
Plectin in Cancer: From Biomarker to Therapeutic Target


Abstract
1. Introduction
2. Plectin Expression in Cancer
2.1. Plectin Cell Surface Mislocalization in Cancer
2.2. Plectin Is a Cancer Biomarker
2.3. Plectin Mutations in Cancer
3. Plectin’s Role in Cancer
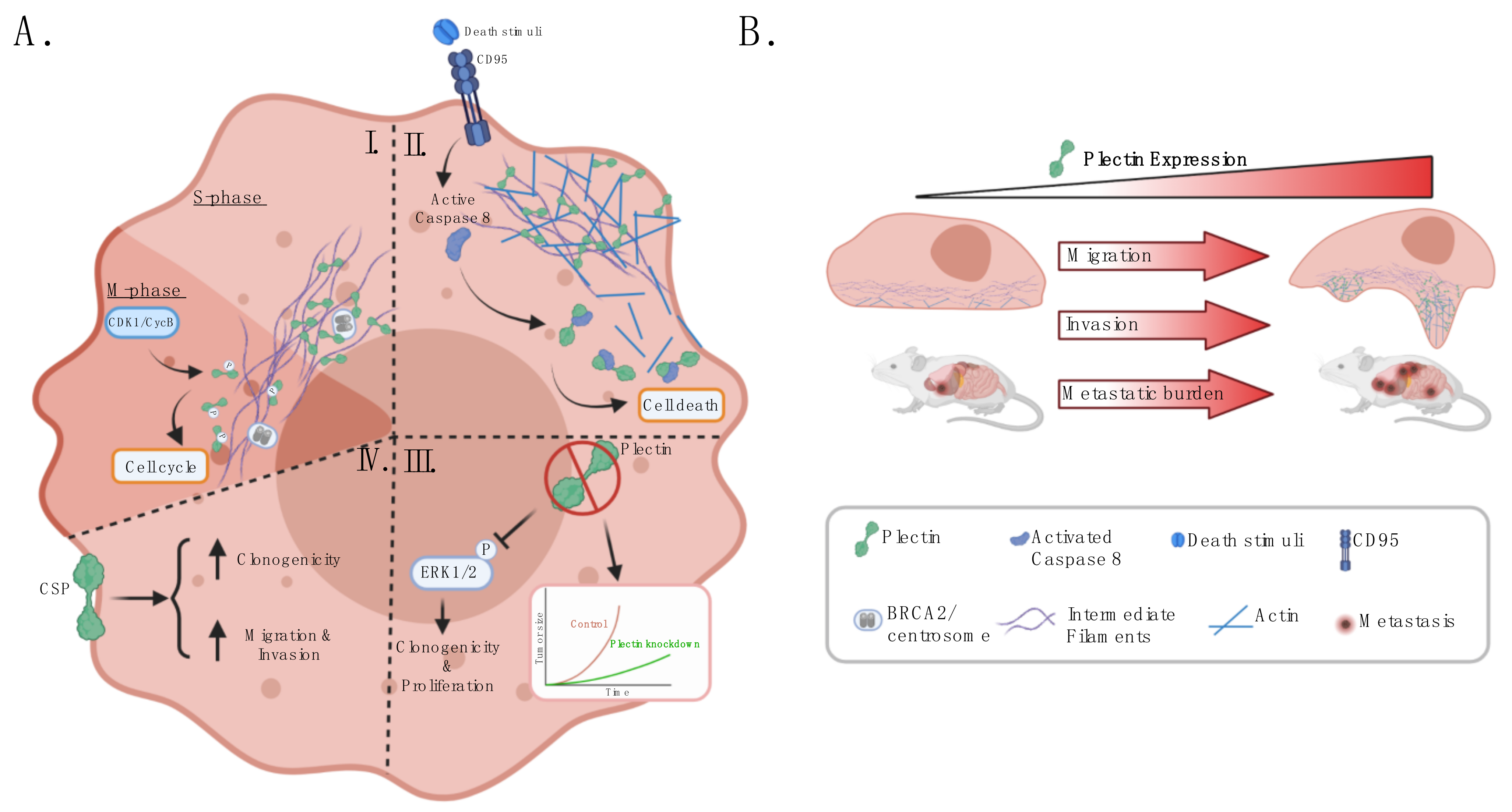
3.1. Plectin Is a Regulator of Cancer Cell Survival and Proliferation
3.2. Plectin Modulates Cancer Cell Migration, Invasion, and Metastatic Potential
3.3. Plectin Cross-Talks with the Tumor Microenvironment and Immune System
3.4. CSP Regulates Malignant Hallmarks
4. Clinical Utility of Plectin
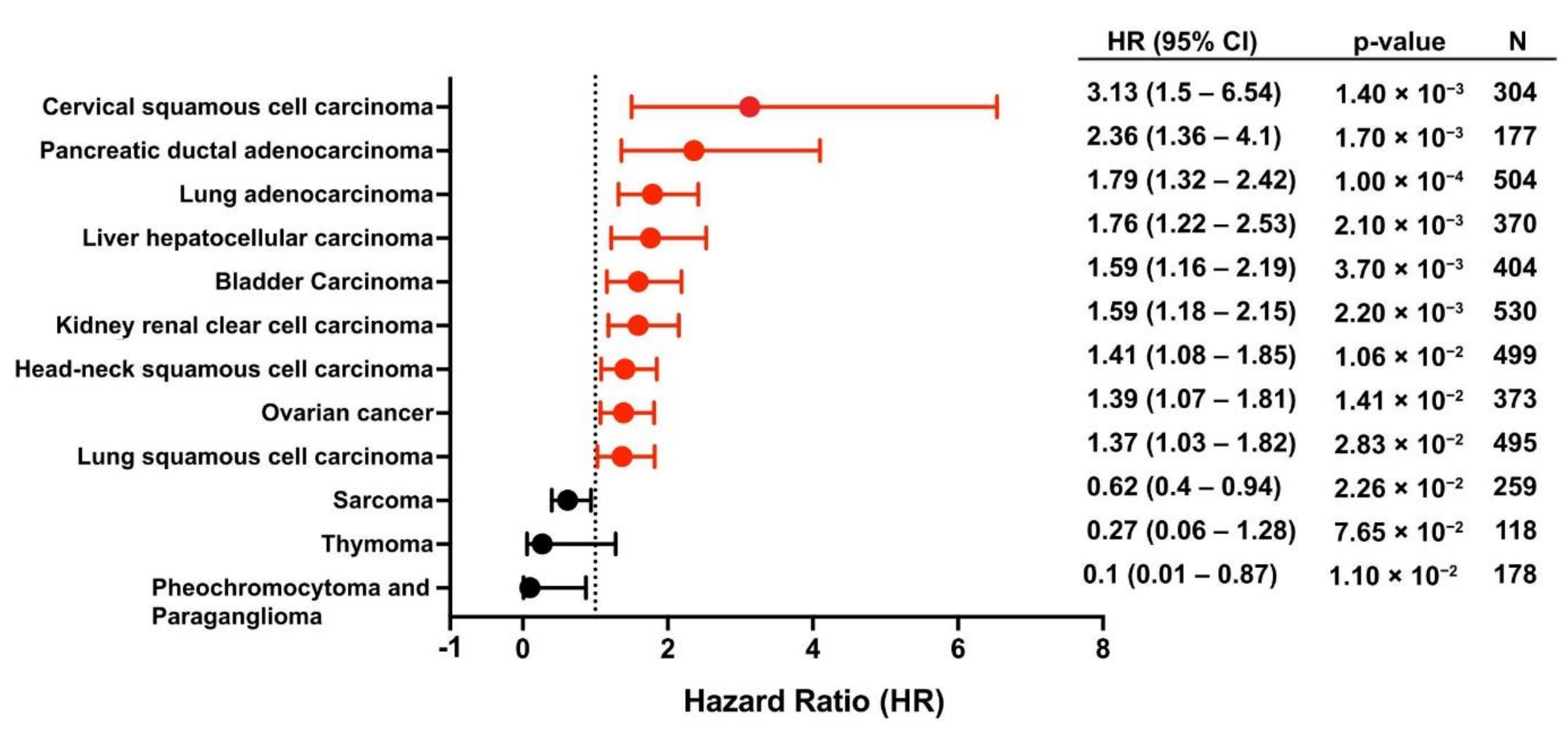
4.1. Plectin as a Risk Factor
4.2. Plectin as a Prognostic Indicator
4.3. Plectin as a Diagnostic Biomarker
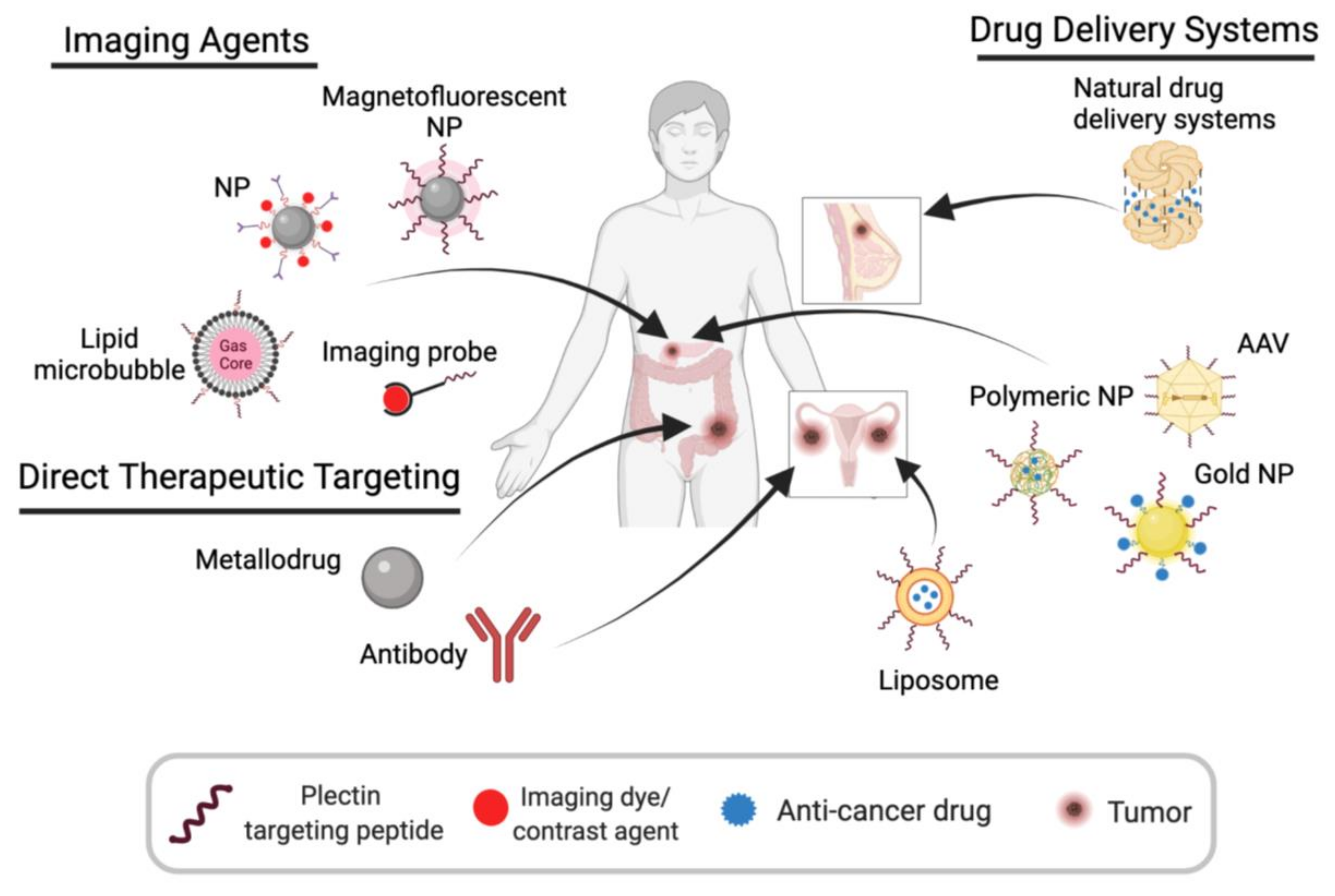
5. Plectin-Targeting Imaging Agents and Therapeutics
5.1. Imaging Agents
5.2. Plectin-Guided Drug Delivery
5.2.1. Polymeric Nanoparticles
5.2.2. Gold Nanoparticles
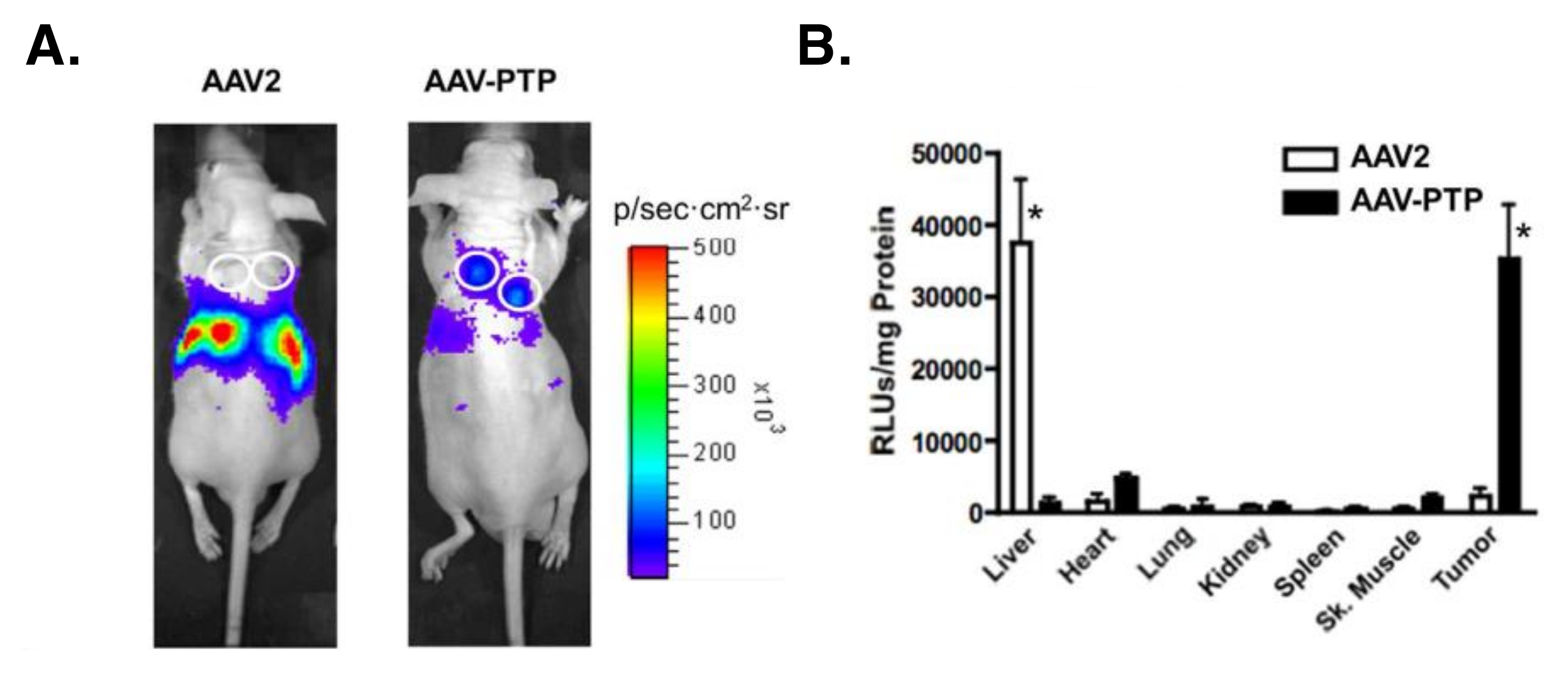
5.2.3. Targeted Adeno-Associated Virus (AAV) Particles
5.2.4. Targeted Liposomes
5.2.5. Natural Protein Drug Delivery Systems
5.3. Plectin and CSP-Targeted Therapeutics
5.3.1. Metallodrugs
5.3.2. Monoclonal Antibody
6. Role of Plectin and CSP in Drug Sensitivity and Resistance
7. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AML | Acute myeloid leukemia |
| BM | Bone marrow |
| CDK1 | Cyclin-dependent kinase 1 |
| CP | Chronic pancreatitis |
| CSC | Cancer stem cell |
| CSP | Cancer-specific plectin |
| CTC | Circulating tumor cell |
| CycB | Cyclin B |
| DAMPs | Damage-associated molecular patterns |
| Dlc1 | Deleted in liver cancer 1 |
| EMT | Epithelial–mesenchymal transition |
| EPLIN | Epithelial protein lost in neoplasm |
| ESCC | Esophageal squamous cell carcinoma |
| EUS-FNA | Endoscopic ultrasound-guided fine-needle aspiration |
| ExAC | Exosome Aggregation Consortium |
| FUS | Fused in sarcoma |
| GEO | Gene Expression Omnibus |
| GNP | Gold nanoparticle |
| GTEx | Genotype-Tissue Expression |
| HR | Hazard ratio |
| HMGB-1 | High mobility group box protein 1 |
| HNSCC | Head and neck squamous cell carcinoma |
| IF | Intermediate filament |
| IHC | Immunohistochemistry |
| IPMNs | Intraductal papillary mucinous neoplasms |
| miR | microRNA |
| MRI | Magnetic resonance imaging |
| MT | Microtubule |
| NP | Nanoparticle |
| OC | Ovarian Cancer |
| OSCC | Oral squamous cell carcinoma |
| PanIN | Pancreatic intraepithelial neoplasia |
| PDAC | Pancreatic ductal adenocarcinoma |
| PKC | Protein kinase C |
| PTP | Plectin-targeting peptide |
| RACK1 | Receptor of activated protein C kinase 1 |
| RON | Recepteur d’origine nantais |
| ROS | Reactive oxygen species |
| SCC | Squamous cell carcinoma |
| SPECT | Single-photon emission/CT |
| TARGET | Therapeutically Applicable Research to Generate Effective Treatments |
| TCGA | The Cancer Genome Atlas |
| TGCT | Testicular germ cell tumor |
| TMA | Tissue microarray |
| TNF | Tumor necrosis factor |
References
- Wiche, G.; Castañón, M.J. Intermediate Filament Linker Proteins: Plectin and BPAG1. In Encyclopedia of Biological Chemistry III, 3rd ed.; Jez, J., Ed.; Elsevier: Oxford, UK, 2021; pp. 200–219. ISBN 978-0-12-822040-5. [Google Scholar]
- Hu, L.; Huang, Z.; Wu, Z.; Ali, A.; Qian, A. Mammalian Plakins, Giant Cytolinkers: Versatile Biological Functions and Roles in Cancer. Int. J. Mol. Sci. 2018, 19, 974. [Google Scholar] [CrossRef]
- Wiche, G.; Winter, L. Plectin isoforms as organizers of intermediate filament cytoarchitecture. BioArchitecture 2011, 1, 14–20. [Google Scholar] [CrossRef]
- Wiche, G.; Krepler, R.; Artlieb, U.; Pytela, R.; Denk, H. Occurrence and immunolocalization of plectin in tissues. J. Cell Biol. 1983, 97, 887–901. [Google Scholar] [CrossRef] [PubMed]
- Wiche, G.; Krepler, R.; Artlieb, U.; Pytela, R.; Aberer, W. Identification of plectin in different human cell types and immunolocalization at epithelial basal cell surface membranes. Exp. Cell Res. 1984, 155, 43–49. [Google Scholar] [CrossRef]
- Fuchs, P.; Zörer, M.; Rezniczek, G.A.; Spazierer, D.; Oehler, S.; Castañón, M.J.; Hauptmann, R.; Wiche, G. Unusual 5’ transcript complexity of plectin isoforms: Novel tissue-specific exons modulate actin binding activity. Hum. Mol. Genet. 1999, 8, 2461–2472. [Google Scholar] [CrossRef] [PubMed]
- Andrä, K.; Nikolic, B.; Stöcher, M.; Drenckhahn, D.; Wiche, G. Not just scaffolding: Plectin regulates actin dynamics in cultured cells. Genes Dev. 1998, 12, 3442–3451. [Google Scholar] [CrossRef]
- Rezniczek, G.A.; de Pereda, J.M.; Reipert, S.; Wiche, G. Linking Integrin α6β4-based Cell Adhesion to the Intermediate Filament Cytoskeleton: Direct Interaction between the β4 Subunit and Plectin at Multiple Molecular Sites. J. Cell Biol. 1998, 141, 209–225. [Google Scholar] [CrossRef]
- Kostan, J.; Gregor, M.; Walko, G.; Wiche, G. Plectin Isoform-dependent Regulation of Keratin-Integrin α6β4 Anchorage via Ca2+/Calmodulin. J. Biol. Chem. 2009, 284, 18525–18536. [Google Scholar] [CrossRef]
- Katada, K.; Tomonaga, T.; Satoh, M.; Matsushita, K.; Tonoike, Y.; Kodera, Y.; Hanazawa, T.; Nomura, F.; Okamoto, Y. Plectin promotes migration and invasion of cancer cells and is a novel prognostic marker for head and neck squamous cell carcinoma. J. Proteom. 2012, 75, 1803–1815. [Google Scholar] [CrossRef]
- Buckup, M.; Rice, M.A.; Hsu, E.-C.; Garcia-Marques, F.; Liu, S.; Aslan, M.; Bermudez, A.; Huang, J.; Pitteri, S.J.; Stoyanova, T. Plectin is a regulator of prostate cancer growth and metastasis. Oncogene 2020, 40, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.J.; Smith, J.A.; Rezniczek, G.A.; Pan, S.; Chen, R.; Brentnall, T.A.; Wiche, G.; Kelly, K.A. Unexpected gain of function for the scaffolding protein plectin due to mislocalization in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 19414–19419. [Google Scholar] [CrossRef]
- Kelly, K.A.; Bardeesy, N.; Anbazhagan, R.; Gurumurthy, S.; Berger, J.; Alencar, H.; DePinho, R.; Mahmood, U.; Weissleder, R. Targeted Nanoparticles for Imaging Incipient Pancreatic Ductal Adenocarcinoma. PLoS Med. 2008, 5, e85. [Google Scholar] [CrossRef]
- Bausch, D.; Mino-Kenudson, M.; Castillo, C.F.-D.; Warshaw, A.L.; Kelly, K.A.; Thayer, S.P. Plectin-1 is a biomarker of malignant pancreatic intraductal papillary mucinous neoplasms. J. Gastrointest. Surg. 2009, 13, 1948–1954. [Google Scholar] [CrossRef]
- Dasa, S.S.K.; Diakova, G.; Suzuki, R.; Mills, A.; Gutknecht, M.F.; Klibanov, A.L.; Slack-Davis, J.K.; Kelly, K.A. Plectin-targeted liposomes enhance the therapeutic efficacy of a PARP inhibitor in the treatment of ovarian cancer. Theranostics 2018, 8, 2782–2798. [Google Scholar] [CrossRef]
- Raymond, A.C.; Gao, B.; Girard, L.; Minna, J.D.; Udugamasooriya, D.G. Unbiased peptoid combinatorial cell screen identifies plectin protein as a potential biomarker for lung cancer stem cells. Sci. Rep. 2019, 9, 14954. [Google Scholar] [CrossRef]
- Reynolds, F.; Panneer, N.; Tutino, C.M.; Wu, M.; Skrabal, W.R.; Moskaluk, C.; Kelly, K.A. A Functional Proteomic Method for Biomarker Discovery. PLoS ONE 2011, 6, e22471. [Google Scholar] [CrossRef] [PubMed]
- Konkalmatt, P.R.; Deng, D.; Thomas, S.; Wu, M.T.; Logsdon, C.D.; French, B.A.; Kelly, K.A. Plectin-1 Targeted AAV Vector for the Molecular Imaging of Pancreatic Cancer. Front. Oncol. 2013, 3, 84. [Google Scholar] [CrossRef] [PubMed]
- Sanna, V.; Nurra, S.; Pala, N.; Marceddu, S.; Pathania, D.; Neamati, N.; Sechi, M. Targeted Nanoparticles for the Delivery of Novel Bioactive Molecules to Pancreatic Cancer Cells. J. Med. Chem. 2016, 59, 5209–5220. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Tang, Y.; Xie, S.; Zheng, X.; Zhang, S.; Mao, J.; Wang, B.; Hou, Y.; Hu, L.; Chai, K.; et al. Chimeric peptide supramolecular nanoparticles for plectin-1 targeted miRNA-9 delivery in pancreatic cancer. Theranostics 2020, 10, 1151–1165. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhao, Z.; Liu, H.; Fetse, J.P.; Jain, A.; Lin, C.-Y.; Cheng, K. Development of a Tumor-Responsive Nanopolyplex Targeting Pancreatic Cancer Cells and Stroma. ACS Appl. Mater. Interfaces 2019, 11, 45390–45403. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, B.; Xing, X.; Liu, F.; Cheng, Y.; Shi, D. Surface engineered antifouling optomagnetic SPIONs for bimodal targeted imaging of pancreatic cancer cells. Int. J. Nanomed. 2014, 9, 1601–1615. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhou, H.; Li, X.; Duan, N.; Hu, S.; Liu, Y.; Yue, Y.; Song, L.; Zhang, Y.; Li, D.; et al. Plectin-1 Targeted Dual-modality Nanoparticles for Pancreatic Cancer Imaging. EBioMedicine 2018, 30, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Pal, K.; Al-Suraih, F.; Rodriguez, R.G.; Dutta, S.K.; Wang, E.; Kwak, H.S.; Caulfield, T.R.; Coffer, J.L.; Bhattacharya, S. Multifaceted peptide assisted one-pot synthesis of gold nanoparticles for plectin-1 targeted gemcitabine delivery in pancreatic cancer. Nanoscale 2017, 9, 15622–15634. [Google Scholar] [CrossRef] [PubMed]
- Wernitznig, D.; Meier-Menches, S.M.; Cseh, K.; Theiner, S.; Wenisch, D.; Schweikert, A.; Jakupec, M.A.; Koellensperger, G.; Wernitznig, A.; Sommergruber, W.; et al. Plecstatin-1 induces an immunogenic cell death signature in colorectal tumour spheroids. Metallomics 2020, 10, 2121–2133. [Google Scholar] [CrossRef]
- Meier, S.M.; Kreutz, M.S.D.; Winter, L.; Klose, M.H.M.; Cseh, M.S.K.; Weiss, M.S.T.; Bileck, A.; Alte, M.S.B.; Mader, J.C.; Jana, S.; et al. An Organoruthenium Anticancer Agent Shows Unexpected Target Selectivity For Plectin. Angew. Chem. Int. Ed. 2017, 56, 8267–8271. [Google Scholar] [CrossRef]
- Meier-Menches, S.M.; Zappe, K.; Bileck, A.; Kreutz, D.; Tahir, A.; Cichna-Markl, M.; Gerner, C. Time-dependent shotgun proteomics revealed distinct effects of an organoruthenium prodrug and its activation product on colon carcinoma cells. Metallomics 2018, 11, 118–127. [Google Scholar] [CrossRef]
- Yuan, Y.; Du, C.; Sun, C.; Zhu, J.; Wu, S.; Zhang, Y.; Ji, T.; Lei, J.; Yang, Y.; Gao, N.; et al. Chaperonin-GroEL as a Smart Hydrophobic Drug Delivery and Tumor Targeting Molecular Machine for Tumor Therapy. Nano Lett. 2018, 18, 921–928. [Google Scholar] [CrossRef]
- Bartha, Á.; Győrffy, B. TNMplot.com: A Web Tool for the Comparison of Gene Expression in Normal, Tumor and Metastatic Tissues. Int. J. Mol. Sci. 2021, 22, 2622. [Google Scholar] [CrossRef]
- Bray, F.; Me, J.F.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Basturk, O.; Hong, S.-M.; Wood, L.D.; Adsay, V.; Albores-Saavedra, J.; Biankin, A.; Brosens, L.A.; Fukushima, N.; Goggins, M.; Hruban, R.H.; et al. A Revised Classification System and Recommendations From the Baltimore Consensus Meeting for Neoplastic Precursor Lesions in the Pancreas. Am. J. Surg. Pathol. 2015, 39, 1730–1741. [Google Scholar] [CrossRef]
- Bausch, D.; Thomas, S.; Mino-Kenudson, M.; Fernández-Del, C.C.; Bauer, T.W.; Williams, M.; Warshaw, A.L.; Thayer, S.P.; Kelly, K.A. Plectin-1 as a Novel Biomarker for Pancreatic Cancer. Clin. Cancer Res. 2010, 17, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Oto, A.; Eltorky, M.A.; Dave, A.; Ernst, R.D.; Chen, K.; Rampy, B.; Chaljub, G.; Nealon, W. Mimicks of Pancreatic Malignancy in Patients with Chronic Pancreatitis: Correlation of Computed Tomography Imaging Features with Histopathologic Findings. Curr. Probl. Diagn. Radiol. 2006, 35, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.E.; Burtness, B.; Leemans, C.R.; Lui, V.W.Y.; Bauman, J.E.; Grandis, J.R. Head and neck squamous cell carcinoma. Nat. Rev. Dis. Prim. 2020, 6, 92. [Google Scholar] [CrossRef]
- Flores, I.L.; Kawahara, R.; Miguel, M.C.; Granato, D.C.; Domingues, R.R.; Macedo, C.C.S.; Carnielli, C.M.; Yokoo, S.; Rodrigues, P.C.; Monteiro, B.; et al. EEF1D modulates proliferation and epithelial–mesenchymal transition in oral squamous cell carcinoma. Clin. Sci. 2016, 130, 785–799. [Google Scholar] [CrossRef]
- Rodrigues, P.C.; Sawazaki-Calone, I.; De Oliveira, C.E.; Macedo, C.C.S.; Dourado, M.R.; Cervigne, N.K.; Miguel, M.C.; Carmo, A.F.D.; Lambert, D.W.; Graner, E.; et al. Fascin promotes migration and invasion and is a prognostic marker for oral squamous cell carcinoma. Oncotarget 2017, 8, 74736–74754. [Google Scholar] [CrossRef]
- Rikardsen, O.G.; Magnussen, S.N.; Svineng, G.; Hadler-Olsen, E.; Uhlin-Hansen, L.; Steigen, S.E. Plectin as a prognostic marker in non-metastatic oral squamous cell carcinoma. BMC Oral Health 2015, 15, 98. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Zhang, Y.; Wang, X.; Huang, J.; Guo, W.; Wei, P.; Li, G.; Wang, Z.; Huang, Z.; Zhang, L. Putative biomarkers of malignant transformation of sinonasal inverted papilloma into squamous cell carcinoma. J. Int. Med Res. 2019, 47, 2371–2380. [Google Scholar] [CrossRef]
- Burch, T.C.; Watson, M.T.; Nyalwidhe, J.O. Variable Metastatic Potentials Correlate with Differential Plectin and Vimentin Expression in Syngeneic Androgen Independent Prostate Cancer Cells. PLoS ONE 2013, 8, e65005. [Google Scholar] [CrossRef]
- Mittal, P.; Klingler-Hoffmann, M.; Arentz, G.; Winderbaum, L.; Lokman, N.; Zhang, C.; Anderson, L.; Scurry, J.; Leung, Y.; Stewart, C.J.; et al. Lymph node metastasis of primary endometrial cancers: Associated proteins revealed by MALDI imaging. Proteomics 2016, 16, 1793–1801. [Google Scholar] [CrossRef]
- Lee, K.Y.; Liu, Y.-H.; Ho, C.-C.; Pei, R.-J.; Yeh, K.-T.; Cheng, C.-C.; Lai, Y.-S. An early evaluation of malignant tendency with plectin expression in human colorectal adenoma and adenocarcinoma. J. Med. 2004, 35, 141–149. [Google Scholar] [PubMed]
- Nagle, R.B.; Hao, J.; Knox, J.D.; Dalkin, B.L.; Clark, V.; Cress, A. Expression of hemidesmosomal and extracellular matrix proteins by normal and malignant human prostate tissue. Am. J. Pathol. 1995, 146, 1498–1507. [Google Scholar]
- Pawar, H.; Kashyap, M.K.; Sahasrabuddhe, N.A.; Renuse, S.; Harsha, H.C.; Kumar, P.; Sharma, J.; Kandasamy, K.; Marimuthu, A.; Nair, B.; et al. Quantitative tissue proteomics of esophageal squamous cell carcinoma for novel biomarker discovery. Cancer Biol. Ther. 2011, 12, 510–522. [Google Scholar] [CrossRef]
- Cole, L.M.; Bluff, J.E.; Carolan, V.A.; Paley, M.N.; Tozer, G.M.; Clench, M.R. MALDI-MSI and label-free LC-ESI-MS/MS shotgun proteomics to investigate protein induction in a murine fibrosarcoma model following treatment with a vascular disrupting agent. Proteomics 2014, 14, 890–903. [Google Scholar] [CrossRef] [PubMed]
- Cole, L.M.; Mahmoud, K.; Haywood-Small, S.; Tozer, G.M.; Smith, D.; Clench, M.R. Recombinant “ IMS TAG” proteins—A new method for validating bottom-up matrix-assisted laser desorption/ionisation ion mobility separation mass spectrometry imaging. Rapid Commun. Mass Spectrom. 2013, 27, 2355–2362. [Google Scholar] [CrossRef]
- Liu, Y.-H.; Ho, C.-C.; Cheng, C.-C.; Pei, R.-J.; Hsu, Y.-H.; Yeh, K.-T.; Tsai, M.-C.; Lai, Y.-S. Pleomorphism of cancer cells with the expression of plectin and concept of filament bundles in human hepatocellular carcinoma. Res. Commun. Mol. Pathol. Pharmacol. 2011, 120, 43–54. [Google Scholar]
- Cheng, C.-C.; Lai, Y.-C.C.; Lai, Y.-S.; Hsu, Y.-H.; Chao, W.-T.; Sia, K.-C.; Tseng, Y.-H.; Liu, Y.-H. Transient knockdown-mediated deficiency in plectin alters hepatocellular motility in association with activated FAK and Rac1-GTPase. Cancer Cell Int. 2015, 15, 29. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.-C.; Liu, Y.-H.; Ho, C.-C.; Chao, W.-T.; Pei, R.-J.; Hsu, Y.-H.; Yeh, K.-T.; Ho, L.-C.; Tsai, M.-C.; Lai, Y.-S. The influence of plectin deficiency on stability of cytokeratin18 in hepatocellular carcinoma. J. Mol. Histol. 2007, 39, 209–216. [Google Scholar] [CrossRef]
- Liu, Y.-H.; Ho, C.-C.; Cheng, C.-C.; Chao, W.-T.; Pei, R.-J.; Hsu, Y.-H.; Lai, Y.-S. Cytokeratin 18-mediated disorganization of intermediate filaments is induced by degradation of plectin in human liver cells. Biochem. Biophys. Res. Commun. 2011, 407, 575–580. [Google Scholar] [CrossRef]
- Liu, Y.-H.; Cheng, C.-C.; Ho, C.-C.; Chao, W.-T.; Pei, R.-J.; Hsu, Y.-H.; Yeh, K.-T.; Ho, L.-C.; Tsai, M.-C.; Lai, Y.-S. Degradation of plectin with modulation of cytokeratin 18 in human liver cells during staurosporine-induced apoptosis. In Vivo 2008, 22, 543–548. [Google Scholar]
- Lee, H.-J.; Na, K.; Kwon, M.-S.; Kim, H.; Kim, K.S.; Paik, Y.-K. Quantitative analysis of phosphopeptides in search of the disease biomarker from the hepatocellular carcinoma specimen. Proteomics 2009, 9, 3395–3408. [Google Scholar] [CrossRef]
- Song, C.; Wang, F.; Ye, M.; Cheng, K.; Chen, R.; Zhu, J.; Tan, Y.; Wang, H.; Figeys, D.; Zou, H. Improvement of the Quantification Accuracy and Throughput for Phosphoproteome Analysis by a Pseudo Triplex Stable Isotope Dimethyl Labeling Approach. Anal. Chem. 2011, 83, 7755–7762. [Google Scholar] [CrossRef]
- Dumas, V.; Kanitakis, J.; Charvat, S.; Euvrard, S.; Faure, M.; Claudy, A. Expression of basement membrane antigens and matrix metalloproteinases 2 and 9 in cutaneous basal and squamous cell carcinomas. Anticancer. Res. 2000, 19, 2929–2938. [Google Scholar]
- Bouameur, J.-E.; Favre, B.; Borradori, L. Plakins, a Versatile Family of Cytolinkers: Roles in Skin Integrity and in Human Diseases. J. Investig. Dermatol. 2014, 134, 885–894. [Google Scholar] [CrossRef] [PubMed]
- Harryman, W.; Pond, E.; Singh, P.; Little, A.S.; Eschbacher, J.M.; Nagle, R.B.; Cress, A.E. Laminin-binding integrin gene copy number alterations in distinct epithelial-type cancers. Am. J. Transl. Res. 2016, 8, 940–954. [Google Scholar] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data: Figure 1. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Li, T.; Fu, J.; Zeng, Z.; Cohen, D.; Li, J.; Chen, Q.; Li, B.; Liu, X.S. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 2020, 48, W509–W514. [Google Scholar] [CrossRef]
- Hoffman, Y.; Bublik, D.R.; Pilpel, Y.; Oren, M. miR-661 downregulates both Mdm2 and Mdm4 to activate p53. Cell Death Differ. 2013, 21, 302–309. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, C.; Zhao, Y.; Feng, Z. MicroRNA Control of p53. J. Cell. Biochem. 2016, 118, 7–14. [Google Scholar] [CrossRef]
- Zhu, T.; Yuan, J.; Wang, Y.; Gong, C.; Xie, Y.; Li, H. MiR-661 contributed to cell proliferation of human ovarian cancer cells by repressing INPP5J expression. Biomed. Pharmacother. 2015, 75, 123–128. [Google Scholar] [CrossRef]
- Liu, F.; Cai, Y.; Rong, X.; Chen, J.; Zheng, D.; Chen, L.; Zhang, J.; Luo, R.; Zhao, P.; Ruan, J. MiR-661 promotes tumor invasion and metastasis by directly inhibiting RB1 in non small cell lung cancer. Mol. Cancer 2017, 16, 122. [Google Scholar] [CrossRef]
- Thomsen, C.; Udhane, S.S.; Runnberg, R.; Wiche, G.; Ståhlberg, A.; Åman, P. Fused in sarcoma (FUS) interacts with the cytolinker protein plectin: Implications for FUS subcellular localization and function. Exp. Cell Res. 2012, 318, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Osmanagic-Myers, S.; Wiche, G. Plectin-RACK1 (Receptor for Activated C Kinase 1) Scaffolding. J. Biol. Chem. 2004, 279, 18701–18710. [Google Scholar] [CrossRef]
- Osmanagic-Myers, S.; Gregor, M.; Walko, G.; Burgstaller, G.; Reipert, S.; Wiche, G. Plectin-controlled keratin cytoarchitecture affects MAP kinases involved in cellular stress response and migration. J. Cell Biol. 2006, 174, 557–568. [Google Scholar] [CrossRef] [PubMed]
- Niwa, T.; Saito, H.; Imajoh-Ohmi, S.; Kaminishi, M.; Seto, Y.; Miki, Y.; Nakanishi, A. BRCA2 interacts with the cytoskeletal linker protein plectin to form a complex controlling centrosome localization. Cancer Sci. 2009, 100, 2115–2125. [Google Scholar] [CrossRef] [PubMed]
- Fish, L.; Khoroshkin, M.; Navickas, A.; Garcia, K.; Culbertson, B.; Hänisch, B.; Zhang, S.; Nguyen, H.C.B.; Soto, L.M.; Dermit, M.; et al. A prometastatic splicing program regulated by SNRPA1 interactions with structured RNA elements. Science 2021, 372, eabc7531. [Google Scholar] [CrossRef]
- Yu, P.T.; Babicky, M.; Jaquish, D.; French, R.; Marayuma, K.; Mose, E.; Niessen, S.; Hoover, H.; Shields, D.; Cheresh, D.; et al. The RON-receptor regulates pancreatic cancer cell migration through phosphorylation-dependent breakdown of the hemidesmosome. Int. J. Cancer 2012, 131, 1744–1754. [Google Scholar] [CrossRef] [PubMed]
- Boczonadi, V.; McInroy, L.; Määttä, A. Cytolinker cross-talk: Periplakin N-terminus interacts with plectin to regulate keratin organisation and epithelial migration. Exp. Cell Res. 2007, 313, 3579–3591. [Google Scholar] [CrossRef]
- Sabbir, M.G.; Dillon, R.; Mowat, M.R.A. Dlc1 interaction with non-muscle myosin heavy chain II-A (Myh9) and Rac1 activation. Biol. Open 2016, 5, 452–460. [Google Scholar] [CrossRef][Green Version]
- Kasai, N.; Kadeer, A.; Kajita, M.; Saitoh, S.; Ishikawa, S.; Maruyama, T.; Fujita, Y. The paxillin-plectin-EPLIN complex promotes apical elimination of RasV12-transformed cells by modulating HDAC6-regulated tubulin acetylation. Sci. Rep. 2018, 8, 2097. [Google Scholar] [CrossRef]
- Kadeer, A.; Maruyama, T.; Kajita, M.; Morita, T.; Sasaki, A.; Ohoka, A.; Ishikawa, S.; Ikegawa, M.; Shimada, T.; Fujita, Y. Plectin is a novel regulator for apical extrusion of RasV12-transformed cells. Sci. Rep. 2017, 7, srep44328. [Google Scholar] [CrossRef] [PubMed]
- Foisner, R.; Malecz, N.; Dressel, N.; Stadler, C.; Wiche, G. M-phase-specific phosphorylation and structural rearrangement of the cytoplasmic cross-linking protein plectin involve p34cdc2 kinase. Mol. Biol. Cell 1996, 7, 273–288. [Google Scholar] [CrossRef]
- Gregor, M.; Osmanagic-Myers, S.; Burgstaller, G.; Wolfram, M.; Fischer, I.; Walko, G.; Resch, G.P.; Jörgl, A.; Herrmann, H.; Wiche, G. Mechanosensing through focal adhesion-anchored intermediate filaments. FASEB J. 2013, 28, 715–729. [Google Scholar] [CrossRef] [PubMed]
- Burgstaller, G.; Gregor, M.; Winter, L.; Wiche, G. Keeping the Vimentin Network under Control: Cell–Matrix Adhesion–associated Plectin 1f Affects Cell Shape and Polarity of Fibroblasts. Mol. Biol. Cell 2010, 21, 3362–3375. [Google Scholar] [CrossRef] [PubMed]
- Stegh, A.H.; Herrmann, H.; Lampel, S.; Weisenberger, D.; Andrä, K.; Seper, M.; Wiche, G.; Krammer, P.H.; Peter, M.E. Identification of the Cytolinker Plectin as a Major Early In Vivo Substrate for Caspase 8 during CD95- and Tumor Necrosis Factor Receptor-Mediated Apoptosis. Mol. Cell. Biol. 2000, 20, 5665–5679. [Google Scholar] [CrossRef]
- Raymond, K.; Kreft, M.; Song, J.-Y.; Janssen, H.; Sonnenberg, A. Dual Role of α6β4 Integrin in Epidermal Tumor Growth: Tumor-suppressive Versus Tumor-promoting Function. Mol. Biol. Cell 2007, 18, 4210–4221. [Google Scholar] [CrossRef][Green Version]
- Wiche, G.; Osmanagic-Myers, S.; Castañón, M.J. Networking and anchoring through plectin: A key to IF functionality and mechanotransduction. Curr. Opin. Cell Biol. 2015, 32, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Jiu, Y.; Lehtimäki, J.; Tojkander, S.; Cheng, F.; Jäälinoja, H.; Liu, X.; Varjosalo, M.; Eriksson, J.E.; Lappalainen, P. Bidirectional Interplay between Vimentin Intermediate Filaments and Contractile Actin Stress Fibers. Cell Rep. 2015, 11, 1511–1518. [Google Scholar] [CrossRef]
- Litjens, S.H.; de Pereda, J.M.; Sonnenberg, A. Current insights into the formation and breakdown of hemidesmosomes. Trends Cell Biol. 2006, 16, 376–383. [Google Scholar] [CrossRef]
- Yoneyama, M.S.; Hatakeyama, S.; Habuchi, T.; Inoue, T.; Nakamura, T.; Funyu, T.; Wiche, G.; Ohyama, C.; Tsuboi, S. Vimentin intermediate filament and plectin provide a scaffold for invadopodia, facilitating cancer cell invasion and extravasation for metastasis. Eur. J. Cell Biol. 2014, 93, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Takkunen, M.; Hukkanen, M.; Liljeström, M.; Grenman, R.; Virtanen, I. Podosome-like structures of non-invasive carcinoma cells are replaced in epithelial-mesenchymal transition by actin comet-embedded invadopodia. J. Cell. Mol. Med. 2009, 14, 1569–1593. [Google Scholar] [CrossRef]
- Takkunen, M.; Grenman, R.; Hukkanen, M.; Korhonen, M.; De Herreros, A.G.; Virtanen, I. Snail-dependent and -independent Epithelial-Mesenchymal Transition in Oral Squamous Carcinoma Cells. J. Histochem. Cytochem. 2006, 54, 1263–1275. [Google Scholar] [CrossRef] [PubMed]
- McInroy, L.; Määttä, A. Plectin regulates invasiveness of SW480 colon carcinoma cells and is targeted to podosome-like adhesions in an isoform-specific manner. Exp. Cell Res. 2011, 317, 2468–2478. [Google Scholar] [CrossRef] [PubMed]
- Dmello, C.; Sawant, S.; Alam, H.; Gangadaran, P.; Tiwari, R.; Dongre, H.; Rana, N.; Barve, S.; Costea, D.E.; Chaukar, D.; et al. Vimentin-mediated regulation of cell motility through modulation of beta4 integrin protein levels in oral tumor derived cells. Int. J. Biochem. Cell Biol. 2015, 70, 161–172. [Google Scholar] [CrossRef]
- Wang, C.-I.; Wang, C.-L.; Wu, Y.-C.; Feng, H.-P.; Liu, P.-J.; Chang, Y.-S.; Yu, J.-S.; Yu, C.-J. Quantitative Proteomics Reveals a Novel Role of Karyopherin Alpha 2 in Cell Migration through the Regulation of Vimentin–pErk Protein Complex Levels in Lung Cancer. J. Proteome Res. 2015, 14, 1739–1751. [Google Scholar] [CrossRef]
- Glen, A.; Evans, C.A.; Gan, C.S.; Cross, S.; Hamdy, F.C.; Gibbins, J.; Lippitt, J.; Eaton, C.L.; Noirel, J.; Wright, P.; et al. Eight-plex iTRAQ analysis of variant metastatic human prostate cancer cells identifies candidate biomarkers of progression: An exploratory study. Prostate 2010, 70, 1313–1332. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, P.R.; Charles, S.E.; D’Souza, Z.C.; Vaidya, M.M. Hemidesmosomal linker proteins regulate cell motility, invasion and tumorigenicity in oral squamous cell carcinoma derived cells. Exp. Cell Res. 2017, 360, 125–137. [Google Scholar] [CrossRef]
- Liu, Y.-H.; Cheng, C.-C.; Ho, C.-C.; Chao, W.-T.; Pei, R.-J.; Hsu, Y.-H.; Ho, L.-C.; Shiu, B.-H.; Lai, Y.-S. Plectin deficiency on cytoskeletal disorganization and transformation of human liver cells in vitro. Med Mol. Morphol. 2011, 44, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.-C.; Chao, W.-T.; Liao, C.-C.; Tseng, Y.-H.; Lai, Y.-C.C.; Lai, Y.-S.; Hsu, Y.-H.; Liu, Y.-H. Plectin deficiency in liver cancer cells promotes cell migration and sensitivity to sorafenib treatment. Cell Adhes. Migr. 2017, 12, 19–27. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Abrahamsberg, C.; Fuchs, P.; Osmanagic-Myers, S.; Fischer, I.; Propst, F.; Elbe-Bürger, A.; Wiche, G. Targeted ablation of plectin isoform 1 uncovers role of cytolinker proteins in leukocyte recruitment. Proc. Natl. Acad. Sci. USA 2005, 102, 18449–18454. [Google Scholar] [CrossRef] [PubMed]
- Von Der Heide, E.K.; Neumann, M.; Vosberg, S.; James, A.R.; Schroeder, M.P.; Ortiz-Tanchez, J.; Isaakidis, K.; Schlee, C.; Luther, M.; Jöhrens, K.; et al. Molecular alterations in bone marrow mesenchymal stromal cells derived from acute myeloid leukemia patients. Leukemia 2016, 31, 1069–1078. [Google Scholar] [CrossRef]
- Puiffe, M.-L.; Le Page, C.; Filali-Mouhim, A.; Zietarska, M.; Ouellet, V.; Tonin, P.N.; Chevrette, M.; Provencher, D.M.; Mes-Masson, A.-M. Characterization of ovarian cancer ascites on cell invasion, proliferation, spheroid formation, gene expression in an in vitro model of epithelial ovarian cancer. Neoplasia 2007, 9, 820–829. [Google Scholar] [CrossRef] [PubMed]
- Osman, A.; Afify, S.M.; Hassan, G.; Fu, X.; Seno, A.; Seno, M. Revisiting Cancer Stem Cells as the Origin of Cancer-Associated Cells in the Tumor Microenvironment: A Hypothetical View from the Potential of iPSCs. Cancers 2020, 12, 879. [Google Scholar] [CrossRef] [PubMed]
- Samardzija, C.; Greening, D.W.; Escalona, R.; Chen, M.; Bilandzic, M.; Luwor, R.; Kannourakis, G.; Findlay, J.K.; Ahmed, N. Knockdown of stem cell regulator Oct4A in ovarian cancer reveals cellular reprogramming associated with key regulators of cytoskeleton-extracellular matrix remodelling. Sci. Rep. 2017, 7, 46312. [Google Scholar] [CrossRef]
- Wesley, T.; Berzins, S.; Kannourakis, G.; Ahmed, N. The attributes of plakins in cancer and disease: Perspectives on ovarian cancer progression, chemoresistance and recurrence. Cell Commun. Signal. 2021, 19, 55. [Google Scholar] [CrossRef]
- Perez, S.M.; Dimastromatteo, J.; Landen, C.N.; Kelly, K.A. A Novel Monoclonal Antibody Targeting Cancer-Specific Plectin Has Potent Antitumor Activity in Ovarian Cancer. Cells 2021, 10, 2218. [Google Scholar] [CrossRef]
- Litchfield, K.; Thomsen, H.; Mitchell, J.S.; Sundquist, J.; Houlston, R.; Hemminki, K.; Turnbull, C. Quantifying the heritability of testicular germ cell tumour using both population-based and genomic approaches. Sci. Rep. 2015, 5, 13889. [Google Scholar] [CrossRef]
- Paumard-Hernández, B.; Calvete, O.; Pérez, L.I.; Tejero, H.; Al-Shahrour, F.; Pita, G.; Barroso, A.; Triviño, J.C.; Urioste, M.; Valverde, C.; et al. Whole exome sequencing identifies PLEC, EXO5 and DNAH7 as novel susceptibility genes in testicular cancer. Int. J. Cancer 2018, 143, 1954–1962. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Wu, Y.; Guan, X.; Liang, Y.; Yao, X.; Tan, D.; Bai, Y.; Xiong, G.; Yang, K. Germline copy number loss of UGT2B28 and gain of PLEC contribute to increased human esophageal squamous cell carcinoma risk in Southwest China. Am. J. Cancer Res. 2015, 5, 3056–3071. [Google Scholar] [PubMed]
- Nagy, Á.; Munkácsy, G.; Győrffy, B. Pancancer survival analysis of cancer hallmark genes. Sci. Rep. 2021, 11, 6047. [Google Scholar] [CrossRef]
- Koorstra, J.-B.M.; Hustinx, S.R.; Offerhaus, G.J.A.; Maitra, A. Pancreatic Carcinogenesis. Pancreatology 2008, 8, 110–125. [Google Scholar] [CrossRef]
- Moris, M.; Dawson, D.W.; Jiang, J.; Lewis, J.; Nassar, A.; Takeuchi, K.K.; Lay, A.R.; Zhai, Q.; Donahue, T.R.; Kelly, K.A.; et al. Plectin-1 as a Biomarker of Malignant Progression in Intraductal Papillary Mucinous Neoplasms. Pancreas 2016, 45, 1353–1358. [Google Scholar] [CrossRef]
- Brugge, W.R.; Lewandrowski, K.; Lee-Lewandrowski, E.; Centeno, B.A.; Szydlo, T.; Regan, S.; del Castillo, C.F.; Warshaw, A.L. Diagnosis of pancreatic cystic neoplasms: A report of the cooperative pancreatic cyst study. Gastroenterology 2004, 126, 1330–1336. [Google Scholar] [CrossRef]
- Ogura, T.; Yamao, K.; Sawaki, A.; Mizuno, N.; Hara, K.; Hijioka, S.; Niwa, Y.; Tajika, M.; Kondo, S.; Shimizu, Y.; et al. Clinical impact of K-ras mutation analysis in EUS-guided FNA specimens from pancreatic masses. Gastrointest. Endosc. 2012, 75, 769–774. [Google Scholar] [CrossRef] [PubMed]
- Park, J.K.; Paik, W.H.; Song, B.J.; Ryu, J.K.; Kim, M.A.; Park, J.M.; Lee, S.H.; Kim, Y.-T. Additional K-ras mutation analysis and Plectin-1 staining improve the diagnostic accuracy of pancreatic solid mass in EUS-guided fine needle aspiration. Oncotarget 2017, 8, 64440–64448. [Google Scholar] [CrossRef]
- Ignatiadis, M.; Sledge, G.W.; Jeffrey, S.S. Liquid biopsy enters the clinic—Implementation issues and future challenges. Nat. Rev. Clin. Oncol. 2021, 18, 297–312. [Google Scholar] [CrossRef] [PubMed]
- Song, B.G.; Kwon, W.; Kim, H.; Lee, E.M.; Han, Y.M.; Kim, H.; Byun, Y.; Lee, K.B.; Lee, K.H.; Lee, K.T.; et al. Detection of Circulating Tumor Cells in Resectable Pancreatic Ductal Adenocarcinoma: A Prospective Evaluation as a Prognostic Marker. Front. Oncol. 2021, 10, 616440. [Google Scholar] [CrossRef] [PubMed]
- Cromey, B.; McDaniel, A.; Matsunaga, T.; Vagner, J.; Kieu, K.Q. Pancreatic cancer cell detection by targeted lipid microbubbles and multiphoton imaging. J. Biomed. Opt. 2018, 23, 046501. [Google Scholar] [CrossRef] [PubMed]
- Harpel, K.; Baker, R.D.; Amirsolaimani, B.; Mehravar, S.; Vagner, J.; Matsunaga, T.O.; Banerjee, B.; Kieu, K. Imaging of targeted lipid microbubbles to detect cancer cells using third harmonic generation microscopy. Biomed. Opt. Express 2016, 7, 2849–2860. [Google Scholar] [CrossRef]
- Ueda, M.; Fukushima, T.; Ogawa, K.; Kimura, H.; Ono, M.; Yamaguchi, T.; Ikehara, Y.; Saji, H. Synthesis and evaluation of a radioiodinated peptide probe targeting αvβ6 integrin for the detection of pancreatic ductal adenocarcinoma. Biochem. Biophys. Res. Commun. 2014, 445, 661–666. [Google Scholar] [CrossRef]
- Li, Z.; Lin, P.; Gao, C.; Peng, C.; Liu, S.; Gao, H.; Wang, B.; Wang, J.; Niu, J.; Niu, W. Integrin β6 acts as an unfavorable prognostic indicator and promotes cellular malignant behaviors via ERK-ETS1 pathway in pancreatic ductal adenocarcinoma (PDAC). Tumor Biol. 2015, 37, 5117–5131. [Google Scholar] [CrossRef]
- Wang, Q.; Yan, H.; Jin, Y.; Wang, Z.; Huang, W.; Qiu, J.; Kang, F.; Wang, K.; Zhao, X.; Tian, J. A novel plectin/integrin-targeted bispecific molecular probe for magnetic resonance/near-infrared imaging of pancreatic cancer. Biomaterials 2018, 183, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.; Yoo, D.; Ling, D.; Cho, M.H.; Hyeon, T.; Cheon, J. Iron Oxide Based Nanoparticles for Multimodal Imaging and Magnetoresponsive Therapy. Chem. Rev. 2015, 115, 10637–10689. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.; Chang, H.; Li, H.; Wang, S. Induction of reactive oxygen species: An emerging approach for cancer therapy. Apoptosis 2017, 22, 1321–1335. [Google Scholar] [CrossRef] [PubMed]
- Pathania, D.; Sechi, M.; Palomba, M.; Sanna, V.; Berrettini, F.; Sias, A.; Taheri, L.; Neamati, N. Design and discovery of novel quinazolinedione-based redox modulators as therapies for pancreatic cancer. Biochim. Biophys. Acta BBA-Gen. Subj. 2014, 1840, 332–343. [Google Scholar] [CrossRef] [PubMed]
- Zhan, H.-X.; Zhou, B.; Cheng, Y.-G.; Xu, J.-W.; Wang, L.; Zhang, G.-Y.; Hu, S.-Y. Crosstalk between stromal cells and cancer cells in pancreatic cancer: New insights into stromal biology. Cancer Lett. 2017, 392, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Rupaimoole, R.; Calin, G.; Lopez-Berestein, G.; Sood, A.K. miRNA Deregulation in Cancer Cells and the Tumor Microenvironment. Cancer Discov. 2016, 6, 235–246. [Google Scholar] [CrossRef]
- Li, Z.; Rana, T.M. Therapeutic targeting of microRNAs: Current status and future challenges. Nat. Rev. Drug Discov. 2014, 13, 622–638. [Google Scholar] [CrossRef]
- Chen, W.; Zhou, Y.; Zhi, X.; Ma, T.; Liu, H.; Chen, B.W.; Zheng, X.; Xie, S.; Zhao, B.; Feng, X.; et al. Delivery of miR-212 by chimeric peptide-condensed supramolecular nanoparticles enhances the sensitivity of pancreatic ductal adenocarcinoma to doxorubicin. Biomaterials 2018, 192, 590–600. [Google Scholar] [CrossRef]
- Raj, S.; Khurana, S.; Choudhari, R.; Kesari, K.K.; Kamal, M.A.; Garg, N.; Ruokolainen, J.; Das, B.C.; Kumar, D. Specific targeting cancer cells with nanoparticles and drug delivery in cancer therapy. Semin. Cancer Biol. 2019, 69, 166–177. [Google Scholar] [CrossRef] [PubMed]
- Hacker, U.T.; Bentler, M.; Kaniowska, D.; Morgan, M.; Büning, H. Towards Clinical Implementation of Adeno-Associated Virus (AAV) Vectors for Cancer Gene Therapy: Current Status and Future Perspectives. Cancers 2020, 12, 1889. [Google Scholar] [CrossRef]
- Saraf, S.; Jain, A.; Tiwari, A.; Verma, A.; Panda, P.K.; Jain, S.K. Advances in liposomal drug delivery to cancer: An overview. J. Drug Deliv. Sci. Technol. 2020, 56, 101549. [Google Scholar] [CrossRef]
- Ndagi, U.; Mhlongo, N.; Soliman, M.E. Metal complexes in cancer therapy—An update from drug design perspective. Drug Des. Dev. Ther. 2017, 11, 599–616. [Google Scholar] [CrossRef] [PubMed]
- Klose, M.H.M.; Theiner, S.; Kornauth, C.; Meier-Menches, S.M.; Heffeter, P.; Berger, W.; Koellensperger, G.; Keppler, B.K. Bioimaging of isosteric osmium and ruthenium anticancer agents by LA-ICP-MS. Metallomics 2018, 10, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Carter, P.J.; Lazar, G.A. Next generation antibody drugs: Pursuit of the ‘high-hanging fruit’. Nat. Rev. Drug Discov. 2017, 17, 197–223. [Google Scholar] [CrossRef]
- Jeon, S.-M.; Shin, E.-A. Exploring vitamin D metabolism and function in cancer. Exp. Mol. Med. 2018, 50, 1–14. [Google Scholar] [CrossRef]
- Pálmer, H.G.; Sánchez-Carbayo, M.; Ordóñez-Morán, P.; Larriba, M.J.; Cordón-Cardó, C.; Muñoz, A. Genetic signatures of differentiation induced by 1alpha,25-dihydroxyvitamin D3 in human colon cancer cells. Cancer Res. 2003, 63, 7799–7806. [Google Scholar]
- Júnior, L.A.L.; Cucielo, M.S.; Domeniconi, R.F.; dos Santos, L.D.; Silveira, H.S.; Nunes, I.D.S.; Martinez, M.; Martinez, F.E.; Fávaro, W.J.; Chuffa, L.G.D.A. P-MAPA and IL-12 Differentially Regulate Proteins Associated with Ovarian Cancer Progression: A Proteomic Study. ACS Omega 2019, 4, 21761–21777. [Google Scholar] [CrossRef]
- Ahmed, N.; Greening, D.; Samardzija, C.; Escalona, R.M.; Chen, M.; Findlay, J.K.; Kannourakis, G. Unique proteome signature of post-chemotherapy ovarian cancer ascites-derived tumor cells. Sci. Rep. 2016, 6, 30061. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Name | Cancer Type | Function of Protein | Effect of Interaction | Reference |
|---|---|---|---|---|
| Dlc1 | Ovarian cancer | Tumor suppressor gene encoding a GTPase-activating protein | Plectin binds to transcriptional isoforms of Dlc1. Loss of Dlc1 increased focal adhesions and stress fiber formation. | [70] |
| KPNA2 | Lung cancer | Imports proteins to the nucleus | An increase in complex formation is seen in lung adenocarcinoma cells with higher metastatic potential. | [86] |
| Periplakin | Breast cancer | Cytolinker plakin protein | Plectin isoforms plectin-1f and -1k interact with the N-terminus of periplakin at cell borders. | [69] |
| RON | Pancreatic cancer | Receptor tyrosine kinase | Upon binding its ligand, RON translocates to the cell surface and interacts with plectin at lamellipodia, disrupting the plectin–integrin β4 interaction via phosphorylation of PI3K, thus increasing cancer cell migration. | [68] |
| SNRPA1 | Breast cancer | Mediator of alternative splicing | Exon 31 of plectin was identified as a SNRPA1 target, resulting in a rod domain-containing isoform that is upregulated in metastatic breast cancer tumors. Ablation of this plectin SNRPA1-mediated isoform resulted in reduced invasion and metastatic capacity. | [67] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perez, S.M.; Brinton, L.T.; Kelly, K.A. Plectin in Cancer: From Biomarker to Therapeutic Target. Cells 2021, 10, 2246. https://doi.org/10.3390/cells10092246
Perez SM, Brinton LT, Kelly KA. Plectin in Cancer: From Biomarker to Therapeutic Target. Cells. 2021; 10(9):2246. https://doi.org/10.3390/cells10092246
Chicago/Turabian StylePerez, Samantha M., Lindsey T. Brinton, and Kimberly A. Kelly. 2021. "Plectin in Cancer: From Biomarker to Therapeutic Target" Cells 10, no. 9: 2246. https://doi.org/10.3390/cells10092246
APA StylePerez, S. M., Brinton, L. T., & Kelly, K. A. (2021). Plectin in Cancer: From Biomarker to Therapeutic Target. Cells, 10(9), 2246. https://doi.org/10.3390/cells10092246