
Plectin-Mediated Intermediate Filament Functions: Why Isoforms Matter


{kind=link}
{kind=link}
{kind=link}
Abstract
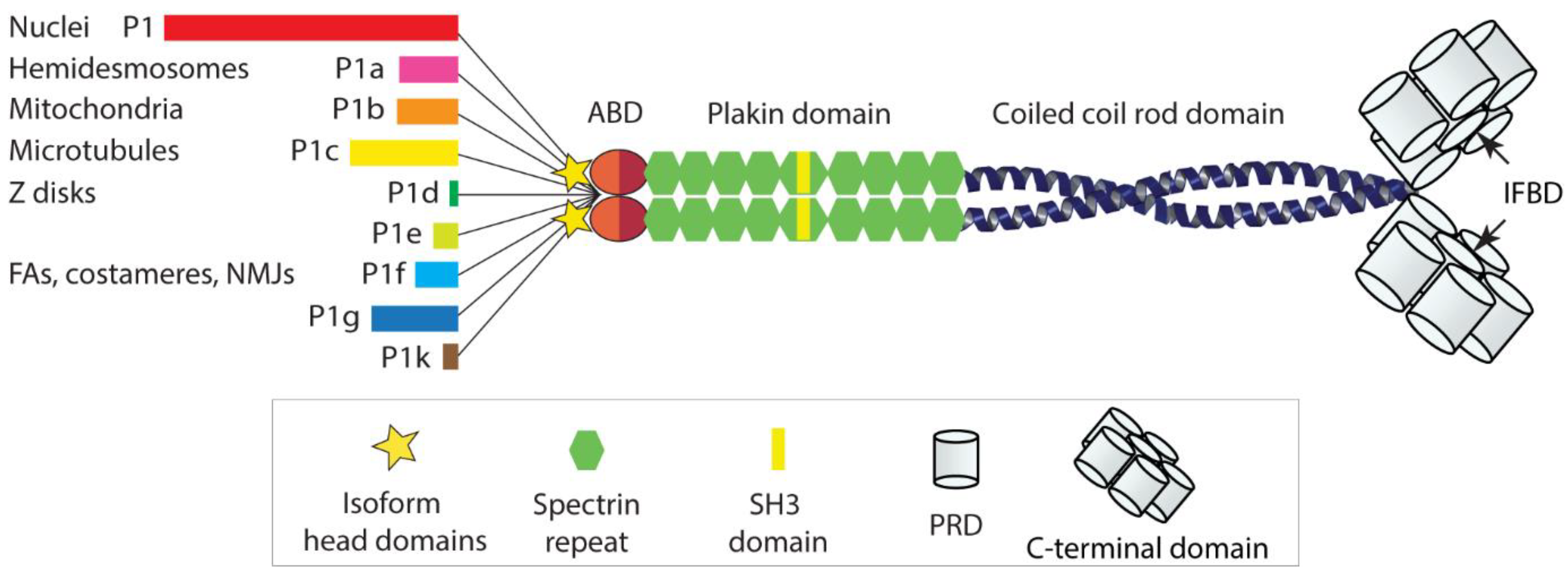
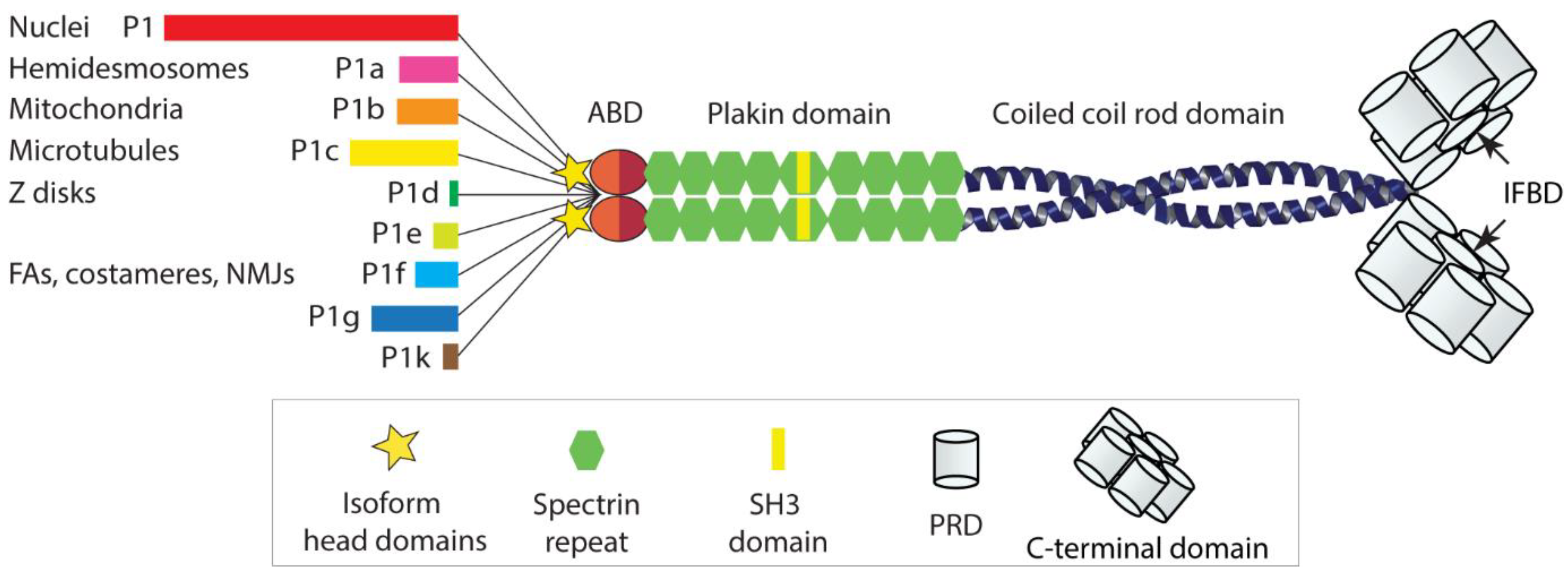
:1. Introduction
2. IFs and Plectin as Modulators of Mechanical Cell Properties
3. Plectin Mechanically Stabilizes IF Networks and Their Docking Platforms
4. Modulation and Compartmentalization of the Actomyosin Machinery through Plectin-Mediated IF Network Recruitment
4.1. Fibroblasts and Keratinocytes
4.2. Endothelial Cells
4.3. Neuromuscular Synapse
4.4. Mechanotransduction to the Nucleus
4.5. Invadopodia
4.6. Mitosis
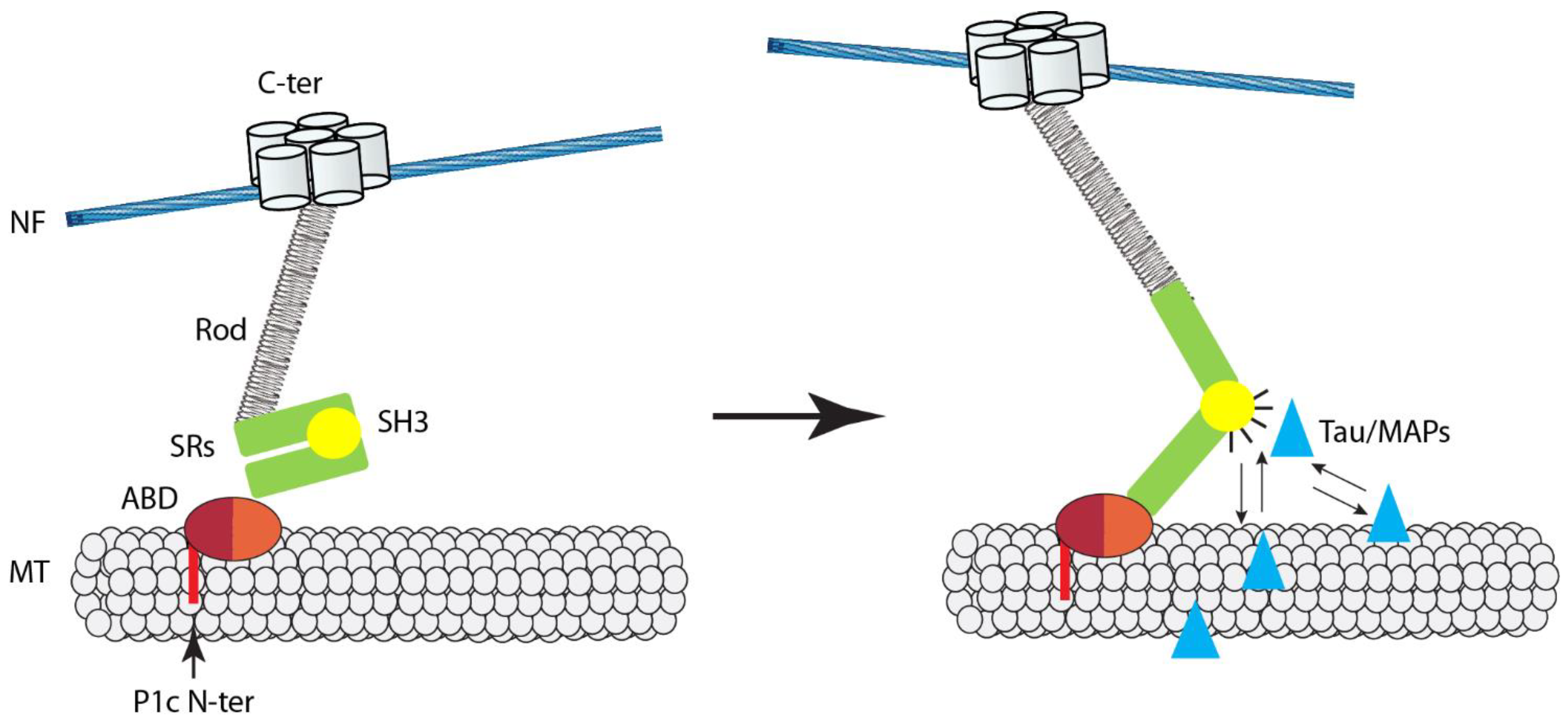
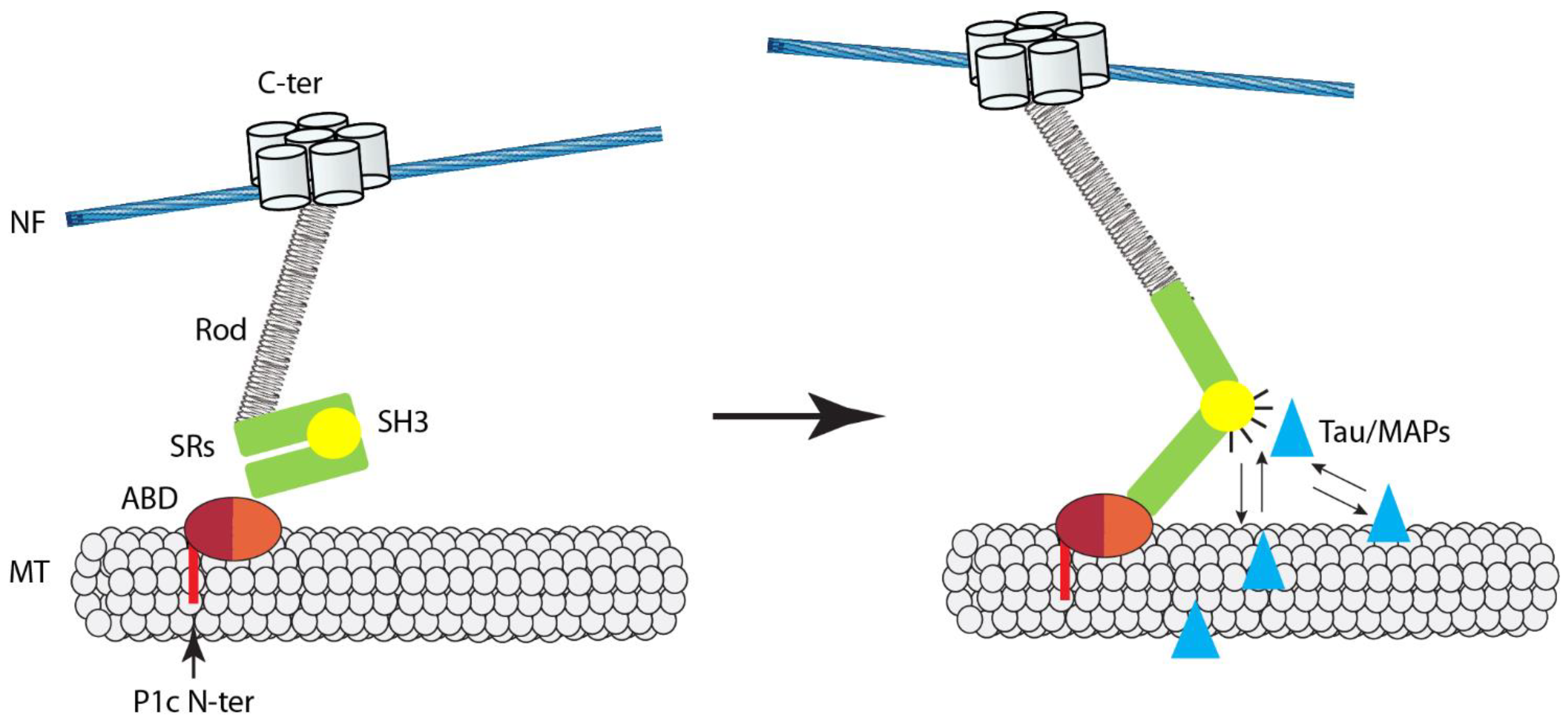
5. The Microtubule Connection
6. Plectin and IF Networks as Mutually Dependent Partners
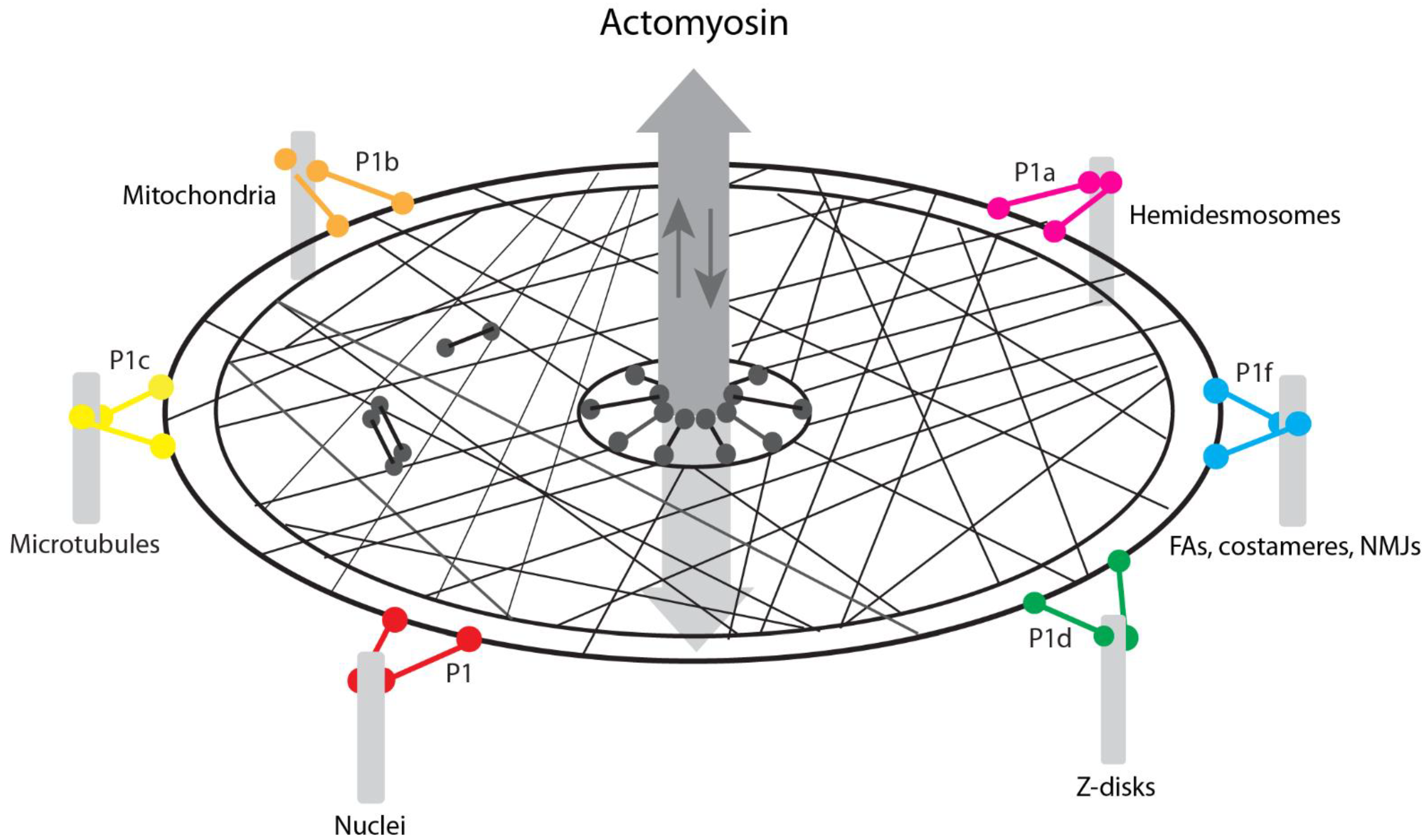
7. Conclusions, Working Model, and Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bouameur, J.E.; Favre, B.; Borradori, L. Plakins, a versatile family of cytolinkers: Roles in skin integrity and in human diseases. J. Investig. Dermatol. 2014, 134, 885–894. [Google Scholar] [CrossRef] [Green Version]
- Wiche, G.; Herrmann, H.; Leichtfried, F.; Pytela, R. Plectin: A high-molecular-weight cytoskeletal polypeptide component that copurifies with intermediate filaments of the vimentin type. Cold Spring Harb. Symp. Quant. Biol. 1982, 46 Pt 1, 475–482. [Google Scholar] [CrossRef]
- Wiche, G. Role of plectin in cytoskeleton organization and dynamics. J. Cell Sci. 1998, 111 Pt 17, 2477–2486. [Google Scholar] [CrossRef]
- Wiche, G.; Winter, L. Plectin isoforms as organizers of intermediate filament cytoarchitecture. Bioarchitecture 2011, 1, 14–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castañón, M.J.; Walko, G.; Winter, L.; Wiche, G. Plectin-intermediate filament partnership in skin, skeletal muscle, and peripheral nerve. Histochem. Cell Biol. 2013, 140, 33–53. [Google Scholar] [CrossRef] [Green Version]
- Wiche, G.; Osmanagic-Myers, S.; Castañón, M.J. Networking and anchoring through plectin: A key to IF functionality and mechanotransduction. Curr. Opin. Cell Biol. 2015, 32, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Ortega, E.; Buey, R.M.; Sonnenberg, A.; de Pereda, J.M. The structure of the plakin domain of plectin reveals a non-canonical SH3 domain interacting with its fourth spectrin repeat. J. Biol. Chem. 2011, 286, 12429–12438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikolic, B.; Mac Nulty, E.; Mir, B.; Wiche, G. Basic amino acid residue cluster within nuclear targeting sequence motif is essential for cytoplasmic plectin-vimentin network junctions. J. Cell Biol. 1996, 134, 1455–1467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuchs, P.; Zörer, M.; Rezniczek, G.A.; Spazierer, D.; Oehler, S.; Castañón, M.J.; Hauptmann, R.; Wiche, G. Unusual 5’ transcript complexity of plectin isoforms: Novel tissue-specific exons modulate actin binding activity. Hum. Mol. Genet. 1999, 8, 2461–2472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rezniczek, G.A.; Konieczny, P.; Nikolic, B.; Reipert, S.; Schneller, D.; Abrahamsberg, C.; Davies, K.E.; Winder, S.J.; Wiche, G. Plectin 1f scaffolding at the sarcolemma of dystrophic (mdx) muscle fibers through multiple interactions with beta-dystroglycan. J. Cell Biol. 2007, 176, 965–977. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, P.; Zörer, M.; Reipert, S.; Rezniczek, G.A.; Propst, F.; Walko, G.; Fischer, I.; Bauer, J.; Leschnik, M.W.; Lüscher, B.; et al. Targeted inactivation of a developmentally regulated neural plectin isoform (plectin 1c) in mice leads to reduced motor nerve conduction velocity. J. Biol. Chem. 2009, 284, 26502–26509. [Google Scholar] [CrossRef] [Green Version]
- Rezniczek, G.A.; Abrahamsberg, C.; Fuchs, P.; Spazierer, D.; Wiche, G. Plectin 5’-transcript diversity: Short alternative sequences determine stability of gene products, initiation of translation and subcellular localization of isoforms. Hum. Mol. Genet. 2003, 12, 3181–3194. [Google Scholar] [CrossRef] [PubMed]
- Konieczny, P.; Fuchs, P.; Reipert, S.; Kunz, W.S.; Zeold, A.; Fischer, I.; Paulin, D.; Schroder, R.; Wiche, G. Myofiber integrity depends on desmin network targeting to Z-disks and costameres via distinct plectin isoforms. J. Cell Biol. 2008, 181, 667–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walko, G.; Castañón, M.J.; Wiche, G. Molecular architecture and function of the hemidesmosome. Cell Tissue Res. 2015, 360, 363–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiche, G.; Castañón, M.J. Intermediate Filament Linker Proteins: Plectin and BPAG1. In Encyclopedia of Biological Chemistry, 3rd ed.; Jez, J., Ed.; Elsevier: New York, NY, USA, 2021; pp. 200–219. [Google Scholar]
- Winter, L.; Wiche, G. The many faces of plectin and plectinopathies: Pathology and mechanisms. Acta Neuropathol. 2013, 125, 77–93. [Google Scholar] [CrossRef] [PubMed]
- Block, J.; Witt, H.; Candelli, A.; Peterman, E.J.; Wuite, G.J.; Janshoff, A.; Köster, S. Nonlinear Loading-Rate-Dependent Force Response of Individual Vimentin Intermediate Filaments to Applied Strain. Phys. Rev. Lett. 2017, 118, 048101. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, C.; Forsting, J.; Schepers, A.V.; Kraxner, J.; Bauch, S.; Witt, H.; Klumpp, S.; Köster, S. Lateral Subunit Coupling Determines Intermediate Filament Mechanics. Phys. Rev. Lett. 2019, 123, 188102. [Google Scholar] [CrossRef]
- Block, J.; Witt, H.; Candelli, A.; Danes, J.C.; Peterman, E.J.G.; Wuite, G.J.L.; Janshoff, A.; Köster, S. Viscoelastic properties of vimentin originate from nonequilibrium conformational changes. Sci. Adv. 2018, 4, eaat1161. [Google Scholar] [CrossRef] [Green Version]
- Golde, T.; Huster, C.; Glaser, M.; Handler, T.; Herrmann, H.; Kas, J.A.; Schnauss, J. Glassy dynamics in composite biopolymer networks. Soft Matter. 2018, 14, 7970–7978. [Google Scholar] [CrossRef] [Green Version]
- Ramms, L.; Fabris, G.; Windoffer, R.; Schwarz, N.; Springer, R.; Zhou, C.; Lazar, J.; Stiefel, S.; Hersch, N.; Schnakenberg, U.; et al. Keratins as the main component for the mechanical integrity of keratinocytes. Proc. Natl. Acad. Sci. USA 2013, 110, 18513–18518. [Google Scholar] [CrossRef] [Green Version]
- Latorre, E.; Kale, S.; Casares, L.; Gomez-Gonzalez, M.; Uroz, M.; Valon, L.; Nair, R.V.; Garreta, E.; Montserrat, N.; Del Campo, A.; et al. Active superelasticity in three-dimensional epithelia of controlled shape. Nature 2018, 563, 203–208. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Li, Y.; Hao, Y.; Zheng, T.; Gupta, S.K.; Parada, G.A.; Wu, H.; Lin, S.; Wang, S.; Zhao, X.; et al. High stretchability, strength, and toughness of living cells enabled by hyperelastic vimentin intermediate filaments. Proc. Natl. Acad. Sci. USA 2019, 116, 17175–17180. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Yue, J.; Wu, X. Spectraplakin family proteins-cytoskeletal crosslinkers with versatile roles. J. Cell Sci. 2017, 130, 2447–2457. [Google Scholar] [CrossRef] [Green Version]
- Johnson, C.P.; Tang, H.Y.; Carag, C.; Speicher, D.W.; Discher, D.E. Forced unfolding of proteins within cells. Science 2007, 317, 663–666. [Google Scholar] [CrossRef] [Green Version]
- Choi, H.J.; Weis, W.I. Crystal structure of a rigid four-spectrin-repeat fragment of the human desmoplakin plakin domain. J. Mol. Biol. 2011, 409, 800–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daday, C.; Kolsek, K.; Gräter, F. The mechano-sensing role of the unique SH3 insertion in plakin domains revealed by Molecular Dynamics simulations. Sci. Rep. 2017, 7, 11669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suman, S.K.; Daday, C.; Ferraro, T.; Vuong-Brender, T.; Tak, S.; Quintin, S.; Robin, F.; Gräter, F.; Labouesse, M. The plakin domain of C. elegans VAB-10/plectin acts as a hub in a mechanotransduction pathway to promote morphogenesis. Development 2019, 146, dev183780. [Google Scholar] [CrossRef] [Green Version]
- Lunter, P.C.; Wiche, G. Direct binding of plectin to Fer kinase and negative regulation of its catalytic activity. Biochem. Biophys. Res. Commun. 2002, 296, 904–910. [Google Scholar] [CrossRef]
- Osmanagic-Myers, S.; Wiche, G. Plectin-RACK1 (receptor for activated C kinase 1) scaffolding: A novel mechanism to regulate protein kinase C activity. J. Biol. Chem. 2004, 279, 18701–18710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osmanagic-Myers, S.; Gregor, M.; Walko, G.; Burgstaller, G.; Reipert, S.; Wiche, G. Plectin-controlled keratin cytoarchitecture affects MAP kinases involved in cellular stress response and migration. J. Cell Biol. 2006, 174, 557–568. [Google Scholar] [CrossRef] [Green Version]
- Gregor, M.; Zeöld, A.; Oehler, S.; Andrä Marobela, K.; Fuchs, P.; Weigel, G.; Hardie, D.G.; Wiche, G. Plectin scaffolds recruit energy-controlling AMP-activated protein kinase (AMPK) in differentiated myofibres. J. Cell Sci. 2006, 119, 1864–1875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janda, L.; Damborsky, J.; Rezniczek, G.A.; Wiche, G. Plectin repeats and modules: Strategic cysteines and their presumed impact on cytolinker functions. Bioessays 2001, 23, 1064–1069. [Google Scholar] [CrossRef]
- Spurny, R.; Abdoulrahman, K.; Janda, L.; Runzler, D.; Kohler, G.; Castanon, M.J.; Wiche, G. Oxidation and nitrosylation of cysteines proximal to the intermediate filament (IF)-binding site of plectin: Effects on structure and vimentin binding and involvement in IF collapse. J. Biol. Chem. 2007, 282, 8175–8187. [Google Scholar] [CrossRef] [Green Version]
- Foisner, R.; Wiche, G. Structure and hydrodynamic properties of plectin molecules. J. Mol. Biol. 1987, 198, 515–531. [Google Scholar] [CrossRef]
- Mayans, O.; Wuerges, J.; Canela, S.; Gautel, M.; Wilmanns, M. Structural evidence for a possible role of reversible disulphide bridge formation in the elasticity of the muscle protein titin. Structure 2001, 9, 331–340. [Google Scholar] [CrossRef] [Green Version]
- Alegre-Cebollada, J.; Kosuri, P.; Giganti, D.; Eckels, E.; Rivas-Pardo, J.A.; Hamdani, N.; Warren, C.M.; Solaro, R.J.; Linke, W.A.; Fernandez, J.M. S-glutathionylation of cryptic cysteines enhances titin elasticity by blocking protein folding. Cell 2014, 156, 1235–1246. [Google Scholar] [CrossRef] [Green Version]
- Block, J.; Schroeder, V.; Pawelzyk, P.; Willenbacher, N.; Köster, S. Physical properties of cytoplasmic intermediate filaments. Biochim. Biophys. Acta 2015, 1853, 3053–3064. [Google Scholar] [CrossRef] [Green Version]
- Kreplak, L.; Bar, H.; Leterrier, J.F.; Herrmann, H.; Aebi, U. Exploring the mechanical behavior of single intermediate filaments. J. Mol. Biol. 2005, 354, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Walko, G.; Vukasinovic, N.; Gross, K.; Fischer, I.; Sibitz, S.; Fuchs, P.; Reipert, S.; Jungwirth, U.; Berger, W.; Salzer, U.; et al. Targeted proteolysis of plectin isoform 1a accounts for hemidesmosome dysfunction in mice mimicking the dominant skin blistering disease EBS-Ogna. PLoS Genet. 2011, 7, e1002396. [Google Scholar] [CrossRef] [Green Version]
- Rezniczek, G.A.; Winter, L.; Walko, G.; Wiche, G. Functional and Genetic Analysis of Plectin in Skin and Muscle. Methods Enzymol. 2016, 569, 235–259. [Google Scholar] [CrossRef]
- Gregor, M.; Osmanagic-Myers, S.; Burgstaller, G.; Wolfram, M.; Fischer, I.; Walko, G.; Resch, G.P.; Jörgl, A.; Herrmann, H.; Wiche, G. Mechanosensing through focal adhesion-anchored intermediate filaments. FASEB J. 2014, 28, 715–729. [Google Scholar] [CrossRef]
- Jirouskova, M.; Nepomucka, K.; Oyman-Eyrilmez, G.; Kalendova, A.; Havelkova, H.; Sarnova, L.; Chalupsky, K.; Schuster, B.; Benada, O.; Miksatkova, P.; et al. Plectin controls biliary tree architecture and stability in cholestasis. J. Hepatol. 2018, 68, 1006–1017. [Google Scholar] [CrossRef]
- Mihailovska, E.; Raith, M.; Valencia, R.G.; Fischer, I.; Al Banchaabouchi, M.; Herbst, R.; Wiche, G. Neuromuscular synapse integrity requires linkage of acetylcholine receptors to postsynaptic intermediate filament networks via rapsyn-plectin 1f complexes. Mol. Biol. Cell 2014, 25, 4130–4149. [Google Scholar] [CrossRef] [PubMed]
- Winter, L.; Staszewska, I.; Mihailovska, E.; Fischer, I.; Goldmann, W.H.; Schroder, R.; Wiche, G. Chemical chaperone ameliorates pathological protein aggregation in plectin-deficient muscle. J. Clin. Investig. 2014, 124, 1144–1157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staszewska, I.; Fischer, I.; Wiche, G. Plectin isoform 1-dependent nuclear docking of desmin networks affects myonuclear architecture and expression of mechanotransducers. Hum. Mol. Genet. 2015, 24, 7373–7389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gache, Y.; Chavanas, S.; Lacour, J.P.; Wiche, G.; Owaribe, K.; Meneguzzi, G.; Ortonne, J.P. Defective expression of plectin/HD1 in epidermolysis bullosa simplex with muscular dystrophy. J. Clin. Investig. 1996, 97, 2289–2298. [Google Scholar] [CrossRef] [Green Version]
- Uematsu, J.; Nishizawa, Y.; Sonnenberg, A.; Owaribe, K. Demonstration of type II hemidesmosomes in a mammary gland epithelial cell line, BMGE-H. J. Biochem. 1994, 115, 469–476. [Google Scholar] [CrossRef]
- Wang, W.; Zuidema, A.; Te Molder, L.; Nahidiazar, L.; Hoekman, L.; Schmidt, T.; Coppola, S.; Sonnenberg, A. Hemidesmosomes modulate force generation via focal adhesions. J. Cell Biol. 2020, 219. [Google Scholar] [CrossRef] [PubMed]
- Andrä, K.; Kornacker, I.; Jörgl, A.; Zörer, M.; Spazierer, D.; Fuchs, P.; Fischer, I.; Wiche, G. Plectin-isoform-specific rescue of hemidesmosomal defects in plectin (-/-) keratinocytes. J. Investig. Dermatol. 2003, 120, 189–197. [Google Scholar] [CrossRef] [Green Version]
- Palmisano, M.G.; Bremner, S.N.; Hornberger, T.A.; Meyer, G.A.; Domenighetti, A.A.; Shah, S.B.; Kiss, B.; Kellermayer, M.; Ryan, A.F.; Lieber, R.L. Skeletal muscle intermediate filaments form a stress-transmitting and stress-signaling network. J. Cell Sci. 2015, 128, 219–224. [Google Scholar] [CrossRef] [Green Version]
- Winter, L.; Abrahamsberg, C.; Wiche, G. Plectin isoform 1b mediates mitochondrion-intermediate filament network linkage and controls organelle shape. J. Cell Biol. 2008, 181, 903–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winter, L.; Kuznetsov, A.V.; Grimm, M.; Zeold, A.; Fischer, I.; Wiche, G. Plectin isoform P1b and P1d deficiencies differentially affect mitochondrial morphology and function in skeletal muscle. Hum. Mol. Genet. 2015, 24, 4530–4544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walko, G.; Wögenstein, K.L.; Winter, L.; Fischer, I.; Feltri, M.L.; Wiche, G. Stabilization of the dystroglycan complex in Cajal bands of myelinating Schwann cells through plectin-mediated anchorage to vimentin filaments. Glia 2013, 61, 1274–1287. [Google Scholar] [CrossRef]
- Burgstaller, G.; Gregor, M.; Winter, L.; Wiche, G. Keeping the vimentin network under control: Cell-matrix adhesion-associated plectin 1f affects cell shape and polarity of fibroblasts. Mol. Biol. Cell 2010, 21, 3362–3375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Pascalis, C.; Perez-Gonzalez, C.; Seetharaman, S.; Boeda, B.; Vianay, B.; Burute, M.; Leduc, C.; Borghi, N.; Trepat, X.; Etienne-Manneville, S. Intermediate filaments control collective migration by restricting traction forces and sustaining cell-cell contacts. J. Cell Biol. 2018, 217, 3031–3044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiche, G.; Krepler, R.; Artlieb, U.; Pytela, R.; Denk, H. Occurrence and immunolocalization of plectin in tissues. J. Cell Biol. 1983, 97, 887–901. [Google Scholar] [CrossRef] [Green Version]
- Seifert, G.J.; Lawson, D.; Wiche, G. Immunolocalization of the intermediate filament-associated protein plectin at focal contacts and actin stress fibers. Eur. J. Cell Biol. 1992, 59, 138–147. [Google Scholar] [PubMed]
- Andrä, K.; Lassmann, H.; Bittner, R.; Shorny, S.; Fässler, R.; Propst, F.; Wiche, G. Targeted inactivation of plectin reveals essential function in maintaining the integrity of skin, muscle, and heart cytoarchitecture. Genes Dev. 1997, 11, 3143–3156. [Google Scholar] [CrossRef] [Green Version]
- Andrä, K.; Nikolic, B.; Stöcher, M.; Drenckhahn, D.; Wiche, G. Not just scaffolding: Plectin regulates actin dynamics in cultured cells. Genes Dev. 1998, 12, 3442–3451. [Google Scholar] [CrossRef] [Green Version]
- Osmanagic-Myers, S.; Rus, S.; Wolfram, M.; Brunner, D.; Goldmann, W.H.; Bonakdar, N.; Fischer, I.; Reipert, S.; Zuzuarregui, A.; Walko, G.; et al. Plectin reinforces vascular integrity by mediating crosstalk between the vimentin and the actin networks. J. Cell Sci. 2015, 128, 4138–4150. [Google Scholar] [CrossRef] [Green Version]
- Kostan, J.; Gregor, M.; Walko, G.; Wiche, G. Plectin isoform-dependent regulation of keratin-integrin alpha6beta4 anchorage via Ca2+/calmodulin. J. Biol. Chem. 2009, 284, 18525–18536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valencia, R.G.; Walko, G.; Janda, L.; Novacek, J.; Mihailovska, E.; Reipert, S.; Andra-Marobela, K.; Wiche, G. Intermediate filament-associated cytolinker plectin 1c destabilizes microtubules in keratinocytes. Mol. Biol. Cell 2013, 24, 768–784. [Google Scholar] [CrossRef] [PubMed]
- Spurny, R.; Gregor, M.; Castanon, M.J.; Wiche, G. Plectin deficiency affects precursor formation and dynamics of vimentin networks. Exp. Cell Res. 2008, 314, 3570–3580. [Google Scholar] [CrossRef] [PubMed]
- Seltmann, K.; Roth, W.; Kroger, C.; Loschke, F.; Lederer, M.; Huttelmaier, S.; Magin, T.M. Keratins mediate localization of hemidesmosomes and repress cell motility. J. Investig. Dermatol. 2013, 133, 181–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.G.; Kostan, J.; Drepper, F.; Knapp, B.; de Almeida Ribeiro, E., Jr.; Konarev, P.V.; Grishkovskaya, I.; Wiche, G.; Gregor, M.; Svergun, D.I.; et al. Structural insights into Ca2+-calmodulin regulation of Plectin 1a-integrin beta4 interaction in hemidesmosomes. Structure 2015, 23, 558–570. [Google Scholar] [CrossRef] [Green Version]
- Rezniczek, G.A.; de Pereda, J.M.; Reipert, S.; Wiche, G. Linking integrin alpha6beta4-based cell adhesion to the intermediate filament cytoskeleton: Direct interaction between the beta4 subunit and plectin at multiple molecular sites. J. Cell Biol. 1998, 141, 209–225. [Google Scholar] [CrossRef]
- Svitkina, T.M.; Verkhovsky, A.B.; Borisy, G.G. Plectin sidearms mediate interaction of intermediate filaments with microtubules and other components of the cytoskeleton. J. Cell Biol. 1996, 135, 991–1007. [Google Scholar] [CrossRef] [Green Version]
- Homan, S.M.; Martinez, R.; Benware, A.; LaFlamme, S.E. Regulation of the association of alpha 6 beta 4 with vimentin intermediate filaments in endothelial cells. Exp. Cell Res. 2002, 281, 107–114. [Google Scholar] [CrossRef]
- Quinlan, R.A.; Schwarz, N.; Windoffer, R.; Richardson, C.; Hawkins, T.; Broussard, J.A.; Green, K.J.; Leube, R.E. A rim-and-spoke hypothesis to explain the biomechanical roles for cytoplasmic intermediate filament networks. J. Cell Sci. 2017, 130, 3437–3445. [Google Scholar] [CrossRef] [Green Version]
- Dai, Z.; Luo, X.; Xie, H.; Peng, H.B. The actin-driven movement and formation of acetylcholine receptor clusters. J. Cell Biol. 2000, 150, 1321–1334. [Google Scholar] [CrossRef]
- Antolik, C.; Catino, D.H.; O’Neill, A.M.; Resneck, W.G.; Ursitti, J.A.; Bloch, R.J. The actin binding domain of ACF7 binds directly to the tetratricopeptide repeat domains of rapsyn. Neuroscience 2007, 145, 56–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almeida, F.V.; Walko, G.; McMillan, J.R.; McGrath, J.A.; Wiche, G.; Barber, A.H.; Connelly, J.T. The cytolinker plectin regulates nuclear mechanotransduction in keratinocytes. J. Cell Sci. 2015, 128, 4475–4486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ketema, M.; Kreft, M.; Secades, P.; Janssen, H.; Sonnenberg, A. Nesprin-3 connects plectin and vimentin to the nuclear envelope of Sertoli cells but is not required for Sertoli cell function in spermatogenesis. Mol. Biol. Cell 2013, 24, 2454–2466. [Google Scholar] [CrossRef]
- Laly, A.C.; Sliogeryte, K.; Pundel, O.J.; Ross, R.; Keeling, M.C.; Avisetti, D.; Waseem, A.; Gavara, N.; Connelly, J.T. The keratin network of intermediate filaments regulates keratinocyte rigidity sensing and nuclear mechanotransduction. Sci. Adv. 2021, 7, eabd6187. [Google Scholar] [CrossRef] [PubMed]
- Patteson, A.E.; Pogoda, K.; Byfield, F.J.; Mandal, K.; Ostrowska-Podhorodecka, Z.; Charrier, E.E.; Galie, P.A.; Deptula, P.; Bucki, R.; McCulloch, C.A.; et al. Loss of Vimentin Enhances Cell Motility through Small Confining Spaces. Small 2019, 15, e1903180. [Google Scholar] [CrossRef]
- Patteson, A.E.; Vahabikashi, A.; Pogoda, K.; Adam, S.A.; Mandal, K.; Kittisopikul, M.; Sivagurunathan, S.; Goldman, A.; Goldman, R.D.; Janmey, P.A. Vimentin protects cells against nuclear rupture and DNA damage during migration. J. Cell Biol. 2019, 218, 4079–4092. [Google Scholar] [CrossRef]
- Vannier, C.; Pesty, A.; San-Roman, M.J.; Schmidt, A.A. The Bin/amphiphysin/Rvs (BAR) domain protein endophilin B2 interacts with plectin and controls perinuclear cytoskeletal architecture. J. Biol. Chem. 2013, 288, 27619–27637. [Google Scholar] [CrossRef] [Green Version]
- Abrahamsberg, C.; Fuchs, P.; Osmanagic-Myers, S.; Fischer, I.; Propst, F.; Elbe-Burger, A.; Wiche, G. Targeted ablation of plectin isoform 1 uncovers role of cytolinker proteins in leukocyte recruitment. Proc. Natl. Acad. Sci. USA 2005, 102, 18449–18454. [Google Scholar] [CrossRef] [Green Version]
- Feng, X.; Coulombe, P.A. A role for disulfide bonding in keratin intermediate filament organization and dynamics in skin keratinocytes. J. Cell Biol. 2015, 209, 59–72. [Google Scholar] [CrossRef] [Green Version]
- Sutoh Yoneyama, M.; Hatakeyama, S.; Habuchi, T.; Inoue, T.; Nakamura, T.; Funyu, T.; Wiche, G.; Ohyama, C.; Tsuboi, S. Vimentin intermediate filament and plectin provide a scaffold for invadopodia, facilitating cancer cell invasion and extravasation for metastasis. Eur. J. Cell Biol. 2014, 93, 157–169. [Google Scholar] [CrossRef]
- van den Dries, K.; Nahidiazar, L.; Slotman, J.A.; Meddens, M.B.M.; Pandzic, E.; Joosten, B.; Ansems, M.; Schouwstra, J.; Meijer, A.; Steen, R.; et al. Modular actin nano-architecture enables podosome protrusion and mechanosensing. Nat. Commun. 2019, 10, 5171. [Google Scholar] [CrossRef] [Green Version]
- van den Dries, K.; Linder, S.; Maridonneau-Parini, I.; Poincloux, R. Probing the mechanical landscape-new insights into podosome architecture and mechanics. J. Cell Sci. 2019, 132, jcs236828. [Google Scholar] [CrossRef]
- Schoumacher, M.; Goldman, R.D.; Louvard, D.; Vignjevic, D.M. Actin, microtubules, and vimentin intermediate filaments cooperate for elongation of invadopodia. J. Cell Biol. 2010, 189, 541–556. [Google Scholar] [CrossRef] [Green Version]
- McInroy, L.; Määttä, A. Plectin regulates invasiveness of SW480 colon carcinoma cells and is targeted to podosome-like adhesions in an isoform-specific manner. Exp. Cell Res. 2011, 317, 2468–2478. [Google Scholar] [CrossRef]
- Shin, S.J.; Smith, J.A.; Rezniczek, G.A.; Pan, S.; Chen, R.; Brentnall, T.A.; Wiche, G.; Kelly, K.A. Unexpected gain of function for the scaffolding protein plectin due to mislocalization in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 19414–19419. [Google Scholar] [CrossRef] [Green Version]
- Hoshino, D.; Kirkbride, K.C.; Costello, K.; Clark, E.S.; Sinha, S.; Grega-Larson, N.; Tyska, M.J.; Weaver, A.M. Exosome secretion is enhanced by invadopodia and drives invasive behavior. Cell Rep. 2013, 5, 1159–1168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serres, M.P.; Samwer, M.; Truong Quang, B.A.; Lavoie, G.; Perera, U.; Gorlich, D.; Charras, G.; Petronczki, M.; Roux, P.P.; Paluch, E.K. F-Actin Interactome Reveals Vimentin as a Key Regulator of Actin Organization and Cell Mechanics in Mitosis. Dev. Cell 2020, 52, 210–222.e7. [Google Scholar] [CrossRef] [Green Version]
- Koszka, C.; Leichtfried, F.E.; Wiche, G. Identification and spatial arrangement of high molecular weight proteins (Mr 300 000-330 000) co-assembling with microtubules from a cultured cell line (rat glioma C6). Eur. J. Cell Biol. 1985, 38, 149–156. [Google Scholar] [PubMed]
- Herrmann, H.; Wiche, G. Plectin and IFAP-300K are homologous proteins binding to microtubule-associated proteins 1 and 2 and to the 240-kilodalton subunit of spectrin. J. Biol. Chem. 1987, 262, 1320–1325. [Google Scholar] [CrossRef]
- Valencia, R.G.; Mihailovska, E.; Winter, L.; Bauer, K.; Fischer, I.; Walko, G.; Jorgacevski, J.; Potokar, M.; Zorec, R.; Wiche, G. Plectin dysfunction in neurons leads to tau accumulation on microtubules affecting neuritogenesis, organelle trafficking, pain sensitivity and memory. Neuropathol. Appl. Neurobiol. 2021, 47, 73–95. [Google Scholar] [CrossRef] [PubMed]
- Baas, P.W.; Rao, A.N.; Matamoros, A.J.; Leo, L. Stability properties of neuronal microtubules. Cytoskeleton 2016, 73, 442–460. [Google Scholar] [CrossRef] [Green Version]
- Qiang, L.; Sun, X.; Austin, T.O.; Muralidharan, H.; Jean, D.C.; Liu, M.; Yu, W.; Baas, P.W. Tau Does Not Stabilize Axonal Microtubules but Rather Enables Them to Have Long Labile Domains. Curr. Biol. 2018, 28, 2181–2189.e4. [Google Scholar] [CrossRef] [Green Version]
- Dehmelt, L.; Halpain, S. The MAP2/Tau family of microtubule-associated proteins. Genome Biol. 2005, 6, 204. [Google Scholar] [CrossRef] [Green Version]
- Raith, M.; Valencia, R.G.; Fischer, I.; Orthofer, M.; Penninger, J.M.; Spuler, S.; Rezniczek, G.A.; Wiche, G. Linking cytoarchitecture to metabolism: Sarcolemma-associated plectin affects glucose uptake by destabilizing microtubule networks in mdx myofibers. Skelet. Muscle 2013, 3, 14. [Google Scholar] [CrossRef] [Green Version]
- Gu, Y.; Oyama, F.; Ihara, Y. Tau is widely expressed in rat tissues. J. Neurochem. 1996, 67, 1235–1244. [Google Scholar] [CrossRef]
- Eckes, B.; Dogic, D.; Colucci-Guyon, E.; Wang, N.; Maniotis, A.; Ingber, D.; Merckling, A.; Langa, F.; Aumailley, M.; Delouvee, A.; et al. Impaired mechanical stability, migration and contractile capacity in vimentin-deficient fibroblasts. J. Cell Sci. 1998, 111 Pt 13, 1897–1907. [Google Scholar] [CrossRef]
- Eckes, B.; Colucci-Guyon, E.; Smola, H.; Nodder, S.; Babinet, C.; Krieg, T.; Martin, P. Impaired wound healing in embryonic and adult mice lacking vimentin. J. Cell Sci. 2000, 113 Pt 13, 2455–2462. [Google Scholar] [CrossRef]
- Seltmann, K.; Cheng, F.; Wiche, G.; Eriksson, J.E.; Magin, T.M. Keratins Stabilize Hemidesmosomes through Regulation of beta4-Integrin Turnover. J. Investig. Dermatol. 2015, 135, 1609–1620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colburn, Z.T.; Jones, J.C.R. Complexes of alpha6beta4 integrin and vimentin act as signaling hubs to regulate epithelial cell migration. J. Cell Sci. 2018, 131, jcs214593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, M.V.; Garcia, M.L.; Miyazaki, Y.; Gotow, T.; Yuan, A.; Mattina, S.; Ward, C.M.; Calcutt, N.A.; Uchiyama, Y.; Nixon, R.A.; et al. Gene replacement in mice reveals that the heavily phosphorylated tail of neurofilament heavy subunit does not affect axonal caliber or the transit of cargoes in slow axonal transport. J. Cell Biol. 2002, 158, 681–693. [Google Scholar] [CrossRef] [PubMed]
- Rice, S.J.; Tselepi, M.; Sorial, A.K.; Aubourg, G.; Shepherd, C.; Almarza, D.; Skelton, A.J.; Pangou, I.; Deehan, D.; Reynard, L.N.; et al. Prioritization of PLEC and GRINA as Osteoarthritis Risk Genes Through the Identification and Characterization of Novel Methylation Quantitative Trait Loci. Arthritis Rheumatol. 2019, 71, 1285–1296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorial, A.K.; Hofer, I.M.J.; Tselepi, M.; Cheung, K.; Parker, E.; Deehan, D.J.; Rice, S.J.; Loughlin, J. Multi-tissue epigenetic analysis of the osteoarthritis susceptibility locus mapping to the plectin gene PLEC. Osteoarthr. Cartil. 2020, 28, 1448–1458. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wiche, G. Plectin-Mediated Intermediate Filament Functions: Why Isoforms Matter. Cells 2021, 10, 2154. https://doi.org/10.3390/cells10082154
Wiche G. Plectin-Mediated Intermediate Filament Functions: Why Isoforms Matter. Cells. 2021; 10(8):2154. https://doi.org/10.3390/cells10082154
Chicago/Turabian StyleWiche, Gerhard. 2021. "Plectin-Mediated Intermediate Filament Functions: Why Isoforms Matter" Cells 10, no. 8: 2154. https://doi.org/10.3390/cells10082154
APA StyleWiche, G. (2021). Plectin-Mediated Intermediate Filament Functions: Why Isoforms Matter. Cells, 10(8), 2154. https://doi.org/10.3390/cells10082154