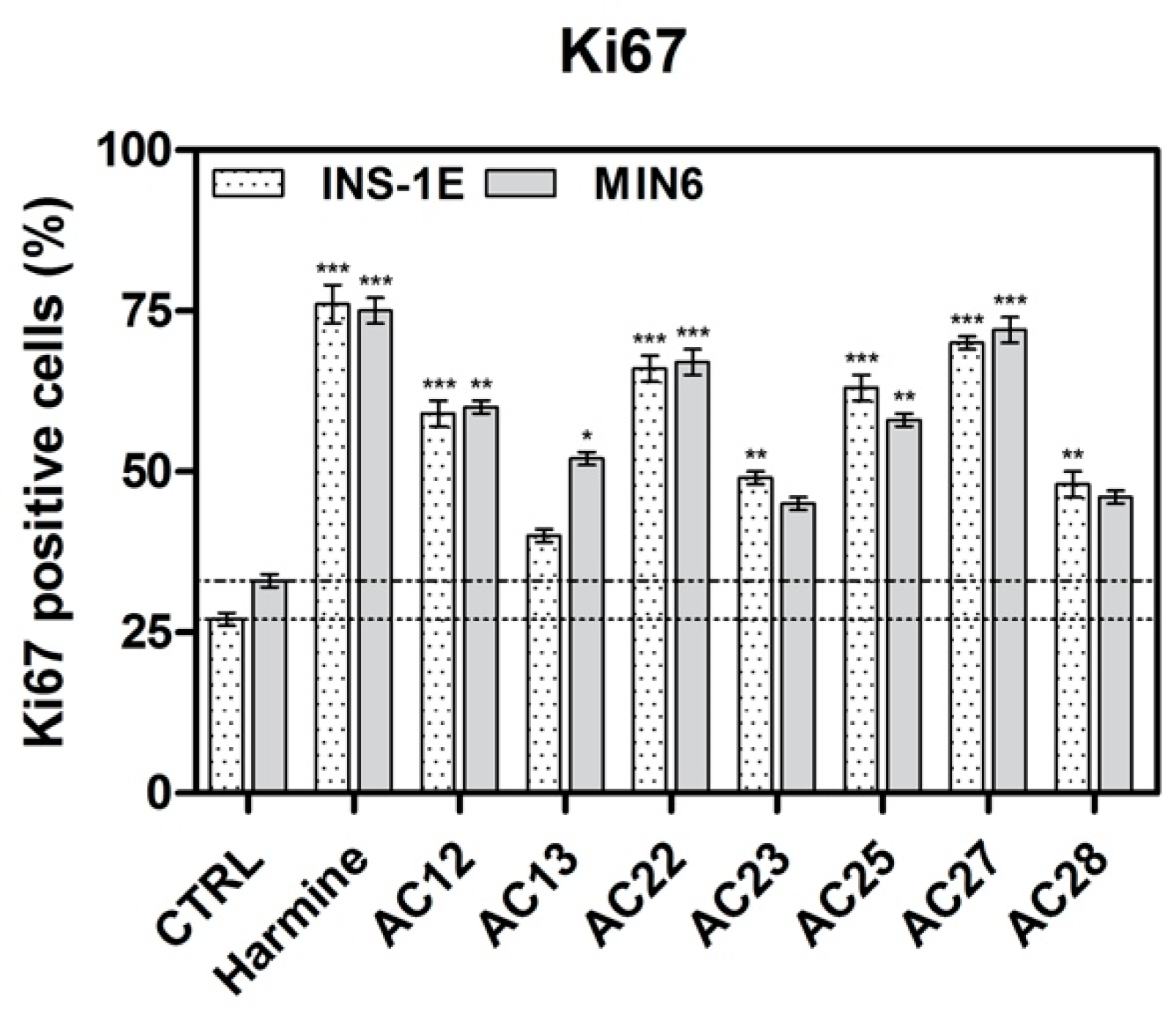
3.4. DYRK1A Inhibition Mediated by the Investigated Compounds Increases β-Cell Proliferation
3.5. The Influence of DYRK1A Inhibitors on the INS-1E and MIN6 Functionality
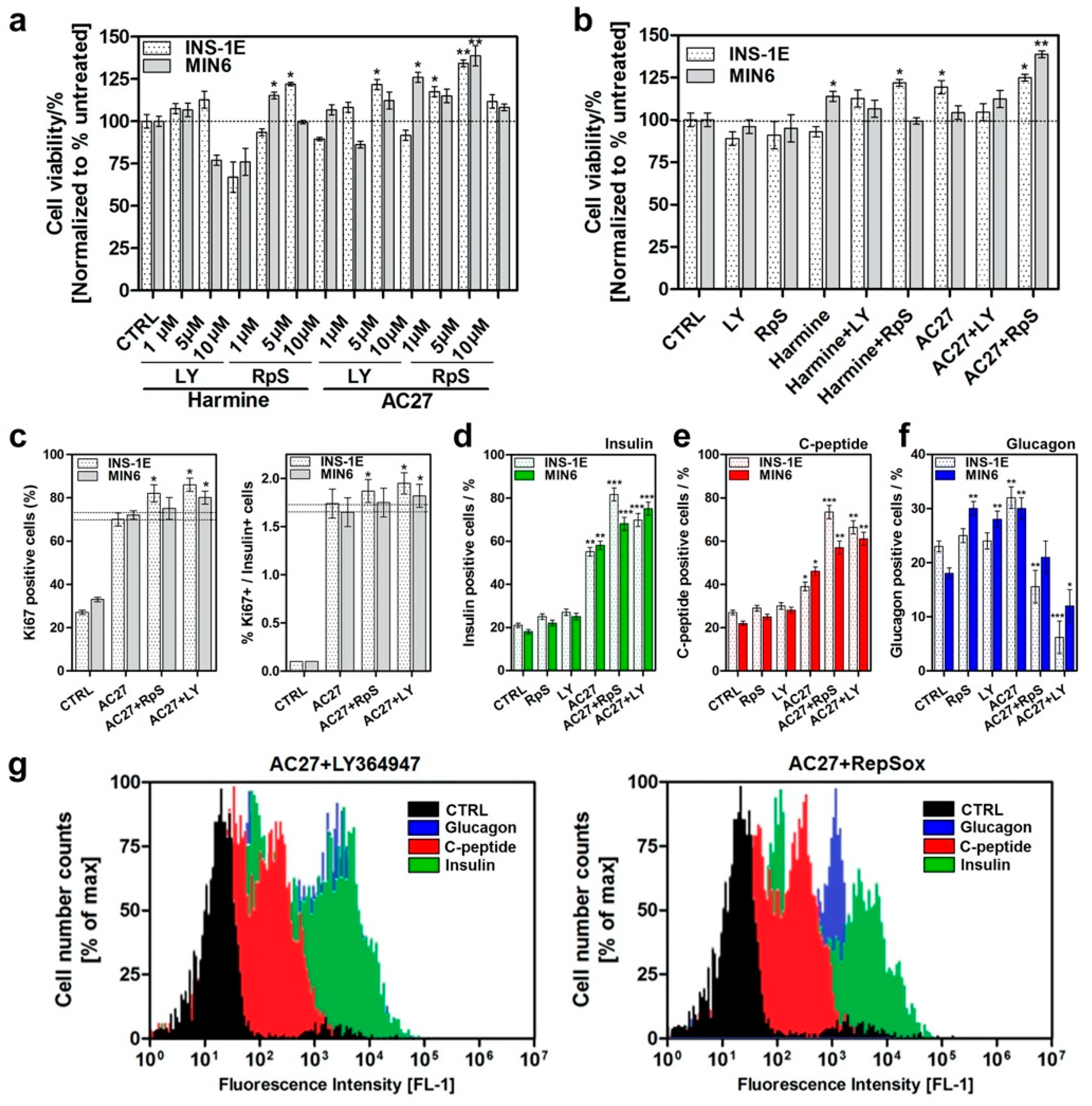
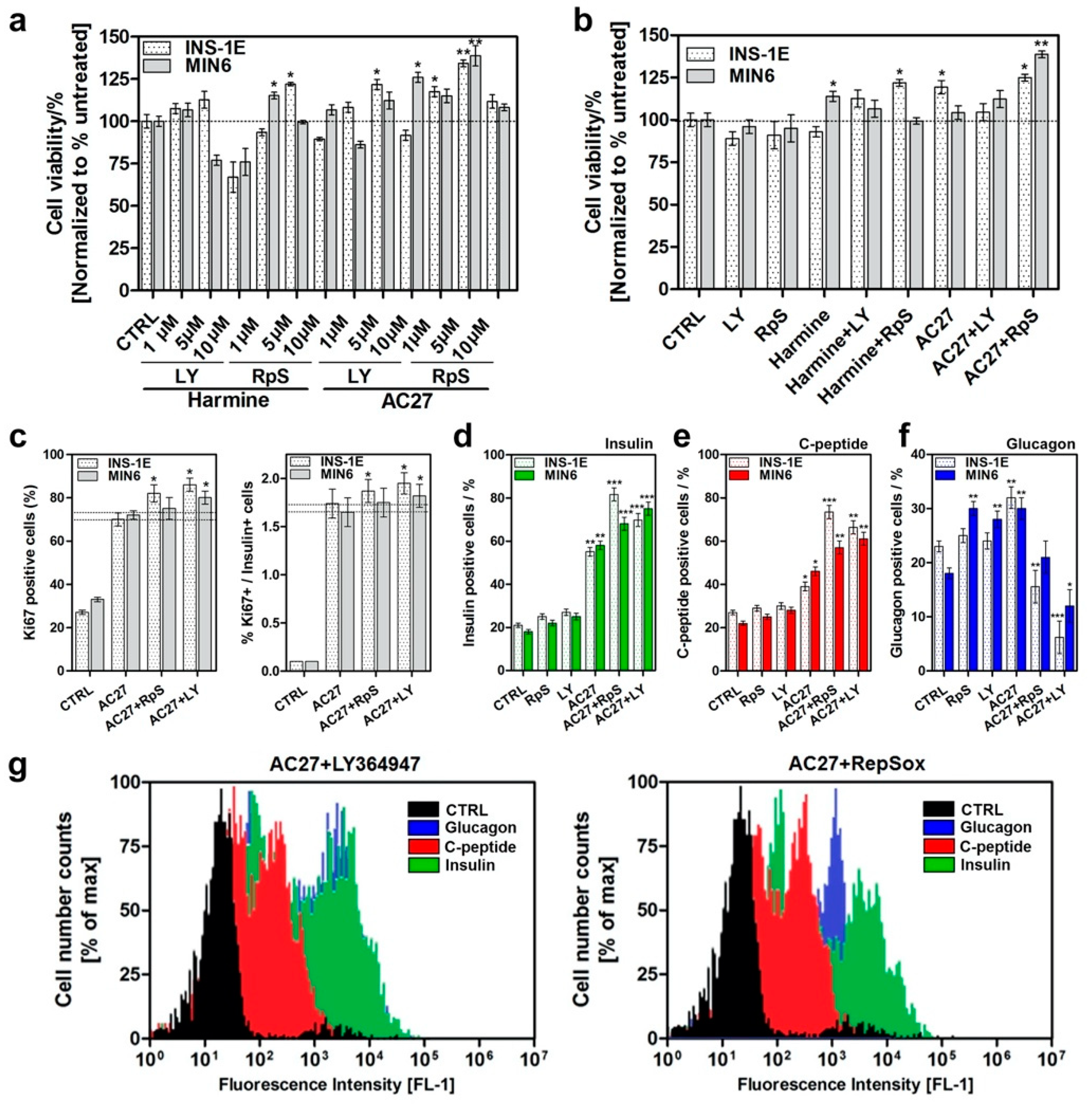
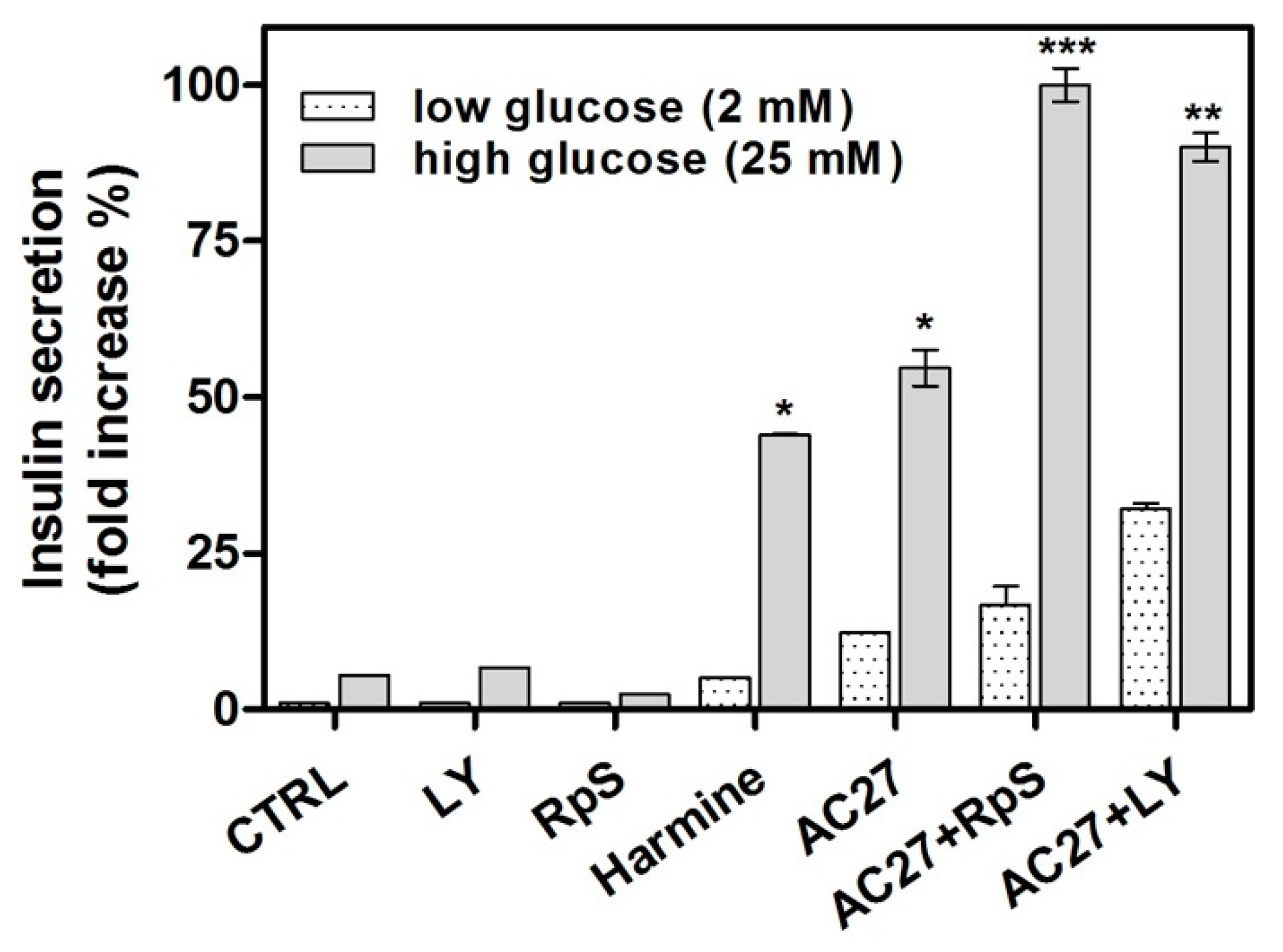
3.6. The Combination of AC27 with TGF-β Inhibitors Improves the Functionality of β-Cells In Vitro
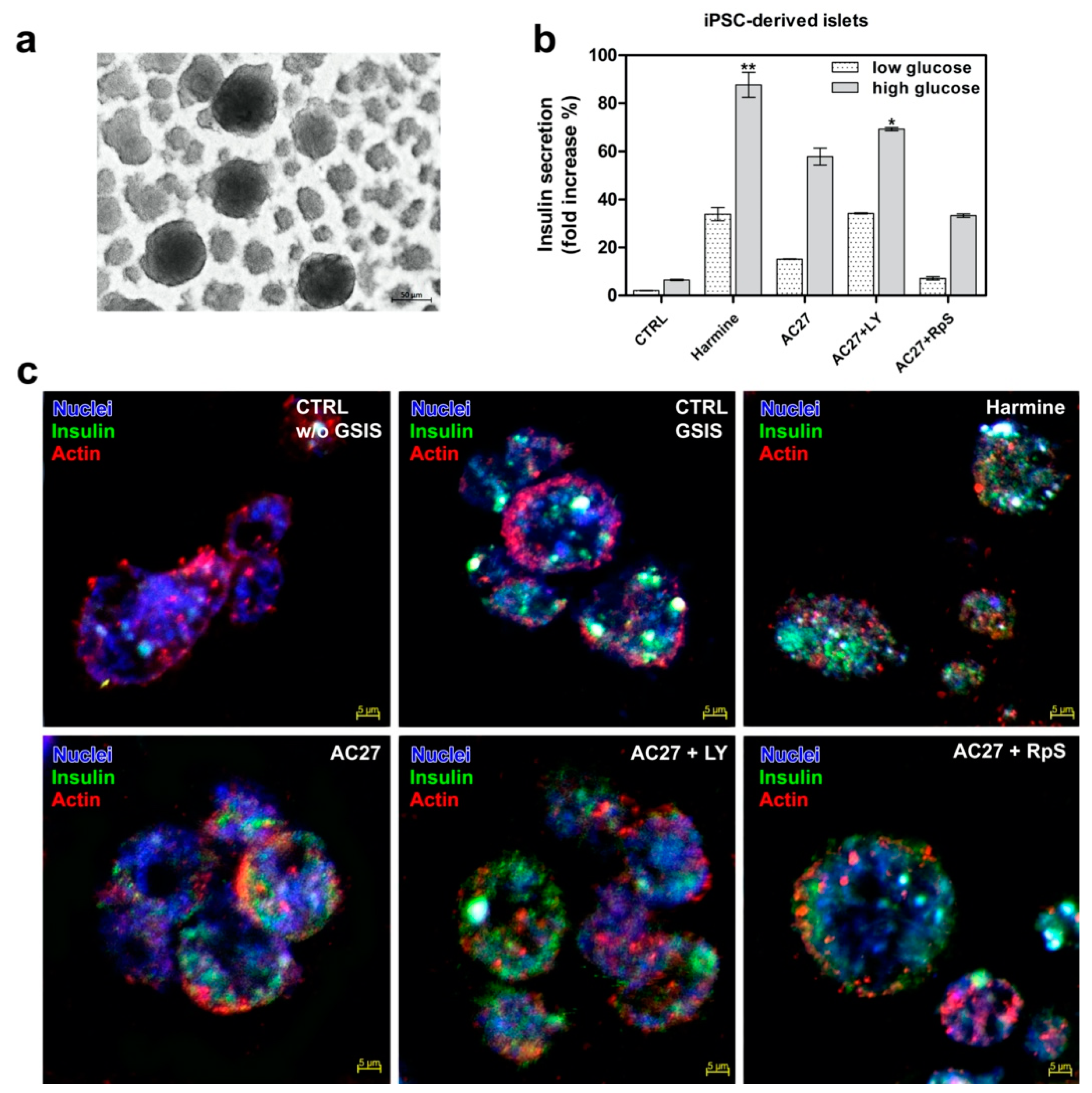
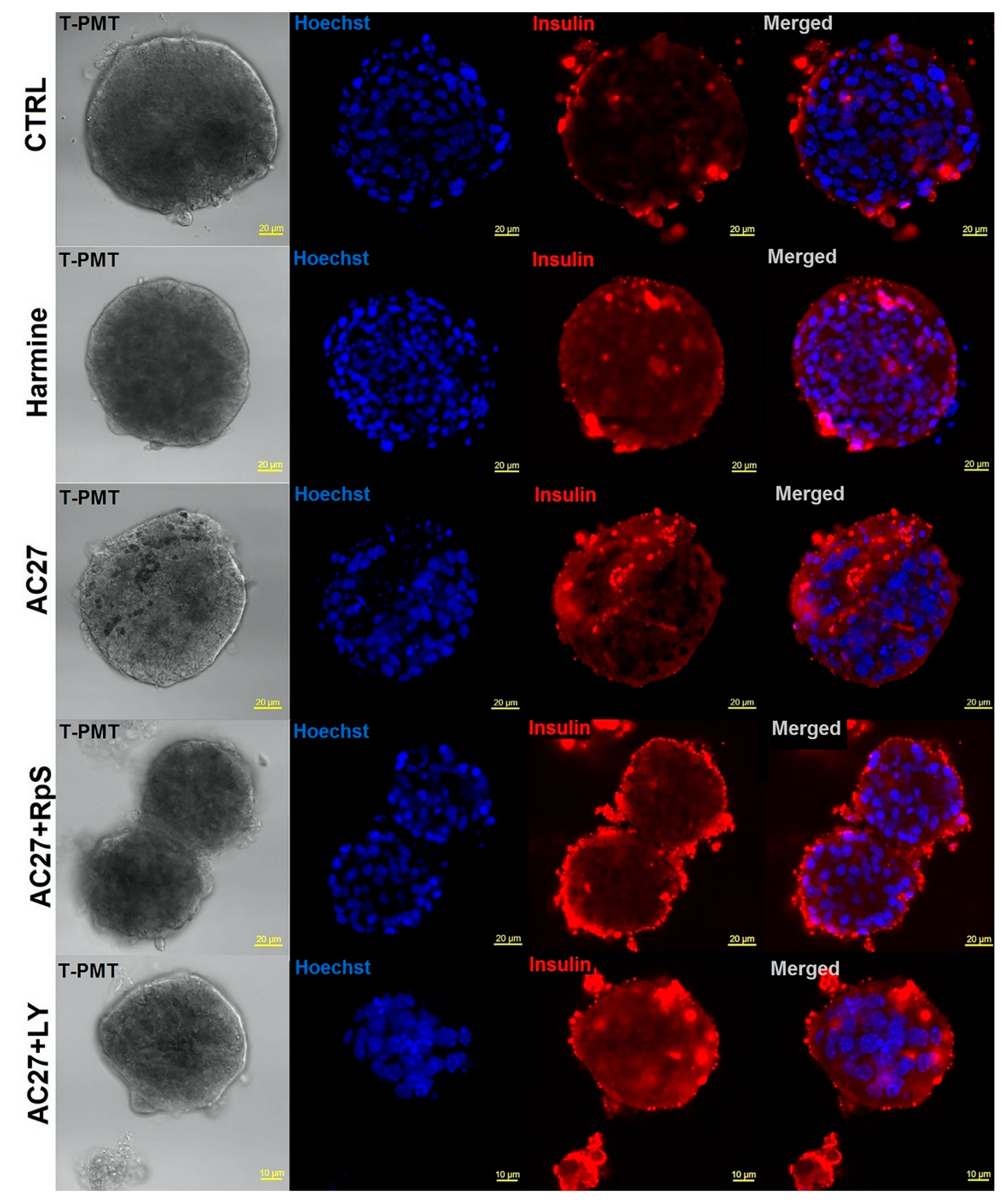
3.7. DYRK1A Inhibitors Activity in the Advanced Pancreatic Islet Models (Hipsc, Isolated Islets) Hipsc
3.8. Isolated Mouse Islets as an Advanced Model for Preclinical Research
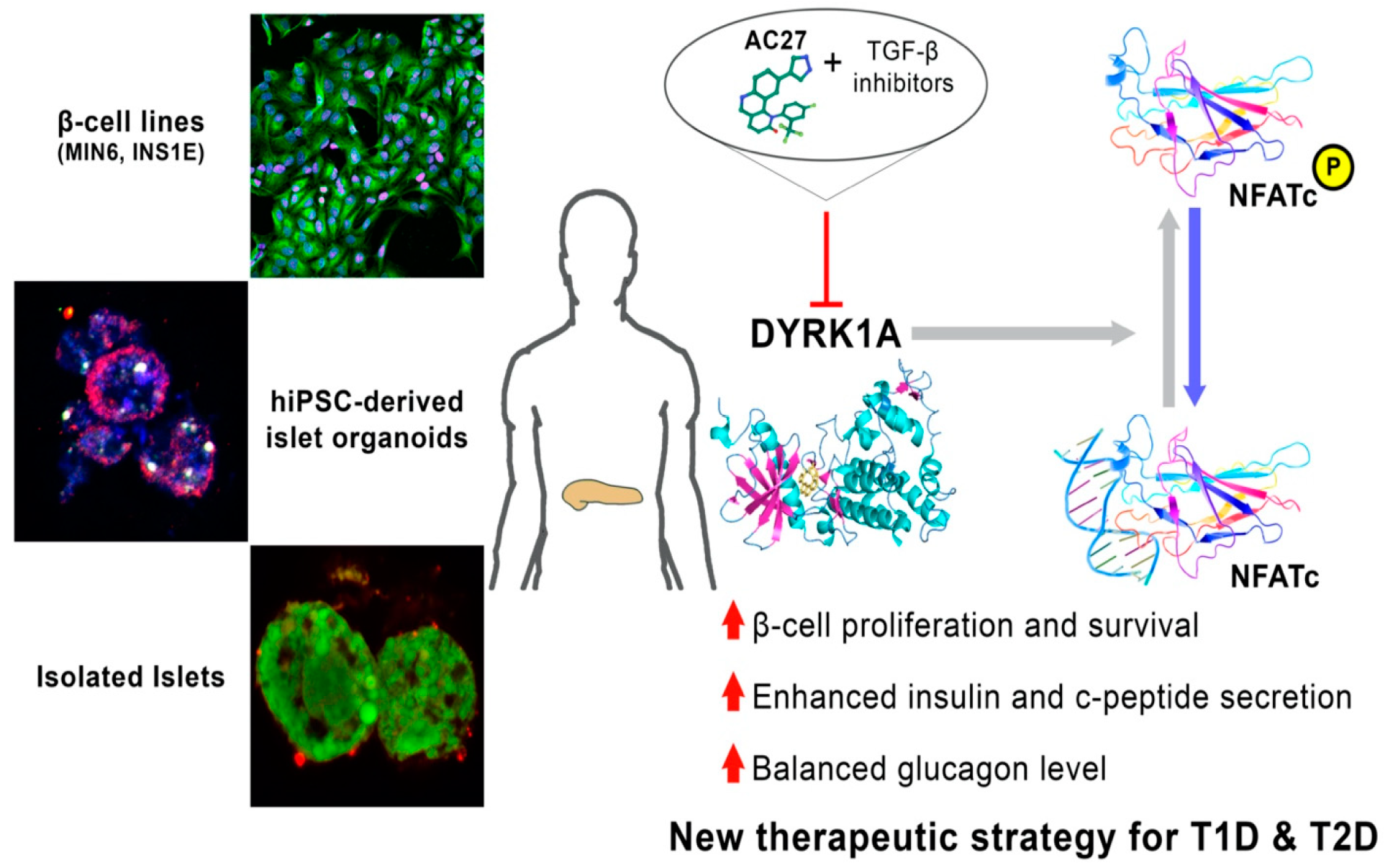
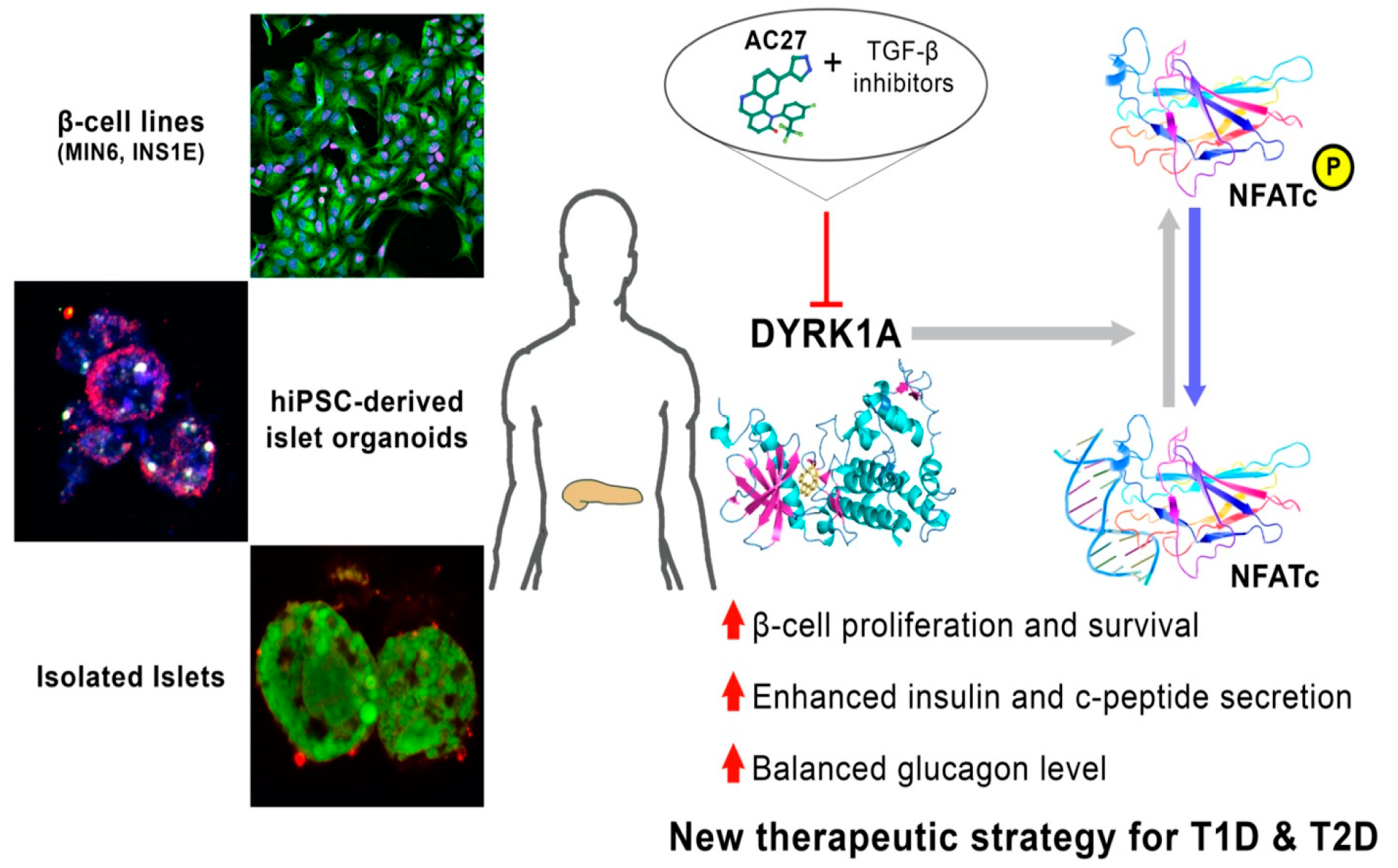
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, C.; Cohrs, C.M.; Stertmann, J.; Bozsak, R.; Speier, S. Human beta cell mass and function in diabetes: Recent advances in knowledge and technologies to understand disease pathogenesis. Mol. Metab. 2017, 6, 943–957. [Google Scholar] [CrossRef]
- Karakose, E.; Ackeifi, C.; Wang, P.; Stewart, A.F. Advances in drug discovery for human beta cell regeneration. Diabetologia 2018, 61, 1693–1699. [Google Scholar] [CrossRef] [Green Version]
- Zhang, E.; Al-Amily, I.M.; Mohammed, S.; Luan, C.; Asplund, O.; Ahmed, M.; Ye, Y.; Ben-Hail, D.; Soni, A.; Vishnu, N. Preserving insulin secretion in diabetes by inhibiting VDAC1 overexpression and surface translocation in β cells. Cell Metab. 2019, 29, 64–77. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Balhuizen, A.; Amisten, S.; Lundquist, I.; Salehi, A. Insulinotropic and antidiabetic effects of 17β-estradiol and the GPR30 agonist G-1 on human pancreatic islets. Endocrinology 2011, 152, 2568–2579. [Google Scholar] [CrossRef]
- Kumar, K.; Wang, P.; Sanchez, R.; Swartz, E.A.; Stewart, A.F.; DeVita, R.J. Development of kinase-selective, harmine-based DYRK1A inhibitors that induce pancreatic human β-cell proliferation. J. Med. Chem. 2018, 61, 7687–7699. [Google Scholar] [CrossRef] [PubMed]
- Göckler, N.; Jofre, G.; Papadopoulos, C.; Soppa, U.; Tejedor, F.J.; Becker, W. Harmine specifically inhibits protein kinase DYRK1A and interferes with neurite formation. FEBS J. 2009, 276, 6324–6337. [Google Scholar] [CrossRef] [Green Version]
- Dakic, V.; de Moraes Maciel, R.; Drummond, H.; Nascimento, J.M.; Trindade, P.; Rehen, S.K. Harmine stimulates proliferation of human neural progenitors. PeerJ 2016, 4, e2727. [Google Scholar] [CrossRef]
- Wang, P.; Alvarez-Perez, J.-C.; Felsenfeld, D.P.; Liu, H.; Sivendran, S.; Bender, A.; Kumar, A.; Sanchez, R.; Scott, D.K.; Garcia-Ocaña, A. A high-throughput chemical screen reveals that harmine-mediated inhibition of DYRK1A increases human pancreatic beta cell replication. Nat. Med. 2015, 21, 383–388. [Google Scholar] [CrossRef] [Green Version]
- Kumar, K.; Wang, P.; Swartz, E.A.; Khamrui, S.; Secor, C.; Lazarus, M.B.; Sanchez, R.; F Stewart, A.; DeVita, R.J. Structure–Activity Relationships and Biological Evaluation of 7-Substituted Harmine Analogs for Human β-Cell Proliferation. Molecules 2020, 25, 1983. [Google Scholar] [CrossRef] [Green Version]
- Kumar, K.; Wang, P.; Wilson, J.; Zlatanic, V.; Berrouet, C.; Khamrui, S.; Secor, C.; Swartz, E.A.; Lazarus, M.; Sanchez, R. Synthesis and Biological Validation of a Harmine-based, Central Nervous System (CNS)-Avoidant, Selective, Human β-Cell Regenerative Dual-Specificity Tyrosine Phosphorylation-Regulated Kinase A (DYRK1A) Inhibitor. J. Med. Chem. 2020, 63, 2986–3003. [Google Scholar] [CrossRef]
- Fant, X.; Durieu, E.; Chicanne, G.; Payrastre, B.; Sbrissa, D.; Shisheva, A.; Limanton, E.; Carreaux, F.; Bazureau, J.-P.; Meijer, L. CLK/DYRK kinases inhibitor Leucettine L41 induces mTOR-dependent autophagy. Implication for Alzheimers’ disease. Mol. Pharmacol. 2013. [Google Scholar] [CrossRef]
- Tahtouh, T.; Elkins, J.M.; Filippakopoulos, P.; Soundararajan, M.; Burgy, G.; Durieu, E.; Cochet, C.; Schmid, R.S.; Lo, D.C.; Delhommel, F. Selectivity, cocrystal structures, and neuroprotective properties of leucettines, a family of protein kinase inhibitors derived from the marine sponge alkaloid leucettamine B. J. Med. Chem. 2012, 55, 9312–9330. [Google Scholar] [CrossRef] [PubMed]
- Abdolazimi, Y.; Zhao, Z.; Lee, S.; Xu, H.; Allegretti, P.; Horton, T.M.; Yeh, B.; Moeller, H.P.; Nichols, R.J.; McCutcheon, D. CC-401 promotes β-cell replication via pleiotropic consequences of DYRK1A/B inhibition. Endocrinology 2018, 159, 3143–3157. [Google Scholar] [CrossRef] [Green Version]
- Dirice, E.; Walpita, D.; Vetere, A.; Meier, B.C.; Kahraman, S.; Hu, J.; Dančík, V.; Burns, S.M.; Gilbert, T.J.; Olson, D.E. Inhibition of DYRK1A stimulates human β-cell proliferation. Diabetes 2016, 65, 1660–1671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogawa, Y.; Nonaka, Y.; Goto, T.; Ohnishi, E.; Hiramatsu, T.; Kii, I.; Yoshida, M.; Ikura, T.; Onogi, H.; Shibuya, H. Development of a novel selective inhibitor of the Down syndrome-related kinase Dyrk1A. Nat. Commun. 2010, 1, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Masaki, S.; Kii, I.; Sumida, Y.; Kato-Sumida, T.; Ogawa, Y.; Ito, N.; Nakamura, M.; Sonamoto, R.; Kataoka, N.; Hosoya, T. Design and synthesis of a potent inhibitor of class 1 DYRK kinases as a suppressor of adipogenesis. Bioorganic Med. Chem. 2015, 23, 4434–4441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Z.; Tremblay, M.S.; Wu, T.Y.-H.; Ding, Q.; Hao, X.; Baaten, J.; Hill, R.; Liu, B.; Meeusen, S.; Zou, Y. Discovery of 5-(3,4-Difluorophenyl)-3-(pyrazol-4-yl)-7-azaindole (GNF3809) for β-Cell Survival in Type 1 Diabetes. ACS Omega 2019, 4, 3571–3581. [Google Scholar] [CrossRef]
- Liu, Y.A.; Jin, Q.; Ding, Q.; Hao, X.; Mo, T.; Yan, S.; Zou, Y.; Huang, Z.; Zhang, X.; Gao, W. A Dual Inhibitor of DYRK1A and GSK3β for β-Cell Proliferation: Aminopyrazine Derivative GNF4877. ChemMedChem 2020, 15, 1562–1570. [Google Scholar] [CrossRef]
- Liu, Y.A.; Jin, Q.; Zou, Y.; Ding, Q.; Yan, S.; Wang, Z.; Hao, X.; Nguyen, B.; Zhang, X.; Pan, J. Selective DYRK1A inhibitor for the treatment of Type 1 Diabetes: Discovery of 6-azaindole derivative GNF2133. J. Med. Chem. 2020, 63, 2958–2973. [Google Scholar] [CrossRef]
- Kumar, K.; Suebsuwong, C.; Wang, P.; Garcia-Ocana, A.; Stewart, A.F.; DeVita, R.J. DYRK1A Inhibitors as Potential Therapeutics for β-Cell Regeneration for Diabetes. J. Med. Chem. 2021, 64, 2901–2922. [Google Scholar] [CrossRef]
- Jarhad, D.B.; Mashelkar, K.K.; Kim, H.-R.; Noh, M.; Jeong, L.S. Dual-specificity tyrosine phosphorylation-regulated kinase 1A (DYRK1A) inhibitors as potential therapeutics. J. Med. Chem. 2018, 61, 9791–9810. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.L.; Fruit, C.; Hérault, Y.; Meijer, L.; Besson, T. Dual-specificity tyrosine phosphorylation-regulated kinase 1A (DYRK1A) inhibitors: A survey of recent patent literature. Expert Opin. Ther. Pat. 2017, 27, 1183–1199. [Google Scholar] [CrossRef]
- Belgardt, B.-F.; Lammert, E. DYRK1A: A Promising Drug Target for Islet Transplant–Based Diabetes Therapies. Diabetes 2016, 65, 1496–1498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, W.; Sippl, W. Activation, regulation, and inhibition of DYRK1A. FEBS J. 2011, 278, 246–256. [Google Scholar] [CrossRef]
- Qian, W.; Jin, N.; Shi, J.; Yin, X.; Jin, X.; Wang, S.; Cao, M.; Iqbal, K.; Gong, C.-X.; Liu, F. Dual-specificity tyrosine phosphorylation-regulated kinase 1A (Dyrk1A) enhances tau expression. J. Alzheimer’s Dis. 2013, 37, 529–538. [Google Scholar] [CrossRef]
- Himpel, S.; Panzer, P.; Eirmbter, K.; Czajkowska, H.; Sayed, M.; Packman, L.C.; Blundell, T.; Kentrup, H.; Grötzinger, J.; Joost, H.-G. Identification of the autophosphorylation sites and characterization of their effects in the protein kinase DYRK1A. Biochem. J. 2001, 359, 497–505. [Google Scholar] [CrossRef]
- Wiechmann, S.; Czajkowska, H.; de Graaf, K.; Grötzinger, J.; Joost, H.-G.; Becker, W. Unusual function of the activation loop in the protein kinase DYRK1A. Biochem. Biophys. Res. Commun. 2003, 302, 403–408. [Google Scholar] [CrossRef]
- Stotani, S.; Giordanetto, F.; Medda, F. DYRK1A inhibition as potential treatment for Alzheimer’s disease. Future Med. Chem. 2016, 8, 681–696. [Google Scholar] [CrossRef] [PubMed]
- Janel, N.; Sarazin, M.; Corlier, F.; Corne, H.; De Souza, L.C.; Hamelin, L.; Aka, A.; Lagarde, J.; Blehaut, H.; Hindié, V. Plasma DYRK1A as a novel risk factor for Alzheimer’s disease. Transl. Psychiatry 2014, 4, e425. [Google Scholar] [CrossRef] [Green Version]
- Kargbo, R.B. Selective DYRK1A Inhibitor for the Treatment of Neurodegenerative Diseases: Alzheimer, Parkinson, Huntington, and Down Syndrome. ACS Med. Chem. Lett. 2020, 11, 1795–1796. [Google Scholar] [CrossRef] [PubMed]
- Rachdi, L.; Kariyawasam, D.; Aïello, V.; Herault, Y.; Janel, N.; Delabar, J.-M.; Polak, M.; Scharfmann, R. Dyrk1A induces pancreatic β cell mass expansion and improves glucose tolerance. Cell Cycle 2014, 13, 2221–2229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, J.; Lee, H.; Argiropoulos, B.; Dorrani, N.; Mann, J.; Martinez-Agosto, J.A.; Gomez-Ospina, N.; Gallant, N.; Bernstein, J.A.; Hudgins, L. DYRK1A haploinsufficiency causes a new recognizable syndrome with microcephaly, intellectual disability, speech impairment, and distinct facies. Eur. J. Hum. Genet. 2015, 23, 1473–1481. [Google Scholar] [CrossRef] [PubMed]
- Rachdi, L.; Kariyawasam, D.; Guez, F.; Aïello, V.; Arbonés, M.L.; Janel, N.; Delabar, J.-M.; Polak, M.; Scharfmann, R. Dyrk1a haploinsufficiency induces diabetes in mice through decreased pancreatic beta cell mass. Diabetologia 2014, 57, 960–969. [Google Scholar] [CrossRef]
- Shen, W.; Taylor, B.; Jin, Q.; Nguyen-Tran, V.; Meeusen, S.; Zhang, Y.-Q.; Kamireddy, A.; Swafford, A.; Powers, A.F.; Walker, J. Inhibition of DYRK1A and GSK3B induces human β-cell proliferation. Nat. Commun. 2015, 6, 1–11. [Google Scholar] [CrossRef]
- Wang, P.; Karakose, E.; Liu, H.; Swartz, E.; Ackeifi, C.; Zlatanic, V.; Wilson, J.; González, B.J.; Bender, A.; Takane, K.K. Combined inhibition of DYRK1A, SMAD, and trithorax pathways synergizes to induce robust replication in adult human beta cells. Cell Metab. 2019, 29, 638–652. [Google Scholar] [CrossRef] [Green Version]
- Brown, M.L.; Schneyer, A.L. Emerging roles for the TGFβ family in pancreatic β-cell homeostasis. Trends Endocrinol. Metab. 2010, 21, 441–448. [Google Scholar] [CrossRef] [Green Version]
- Dhawan, S.; Dirice, E.; Kulkarni, R.N.; Bhushan, A. Inhibition of TGF-β signaling promotes human pancreatic β-cell replication. Diabetes 2016, 65, 1208–1218. [Google Scholar] [CrossRef] [Green Version]
- Puri, S.; Roy, N.; Russ, H.A.; Leonhardt, L.; French, E.K.; Roy, R.; Bengtsson, H.; Scott, D.K.; Stewart, A.F.; Hebrok, M. Replication confers β cell immaturity. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Lechner, C.; Flaßhoff, M.; Falke, H.; Preu, L.; Loaëc, N.; Meijer, L.; Knapp, S.; Chaikuad, A.; Kunick, C. [b]-Annulated Halogen-Substituted Indoles as Potential DYRK1A Inhibitors. Molecules 2019, 24, 4090. [Google Scholar] [CrossRef] [Green Version]
- Fruit, C.; Couly, F.; Bhansali, R.; Rammohan, M.; Lindberg, M.F.; Crispino, J.D.; Meijer, L.; Besson, T. Biological Characterization of 8-Cyclopropyl-2-(pyridin-3-yl) thiazolo [5, 4-f] quinazolin-9 (8H)-one, a Promising Inhibitor of DYRK1A. Pharmaceuticals 2019, 12, 185. [Google Scholar] [CrossRef] [Green Version]
- Foucourt, A.; Hédou, D.; Dubouilh-Benard, C.; Désiré, L.; Casagrande, A.-S.; Leblond, B.; Loäec, N.; Meijer, L.; Besson, T. Design and synthesis of thiazolo [5, 4-f] quinazolines as DYRK1A inhibitors, Part I. Molecules 2014, 19, 15546–15571. [Google Scholar] [CrossRef] [Green Version]
- Grabher, P.; Durieu, E.; Kouloura, E.; Halabalaki, M.; Skaltsounis, L.A.; Meijer, L.; Hamburger, M.; Potterat, O. Library-based discovery of DYRK1A/CLK1 inhibitors from natural product extracts. Planta Med. 2012, 78, 951–956. [Google Scholar] [CrossRef] [PubMed]
- Bruel, A.; Bénéteau, R.; Chabanne, M.; Lozach, O.; Le Guevel, R.; Ravache, M.; Bénédetti, H.; Meijer, L.; Logé, C.; Robert, J.-M. Synthesis of new pyridazino [4, 5-b] indol-4-ones and pyridazin-3 (2H)-one analogs as DYRK1A inhibitors. Bioorganic Med. Chem. Lett. 2014, 24, 5037–5040. [Google Scholar] [CrossRef] [PubMed]
- Czarna, A.; Wang, J.; Zelencova, D.; Liu, Y.; Deng, X.; Choi, H.G.; Zhang, T.; Zhou, W.; Chang, J.W.; Kildalsen, H. Novel scaffolds for Dual specificity tyrosine-phosphorylation-regulated kinase (DYRK1A) inhibitors. J. Med. Chem. 2018, 61, 7560–7572. [Google Scholar] [CrossRef] [Green Version]
- Merglen, A.; Theander, S.; Rubi, B.; Chaffard, G.; Wollheim, C.B.; Maechler, P. Glucose sensitivity and metabolism-secretion coupling studied during two-year continuous culture in INS-1E insulinoma cells. Endocrinology 2004, 145, 667–678. [Google Scholar] [CrossRef]
- Cook, P.F.; Neville Jr, M.E.; Vrana, K.E.; Hartl, F.T.; Roskoski Jr, R. Adenosine cyclic 3’, 5’-monophosphate dependent protein kinase: Kinetic mechanism for the bovine skeletal muscle catalytic subunit. Biochemistry 1982, 21, 5794–5799. [Google Scholar] [CrossRef]
- Fernandez-Martinez, P.; Zahonero, C.; Sanchez-Gomez, P. DYRK1A: The double-edged kinase as a protagonist in cell growth and tumorigenesis. Mol. Cell. Oncol. 2015, 2, e970048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Wang, K.; Chen, S.; Sun, Q.; Zhang, Y.; Chen, L.; Sun, X. NFATc1 phosphorylation by DYRK1A increases its protein stability. PLoS ONE 2017, 12, e0172985. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Wang, X.; Su, Z.; Fei, H.; Liu, X.; Pan, Q. The novel mTOR inhibitor Torin-2 induces autophagy and downregulates the expression of UHRF1 to suppress hepatocarcinoma cell growth. Oncol. Rep. 2015, 34, 1708–1716. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Xu, C.; Kirubakaran, S.; Zhang, X.; Hur, W.; Liu, Y.; Kwiatkowski, N.P.; Wang, J.; Westover, K.D.; Gao, P. Characterization of Torin2, an ATP-competitive inhibitor of mTOR, ATM, and ATR. Cancer Res. 2013, 73, 2574–2586. [Google Scholar] [CrossRef] [Green Version]
- Gerdes, J.S.T.; Scholzen, T. The Ki-67 protein: From the known and the unknown. J. Cell Physiol. 2000, 182, 311–322. [Google Scholar]
- Skelin, M.; Rupnik, M.; Cencič, A. Pancreatic beta cell lines and their applications in diabetes mellitus research. ALTEX-Altern. Anim. Exp. 2010, 27, 105–113. [Google Scholar] [CrossRef] [Green Version]
- Stewart, A.F. Human Beta Cell Regenerative Drug Therapy for Diabetes: Past Achievements and Future Challenges. Front. Endocrinol. 2021, 12, 776. [Google Scholar]
- Stępniewski, J.; Tomczyk, M.; Andrysiak, K.; Kraszewska, I.; Martyniak, A.; Langrzyk, A.; Kulik, K.; Wiśniewska, E.; Jeż, M.; Florczyk-Soluch, U. Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes, in Contrast to Adipose Tissue-Derived Stromal Cells, Efficiently Improve Heart Function in Murine Model of Myocardial Infarction. Biomedicines 2020, 8, 578. [Google Scholar] [CrossRef] [PubMed]
- Jeż, M.; Martyniak, A.; Andrysiak, K.; Mucha, O.; Szade, K.; Kania, A.; Chrobok, Ł.; Palus-Chramiec, K.; Sanetra, A.M.; Lewandowski, M.H. Role of Heme-Oxygenase-1 in Biology of Cardiomyocytes Derived from Human Induced Pluripotent Stem Cells. Cells 2021, 10, 522. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, S.; Manenti, F.; Chimienti, R.; Nano, R.; Ottoboni, L.; Ruffini, F.; Martino, G.; Ravassard, P.; Piemonti, L.; Sordi, V. Differentiation of Sendai Virus-Reprogrammed iPSC into β Cells, Compared with Human Pancreatic Islets and Immortalized β Cell Line. Cell Transplant. 2018, 27, 1548–1560. [Google Scholar] [CrossRef]
- Kaneto, H.; Miyatsuka, T.; Kawamori, D.; Yamamoto, K.; Kato, K.; Shiraiwa, T.; Katakami, N.; Yamasaki, Y.; Matsuhisa, M.; Matsuoka, T.-A. PDX-1 and MafA play a crucial role in pancreatic β-cell differentiation and maintenance of mature β-cell function. Endocr. J. 2007, 55, 235. [Google Scholar] [CrossRef] [Green Version]
- Øster, A.; Jensen, J.; Serup, P.; Galante, P.; Madsen, O.D.; Larsson, L.-I. Rat endocrine pancreatic development in relation to two homeobox gene products (Pdx-1 and Nkx 6.1). J. Histochem. Cytochem. 1998, 46, 707–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hohmeier, H.E.; Zhang, L.; Taylor, B.; Stephens, S.; Lu, D.; McNamara, P.; Laffitte, B.; Newgard, C.B. Identification of a small molecule that stimulates human β-cell proliferation and insulin secretion, and protects against cytotoxic stress in rat insulinoma cells. PLoS ONE 2020, 15, e0224344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zenz, S.; Mader, J.K.; Regittnig, W.; Brunner, M.; Korsatko, S.; Boulgaropoulos, B.; Magnes, C.; Raml, R.; Narath, S.H.; Eller, P. Impact of C-peptide status on the response of glucagon and endogenous glucose production to induced hypoglycemia in T1DM. J. Clin. Endocrinol. Metab. 2018, 103, 1408–1417. [Google Scholar] [CrossRef] [Green Version]
- Qader, S.; Lundquist, I.; Ekelund, M.; Hakanson, R.; Salehi, A. Ghrelin activates neuronal constitutive nitric oxide synthase in pancreatic islet cells while inhibiting insulin release and stimulating glucagon release. Regul. Pept. 2005, 128, 51–56. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Wang, J.; Kang, S.A.; Thoreen, C.C.; Hur, W.; Ahmed, T.; Sabatini, D.M.; Gray, N.S. Discovery of 9-(6-Aminopyridin-3-yl)-1-(3-(trifluoromethyl) phenyl) benzo [h][1, 6] naphthyridin-2 (1 H)-one (Torin2) as a Potent, Selective, and Orally Available Mammalian Target of Rapamycin (mTOR) Inhibitor for Treatment of Cancer. J. Med. Chem. 2011, 54, 1473–1480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malik, R.; Rannikmäe, K.; Traylor, M.; Georgakis, M.K.; Sargurupremraj, M.; Markus, H.S.; Hopewell, J.C.; Debette, S.; Sudlow, C.L.; Dichgans, M. Genome-wide meta-analysis identifies 3 novel loci associated with stroke. Ann. Neurol. 2018, 84, 934–939. [Google Scholar] [CrossRef] [PubMed]
- Salehi, A.; Meidute Abaraviciene, S.; Jimenez-Feltstrom, J.; Östenson, C.-G.; Efendic, S.; Lundquist, I. Excessive islet NO generation in type 2 diabetic GK rats coincides with abnormal hormone secretion and is counteracted by GLP-1. PLoS ONE 2008, 3, e2165. [Google Scholar]
- Muhammed, S.J.; Lundquist, I.; Salehi, A. Pancreatic β-cell dysfunction, expression of iNOS and the effect of phosphodiesterase inhibitors in human pancreatic islets of type 2 diabetes. Diabetes Obes. Metab. 2012, 14, 1010–1019. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | MST | Activity Assay |
|---|---|---|
| Kd [μM] | Ki [μM] | |
| AC12 | 0.38 ± 0.06 | 0.10 ± 0.03 |
| AC13 | 0.9 ± 0.2 | 0.15 ± 0.04 |
| AC22 | 2.6 ± 0.4 | 1.6 ± 0.4 |
| AC23 | 12.9 ± 2.6 | 1.2 ± 0.4 |
| AC25 | 1.1 ± 0.6 | 1.0 ± 0.3 |
| AC27 | 1.8 ± 0.5 | 0.2 ± 0.3 |
| AC28 | 27 ± 17 | 3 ± 1 |
| Torin2 | 0.3 ± 0.1 | 4.6 ± 1.8 |
| Harmine | 0.9 ± 0.3 | 0.11 ± 0.03 |
| Harmine | AC27 | ||
|---|---|---|---|
| Biochemistry | Ki | 0.11 ± 0.03 | 0.17 ± 0.26 |
| Kd | 0.9 ± 0.294 | 1.83 ± 0.543 | |
| specificity [44] | no | yes | |
| off-targets [44] | yes | no | |
| Cell lines | toxicity | no | no |
| Ki67+ | ↑↑ | ↑ | |
| Ki67+/insulin+ | ↑ | ↑ | |
| insulin+ | ↑ | ↑↑ | |
| glucagon+ | ↑↑ | ↑ | |
| C-peptide+ | ↑ | ↑↑ | |
| hiPSC | insulin secretion (ELISA) | ↑↑ | ↑ |
| Isolated islets | insulin secretion (ELISA) | ↑ | ↑↑ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barzowska, A.; Pucelik, B.; Pustelny, K.; Matsuda, A.; Martyniak, A.; Stępniewski, J.; Maksymiuk, A.; Dawidowski, M.; Rothweiler, U.; Dulak, J.; et al. DYRK1A Kinase Inhibitors Promote β-Cell Survival and Insulin Homeostasis. Cells 2021, 10, 2263. https://doi.org/10.3390/cells10092263
Barzowska A, Pucelik B, Pustelny K, Matsuda A, Martyniak A, Stępniewski J, Maksymiuk A, Dawidowski M, Rothweiler U, Dulak J, et al. DYRK1A Kinase Inhibitors Promote β-Cell Survival and Insulin Homeostasis. Cells. 2021; 10(9):2263. https://doi.org/10.3390/cells10092263
Chicago/Turabian StyleBarzowska, Agata, Barbara Pucelik, Katarzyna Pustelny, Alex Matsuda, Alicja Martyniak, Jacek Stępniewski, Anna Maksymiuk, Maciej Dawidowski, Ulli Rothweiler, Józef Dulak, and et al. 2021. "DYRK1A Kinase Inhibitors Promote β-Cell Survival and Insulin Homeostasis" Cells 10, no. 9: 2263. https://doi.org/10.3390/cells10092263
APA StyleBarzowska, A., Pucelik, B., Pustelny, K., Matsuda, A., Martyniak, A., Stępniewski, J., Maksymiuk, A., Dawidowski, M., Rothweiler, U., Dulak, J., Dubin, G., & Czarna, A. (2021). DYRK1A Kinase Inhibitors Promote β-Cell Survival and Insulin Homeostasis. Cells, 10(9), 2263. https://doi.org/10.3390/cells10092263