
Simple Method for Establishing Primary Leporidae Skin Fibroblast Cultures

 ,
,  ,
,  ,
,  and
and
Abstract
1. Introduction
2. Material and Methods
2.1. Tissue Sample Collection
2.2. Reagents
2.3. Preparation of Fibroblasts from Skin Biopsy
- (1)
- Wash the original tissue fragments to remove impurities by placing the sample in a 50-milliliter Falcon tube containing 30 mL DMEM/HAMS F12 medium with L-Glutamine and HEPES (3.5 g/L) (CORNING), 5× Antibiotic–Antimicotic solution (200 units/mL of penicillin, 200 µg/mL of streptomycin, 0.5 µg/mL of Gibco amphotericin B and 50 µg/mL of gentamicin); immediately proceed with vigorous washing, using a shaking robot or just manual shaking. Change medium seven times after 1-min-long agitation.
- (2)
- Place the original tissue fragments in 50-milliliter Falcon tubes (or alternatively in T25 bottles, or Petri dishes) and fill with DMEM medium prepared as described below.
- (3)
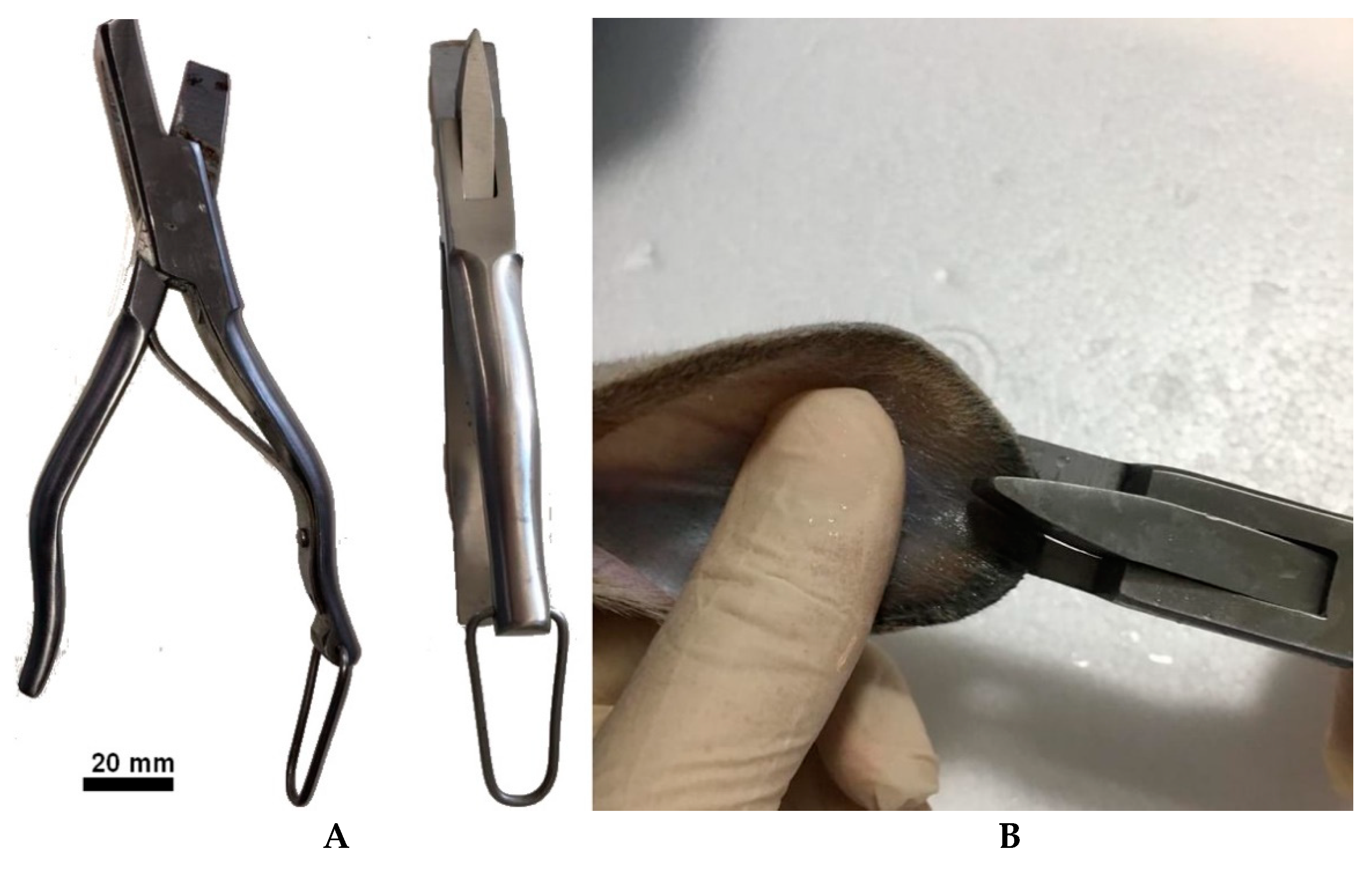
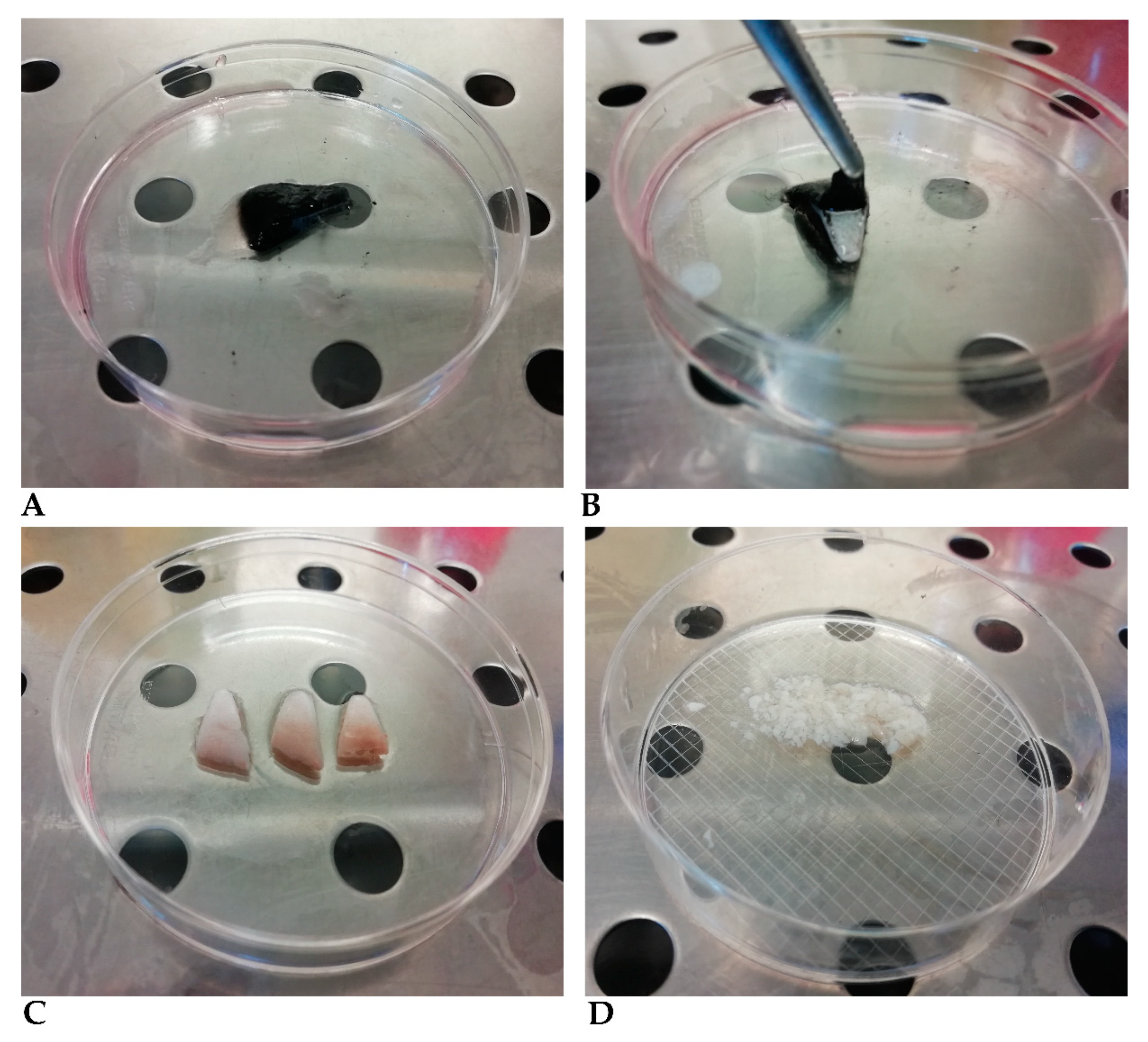
- Remove the medium and add a volume of 0.2 mg/mL dispase II prepared in DMEM medium enough to cover the tissue fragments. The epidermis–dermis separation process takes a variable time depending on the dimension, shape, and thickness of the fragments, generally 1–2 h at 37 °C. We recommend shortening this procedure as much as possible, since hair and epidermis are contaminated by bacteria, fungi, and yeasts, even after washing, and must be removed from the preparation as soon as possible. The operator must, therefore, test the detachment every hour, although cells may be left overnight in this medium with no adverse effects. Remove the epidermis completely using two tweezers (Figure 2B,C) and wash the epidermis fragment with DMEM medium as often as necessary until no hairs are visible (Figure 2C).
- (4)
- Cut the original fragments into smaller fragments less than 5 mm wide, using a scalpel (Figure 2D).
- (5)
- Incubate the fragments with the trypsin dissociation solution (the smallest volume that covers the fragments), previously warmed to 37 °C, in 50-milliliter tubes, T25 culture flasks or Petri dishes, and incubate at 37 °C for 10 min. The dissociation solution becomes cloudy as cells detach. Caution must be taken at this step since trypsin activity continues beyond 30 min, destroying the cells.
- (6)
- Remove the enzymatic digestion solution containing the cells and centrifuge at 150× g for 10 min. Resuspend the cells in a conical tube containing DMEM/HAMS F12 medium with L-Glutamine AND HEPES (3.5 g/L), 5× antibiotic-antimycotic solution (100 units/mL of penicillin, 100 µg/mL of streptomycin, 0.5 µg/mL of Gibco amphotericin B, and 25 µg/mL of gentamicin) and 10% of fetal bovine serum (Sigma-Aldrich, St. Louis, MO, USA).
- (7)
- Add fresh dissociation solution to the tissue fragments and repeat steps (5) and (6) as many times as necessary.
- (8)
- Seed the cells in culture flasks. Incubate at 37 °C with 5% CO2.
- (9)
- Remove medium after 2–4 h incubation, wash the cells with DMEM, and add fresh medium.
- (10)
- Remove medium again after 2–4 h of incubation. Wash the cell layer with DMEM and add fresh medium.
2.4. Subculturing and Harvesting Primary Fibroblast Cells
- (1)
- Remove the medium and wash cells twice with sterile PBS.
- (2)
- Incubate cells with trypsin-EDTA (0.25%), enough to cover the cell layer, and incubate at 37 °C until cell detachment.
- (3)
- Centrifuge the cell suspension at 150× g for 10 min and recover the pellet.
- (4)
- Cells are counted in a Neubauer hematocytometer and plated at 1–2 × 104 cells/cm2
2.5. Viability Assay Using Trypan Blue Dye
2.6. Freezing and Thawing Fibroblast Cells
- (1)
- Trypsinise cells with 70–80% confluence, as described above in Section 2.4 (steps (1)–(3)).
- (2)
- Centrifuge at 150× g for 10 min and resuspend the cell pellet with 10% DMEM, 10% DMSO, and 80% FBS (v/v).
- (3)
- Aliquot the cell suspension in cryogenic storage tubes at a density of 1–3 × 106 cells/vial. We have obtained an average of two vials for every three T25 flasks with 90% confluence.
- (4)
- Freeze cells at −80 °C overnight and then transfer them into liquid nitrogen.
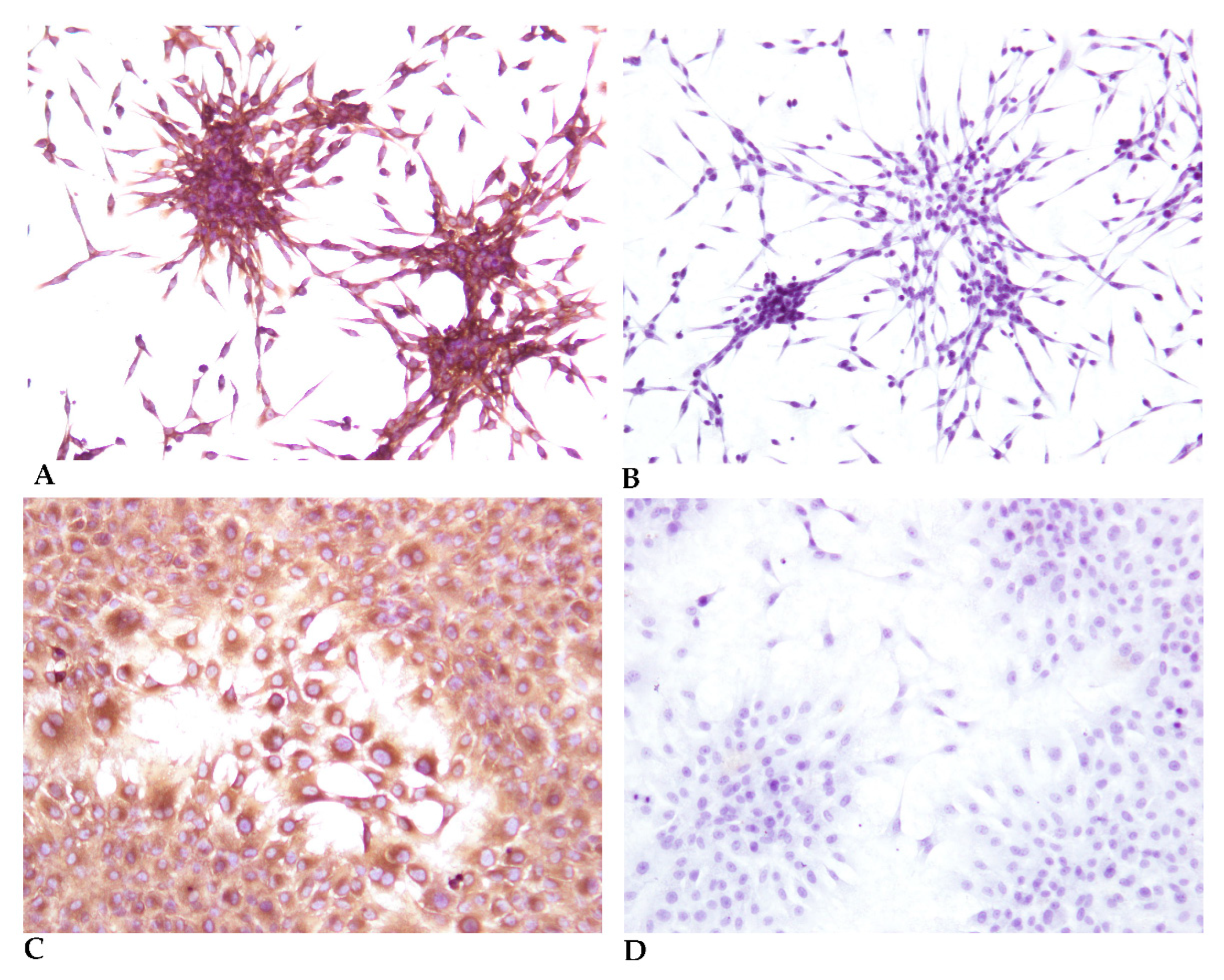
2.7. Fibroblast Marker by Immunocytochemistry
- (1)
- Grow cells in chamber slide system (e.g., (Thermo Scientific™ Nunc™ Lab-Tek™ Chamber Slide System)) until 95% cell confluence is reached.
- (2)
- Remove the medium and wash 2 times with PBS.
- (3)
- Fix cells by adding 100 μL of 100% acetone (we used Acetone, for GC residue analysis (Scharlab, Barcelona, Spain)) to each well and incubate for 10 min. Remove the acetone.
- (4)
- Remove the slides from the chamber slide system. Rehydrate the cells in Dako EnVision FLEX Wash Buffer (Agilent, Santa Clara, CA, USA) with 1% of Triton X-100 for 5 min.
- (5)
- Wash the cells with Dako EnVision FLEX Wash Buffer.
- (6)
- Delimitate the cell wells with a Dako Pen.
- (7)
- Incubate the cells with EnVision FLEX Peroxidase-Blocking Reagent for 10 min.
- (8)
- Wash the cells with Dako EnVision FLEX Wash Buffer.
- (9)
- Incubate the cells at RT with Monoclonal Mouse Anti-Human Cytokeratin Clones AE1/AE3 (Dako, M3515) and Monoclonal Mouse Anti-Vimentin Clone V9 (Dako, M0725), both diluted 1:100 in EnVision FLEX Antibody Diluent.
- (10)
- Wash the slides with Dako EnVision FLEX Wash Buffer, 2 times for 5 min, with manual shaking. Change the buffer between washes.
- (11)
- Incubate with Envision FLEX/HRP for 30 min (Dako, SM802) at RT.
- (12)
- Wash the slides with Dako EnVision FLEX Wash Buffer, 2 times for 5 min, with manual shaking. Change the buffer between washes.
- (13)
- Incubate with Envision FLEX DAB and Chromogen dilution in the respective buffer, for 3–5 min at RT.
- (14)
- Wash in distilled water.
- (15)
- Nuclei counterstaining with hematoxylin solution for 5 min at RT.
- (16)
- Dehydrate the cells by incubation of ethanol in the following sequence: 70, 80%, 95, and 100%, 1 min each, followed by 2 baths in xylene, 1 min each.
- (17)
- Mount the slides with Slide Mounting Media.
3. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, Y.; Chen, S.; Yan, Z.; Pei, M. A prospect of cell immortalization combined with matrix microenvironmental optimization strategy for tissue engineering and regeneration. Cell Biosci. 2019, 9, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Hausmann, C.; Zoschke, C.; Wolff, C.; Darvin, M.E.; Sochorová, M.; Kováčik, A.; Wanjiku, B.; Schumacher, F.; Tigges, J.; Kleuser, B.; et al. Fibroblast origin shapes tissue homeostasis, epidermal differentiation, and drug uptake. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Rogers, K.M.; Black, D.H.; Eberle, R. Primary mouse dermal fibroblast cell cultures as an in vitro model system for the differential pathogenicity of cross-species herpesvirus papio 2 infections. Arch. Virol. 2006, 152, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Pham, P.H.; Leacy, A.; Deng, L.; Nagy, É.; Susta, L. Isolation of Ontario aquatic bird bornavirus 1 and characterization of its replication in immortalized avian cell lines. Virol. J. 2020, 17, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Coelho, J.C.; Giugliani, R. Fibroblasts of skin fragments as a tool for the investigation of genetic diseases: Technical recommendations. Genet. Mol. Biol. 2000, 23, 269–271. [Google Scholar] [CrossRef][Green Version]
- Vangipuram, M.; Ting, D.; Kim, S.; Diaz, R.; Schuele, B. Skin Punch Biopsy Explant Culture for Derivation of Primary Human Fibroblasts. J. Vis. Exp. 2013, e3779. [Google Scholar] [CrossRef] [PubMed]
- Takashima, A. Establishment of Fibroblast Cultures. Curr. Protoc. Cell Biol. 1998, 2.1.1–2.1.12. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Gasser, S. Generating Primary Fibroblast Cultures from Mouse Ear and Tail Tissues. J. Vis. Exp. 2016, e53565. [Google Scholar] [CrossRef] [PubMed]
- Ishak, M.F.; Manira, M.; Ng, M.H.; Khairul, B.; Gargy, L.; Aminuddin, B.S.; Ruszymah, B.H. Long Term Effect of Cryopreservation on Primary Human Skin Cells. Sains Malays. 2019, 48, 137–144. [Google Scholar] [CrossRef]
- Siengdee, P.; Klinhom, S.; Thitaram, C.; Nganvongpanit, K. Isolation and culture of primary adult skin fibroblasts from the Asian elephant (Elephas maximus). PeerJ 2018, 6, e4302. [Google Scholar] [CrossRef] [PubMed]
- Villegas, J.; McPhaul, M. Establishment and Culture of Human Skin Fibroblasts. Curr. Protoc. Mol. Biol. 2005, 71, 28.3.1–28.3.9. [Google Scholar] [CrossRef] [PubMed]
- Moresco, K.A.; Stallknecht, D.E.; Swayne, D. Evaluation and Attempted Optimization of Avian Embryos and Cell Culture Methods for Efficient Isolation and Propagation of Low Pathogenicity Avian Influenza Viruses. Avian Dis. 2010, 54, 622–626. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Feng, M.; Zhao, X.; Dai, X.; Xiang, B.; Gao, P.; Li, Y.; Li, Y.; Ren, T. Newcastle disease virus infection in chicken embryonic fibroblasts but not duck embryonic fibroblasts is associated with elevated host innate immune response. Virol. J. 2016, 13, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kondo, A. Growth characteristics of rabies virus in primary chick embryo cells. Virology 1965, 27, 199–204. [Google Scholar] [CrossRef]
- Ono, E.; Amagai, K.; Taharaguchi, S.; Tomioka, Y.; Yoshino, S.; Watanabe, Y.; Cherel, P.; Houdebine, L.-M.; Adam, M.; Eloit, M.; et al. Transgenic mice expressing a soluble form of porcine nectin-1/herpesvirus entry mediator C as a model for pseudorabies-resistant livestock. Proc. Natl. Acad. Sci. USA 2004, 101, 16150–16155. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Gupta, N.; Ahlawat, S.; Kumar, A.; Taneja, R.; Sharma, R.; Sunder, S.; Tantia, M. In Vitro Culture of Skin Fibroblast Cells for Potential Cloning by Nuclear Transfer. In Applications of Gene-Based Technologies for Improving Animal Production and Health in Developing Countries; Springer: Dordrecht, The Netherlands, 2005; pp. 631–640. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species Used | Tissue Type | Harvest | Technique | Notes | Reference |
|---|---|---|---|---|---|
| Human | Skin biopsies from anterior surface of the forearm | HAM-F10 cell culture medium with 20% FBS | Adhesion of fragments to the flask surface | Large maintenance costs of culture medium during initial fibroblast growth. | [5] |
| Human | Skin biopsy | DMEM cell culture medium with 20% FBS | Adhesion of fragments to the well bottom | Estimation of 25–35 days to second passage. Large maintenance costs of culture medium during initial fibroblast growth | [6] |
| Human | Skin biopsy | Complete DMEM or complete RPMI | Primary explant method without/with epidermis removal with 0.5% dispase/PBS or 0.3% trypsin/PBS | Confluence is generally reached in ∼3 to 5 weeks. | [7] |
| Mouse | Skin from tail and ear | RPMI: 10% fetal calf serum (FCS), 50 μM 2-mercaptoethanol, 100 μM asparagine, 2 mM glutamine, 1% penicillin-streptomycin solution. | Digestion with collagenase and D-pronase solution | Euthanasia was used. The fragments correspond to epidermis and dermis to ensure the grow of keratinocytes is probable. | [8] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abade dos Santos, F.A.; Carvalho, C.L.; Almeida, I.; Fagulha, T.; Rammos, F.; Barros, S.C.; Henriques, M.; Luís, T.; Duarte, M.D. Simple Method for Establishing Primary Leporidae Skin Fibroblast Cultures. Cells 2021, 10, 2100. https://doi.org/10.3390/cells10082100
Abade dos Santos FA, Carvalho CL, Almeida I, Fagulha T, Rammos F, Barros SC, Henriques M, Luís T, Duarte MD. Simple Method for Establishing Primary Leporidae Skin Fibroblast Cultures. Cells. 2021; 10(8):2100. https://doi.org/10.3390/cells10082100
Chicago/Turabian StyleAbade dos Santos, Fábio A., C. L. Carvalho, Isabel Almeida, Teresa Fagulha, Fernanda Rammos, Sílvia C. Barros, Margarida Henriques, Tiago Luís, and Margarida D. Duarte. 2021. "Simple Method for Establishing Primary Leporidae Skin Fibroblast Cultures" Cells 10, no. 8: 2100. https://doi.org/10.3390/cells10082100
APA StyleAbade dos Santos, F. A., Carvalho, C. L., Almeida, I., Fagulha, T., Rammos, F., Barros, S. C., Henriques, M., Luís, T., & Duarte, M. D. (2021). Simple Method for Establishing Primary Leporidae Skin Fibroblast Cultures. Cells, 10(8), 2100. https://doi.org/10.3390/cells10082100