
Annexin A1 as a Regulator of Immune Response in Cancer

 , and
, and 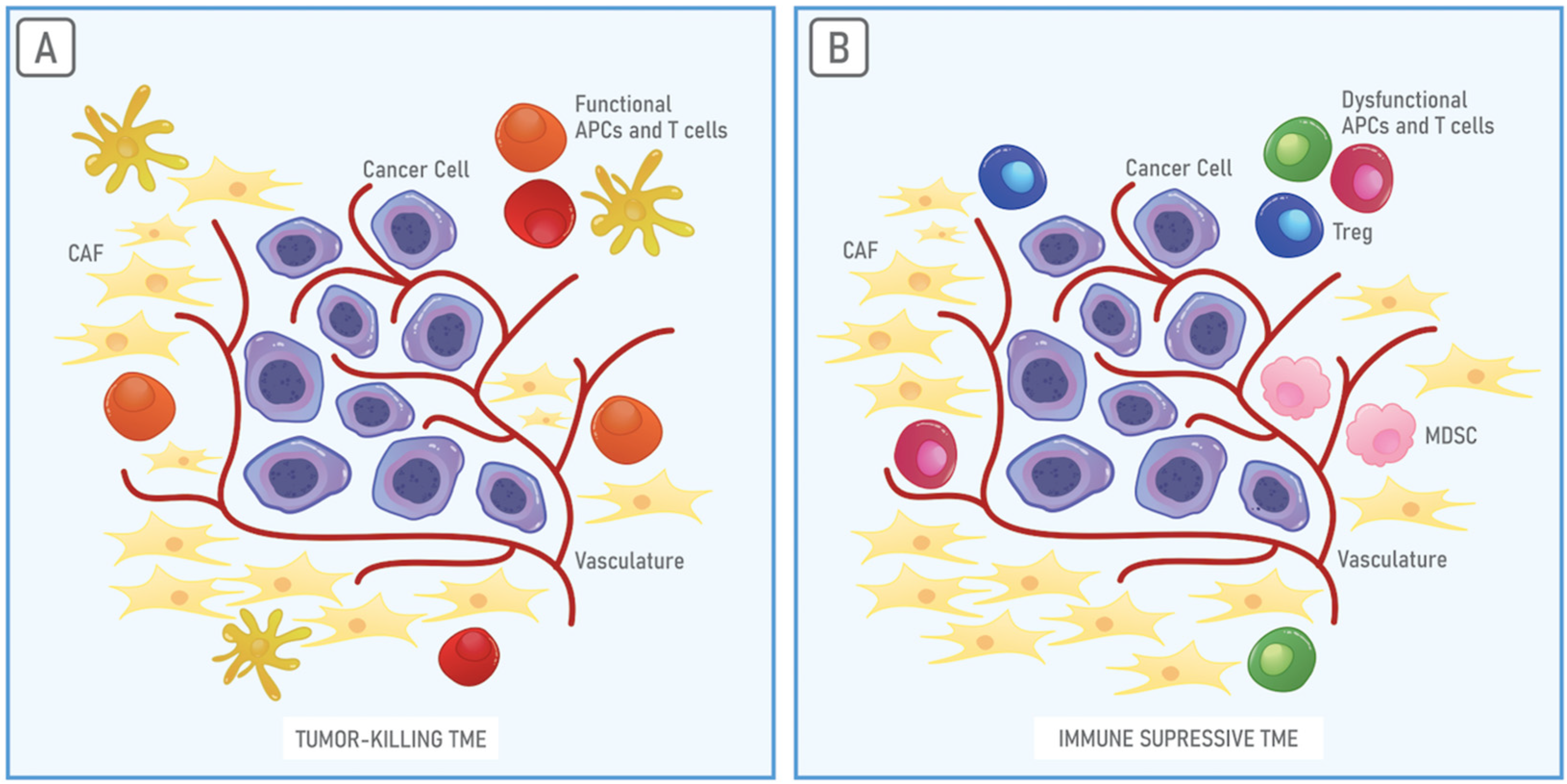{kind=link}
{kind=link}
{kind=link}
{kind=link}
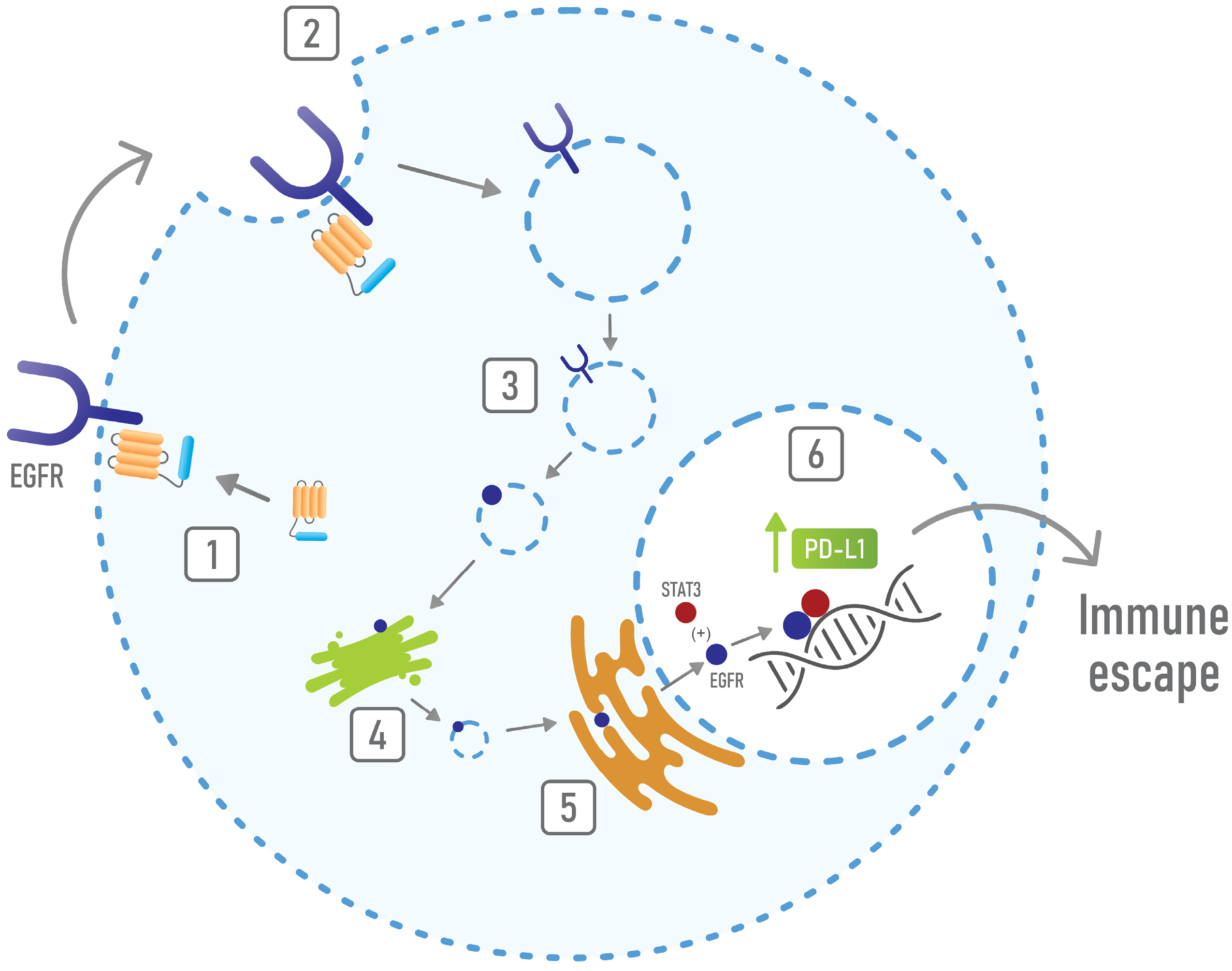{kind=link}
Abstract
1. Introduction
2. Anticancer Immune Response and Cancer Immune Escape
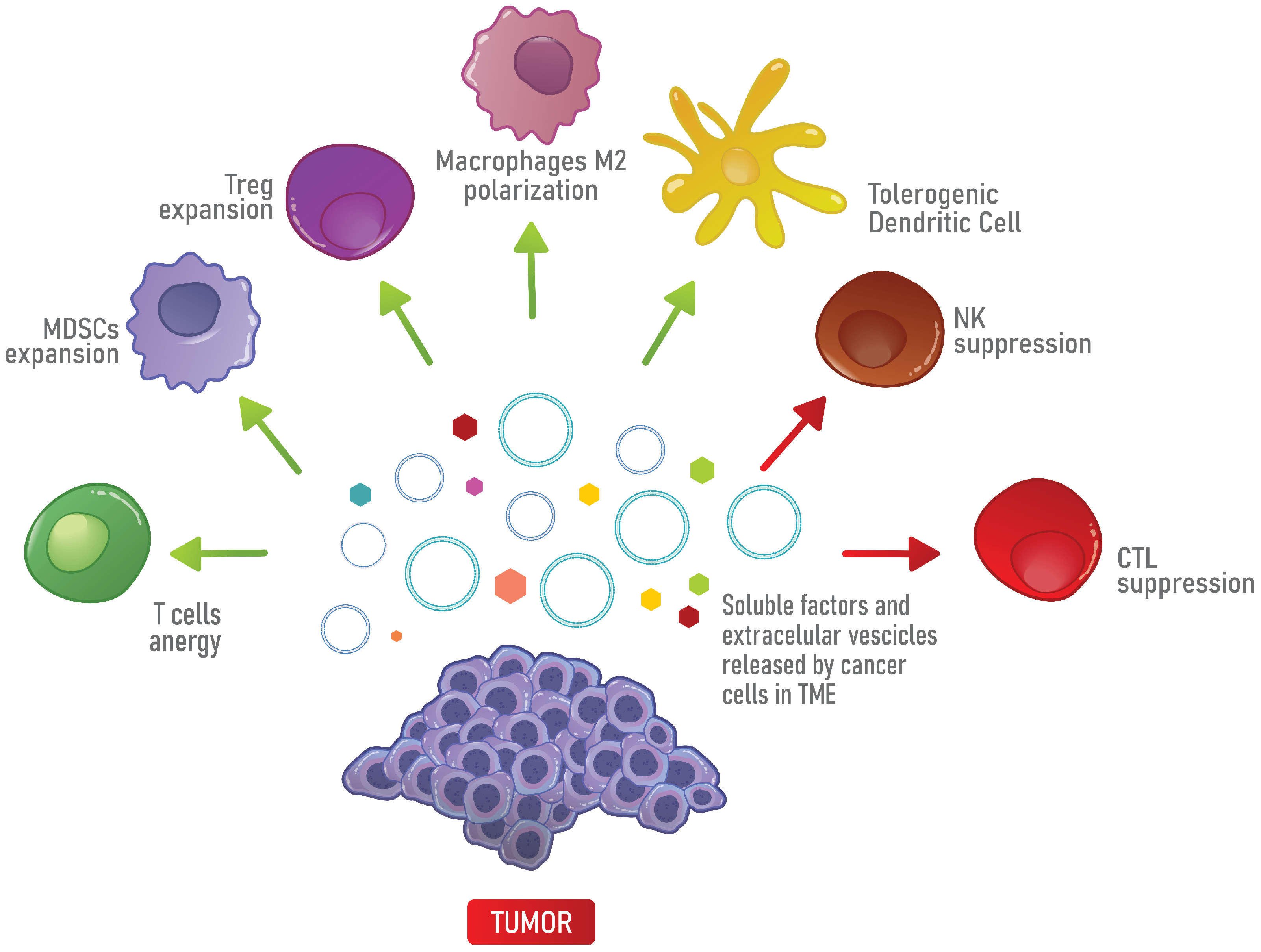
3. The Immunosuppressive Properties of the TME
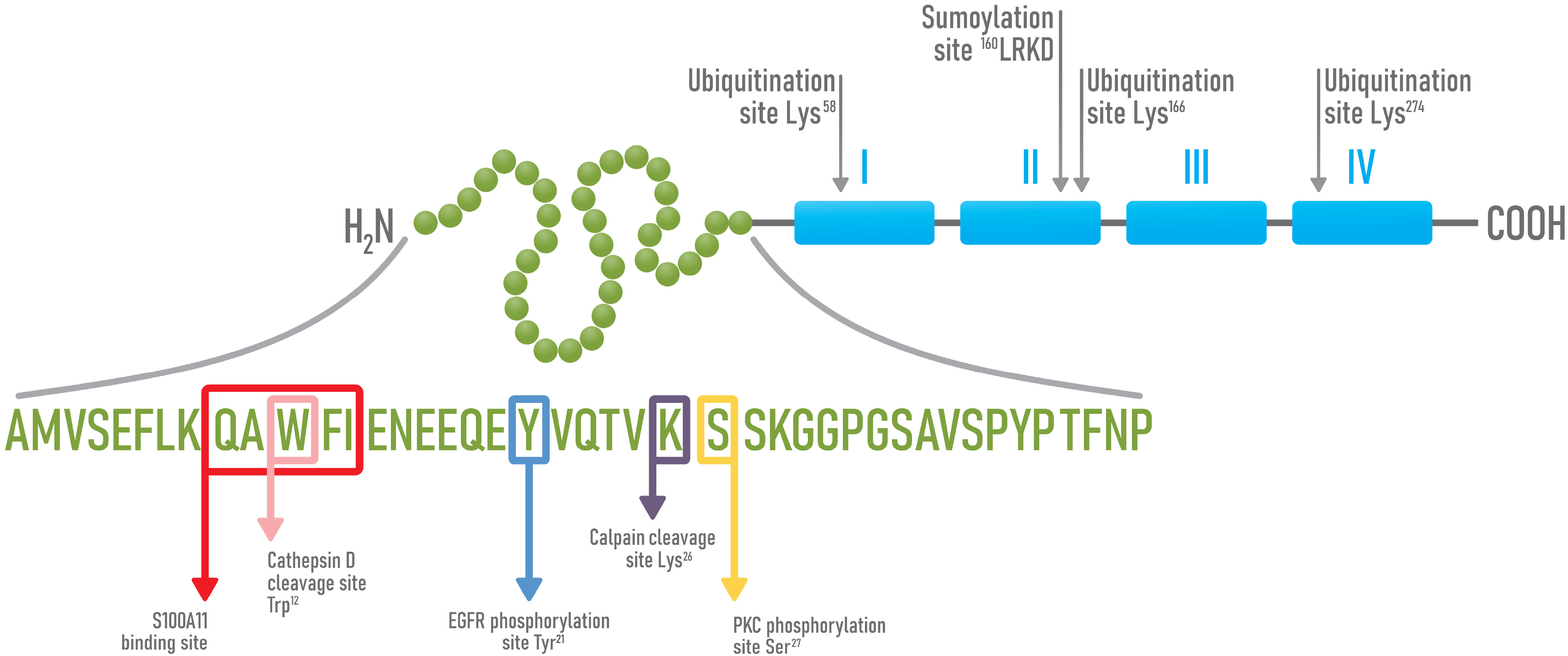
4. Structure and Functions of Annexin A1
4.1. AnxA1 in Cancer
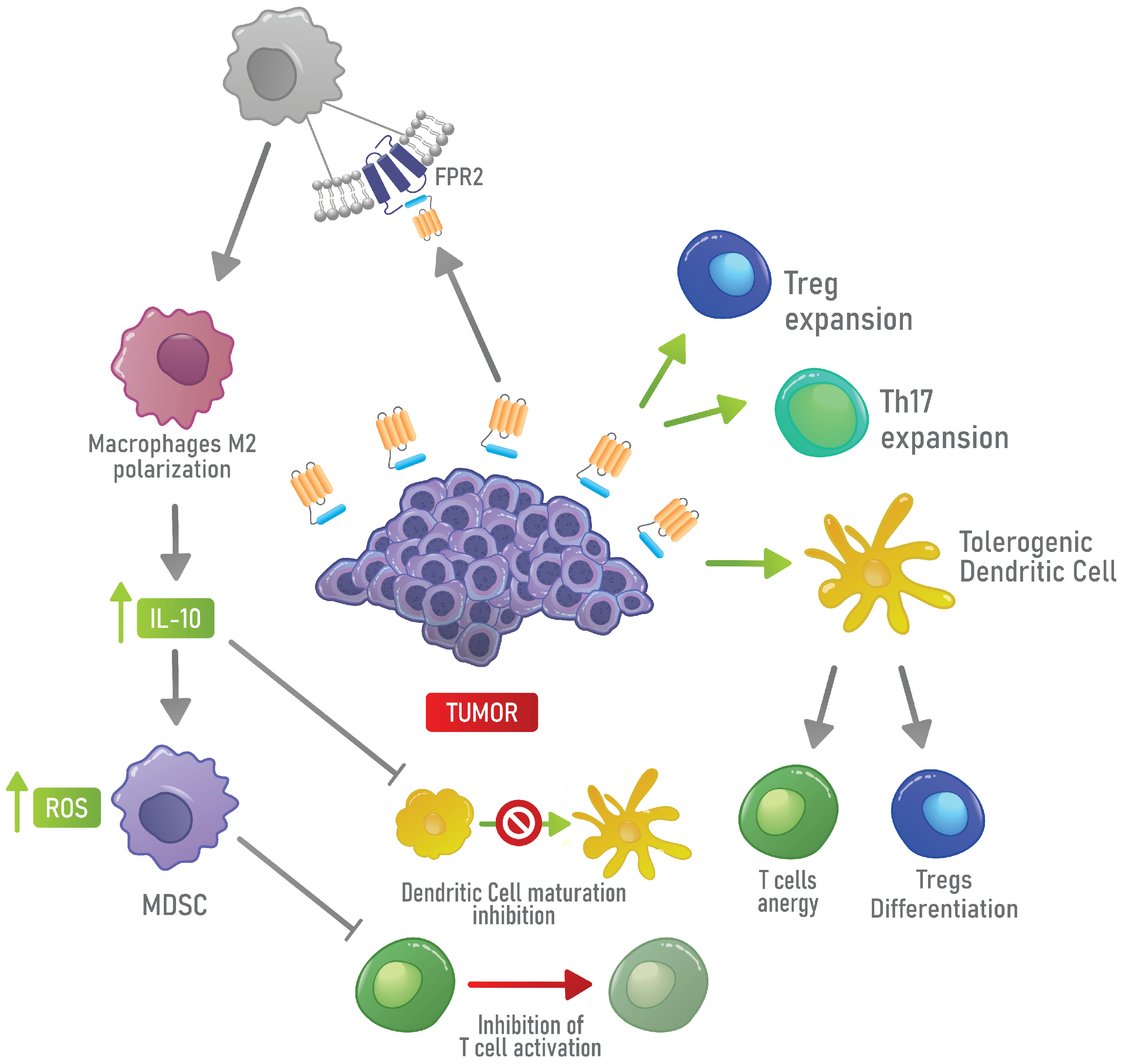
4.2. Immunosuppressive Functions of AnxA1 in Physiological and Cancer Contexts
5. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ABC A1 | ATP A1 binding cassette transport system |
| Ala | alanine |
| AnxA1 | Annexin A1 |
| APC | antigen-presenting cell |
| Asn | asparagine |
| Bad | death promoter associated with Bcl-2 |
| Bcl2 | B cell lymphoma 2 |
| Bregs | regulatory B lymphocytes |
| AP-1 | activating protein-1 |
| APC | antigen presenting cell |
| Arg-1 | arginase-1 |
| CAF | cancer-associated fibroblast |
| CCR8 | C-C motif chemokine receptor 8 |
| CD | cluster of differentiation |
| CTL | cytotoxic T lymphocytes |
| CTLA4 | cytotoxic T-lymphocyte antigen 4 |
| DC | dendritic cell |
| ECM | extracellular matrix |
| EGFR | epidermal growth factor receptor |
| EMT | epithelial–mesenchymal transition |
| ERK | extracellular signal-regulated kinases |
| EV | extracellular vesicle |
| FOXP3 | factor forkhead box P3 |
| FPR | formylated peptide receptor |
| Gly | glycine |
| HGFR | hepatocyte growth factor receptor |
| ICAM | intracellular adhesion molecule |
| iDC | immature DC |
| IFN-γ | interferon-gamma |
| IL | interleukin Ile, isoleucine |
| iNOS | inducible nitric oxide synthase |
| Lys | lysine |
| MDSC | myeloid-derived suppressor cells |
| MHC | major histocompatibility complex |
| MV | microvesicle |
| NFAT | nuclear factor of activated T-cells |
| NF-kB | nuclear factor kappa B |
| NK | natural killer cell |
| NO | nitric oxide |
| PD-L1 | programmed death-ligand 1 |
| PLA2 | phospholipase A2 |
| ROS | reactive oxygen species |
| Ser | serine |
| Smad2 | small mothers against decapentaplegic homolog 2 |
| STAT3 | signal transducer and activator of transcription 3 |
| TAM | tumor-associated macrophages |
| TCR | T cell receptor |
| tDC | tolerogenic DC |
| TGF-β | transforming growth factor-β |
| TH | T helper cell |
| TME | tumor microenvironment |
| TNF-α | tumor necrosis factor α |
| Treg | regulatory T cell |
| Trp | tryptophane |
| VCAM 1 | vascular cell adhesion molecule 1 |
| VEGF | vascular endothelial growth factor |
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Brassart-Pasco, S.; Brezillon, S.; Brassart, B.; Ramont, L.; Oudart, J.B.; Monboisse, J.C. Tumor Microenvironment: Extracellular Matrix Alterations Influence Tumor Progression. Front. Oncol. 2020, 10, 397. [Google Scholar] [CrossRef]
- Chaffer, C.L.; Weinberg, R.A. A perspective on cancer cell metastasis. Science 2011, 331, 1559–1564. [Google Scholar] [CrossRef]
- Blankenstein, T.; Coulie, P.G.; Gilboa, E.; Jaffee, E.M. The determinants of tumour immunogenicity. Nat. Rev. Cancer 2012, 12, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Pradeu, T.; Carosella, E.D. On the definition of a criterion of immunogenicity. Proc. Natl. Acad. Sci. USA 2006, 103, 17858–17861. [Google Scholar] [CrossRef]
- Kitamura, T.; Qian, B.Z.; Pollard, J.W. Immune cell promotion of metastasis. Nat. Rev. Immunol. 2015, 15, 73–86. [Google Scholar] [CrossRef]
- Crusz, S.M.; Balkwill, F.R. Inflammation and cancer: Advances and new agents. Nat. Rev. Clin. Oncol. 2015, 12, 584–596. [Google Scholar] [CrossRef] [PubMed]
- Colotta, F.; Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis 2009, 30, 1073–1081. [Google Scholar] [CrossRef]
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the immune system in cancer: From tumor initiation to metastatic progression. Genes Dev. 2018, 32, 1267–1284. [Google Scholar] [CrossRef]
- Kim, R.; Emi, M.; Tanabe, K. Cancer cell immune escape and tumor progression by exploitation of anti-inflammatory and pro-inflammatory responses. Cancer Biol. Ther. 2005, 4, 924–933. [Google Scholar] [CrossRef]
- Gaudino, S.J.; Kumar, P. Cross-Talk Between Antigen Presenting Cells and T Cells Impacts Intestinal Homeostasis, Bacterial Infections, and Tumorigenesis. Front. Immunol. 2019, 10, 360. [Google Scholar] [CrossRef]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef]
- Matsuzaki, J.; Tsuji, T.; Luescher, I.; Old, L.J.; Shrikant, P.; Gnjatic, S.; Odunsi, K. Nonclassical antigen-processing pathways are required for MHC class II-restricted direct tumor recognition by NY-ESO-1-specific CD4(+) T cells. Cancer Immunol. Res. 2014, 2, 341–350. [Google Scholar] [CrossRef]
- Knutson, K.L.; Disis, M.L.; Salazar, L.G. CD4 regulatory T cells in human cancer pathogenesis. Cancer Immunol. Immunother. 2007, 56, 271–285. [Google Scholar] [CrossRef]
- Dardalhon, V.; Korn, T.; Kuchroo, V.K.; Anderson, A.C. Role of Th1 and Th17 cells in organ-specific autoimmunity. J. Autoimmun. 2008, 31, 252–256. [Google Scholar] [CrossRef]
- Bretscher, P. On Analyzing How the Th1/Th2 Phenotype of an Immune Response Is Determined: Classical Observations Must Not Be Ignored. Front. Immunol. 2019, 10, 1234. [Google Scholar] [CrossRef]
- Emens, L.A. Breast cancer immunobiology driving immunotherapy: Vaccines and immune checkpoint blockade. Expert Rev. Anticancer Ther. 2012, 12, 1597–1611. [Google Scholar] [CrossRef] [PubMed]
- Burkholder, B.; Huang, R.Y.; Burgess, R.; Luo, S.; Jones, V.S.; Zhang, W.; Lv, Z.Q.; Gao, C.Y.; Wang, B.L.; Zhang, Y.M.; et al. Tumor-induced perturbations of cytokines and immune cell networks. Biochim. Biophys. Acta 2014, 1845, 182–201. [Google Scholar] [CrossRef]
- Zambrano-Zaragoza, J.F.; Romo-Martinez, E.J.; Duran-Avelar Mde, J.; Garcia-Magallanes, N.; Vibanco-Perez, N. Th17 cells in autoimmune and infectious diseases. Int. J. Inflam. 2014, 2014, 651503. [Google Scholar] [CrossRef] [PubMed]
- Kryczek, I.; Wei, S.; Szeliga, W.; Vatan, L.; Zou, W. Endogenous IL-17 contributes to reduced tumor growth and metastasis. Blood 2009, 114, 357–359. [Google Scholar] [CrossRef]
- Zou, W.; Restifo, N.P. T(H)17 cells in tumour immunity and immunotherapy. Nat. Rev. Immunol. 2010, 10, 248–256. [Google Scholar] [CrossRef]
- Guery, L.; Hugues, S. Th17 Cell Plasticity and Functions in Cancer Immunity. Biomed. Res. Int. 2015, 2015, 314620. [Google Scholar] [CrossRef]
- Cvetanovich, G.L.; Hafler, D.A. Human regulatory T cells in autoimmune diseases. Curr. Opin. Immunol. 2010, 22, 753–760. [Google Scholar] [CrossRef]
- Martin-Orozco, E.; Norte-Munoz, M.; Martinez-Garcia, J. Regulatory T Cells in Allergy and Asthma. Front. Pediatr. 2017, 5, 117. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Li, P.; Shao, N.; Ma, J.; Ji, M.; Sun, X.; Ma, D.; Ji, C. Aberrant expression of Treg-associated cytokine IL-35 along with IL-10 and TGF-beta in acute myeloid leukemia. Oncol. Lett. 2012, 3, 1119–1123. [Google Scholar] [CrossRef]
- Ohue, Y.; Nishikawa, H. Regulatory T (Treg) cells in cancer: Can Treg cells be a new therapeutic target? Cancer Sci. 2019, 110, 2080–2089. [Google Scholar] [CrossRef]
- Andersen, M.H.; Schrama, D.; Thor Straten, P.; Becker, J.C. Cytotoxic T cells. J. Investig. Dermatol. 2006, 126, 32–41. [Google Scholar] [CrossRef]
- Mills, C.D. M1 and M2 Macrophages: Oracles of Health and Disease. Crit. Rev. Immunol. 2012, 32, 463–488. [Google Scholar] [CrossRef]
- Cai, J.; Zhang, W.; Yang, P.; Wang, Y.; Li, M.; Zhang, C.; Wang, Z.; Hu, H.; Liu, Y.; Li, Q.; et al. Identification of a 6-cytokine prognostic signature in patients with primary glioblastoma harboring M2 microglia/macrophage phenotype relevance. PLoS ONE 2015, 10, e0126022. [Google Scholar] [CrossRef]
- Qi, L.; Yu, H.; Zhang, Y.; Zhao, D.; Lv, P.; Zhong, Y.; Xu, Y. IL-10 secreted by M2 macrophage promoted tumorigenesis through interaction with JAK2 in glioma. Oncotarget 2016, 7, 71673–71685. [Google Scholar] [CrossRef]
- Arlauckas, S.P.; Garren, S.B.; Garris, C.S.; Kohler, R.H.; Oh, J.; Pittet, M.J.; Weissleder, R. Arg1 expression defines immunosuppressive subsets of tumor-associated macrophages. Theranostics 2018, 8, 5842–5854. [Google Scholar] [CrossRef] [PubMed]
- Hao, N.B.; Lu, M.H.; Fan, Y.H.; Cao, Y.L.; Zhang, Z.R.; Yang, S.M. Macrophages in tumor microenvironments and the progression of tumors. Clin. Dev. Immunol. 2012, 2012, 948098. [Google Scholar] [CrossRef]
- Solinas, G.; Germano, G.; Mantovani, A.; Allavena, P. Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J. Leukoc. Biol. 2009, 86, 1065–1073. [Google Scholar] [CrossRef] [PubMed]
- Leek, R.D.; Lewis, C.E.; Whitehouse, R.; Greenall, M.; Clarke, J.; Harris, A.L. Association of macrophage infiltration with angiogenesis and prognosis in invasive breast carcinoma. Cancer Res. 1996, 56, 4625–4629. [Google Scholar] [PubMed]
- Cruvinel Wde, M.; Mesquita, D., Jr.; Araujo, J.A.; Catelan, T.T.; de Souza, A.W.; da Silva, N.P.; Andrade, L.E. Immune system—Part I. Fundamentals of innate immunity with emphasis on molecular and cellular mechanisms of inflammatory response. Rev. Bras. Reumatol. 2010, 50, 434–461. [Google Scholar]
- Hubo, M.; Trinschek, B.; Kryczanowsky, F.; Tuettenberg, A.; Steinbrink, K.; Jonuleit, H. Costimulatory molecules on immunogenic versus tolerogenic human dendritic cells. Front. Immunol. 2013, 4, 82. [Google Scholar] [CrossRef]
- Domogalla, M.P.; Rostan, P.V.; Raker, V.K.; Steinbrink, K. Tolerance through Education: How Tolerogenic Dendritic Cells Shape Immunity. Front. Immunol. 2017, 8, 1764. [Google Scholar] [CrossRef]
- Bharadwaj, U.; Li, M.; Chen, C.; Yao, Q. Mesothelin-induced pancreatic cancer cell proliferation involves alteration of cyclin E via activation of signal transducer and activator of transcription protein 3. Mol. Cancer Res. 2008, 6, 1755–1765. [Google Scholar] [CrossRef]
- Jarnicki, A.G.; Lysaght, J.; Todryk, S.; Mills, K.H. Suppression of antitumor immunity by IL-10 and TGF-beta-producing T cells infiltrating the growing tumor: Influence of tumor environment on the induction of CD4+ and CD8+ regulatory T cells. J. Immunol. 2006, 177, 896–904. [Google Scholar] [CrossRef]
- Wiguna, A.P.; Walden, P. Role of IL-10 and TGF-beta in melanoma. Exp. Dermatol. 2015, 24, 209–214. [Google Scholar] [CrossRef]
- Bonetti, M.I.; Pieri, L.; Domenici, L.; Urbani, S.; Romano, G.; Aldinucci, A.; Ballerini, C.; Monici, M.; Saccardi, R.; Basile, V.; et al. Dendritic cells with lymphocyte-stimulating activity differentiate from human CD133 positive precursors. Blood 2011, 117, 3983–3995. [Google Scholar] [CrossRef][Green Version]
- Saibil, S.D.; Deenick, E.K.; Ohashi, P.S. The sound of silence: Modulating anergy in T lymphocytes. Curr. Opin. Immunol. 2007, 19, 658–664. [Google Scholar] [CrossRef]
- Candolfi, M.; Yagiz, K.; Foulad, D.; Alzadeh, G.E.; Tesarfreund, M.; Muhammad, A.K.; Puntel, M.; Kroeger, K.M.; Liu, C.; Lee, S.; et al. Release of HMGB1 in response to proapoptotic glioma killing strategies: Efficacy and neurotoxicity. Clin. Cancer Res. 2009, 15, 4401–4414. [Google Scholar] [CrossRef]
- Olkhanud, P.B.; Damdinsuren, B.; Bodogai, M.; Gress, R.E.; Sen, R.; Wejksza, K.; Malchinkhuu, E.; Wersto, R.P.; Biragyn, A. Tumor-evoked regulatory B cells promote breast cancer metastasis by converting resting CD4(+) T cells to T-regulatory cells. Cancer Res. 2011, 71, 3505–3515. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, T. Regulatory and effector B cells: Friends or foes? J. Dermatol. Sci. 2019, 93, 2–7. [Google Scholar] [CrossRef]
- Veglia, F.; Perego, M.; Gabrilovich, D. Myeloid-derived suppressor cells coming of age. Nat. Immunol. 2018, 19, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Sendo, S.; Saegusa, J.; Morinobu, A. Myeloid-derived suppressor cells in non-neoplastic inflamed organs. Inflamm. Regen. 2018, 38, 19. [Google Scholar] [CrossRef] [PubMed]
- Beury, D.W.; Carter, K.A.; Nelson, C.; Sinha, P.; Hanson, E.; Nyandjo, M.; Fitzgerald, P.J.; Majeed, A.; Wali, N.; Ostrand-Rosenberg, S. Myeloid-Derived Suppressor Cell Survival and Function Are Regulated by the Transcription Factor Nrf2. J. Immunol. 2016, 196, 3470–3478. [Google Scholar] [CrossRef] [PubMed]
- Anani, W.; Shurin, M.R. Targeting Myeloid-Derived Suppressor Cells in Cancer. In Tumor Immune Microenvironment in Cancer Progression and Cancer Therapy; Kalinski, P., Ed.; Springer International Publishing: Cham, Switzerland, 2017; pp. 105–128. [Google Scholar]
- Purdy, A.K.; Campbell, K.S. Natural killer cells and cancer: Regulation by the killer cell Ig-like receptors (KIR). Cancer Biol. Ther. 2009, 8, 2211–2220. [Google Scholar] [CrossRef]
- Jost, S.; Altfeld, M. Control of human viral infections by natural killer cells. Annu. Rev. Immunol. 2013, 31, 163–194. [Google Scholar] [CrossRef] [PubMed]
- Vivier, E.; Raulet, D.H.; Moretta, A.; Caligiuri, M.A.; Zitvogel, L.; Lanier, L.L.; Yokoyama, W.M.; Ugolini, S. Innate or adaptive immunity? The example of natural killer cells. Science 2011, 331, 44–49. [Google Scholar] [CrossRef]
- Morvan, M.G.; Lanier, L.L. NK cells and cancer: You can teach innate cells new tricks. Nat. Rev. Cancer 2016, 16, 7–19. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, S.; Zhang, B.; Qiao, L.; Zhang, Y.; Zhang, Y. T Cell Dysfunction and Exhaustion in Cancer. Front. Cell Dev. Biol. 2020, 8, 17. [Google Scholar] [CrossRef]
- DeVito, N.C.; Plebanek, M.P.; Theivanthiran, B.; Hanks, B.A. Role of Tumor-Mediated Dendritic Cell Tolerization in Immune Evasion. Front. Immunol. 2019, 10, 2876. [Google Scholar] [CrossRef] [PubMed]
- Correia, A.L.; Bissell, M.J. The tumor microenvironment is a dominant force in multidrug resistance. Drug Resist. Updates 2012, 15, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Jiang, Y.C.; Sun, C.K.; Chen, Q.M. Role of the tumor microenvironment in tumor progression and the clinical applications (Review). Oncol. Rep. 2016, 35, 2499–2515. [Google Scholar] [CrossRef] [PubMed]
- Spill, F.; Reynolds, D.S.; Kamm, R.D.; Zaman, M.H. Impact of the physical microenvironment on tumor progression and metastasis. Curr. Opin. Biotechnol. 2016, 40, 41–48. [Google Scholar] [CrossRef]
- Reina-Campos, M.; Moscat, J.; Diaz-Meco, M. Metabolism shapes the tumor microenvironment. Curr. Opin. Cell Biol. 2017, 48, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Kashyap, A.S.; Schmittnaegel, M.; Rigamonti, N.; Pais-Ferreira, D.; Mueller, P.; Buchi, M.; Ooi, C.H.; Kreuzaler, M.; Hirschmann, P.; Guichard, A.; et al. Optimized antiangiogenic reprogramming of the tumor microenvironment potentiates CD40 immunotherapy. Proc. Natl. Acad. Sci. USA 2020, 117, 541–551. [Google Scholar] [CrossRef]
- Anderson, N.M.; Simon, M.C. The tumor microenvironment. Curr. Biol. 2020, 30, R921–R925. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhao, J.; Zhang, L.; Wei, F.; Lian, Y.; Wu, Y.; Gong, Z.; Zhang, S.; Zhou, J.; Cao, K.; et al. Role of tumor microenvironment in tumorigenesis. J. Cancer 2017, 8, 761–773. [Google Scholar] [CrossRef]
- Huang, W.; Luo, S.; Burgess, R.; Yi, Y.H.; Huang, G.F.; Huang, R.P. New Insights into the Tumor Microenvironment Utilizing Protein Array Technology. Int. J. Mol. Sci. 2018, 19, 559. [Google Scholar] [CrossRef]
- Frantz, C.; Stewart, K.M.; Weaver, V.M. The extracellular matrix at a glance. J. Cell Sci. 2010, 123, 4195–4200. [Google Scholar] [CrossRef] [PubMed]
- Theocharis, A.D.; Skandalis, S.S.; Gialeli, C.; Karamanos, N.K. Extracellular matrix structure. Adv. Drug Deliv. Rev. 2016, 97, 4–27. [Google Scholar] [CrossRef]
- Deryugina, E.I.; Quigley, J.P. Tumor angiogenesis: MMP-mediated induction of intravasation- and metastasis-sustaining neovasculature. Matrix Biol. 2015, 44–46, 94–112. [Google Scholar] [CrossRef]
- Shiga, K.; Hara, M.; Nagasaki, T.; Sato, T.; Takahashi, H.; Takeyama, H. Cancer-Associated Fibroblasts: Their Characteristics and Their Roles in Tumor Growth. Cancers 2015, 7, 2443–2458. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, W.; Sun, X.; Lin, Y.; Chen, W. Cancer-associated fibroblasts induce epithelial-mesenchymal transition through secreted cytokines in endometrial cancer cells. Oncol. Lett. 2018, 15, 5694–5702. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Lei, Y.; Li, J.K.; Du, W.X.; Li, R.G.; Yang, J.; Li, J.; Li, F.; Tan, H.B. Immune cells within the tumor microenvironment: Biological functions and roles in cancer immunotherapy. Cancer Lett. 2020, 470, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Mayer, A.; Haist, M.; Loquai, C.; Grabbe, S.; Rapp, M.; Roth, W.; Vaupel, P.; Schmidberger, H. Role of Hypoxia and the Adenosine System in Immune Evasion and Prognosis of Patients with Brain Metastases of Melanoma: A Multiplex Whole Slide Immunofluorescence Study. Cancers 2020, 12, 3753. [Google Scholar] [CrossRef]
- Nachmany, I.; Bogoch, Y.; Friedlander-Malik, G.; Amar, O.; Bondar, E.; Zohar, N.; Hantisteanu, S.; Fainaru, O.; Lubezky, N.; Klausner, J.M.; et al. The transcriptional profile of circulating myeloid derived suppressor cells correlates with tumor development and progression in mouse. Genes Immun. 2019, 20, 589–598. [Google Scholar] [CrossRef]
- Magnuson, A.M.; Kiner, E.; Ergun, A.; Park, J.S.; Asinovski, N.; Ortiz-Lopez, A.; Kilcoyne, A.; Paoluzzi-Tomada, E.; Weissleder, R.; Mathis, D.; et al. Identification and validation of a tumor-infiltrating Treg transcriptional signature conserved across species and tumor types. Proc. Natl. Acad. Sci. USA 2018, 115, E10672–E10681. [Google Scholar] [CrossRef]
- Gabrilovich, D. Mechanisms and functional significance of tumour-induced dendritic-cell defects. Nat. Rev. Immunol. 2004, 4, 941–952. [Google Scholar] [CrossRef]
- Melief, C.J. Cancer immunotherapy by dendritic cells. Immunity 2008, 29, 372–383. [Google Scholar] [CrossRef]
- Chen, J.; Ganguly, A.; Mucsi, A.D.; Meng, J.; Yan, J.; Detampel, P.; Munro, F.; Zhang, Z.; Wu, M.; Hari, A.; et al. Strong adhesion by regulatory T cells induces dendritic cell cytoskeletal polarization and contact-dependent lethargy. J. Exp. Med. 2017, 214, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Wing, K.; Onishi, Y.; Prieto-Martin, P.; Yamaguchi, T.; Miyara, M.; Fehervari, Z.; Nomura, T.; Sakaguchi, S. CTLA-4 control over Foxp3+ regulatory T cell function. Science 2008, 322, 271–275. [Google Scholar] [CrossRef]
- Fife, B.T.; Bluestone, J.A. Control of peripheral T-cell tolerance and autoimmunity via the CTLA-4 and PD-1 pathways. Immunol. Rev. 2008, 224, 166–182. [Google Scholar] [CrossRef]
- Peng, L.S.; Zhang, J.Y.; Teng, Y.S.; Zhao, Y.L.; Wang, T.T.; Mao, F.Y.; Lv, Y.P.; Cheng, P.; Li, W.H.; Chen, N.; et al. Tumor-Associated Monocytes/Macrophages Impair NK-Cell Function via TGFbeta1 in Human Gastric Cancer. Cancer Immunol. Res. 2017, 5, 248–256. [Google Scholar] [CrossRef]
- Gordon, S.; Taylor, P.R. Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 2005, 5, 953–964. [Google Scholar] [CrossRef]
- Ruffell, B.; Affara, N.I.; Coussens, L.M. Differential macrophage programming in the tumor microenvironment. Trends Immunol. 2012, 33, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.J.; Lin, Y.C.; Yao, P.L.; Yuan, A.; Chen, H.Y.; Shun, C.T.; Tsai, M.F.; Chen, C.H.; Yang, P.C. Tumor-associated macrophages: The double-edged sword in cancer progression. J. Clin. Oncol. 2005, 23, 953–964. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Chen, J.; Zhang, W.; Zhang, R.; Ye, Y.; Liu, P.; Yu, W.; Wei, F.; Ren, X.; Yu, J. Interleukin-6 Trans-Signaling Pathway Promotes Immunosuppressive Myeloid-Derived Suppressor Cells via Suppression of Suppressor of Cytokine Signaling 3 in Breast Cancer. Front. Immunol. 2017, 8, 1840. [Google Scholar] [CrossRef]
- Shvedova, A.A.; Kisin, E.R.; Yanamala, N.; Tkach, A.V.; Gutkin, D.W.; Star, A.; Shurin, G.V.; Kagan, V.E.; Shurin, M.R. MDSC and TGFbeta Are Required for Facilitation of Tumor Growth in the Lungs of Mice Exposed to Carbon Nanotubes. Cancer Res. 2015, 75, 1615–1623. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Pan, P.Y.; Li, Q.; Sato, A.I.; Levy, D.E.; Bromberg, J.; Divino, C.M.; Chen, S.H. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. 2006, 66, 1123–1131. [Google Scholar] [CrossRef]
- Armstrong, D.; Chang, C.Y.; Lazarus, D.R.; Corry, D.; Kheradmand, F. Lung Cancer Heterogeneity in Modulation of Th17/IL17A Responses. Front. Oncol. 2019, 9, 1384. [Google Scholar] [CrossRef] [PubMed]
- Mucida, D.; Park, Y.; Kim, G.; Turovskaya, O.; Scott, I.; Kronenberg, M.; Cheroutre, H. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science 2007, 317, 256–260. [Google Scholar] [CrossRef]
- Garrido-Mesa, N.; Algieri, F.; Rodriguez Nogales, A.; Galvez, J. Functional plasticity of Th17 cells: Implications in gastrointestinal tract function. Int. Rev. Immunol. 2013, 32, 493–510. [Google Scholar] [CrossRef]
- Vecchi, L.; Araújo, T.G.; Azevedo, F.V.P.D.V.; Mota, S.T.S.; Ávila, V.D.M.R.; Ribeiro, M.A.; Goulart, L.R. Phospholipase A2 Drives Tumorigenesis and Cancer Aggressiveness through Its Interaction with Annexin A1. Cells 2021, 10, 1472. [Google Scholar] [CrossRef]
- Parente, L.; Solito, E. Annexin 1: More than an anti-phospholipase protein. Inflamm. Res. 2004, 53, 125–132. [Google Scholar] [CrossRef]
- Gerke, V.; Creutz, C.E.; Moss, S.E. Annexins: Linking Ca2+ signalling to membrane dynamics. Nat. Rev. Mol. Cell Biol. 2005, 6, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, M.H.; Solito, E. Annexin A1: Uncovering the Many Talents of an Old Protein. Int. J. Mol. Sci. 2018, 19, 1045. [Google Scholar] [CrossRef]
- Pepinsky, R.B.; Sinclair, L.K.; Browning, J.L.; Mattaliano, R.J.; Smart, J.E.; Chow, E.P.; Falbel, T.; Ribolini, A.; Garwin, J.L.; Wallner, B.P. Purification and partial sequence analysis of a 37-kDa protein that inhibits phospholipase A2 activity from rat peritoneal exudates. J. Biol. Chem. 1986, 261, 4239–4246. [Google Scholar] [CrossRef]
- Perretti, M.; D’Acquisto, F. Annexin A1 and glucocorticoids as effectors of the resolution of inflammation. Nat. Rev. Immunol. 2009, 9, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Arcone, R.; Arpaia, G.; Ruoppolo, M.; Malorni, A.; Pucci, P.; Marino, G.; Ialenti, A.; Di Rosa, M.; Ciliberto, G. Structural characterization of a biologically active human lipocortin 1 expressed in Escherichia coli. Eur. J. Biochem. 1993, 211, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Flower, R.J. Eleventh Gaddum memorial lecture. Lipocortin and the mechanism of action of the glucocorticoids. Br. J. Pharmacol. 1988, 94, 987–1015. [Google Scholar] [CrossRef]
- Sugimoto, M.A.; Vago, J.P.; Teixeira, M.M.; Sousa, L.P. Annexin A1 and the Resolution of Inflammation: Modulation of Neutrophil Recruitment, Apoptosis, and Clearance. J. Immunol. Res. 2016, 2016, 8239258. [Google Scholar] [CrossRef] [PubMed]
- Purvis, G.S.D.; Solito, E.; Thiemermann, C. Annexin-A1: Therapeutic Potential in Microvascular Disease. Front. Immunol. 2019, 10, 938. [Google Scholar] [CrossRef]
- Shen, X.; Zhang, S.; Guo, Z.; Xing, D.; Chen, W. The crosstalk of ABCA1 and ANXA1: A potential mechanism for protection against atherosclerosis. Mol. Med. 2020, 26, 84. [Google Scholar] [CrossRef]
- Hall, S.C.; Smith, D.M.; Masiarz, F.R.; Soo, V.W.; Tran, H.M.; Epstein, L.B.; Burlingame, A.L. Mass spectrometric and Edman sequencing of lipocortin I isolated by two-dimensional SDS/PAGE of human melanoma lysates. Proc. Natl. Acad. Sci. USA 1993, 90, 1927–1931. [Google Scholar] [CrossRef]
- Bai, F.; Zhang, P.; Fu, Y.; Chen, H.; Zhang, M.; Huang, Q.; Li, D.; Li, B.; Wu, K. Targeting ANXA1 abrogates Treg-mediated immune suppression in triple-negative breast cancer. J. Immunother. Cancer 2020, 8, e000169. [Google Scholar] [CrossRef] [PubMed]
- Rosengarth, A.; Luecke, H. A calcium-driven conformational switch of the N-terminal and core domains of Annexin A1. J. Mol. Biol. 2003, 326, 1317–1325. [Google Scholar] [CrossRef]
- Rosengarth, A.; Gerke, V.; Luecke, H. X-ray structure of full-length Annexin 1 and implications for membrane aggregation. J. Mol. Biol. 2001, 306, 489–498. [Google Scholar] [CrossRef]
- Glenney, J.R., Jr.; Tack, B.F. Amino-terminal sequence of p36 and associated p10: Identification of the site of tyrosine phosphorylation and homology with S-100. Proc. Natl. Acad. Sci. USA 1985, 82, 7884–7888. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, S.R.; Moss, S.E. Functional analysis of the human Annexin I and VI gene promoters. Biochem. J. 1998, 332 Pt 3, 681–687. [Google Scholar] [CrossRef]
- Boudhraa, Z.; Bouchon, B.; Viallard, C.; D’Incan, M.; Degoul, F. Annexin A1 localization and its relevance to cancer. Clin. Sci. 2016, 130, 205–220. [Google Scholar] [CrossRef]
- Gerke, V.; Moss, S.E. Annexins: From structure to function. Physiol. Rev. 2002, 82, 331–371. [Google Scholar] [CrossRef] [PubMed]
- Moss, S.E.; Morgan, R.O. The annexins. Genome Biol. 2004, 5, 219. [Google Scholar] [CrossRef]
- Weyd, H. More than just innate affairs—On the role of annexins in adaptive immunity. Biol. Chem. 2016, 397, 1017–1029. [Google Scholar] [CrossRef] [PubMed]
- Grewal, T.; Hoque, M.; Conway, J.R.W.; Reverter, M.; Wahba, M.; Beevi, S.S.; Timpson, P.; Enrich, C.; Rentero, C. Annexin A6-A multifunctional scaffold in cell motility. Cell Adhes. Migr. 2017, 11, 288–304. [Google Scholar] [CrossRef] [PubMed]
- Boudhraa, Z.; Merle, C.; Mazzocut, D.; Chezal, J.M.; Chambon, C.; Miot-Noirault, E.; Theisen, M.; Bouchon, B.; Degoul, F. Characterization of pro-invasive mechanisms and N-terminal cleavage of ANXA1 in melanoma. Arch. Dermatol. Res. 2014, 306, 903–914. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, H.H. Membrane traffic in the secretory pathway. Cell. Mol. Life Sci. 2008, 65, 2779–2780. [Google Scholar] [CrossRef]
- Han, G.; Tian, Y.; Duan, B.; Sheng, H.; Gao, H.; Huang, J. Association of nuclear Annexin A1 with prognosis of patients with esophageal squamous cell carcinoma. Int. J. Clin. Exp. Pathol. 2014, 7, 751–759. [Google Scholar]
- Futter, C.E.; White, I.J. Annexins and endocytosis. Traffic 2007, 8, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Lim, L.H.; Pervaiz, S. Annexin 1: The new face of an old molecule. FASEB J. 2007, 21, 968–975. [Google Scholar] [CrossRef] [PubMed]
- Hirata, A.; Hirata, F. DNA chain unwinding and annealing reactions of lipocortin (Annexin) I heterotetramer: Regulation by Ca(2+) and Mg(2+). Biochem. Biophys. Res. Commun. 2002, 291, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Hickey, R.; Malkas, L. The biochemical status of the DNA synthesome can distinguish between permanent and temporary cell growth arrest. Cell Growth Differ. 1997, 8, 1359–1369. [Google Scholar] [PubMed]
- Rescher, U.; Zobiack, N.; Gerke, V. Intact Ca(2+)-binding sites are required for targeting of Annexin 1 to endosomal membranes in living HeLa cells. J. Cell Sci. 2000, 113 Pt 22, 3931–3938. [Google Scholar] [CrossRef]
- Sudlow, A.W.; Carey, F.; Forder, R.; Rothwell, N.J. The role of lipocortin-1 in dexamethasone-induced suppression of PGE2 and TNF alpha release from human peripheral blood mononuclear cells. Br. J. Pharmacol. 1996, 117, 1449–1456. [Google Scholar] [CrossRef] [PubMed]
- Sanches, J.M.; Branco, L.M.; Duarte, G.H.B.; Oliani, S.M.; Bortoluci, K.R.; Moreira, V.; Gil, C.D. Annexin A1 Regulates NLRP3 Inflammasome Activation and Modifies Lipid Release Profile in Isolated Peritoneal Macrophages. Cells 2020, 9, 926. [Google Scholar] [CrossRef] [PubMed]
- Seidel, S.; Neymeyer, H.; Kahl, T.; Röschel, T.; Mutig, K.; Flower, R.; Schnermann, J.; Bachmann, S.; Paliege, A. Annexin A1 modulates macula densa function by inhibiting cyclooxygenase 2. Am. J. Physiol. Renal. Physiol. 2012, 303, F845–F854. [Google Scholar] [CrossRef]
- Flower, R.J.; Rothwell, N.J. Lipocortin-1: Cellular mechanisms and clinical relevance. Trends Pharmacol. Sci. 1994, 15, 71–76. [Google Scholar] [CrossRef]
- Flower, R.J.; Blackwell, G.J. Anti-inflammatory steroids induce biosynthesis of a phospholipase A2 inhibitor which prevents prostaglandin generation. Nature 1979, 278, 456–459. [Google Scholar] [CrossRef]
- Leoni, G.; Nusrat, A. Annexin A1: Shifting the balance towards resolution and repair. Biol. Chem. 2016, 397, 971–979. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, B.E.; Yona, S.; Rosignoli, G.; Young, R.E.; Nourshargh, S.; Flower, R.J.; Perretti, M. Annexin 1-deficient neutrophils exhibit enhanced transmigration in vivo and increased responsiveness in vitro. J. Leukoc. Biol. 2005, 78, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Kiani-Esfahani, A.; Kazemi Sheykhshabani, S.; Peymani, M.; Hashemi, M.S.; Ghaedi, K.; Nasr-Esfahani, M.H. Overexpression of Annexin A1 Suppresses Pro-Inflammatory Factors in PC12 Cells Induced by 1-Methyl-4-Phenylpyridinium. Cell J. 2016, 18, 197–204. [Google Scholar] [CrossRef]
- Sakaguchi, M.; Murata, H.; Sonegawa, H.; Sakaguchi, Y.; Futami, J.; Kitazoe, M.; Yamada, H.; Huh, N.H. Truncation of Annexin A1 is a regulatory lever for linking epidermal growth factor signaling with cytosolic phospholipase A2 in normal and malignant squamous epithelial cells. J. Biol. Chem. 2007, 282, 35679–35686. [Google Scholar] [CrossRef]
- Sakaguchi, M.; Huh, N.H. S100A11, a dual growth regulator of epidermal keratinocytes. Amino. Acids 2011, 41, 797–807. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Santamaria-Kisiel, L.; Rintala-Dempsey, A.C.; Shaw, G.S. Calcium-dependent and -independent interactions of the S100 protein family. Biochem. J. 2006, 396, 201–214. [Google Scholar] [CrossRef]
- Blume, K.E.; Soeroes, S.; Keppeler, H.; Stevanovic, S.; Kretschmer, D.; Rautenberg, M.; Wesselborg, S.; Lauber, K. Cleavage of Annexin A1 by ADAM10 during secondary necrosis generates a monocytic “find-me” signal. J. Immunol. 2012, 188, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Movitz, C.; Sjolin, C.; Dahlgren, C. Cleavage of Annexin I in human neutrophils is mediated by a membrane-localized metalloprotease. Biochim. Biophys. Acta 1999, 1416, 101–108. [Google Scholar] [CrossRef]
- Vong, L.; D’Acquisto, F.; Pederzoli-Ribeil, M.; Lavagno, L.; Flower, R.J.; Witko-Sarsat, V.; Perretti, M. Annexin 1 cleavage in activated neutrophils: A pivotal role for proteinase 3. J. Biol. Chem. 2007, 282, 29998–30004. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Creutz, C.E. Role of the amino-terminal domain in regulating interactions of Annexin I with membranes: Effects of amino-terminal truncation and mutagenesis of the phosphorylation sites. Biochemistry 1994, 33, 275–282. [Google Scholar] [CrossRef]
- Zoia, M.A.P.; Azevedo, F.V.P.; Vecchi, L.; Mota, S.T.S.; Rodovalho, V.R.; Cordeiro, A.O.; Correia, L.I.V.; Silva, A.C.A.; Avila, V.M.R.; Araujo, T.G.; et al. Inhibition of Triple-Negative Breast Cancer Cell Aggressiveness by Cathepsin D Blockage: Role of Annexin A1. Int. J. Mol. Sci. 2019, 20, 1337. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.L.; Milne, I.R.; Bagley, C.J.; Gamble, J.R.; Vadas, M.A.; Pitson, S.M.; Khew-Goodall, Y. A proinflammatory role for proteolytically cleaved Annexin A1 in neutrophil transendothelial migration. J. Immunol. 2010, 185, 3057–3063. [Google Scholar] [CrossRef]
- Solito, E.; Christian, H.C.; Festa, M.; Mulla, A.; Tierney, T.; Flower, R.J.; Buckingham, J.C. Post-translational modification plays an essential role in the translocation of Annexin A1 from the cytoplasm to the cell surface. FASEB J. 2006, 20, 1498–1500. [Google Scholar] [CrossRef] [PubMed]
- Solito, E.; Kamal, A.; Russo-Marie, F.; Buckingham, J.C.; Marullo, S.; Perretti, M. A novel calcium-dependent proapoptotic effect of Annexin 1 on human neutrophils. FASEB J. 2003, 17, 1544–1546. [Google Scholar] [CrossRef]
- Frambach, S.; de Haas, R.; Smeitink, J.A.M.; Rongen, G.A.; Russel, F.G.M.; Schirris, T.J.J. Brothers in Arms: ABCA1- and ABCG1-Mediated Cholesterol Efflux as Promising Targets in Cardiovascular Disease Treatment. Pharmacol. Rev. 2020, 72, 152–190. [Google Scholar] [CrossRef]
- Hafiane, A.; Genest, J. ATP binding cassette A1 (ABCA1) mediates microparticle formation during high-density lipoprotein (HDL) biogenesis. Atherosclerosis 2017, 257, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Nandi, S.; Ma, L.; Denis, M.; Karwatsky, J.; Li, Z.; Jiang, X.C.; Zha, X. ABCA1-mediated cholesterol efflux generates microparticles in addition to HDL through processes governed by membrane rigidity. J. Lipid Res. 2009, 50, 456–466. [Google Scholar] [CrossRef] [PubMed]
- Brancaleone, V.; Mitidieri, E.; Flower, R.J.; Cirino, G.; Perretti, M. Annexin A1 mediates hydrogen sulfide properties in the control of inflammation. J. Pharmacol. Exp. Ther. 2014, 351, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Perretti, M.; Christian, H.; Wheller, S.K.; Aiello, I.; Mugridge, K.G.; Morris, J.F.; Flower, R.J.; Goulding, N.J. Annexin I is stored within gelatinase granules of human neutrophil and mobilized on the cell surface upon adhesion but not phagocytosis. Cell Biol. Int. 2000, 24, 163–174. [Google Scholar] [CrossRef]
- Li, Y.; Ye, D. Molecular biology for formyl peptide receptors in human diseases. J. Mol. Med. 2013, 91, 781–789. [Google Scholar] [CrossRef] [PubMed]
- Ye, R.D.; Boulay, F.; Wang, J.M.; Dahlgren, C.; Gerard, C.; Parmentier, M.; Serhan, C.N.; Murphy, P.M. International Union of Basic and Clinical Pharmacology. LXXIII. Nomenclature for the formyl peptide receptor (FPR) family. Pharmacol. Rev. 2009, 61, 119–161. [Google Scholar] [CrossRef]
- Migeotte, I.; Communi, D.; Parmentier, M. Formyl peptide receptors: A promiscuous subfamily of G protein-coupled receptors controlling immune responses. Cytokine Growth Factor Rev. 2006, 17, 501–519. [Google Scholar] [CrossRef] [PubMed]
- Perretti, M.; Chiang, N.; La, M.; Fierro, I.M.; Marullo, S.; Getting, S.J.; Solito, E.; Serhan, C.N. Endogenous lipid- and peptide-derived anti-inflammatory pathways generated with glucocorticoid and aspirin treatment activate the lipoxin A4 receptor. Nat. Med. 2002, 8, 1296–1302. [Google Scholar] [CrossRef]
- Dalpiaz, A.; Spisani, S.; Biondi, C.; Fabbri, E.; Nalli, M.; Ferretti, M.E. Studies on human neutrophil biological functions by means of formyl-peptide receptor agonists and antagonists. Curr. Drug Targets Immune Endocr. Metab. Disord. 2003, 3, 33–42. [Google Scholar] [CrossRef]
- Gavins, F.N.; Hickey, M.J. Annexin A1 and the regulation of innate and adaptive immunity. Front. Immunol. 2012, 3, 354. [Google Scholar] [CrossRef]
- Cooray, S.N.; Gobbetti, T.; Montero-Melendez, T.; McArthur, S.; Thompson, D.; Clark, A.J.; Flower, R.J.; Perretti, M. Ligand-specific conformational change of the G-protein-coupled receptor ALX/FPR2 determines proresolving functional responses. Proc. Natl. Acad. Sci. USA 2013, 110, 18232–18237. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Chen, K.; Xiang, Y.; Yoshimura, T.; Su, S.; Zhu, J.; Bian, X.W.; Wang, J.M. New development in studies of formyl-peptide receptors: Critical roles in host defense. J. Leukoc. Biol. 2016, 99, 425–435. [Google Scholar] [CrossRef]
- Vacchelli, E.; Ma, Y.; Baracco, E.E.; Sistigu, A.; Enot, D.P.; Pietrocola, F.; Yang, H.; Adjemian, S.; Chaba, K.; Semeraro, M.; et al. Chemotherapy-induced antitumor immunity requires formyl peptide receptor 1. Science 2015, 350, 972–978. [Google Scholar] [CrossRef] [PubMed]
- Nagaya, T.; Kawata, K.; Kamekura, R.; Jitsukawa, S.; Kubo, T.; Kamei, M.; Ogasawara, N.; Takano, K.I.; Himi, T.; Ichimiya, S. Lipid mediators foster the differentiation of T follicular helper cells. Immunol. Lett. 2017, 181, 51–57. [Google Scholar] [CrossRef]
- Chen, K.; Xiang, Y.; Huang, J.; Gong, W.; Yoshimura, T.; Jiang, Q.; Tessarollo, L.; Le, Y.; Wang, J.M. The formylpeptide receptor 2 (Fpr2) and its endogenous ligand cathelin-related antimicrobial peptide (CRAMP) promote dendritic cell maturation. J. Biol. Chem. 2014, 289, 17553–17563. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.D.; Kim, J.M.; Jo, S.H.; Lee, H.Y.; Lee, S.Y.; Shim, J.W.; Seo, S.K.; Yun, J.; Bae, Y.S. Functional expression of formyl peptide receptor family in human NK cells. J. Immunol. 2009, 183, 5511–5517. [Google Scholar] [CrossRef] [PubMed]
- McArthur, S.; Gobbetti, T.; Kusters, D.H.; Reutelingsperger, C.P.; Flower, R.J.; Perretti, M. Definition of a Novel Pathway Centered on Lysophosphatidic Acid To Recruit Monocytes during the Resolution Phase of Tissue Inflammation. J. Immunol. 2015, 195, 1139–1151. [Google Scholar] [CrossRef]
- Peshavariya, H.M.; Taylor, C.J.; Goh, C.; Liu, G.S.; Jiang, F.; Chan, E.C.; Dusting, G.J. Annexin peptide Ac2-26 suppresses TNFalpha-induced inflammatory responses via inhibition of Rac1-dependent NADPH oxidase in human endothelial cells. PLoS ONE 2013, 8, e60790. [Google Scholar] [CrossRef]
- Miki, T.; Yano, S.; Hanibuchi, M.; Sone, S. Bone metastasis model with multiorgan dissemination of human small-cell lung cancer (SBC-5) cells in natural killer cell-depleted SCID mice. Oncol. Res. 2000, 12, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Chen, L.Y.; Zuraw, B.L.; Ye, R.D.; Pan, Z.K. Chemoattractant-stimulated NF-kappaB activation is dependent on the low molecular weight GTPase RhoA. J. Biol. Chem. 2001, 276, 40977–40981. [Google Scholar] [CrossRef]
- Han, P.F.; Che, X.D.; Li, H.Z.; Gao, Y.Y.; Wei, X.C.; Li, P.C. Annexin A1 involved in the regulation of inflammation and cell signaling pathways. Chin. J. Traumatol. 2020, 23, 96–101. [Google Scholar] [CrossRef]
- Biaoxue, R.; Xiguang, C.; Shuanying, Y. Annexin A1 in malignant tumors: Current opinions and controversies. Int. J. Biol. Markers 2014, 29, e8–e20. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Serfass, L.; Roy, M.O.; Wong, J.; Bonneau, A.M.; Georges, E. Annexin-I expression modulates drug resistance in tumor cells. Biochem. Biophys. Res. Commun. 2004, 314, 565–570. [Google Scholar] [CrossRef]
- Liu, Y.; Hu, X.; Han, C.; Wang, L.; Zhang, X.; He, X.; Lu, X. Targeting tumor suppressor genes for cancer therapy. BioEssays 2015, 37, 1277–1286. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Li, Y.; Edelweiss, M.; Arun, B.; Rosen, D.; Resetkova, E.; Wu, Y.; Liu, J.; Sahin, A.; Albarracin, C.T. Loss of Annexin A1 expression in breast cancer progression. Appl. Immunohistochem. Mol. Morphol. 2008, 16, 530–534. [Google Scholar] [CrossRef] [PubMed]
- Hnisz, D.; Weintraub, A.S.; Day, D.S.; Valton, A.L.; Bak, R.O.; Li, C.H.; Goldmann, J.; Lajoie, B.R.; Fan, Z.P.; Sigova, A.A.; et al. Activation of proto-oncogenes by disruption of chromosome neighborhoods. Science 2016, 351, 1454–1458. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.W.; Reynolds, S.H.; You, M.; Maronpot, R.M. Role of proto-oncogene activation in carcinogenesis. Environ. Health Perspect. 1992, 98, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Foo, S.L.; Yap, G.; Cui, J.; Lim, L.H.K. Annexin-A1—A Blessing or a Curse in Cancer? Trends Mol. Med. 2019, 25, 315–327. [Google Scholar] [CrossRef]
- Sobral-Leite, M.; Wesseling, J.; Smit, V.T.; Nevanlinna, H.; van Miltenburg, M.H.; Sanders, J.; Hofland, I.; Blows, F.M.; Coulson, P.; Patrycja, G.; et al. Annexin A1 expression in a pooled breast cancer series: Association with tumor subtypes and prognosis. BMC Med. 2015, 13, 156. [Google Scholar] [CrossRef]
- Vecchi, L.; Alves Pereira Zoia, M.; Goss Santos, T.; de Oliveira Beserra, A.; Colaco Ramos, C.M.; Franca Matias Colombo, B.; Paiva Maia, Y.C.; Piana de Andrade, V.; Teixeira Soares Mota, S.; Goncalves de Araujo, T.; et al. Inhibition of the AnxA1/FPR1 autocrine axis reduces MDA-MB-231 breast cancer cell growth and aggressiveness in vitro and in vivo. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 1368–1382. [Google Scholar] [CrossRef]
- Boudhraa, Z.; Rondepierre, F.; Ouchchane, L.; Kintossou, R.; Trzeciakiewicz, A.; Franck, F.; Kanitakis, J.; Labeille, B.; Joubert-Zakeyh, J.; Bouchon, B.; et al. Annexin A1 in primary tumors promotes melanoma dissemination. Clin. Exp. Metastasis 2014, 31, 749–760. [Google Scholar] [CrossRef]
- Lin, Y.; Lin, G.; Fang, W.; Zhu, H.; Chu, K. Increased expression of Annexin A1 predicts poor prognosis in human hepatocellular carcinoma and enhances cell malignant phenotype. Med Oncol. 2014, 31, 327. [Google Scholar] [CrossRef] [PubMed]
- Su, N.; Xu, X.Y.; Chen, H.; Gao, W.C.; Ruan, C.P.; Wang, Q.; Sun, Y.P. Increased expression of Annexin A1 is correlated with K-ras mutation in colorectal cancer. Tohoku J. Exp. Med. 2010, 222, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.X.; Tu, Y.; Zhang, S. FoxM1 promotes glioma cells progression by up-regulating Anxa1 expression. PLoS ONE 2013, 8, e72376. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.F.; Zhang, P.F.; Li, M.Y.; Li, Q.Q.; Chen, Z.C. Identification of Annexin A1 as a proinvasive and prognostic factor for lung adenocarcinoma. Clin. Exp. Metastasis 2011, 28, 413–425. [Google Scholar] [CrossRef]
- Biaoxue, R.; Xiling, J.; Shuanying, Y.; Wei, Z.; Xiguang, C.; Jinsui, W.; Min, Z. Upregulation of Hsp90-beta and Annexin A1 correlates with poor survival and lymphatic metastasis in lung cancer patients. J. Exp. Clin. Cancer Res. 2012, 31, 70. [Google Scholar] [CrossRef]
- Mota, S.T.S.; Vecchi, L.; Alves, D.A.; Cordeiro, A.O.; Guimaraes, G.S.; Campos-Fernandez, E.; Maia, Y.C.P.; Dornelas, B.C.; Bezerra, S.M.; de Andrade, V.P.; et al. Annexin A1 promotes the nuclear localization of the epidermal growth factor receptor in castration-resistant prostate cancer. Int. J. Biochem. Cell Biol. 2020, 127, 105838. [Google Scholar] [CrossRef]
- Sato, Y.; Kumamoto, K.; Saito, K.; Okayama, H.; Hayase, S.; Kofunato, Y.; Miyamoto, K.; Nakamura, I.; Ohki, S.; Koyama, Y.; et al. Up-regulated Annexin A1 expression in gastrointestinal cancer is associated with cancer invasion and lymph node metastasis. Exp. Ther. Med. 2011, 2, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Rodrigo, J.P.; Garcia-Pedrero, J.M.; Fernandez, M.P.; Morgan, R.O.; Suárez, C.; Herrero, A. Annexin A1 expression in nasopharyngeal carcinoma correlates with squamous differentiation. Am. J. Rhinol. 2005, 19, 483–487. [Google Scholar] [CrossRef] [PubMed]
- Petrella, A.; Festa, M.; Ercolino, S.F.; Zerilli, M.; Stassi, G.; Solito, E.; Parente, L. Annexin-1 downregulation in thyroid cancer correlates to the degree of tumor differentiation. Cancer Biol. Ther. 2006, 5, 643–647. [Google Scholar] [CrossRef]
- Garcia Pedrero, J.M.; Fernandez, M.P.; Morgan, R.O.; Herrero Zapatero, A.; Gonzalez, M.V.; Suarez Nieto, C.; Rodrigo, J.P. Annexin A1 down-regulation in head and neck cancer is associated with epithelial differentiation status. Am. J. Pathol. 2004, 164, 73–79. [Google Scholar] [CrossRef]
- Shen, D.; Nooraie, F.; Elshimali, Y.; Lonsberry, V.; He, J.; Bose, S.; Chia, D.; Seligson, D.; Chang, H.R.; Goodglick, L. Decreased expression of Annexin A1 is correlated with breast cancer development and progression as determined by a tissue microarray analysis. Hum. Pathol. 2006, 37, 1583–1591. [Google Scholar] [CrossRef] [PubMed]
- Yom, C.K.; Han, W.; Kim, S.W.; Kim, H.S.; Shin, H.C.; Chang, J.N.; Koo, M.; Noh, D.Y.; Moon, B.I. Clinical significance of Annexin A1 expression in breast cancer. J. Breast Cancer 2011, 14, 262–268. [Google Scholar] [CrossRef]
- de Graauw, M.; van Miltenburg, M.H.; Schmidt, M.K.; Pont, C.; Lalai, R.; Kartopawiro, J.; Pardali, E.; Le Devedec, S.E.; Smit, V.T.; van der Wal, A.; et al. Annexin A1 regulates TGF-beta signaling and promotes metastasis formation of basal-like breast cancer cells. Proc. Natl. Acad. Sci. USA 2010, 107, 6340–6345. [Google Scholar] [CrossRef]
- Anbalagan, D.; Yap, G.; Yuan, Y.; Pandey, V.K.; Lau, W.H.; Arora, S.; Bist, P.; Wong, J.S.; Sethi, G.; Nissom, P.M.; et al. Annexin-A1 regulates microRNA-26b* and microRNA-562 to directly target NF-kappaB and angiogenesis in breast cancer cells. PLoS ONE 2014, 9, e114507. [Google Scholar] [CrossRef]
- Cheng, T.Y.; Wu, M.S.; Lin, J.T.; Lin, M.T.; Shun, C.T.; Huang, H.Y.; Hua, K.T.; Kuo, M.L. Annexin A1 is associated with gastric cancer survival and promotes gastric cancer cell invasiveness through the formyl peptide receptor/extracellular signal-regulated kinase/integrin beta-1-binding protein 1 pathway. Cancer 2012, 118, 5757–5767. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.F.; Huang, W.; Yi, H.M.; Xiao, T.; Li, J.Y.; Feng, J.; Yi, H.; Lu, S.S.; Li, X.H.; Lu, R.H.; et al. Annexin A1-suppressed autophagy promotes nasopharyngeal carcinoma cell invasion and metastasis by PI3K/AKT signaling activation. Cell Death Dis. 2018, 9, 1154. [Google Scholar] [CrossRef]
- Chen, P.; Min, J.; Wu, H.; Zhang, H.; Wang, C.; Tan, G.; Zhang, F. Annexin A1 is a potential biomarker of bone metastasis in small cell lung cancer. Oncol. Lett. 2021, 21, 141. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.H.; Ping, Y.F.; Chen, J.H.; Chen, D.L.; Xu, C.P.; Zheng, J.; Wang, J.M.; Bian, X.W. Production of angiogenic factors by human glioblastoma cells following activation of the G-protein coupled formylpeptide receptor FPR. J. Neurooncol. 2008, 86, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Chen, K.; Chen, J.; Gong, W.; Dunlop, N.M.; Howard, O.M.; Gao, Y.; Bian, X.W.; Wang, J.M. The G-protein-coupled formylpeptide receptor FPR confers a more invasive phenotype on human glioblastoma cells. Br. J. Cancer 2010, 102, 1052–1060. [Google Scholar] [CrossRef]
- Zhou, Y.; Bian, X.; Le, Y.; Gong, W.; Hu, J.; Zhang, X.; Wang, L.; Iribarren, P.; Salcedo, R.; Howard, O.M.; et al. Formylpeptide receptor FPR and the rapid growth of malignant human gliomas. J. Natl. Cancer Inst. 2005, 97, 823–835. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.L.; Ping, Y.F.; Yu, S.C.; Chen, J.H.; Yao, X.H.; Jiang, X.F.; Zhang, H.R.; Wang, Q.L.; Bian, X.W. Downregulating FPR restrains xenograft tumors by impairing the angiogenic potential and invasive capability of malignant glioma cells. Biochem. Biophys. Res. Commun. 2009, 381, 448–452. [Google Scholar] [CrossRef] [PubMed]
- Snapkov, I.; Oqvist, C.O.; Figenschau, Y.; Kogner, P.; Johnsen, J.I.; Sveinbjornsson, B. The role of formyl peptide receptor 1 (FPR1) in neuroblastoma tumorigenesis. BMC Cancer 2016, 16, 490. [Google Scholar] [CrossRef]
- Alldridge, L.C.; Harris, H.J.; Plevin, R.; Hannon, R.; Bryant, C.E. The annexin protein lipocortin 1 regulates the MAPK/ERK pathway. J. Biol. Chem. 1999, 274, 37620–37628. [Google Scholar] [CrossRef]
- Jo, E.J.; Lee, H.Y.; Kim, J.I.; Kang, H.K.; Lee, Y.N.; Kwak, J.Y.; Bae, Y.S. Activation of formyl peptide receptor-like 1 by WKYMVm induces serine phosphorylation of STAT3, which inhibits its tyrosine phosphorylation and nuclear translocation induced by hydrogen peroxide. Life Sci. 2004, 75, 2217–2232. [Google Scholar] [CrossRef]
- Shi, Y.; Lai, X.; Ye, L.; Chen, K.; Cao, Z.; Gong, W.; Jin, L.; Wang, C.; Liu, M.; Liao, Y.; et al. Activated niacin receptor HCA2 inhibits chemoattractant-mediated macrophage migration via Gβγ/PKC/ERK1/2 pathway and heterologous receptor desensitization. Sci. Rep. 2017, 7, 42279. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Qin, W.; Song, M.; Zhang, Y.; Sun, B. Exogenous carbon monoxide inhibits neutrophil infiltration in LPS-induced sepsis by interfering with FPR1 via p38 MAPK but not GRK2. Oncotarget 2016, 7, 34250–34265. [Google Scholar] [CrossRef] [PubMed]
- Cao, G.; Zhang, Z. FPR1 mediates the tumorigenicity of human cervical cancer cells. Cancer Manag. Res. 2018, 10, 5855–5865. [Google Scholar] [CrossRef] [PubMed]
- Khau, T.; Langenbach, S.Y.; Schuliga, M.; Harris, T.; Johnstone, C.N.; Anderson, R.L.; Stewart, A.G. Annexin-1 signals mitogen-stimulated breast tumor cell proliferation by activation of the formyl peptide receptors (FPRs) 1 and 2. FASEB J. 2011, 25, 483–496. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Bao, Z.; Gong, W.; Tang, P.; Yoshimura, T.; Wang, J.M. Regulation of inflammation by members of the formyl-peptide receptor family. J. Autoimmun. 2017, 85, 64–77. [Google Scholar] [CrossRef]
- Cheng, T.Y.; Wu, M.S.; Lin, J.T.; Lin, M.T.; Shun, C.T.; Hua, K.T.; Kuo, M.L. Formyl Peptide receptor 1 expression is associated with tumor progression and survival in gastric cancer. Anticancer. Res. 2014, 34, 2223–2229. [Google Scholar]
- Prevete, N.; Liotti, F.; Visciano, C.; Marone, G.; Melillo, R.M.; de Paulis, A. The formyl peptide receptor 1 exerts a tumor suppressor function in human gastric cancer by inhibiting angiogenesis. Oncogene 2015, 34, 3826–3838. [Google Scholar] [CrossRef]
- Lin, C.Y.; Jeng, Y.M.; Chou, H.Y.; Hsu, H.C.; Yuan, R.H.; Chiang, C.P.; Kuo, M.Y. Nuclear localization of Annexin A1 is a prognostic factor in oral squamous cell carcinoma. J. Surg. Oncol. 2008, 97, 544–550. [Google Scholar] [CrossRef]
- Zhu, F.; Xu, C.; Jiang, Z.; Jin, M.; Wang, L.; Zeng, S.; Teng, L.; Cao, J. Nuclear localization of Annexin A1 correlates with advanced disease and peritoneal dissemination in patients with gastric carcinoma. Anat. Rec. 2010, 293, 1310–1314. [Google Scholar] [CrossRef]
- Kunkel, T.A. The high cost of living. American Association for Cancer Research Special Conference: Endogenous sources of mutations, Fort Myers, Florida, USA, 11–15 November 1998. Trends Genet. 1999, 15, 93–94. [Google Scholar] [CrossRef]
- Kunz, B.A.; Straffon, A.F.; Vonarx, E.J. DNA damage-induced mutation: Tolerance via translesion synthesis. Mutat. Res. 2000, 451, 169–185. [Google Scholar] [CrossRef]
- Makridakis, N.M.; Reichardt, J.K. Translesion DNA Polymerases and Cancer. Front. Genet. 2012, 3, 174. [Google Scholar] [CrossRef] [PubMed]
- Dieckman, L.M.; Freudenthal, B.D.; Washington, M.T. PCNA structure and function: Insights from structures of PCNA complexes and post-translationally modified PCNA. Sub Cell. Biochem. 2012, 62, 281–299. [Google Scholar] [CrossRef]
- Choi, S.; Srivas, R.; Fu, K.Y.; Hood, B.L.; Dost, B.; Gibson, G.A.; Watkins, S.C.; Van Houten, B.; Bandeira, N.; Conrads, T.P.; et al. Quantitative proteomics reveal ATM kinase-dependent exchange in DNA damage response complexes. J. Proteome Res. 2012, 11, 4983–4991. [Google Scholar] [CrossRef] [PubMed]
- Hirata, F.; Thibodeau, L.M.; Hirata, A. Ubiquitination and SUMOylation of Annexin A1 and helicase activity. Biochim. Biophys. Acta 2010, 1800, 899–905. [Google Scholar] [CrossRef]
- Ganesan, T.; Sinniah, A.; Ibrahim, Z.A.; Chik, Z.; Alshawsh, M.A. Annexin A1: A Bane or a Boon in Cancer? A Systematic Review. Molecules 2020, 25, 3700. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Wang, Y.; Zeng, S.; Fu, X.; Wang, L.; Cao, J. Involvement of Annexin A1 in multidrug resistance of K562/ADR cells identified by the proteomic study. Omics 2009, 13, 467–476. [Google Scholar] [CrossRef]
- D’Acunto, C.W.; Gbelcova, H.; Festa, M.; Ruml, T. The complex understanding of Annexin A1 phosphorylation. Cell. Signal. 2014, 26, 173–178. [Google Scholar] [CrossRef]
- Pepinsky, R.B.; Sinclair, L.K. Epidermal growth factor-dependent phosphorylation of lipocortin. Nature 1986, 321, 81–84. [Google Scholar] [CrossRef]
- Skouteris, G.G.; Schröder, C.H. The hepatocyte growth factor receptor kinase-mediated phosphorylation of lipocortin-1 transduces the proliferating signal of the hepatocyte growth factor. J. Biol. Chem. 1996, 271, 27266–27273. [Google Scholar] [CrossRef]
- Shao, G.; Zhou, H.; Zhang, Q.; Jin, Y.; Fu, C. Advancements of Annexin A1 in inflammation and tumorigenesis. OncoTargets Ther. 2019, 12, 3245–3254. [Google Scholar] [CrossRef] [PubMed]
- Moraes, L.A.; Kar, S.; Foo, S.L.; Gu, T.; Toh, Y.Q.; Ampomah, P.B.; Sachaphibulkij, K.; Yap, G.; Zharkova, O.; Lukman, H.M.; et al. Annexin-A1 enhances breast cancer growth and migration by promoting alternative macrophage polarization in the tumour microenvironment. Sci. Rep. 2017, 7, 17925. [Google Scholar] [CrossRef]
- Li, Y.; Cai, L.; Wang, H.; Wu, P.; Gu, W.; Chen, Y.; Hao, H.; Tang, K.; Yi, P.; Liu, M.; et al. Pleiotropic regulation of macrophage polarization and tumorigenesis by formyl peptide receptor-2. Oncogene 2011, 30, 3887–3899. [Google Scholar] [CrossRef]
- Locatelli, I.; Sutti, S.; Jindal, A.; Vacchiano, M.; Bozzola, C.; Reutelingsperger, C.; Kusters, D.; Bena, S.; Parola, M.; Paternostro, C.; et al. Endogenous Annexin A1 is a novel protective determinant in nonalcoholic steatohepatitis in mice. Hepatology 2014, 60, 531–544. [Google Scholar] [CrossRef]
- Ferlazzo, V.; D’Agostino, P.; Milano, S.; Caruso, R.; Feo, S.; Cillari, E.; Parente, L. Anti-inflammatory effects of Annexin-1: Stimulation of IL-10 release and inhibition of nitric oxide synthesis. Int. Immunopharmacol. 2003, 3, 1363–1369. [Google Scholar] [CrossRef]
- Weyd, H.; Abeler-Dorner, L.; Linke, B.; Mahr, A.; Jahndel, V.; Pfrang, S.; Schnolzer, M.; Falk, C.S.; Krammer, P.H. Annexin A1 on the surface of early apoptotic cells suppresses CD8+ T cell immunity. PLoS ONE 2013, 8, e62449. [Google Scholar] [CrossRef] [PubMed]
- Gold, R.; Schmied, M.; Tontsch, U.; Hartung, H.P.; Wekerle, H.; Toyka, K.V.; Lassmann, H. Antigen presentation by astrocytes primes rat T lymphocytes for apoptotic cell death. A model for T-cell apoptosis in vivo. Brain 1996, 119 Pt 2, 651–659. [Google Scholar] [CrossRef][Green Version]
- D’Acquisto, F.; Merghani, A.; Lecona, E.; Rosignoli, G.; Raza, K.; Buckley, C.D.; Flower, R.J.; Perretti, M. Annexin-1 modulates T-cell activation and differentiation. Blood 2007, 109, 1095–1102. [Google Scholar] [CrossRef]
- Ng, F.S.; Wong, K.Y.; Guan, S.P.; Mustafa, F.B.; Kajiji, T.S.; Bist, P.; Biswas, S.K.; Wong, W.S.; Lim, L.H. Annexin-1-deficient mice exhibit spontaneous airway hyperresponsiveness and exacerbated allergen-specific antibody responses in a mouse model of asthma. Clin. Exp. Allergy 2011, 41, 1793–1803. [Google Scholar] [CrossRef] [PubMed]
- Kamal, A.M.; Flower, R.J.; Perretti, M. An overview of the effects of Annexin 1 on cells involved in the inflammatory process. Memórias Inst. Oswaldo Cruz 2005, 100 (Suppl. 1), 39–47. [Google Scholar] [CrossRef]
- Jeppesen, D.K.; Fenix, A.M.; Franklin, J.L.; Higginbotham, J.N.; Zhang, Q.; Zimmerman, L.J.; Liebler, D.C.; Ping, J.; Liu, Q.; Evans, R.; et al. Reassessment of Exosome Composition. Cell 2019, 177, 428–445.e418. [Google Scholar] [CrossRef]
- Lo Cicero, A.; Stahl, P.D.; Raposo, G. Extracellular vesicles shuffling intercellular messages: For good or for bad. Curr. Opin. Cell Biol. 2015, 35, 69–77. [Google Scholar] [CrossRef]
- van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell. Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef]
- Aalberts, M.; van Dissel-Emiliani, F.M.; van Adrichem, N.P.; van Wijnen, M.; Wauben, M.H.; Stout, T.A.; Stoorvogel, W. Identification of distinct populations of prostasomes that differentially express prostate stem cell antigen, Annexin A1, and GLIPR2 in humans. Biol. Reprod. 2012, 86, 82. [Google Scholar] [CrossRef]
- Perretti, M.; Cooper, D.; Dalli, J.; Norling, L.V. Immune resolution mechanisms in inflammatory arthritis. Nat. Rev. Rheumatol. 2017, 13, 87–99. [Google Scholar] [CrossRef]
- Headland, S.E.; Norling, L.V. The resolution of inflammation: Principles and challenges. Semin. Immunol. 2015, 27, 149–160. [Google Scholar] [CrossRef]
- Gasser, O.; Schifferli, J.A. Activated polymorphonuclear neutrophils disseminate anti-inflammatory microparticles by ectocytosis. Blood 2004, 104, 2543–2548. [Google Scholar] [CrossRef] [PubMed]
- Rhys, H.I.; Dell’Accio, F.; Pitzalis, C.; Moore, A.; Norling, L.V.; Perretti, M. Neutrophil Microvesicles from Healthy Control and Rheumatoid Arthritis Patients Prevent the Inflammatory Activation of Macrophages. EBioMedicine 2018, 29, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liu, Y.; Yao, X.; Ping, Y.; Jiang, T.; Liu, Q.; Xu, S.; Huang, J.; Mou, H.; Gong, W.; et al. Annexin 1 released by necrotic human glioblastoma cells stimulates tumor cell growth through the formyl peptide receptor 1. Am. J. Pathol. 2011, 179, 1504–1512. [Google Scholar] [CrossRef]
- Link, C.; Bujupi, F.; Krammer, P.H.; Weyd, H. Annexin-coated particles induce antigen-specific immunosuppression. Autoimmunity 2020, 53, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Ampomah, P.B.; Moraes, L.A.; Lukman, H.M.; Lim, L.H.K. Formyl peptide receptor 2 is regulated by RNA mimics and viruses through an IFN-β-STAT3-dependent pathway. FASEB J. 2018, 32, 1468–1478. [Google Scholar] [CrossRef]
- Oshi, M.; Tokumaru, Y.; Mukhopadhyay, S.; Yan, L.; Matsuyama, R.; Endo, I.; Takabe, K. Annexin A1 Expression Is Associated with Epithelial-Mesenchymal Transition (EMT), Cell Proliferation, Prognosis, and Drug Response in Pancreatic Cancer. Cells 2021, 10, 653. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.H.; Li, Y.; Zhang, J.; Rong, J.; Ye, S. Epidermal growth factor receptor-containing exosomes induce tumor-specific regulatory T cells. Cancer Investig. 2013, 31, 330–335. [Google Scholar] [CrossRef]
- Azuma, K.; Ota, K.; Kawahara, A.; Hattori, S.; Iwama, E.; Harada, T.; Matsumoto, K.; Takayama, K.; Takamori, S.; Kage, M.; et al. Association of PD-L1 overexpression with activating EGFR mutations in surgically resected nonsmall-cell lung cancer. Ann. Oncol. 2014, 25, 1935–1940. [Google Scholar] [CrossRef]
- Akbay, E.A.; Koyama, S.; Carretero, J.; Altabef, A.; Tchaicha, J.H.; Christensen, C.L.; Mikse, O.R.; Cherniack, A.D.; Beauchamp, E.M.; Pugh, T.J.; et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov. 2013, 3, 1355–1363. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Fang, W.; Zhan, J.; Hong, S.; Tang, Y.; Kang, S.; Zhang, Y.; He, X.; Zhou, T.; Qin, T.; et al. Upregulation of PD-L1 by EGFR Activation Mediates the Immune Escape in EGFR-Driven NSCLC: Implication for Optional Immune Targeted Therapy for NSCLC Patients with EGFR Mutation. J. Thorac. Oncol. 2015, 10, 910–923. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Zhang, C.; Zhao, G.; Zhang, X.; Hao, M.; Hassan, S.; Zhang, M.; Zheng, H.; Yang, D.; Liu, L.; et al. IGFBP2 regulates PD-L1 expression by activating the EGFR-STAT3 signaling pathway in malignant melanoma. Cancer Lett. 2020, 477, 19–30. [Google Scholar] [CrossRef]
- Lo, H.W.; Hsu, S.C.; Ali-Seyed, M.; Gunduz, M.; Xia, W.; Wei, Y.; Bartholomeusz, G.; Shih, J.Y.; Hung, M.C. Nuclear interaction of EGFR and STAT3 in the activation of the iNOS/NO pathway. Cancer Cell 2005, 7, 575–589. [Google Scholar] [CrossRef]
- Bronte, V.; Kasic, T.; Gri, G.; Gallana, K.; Borsellino, G.; Marigo, I.; Battistini, L.; Iafrate, M.; Prayer-Galetti, T.; Pagano, F.; et al. Boosting antitumor responses of T lymphocytes infiltrating human prostate cancers. J. Exp. Med. 2005, 201, 1257–1268. [Google Scholar] [CrossRef] [PubMed]
- Brand, T.M.; Iida, M.; Luthar, N.; Starr, M.M.; Huppert, E.J.; Wheeler, D.L. Nuclear EGFR as a molecular target in cancer. Radiother. Oncol. 2013, 108, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Eden, E.R.; Sanchez-Heras, E.; Tsapara, A.; Sobota, A.; Levine, T.P.; Futter, C.E. Annexin A1 Tethers Membrane Contact Sites that Mediate ER to Endosome Cholesterol Transport. Dev. Cell 2016, 37, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Ma, W.; Li, X.; Li, H.; Li, J.; Li, H.; He, F. ANXA1 enhances tumor proliferation and migration by regulating epithelial-mesenchymal transition and IL-6/JAK2/STAT3 pathway in papillary thyroid carcinoma. J. Cancer 2021, 12, 1295–1306. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Araújo, T.G.; Mota, S.T.S.; Ferreira, H.S.V.; Ribeiro, M.A.; Goulart, L.R.; Vecchi, L. Annexin A1 as a Regulator of Immune Response in Cancer. Cells 2021, 10, 2245. https://doi.org/10.3390/cells10092245
Araújo TG, Mota STS, Ferreira HSV, Ribeiro MA, Goulart LR, Vecchi L. Annexin A1 as a Regulator of Immune Response in Cancer. Cells. 2021; 10(9):2245. https://doi.org/10.3390/cells10092245
Chicago/Turabian StyleAraújo, Thaise Gonçalves, Sara Teixeira Soares Mota, Helen Soares Valença Ferreira, Matheus Alves Ribeiro, Luiz Ricardo Goulart, and Lara Vecchi. 2021. "Annexin A1 as a Regulator of Immune Response in Cancer" Cells 10, no. 9: 2245. https://doi.org/10.3390/cells10092245
APA StyleAraújo, T. G., Mota, S. T. S., Ferreira, H. S. V., Ribeiro, M. A., Goulart, L. R., & Vecchi, L. (2021). Annexin A1 as a Regulator of Immune Response in Cancer. Cells, 10(9), 2245. https://doi.org/10.3390/cells10092245