
Epigenetic Regulation in Melanoma: Facts and Hopes

 , , ,
, , ,  , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
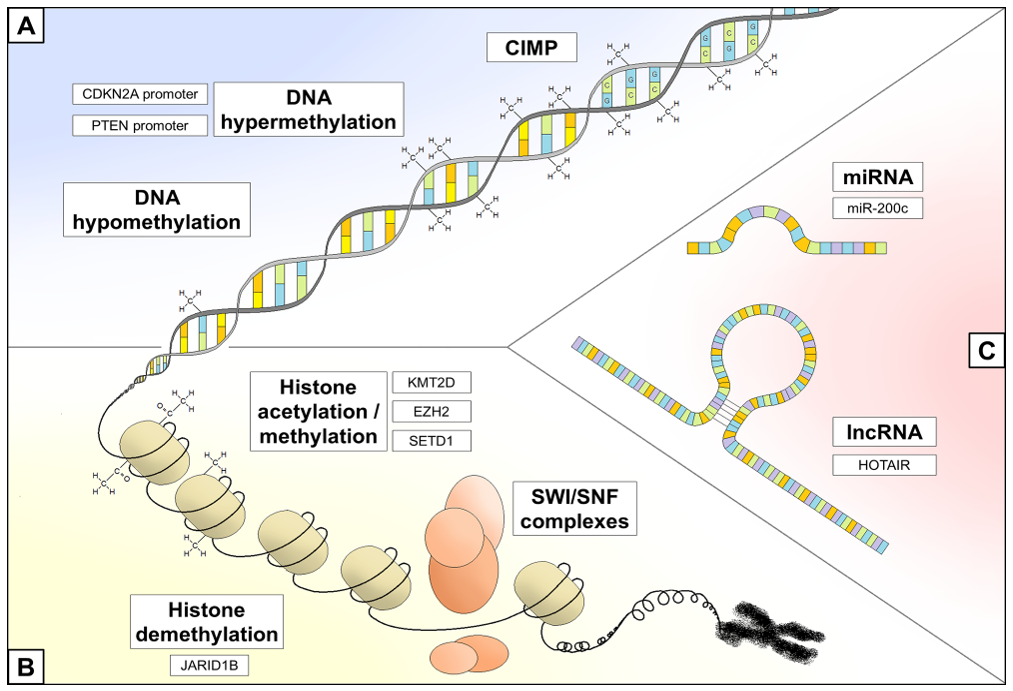
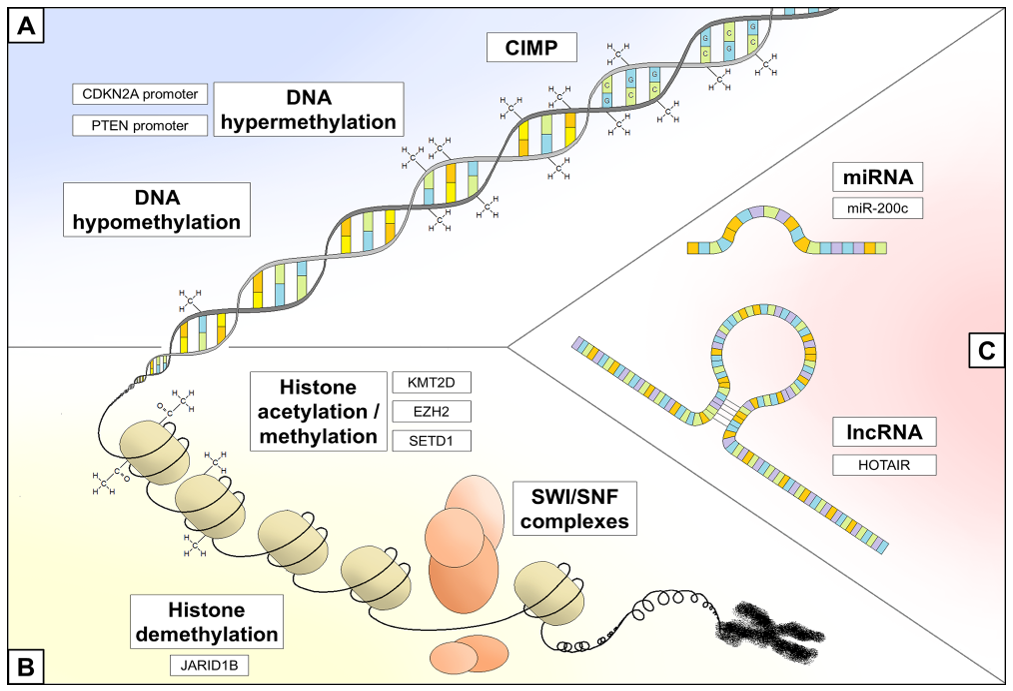
3. Epigenetic Regulation and Melanoma
3.1. DNA Methylation Status
3.2. Chromatin Remodeling
3.3. Non-Coding RNA Regulation
4. Targeting Epigenetic Machinery in Melanoma
4.1. Immunotherapy
4.2. Targeted Therapy
4.3. Chemotherapy/Radiotherapy
5. Future Directions and Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Melanoma|Cancer.Net. Available online: https://www.cancer.net/cancer-types/melanoma/statistics (accessed on 11 June 2021).
- Leonardi, G.C.; Candido, S.; Falzone, L.; Spandidos, D.A.; Libra, M. Cutaneous melanoma and the immunotherapy revolution (Review). Int. J. Oncol. 2020, 57, 609–618. [Google Scholar] [CrossRef]
- Giunta, E.F.; De Falco, V.; Napolitano, S.; Argenziano, G.; Brancaccio, G.; Moscarella, E.; Ciardiello, D.; Ciardiello, F.; Troiani, T. Optimal treatment strategy for metastatic melanoma patients harboring BRAF-V600 mutations. Ther. Adv. Med. Oncol. 2020, 12, 1758835920925219. [Google Scholar] [CrossRef]
- Pham, D.D.M.; Guhan, S.; Tsao, H. KIT and melanoma: Biological insights and clinical implications. Yonsei Med. J. 2020, 61, 562–571. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Couselo, E.; Adelantado, E.Z.; Vélez, C.O.; Soberino-García, J.; Perez-Garcia, J.M. NRAS-mutant melanoma: Current challenges and future prospect. OncoTargets Ther. 2017, 10, 3941–3947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michielin, O.; Atkins, M.B.; Koon, H.B.; Dummer, R.; Ascierto, P.A. Evolving impact of long-term survival results on metastatic melanoma treatment. J. Immunother. Cancer 2020, 8, e000948. [Google Scholar] [CrossRef] [PubMed]
- Winder, M.; Virós, A. Mechanisms of Drug Resistance in Melanoma. Handb. Exp. Pharmacol. 2018, 249, 91–108. [Google Scholar]
- Feinberg, A.P.; Tycko, B. The history of cancer epigenetics. Nat. Rev. Cancer 2004, 4, 143–153. [Google Scholar] [CrossRef]
- Feinberg, A.; Koldobskiy, A.P.F.M.A.; Göndör, A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat. Rev. Genet. 2016, 17, 284–299. [Google Scholar] [CrossRef] [PubMed]
- Jin, B.; Li, Y.; Robertson, K.D. DNA methylation: Superior or subordinate in the epigenetic hierarchy? Genes Cancer 2011, 2, 607–617. [Google Scholar] [CrossRef] [Green Version]
- Deaton, A.; Bird, A. CpG islands and the regulation of transcription. Genes Dev. 2011, 25, 1010–1022. [Google Scholar] [CrossRef] [Green Version]
- Feinberg, A.; Vogelstein, B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nat. Cell Biol. 1983, 301, 89–92. [Google Scholar] [CrossRef]
- Kim, Y.-I.; Giuliano, A.; Hatch, K.D.; Schneider, A.; Nour, M.A.; Dallal, G.E.; Selhub, J.; Mason, J.B. Global DNA hypomethylation increases progressively in cervical dysplasia and carcinoma. Cancer 1994, 74, 893–899. [Google Scholar] [CrossRef]
- Cravo, M.; Pinto, R.; Fidalgo, P.; Chaves, P.; Gloria, L.; Nobre-Leitao, C.; Mira, F.C. Global DNA hypomethylation occurs in the early stages of intestinal type gastric carcinoma. Gut 1996, 39, 434–438. [Google Scholar] [CrossRef] [Green Version]
- Soares, J.; E Pinto, A.; Cunha, C.V.; André, S.; Barão, I.; Sousa, J.M.; Cravo, M. Global DNA hypomethylation in breast carcinoma: Correlation with prognostic factors and tumor progression. Cancer 1999, 85, 112–118. [Google Scholar] [CrossRef]
- Ehrlich, M. DNA hypomethylation in cancer cells. Epigenomics 2009, 1, 239–259. [Google Scholar] [CrossRef] [Green Version]
- Karpf, A.R.; Matsui, S.-I. Genetic disruption of cytosine DNA methyltransferase enzymes induces chromosomal instability in human cancer cells. Cancer Res. 2005, 65, 8635–8639. [Google Scholar] [CrossRef] [Green Version]
- Nakayama, M.; Wada, M.; Harada, T.; Nagayama, J.; Kusaba, H.; Ohshima, K.; Kozuru, M.; Komatsu, H.; Ueda, R.; Kuwano, M. Hypomethylation status of CpG sites at the promoter region and overexpression of the human MDR1 gene in acute myeloid leukemias. Blood 1998, 92, 4296–4307. [Google Scholar] [CrossRef]
- Fraga, M.; Herranz, M.; Espada, J.; Ballestar, E.; Paz, M.F.; Ropero, S.; Erkek, E.; Bozdogan, O.; Peinado, H.; Niveleau, A.; et al. A mouse skin multistage carcinogenesis model reflects the aberrant DNA methylation patterns of human tumors. Cancer Res. 2004, 64, 5527–5534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toyota, M.; Ahuja, N.; Ohe-Toyota, M.; Herman, J.G.; Baylin, S.B.; Issa, J.-P. CpG island methylator phenotype in colorectal cancer. Proc. Natl. Acad. Sci. USA 1999, 96, 8681–8686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanemura, A.; Terando, A.M.; Sim, M.-S.; Van Hoesel, A.Q.; De Maat, M.F.; Morton, D.L.; Hoon, D.S. CpG island methylator phenotype predicts progression of malignant melanoma. Clin. Cancer Res. 2009, 15, 1801–1807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, L.A.; Melotte, V.; de Schrijver, J.; de Maat, M.; Smit, V.T.; Bovée, J.V.; French, P.J.; Brandt, P.A.V.D.; Schouten, L.J.; de Meyer, T.; et al. The CpG island methylator phenotype: What’s in a name? Cancer Res. 2013, 73, 5858–5868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stahl, J.M.; Cheung, M.; Sharma, A.; Trivedi, N.R.; Shanmugam, S.; Robertson, G.P. Loss of PTEN promotes tumor development in malignant melanoma. Cancer Res. 2003, 63, 2881–2890. [Google Scholar]
- Mirmohammadsadegh, A.; Marini, A.; Nambiar, S.; Hassan, M.; Tannapfel, A.; Ruzicka, T.; Hengge, U.R. Epigenetic silencing of the PTEN gene in melanoma. Cancer Res. 2006, 66, 6546–6552. [Google Scholar] [CrossRef] [Green Version]
- Lahtz, C.; Stranzenbach, R.; Fiedler, E.; Helmbold, P.; Dammann, R.H. Methylation of PTEN as a prognostic factor in malignant melanoma of the skin. J. Investig. Dermatol. 2010, 130, 620–622. [Google Scholar] [CrossRef] [Green Version]
- Gonzalgo, M.L.; Bender, C.M.; You, E.H.; Glendening, J.M.; Flores, J.F.; Walker, G.J.; Hayward, N.K.; A Jones, P.; Fountain, J.W. Low frequency of p16/CDKN2A methylation in sporadic melanoma: Comparative approaches for methylation analysis of primary tumors. Cancer Res. 1997, 57, 5336–5347. [Google Scholar]
- Straume, O.; Smeds, J.; Kumar, R.; Hemminki, K.; Akslen, L.A. Significant impact of promoter hypermethylation and the 540 C > T polymorphism of CDKN2A in cutaneous melanoma of the vertical growth phase. Am. J. Pathol. 2002, 161, 229–237. [Google Scholar] [CrossRef]
- Fujimoto, A.; Morita, R.; Hatta, N.; Takehara, K.; Takata, M. p16INK4a inactivation is not frequent in uncultured sporadic primary cutaneous melanoma. Oncogene 1999, 18, 2527–2532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonsson, A.; Tuominen, R.; Grafström, E.; Hansson, J.; Egyhazi, S. High frequency of p16INK4A promoter methylation in NRAS-mutated cutaneous melanoma. J. Investig. Dermatol. 2010, 130, 2809–2817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freedberg, D.E.; Rigas, S.H.; Russak, J.; Gai, W.; Kaplow, M.; Osman, I.; Turner, F.; Randerson-Moor, J.A.; Houghton, A.; Busam, K.; et al. Frequent p16-independent inactivation of p14ARF in human melanoma. J. Natl. Cancer Inst. 2008, 100, 784–795. [Google Scholar] [CrossRef] [Green Version]
- Spugnardi, M.; Tommasi, S.; Dammann, R.; Pfeifer, G.P.; Hoon, D.S.B. Epigenetic inactivation of RAS association domain family protein 1 (RASSF1A) in malignant cutaneous melanoma. Cancer Res. 2003, 63, 1639–1643. [Google Scholar] [PubMed]
- Gao, L.; Smit, M.A.; Oord, J.J.V.D.; Goeman, J.J.; Verdegaal, E.M.E.; Van Der Burg, S.H.; Stas, M.; Beck, S.; Gruis, N.A.; Tensen, C.; et al. Genome-wide promoter methylation analysis identifies epigenetic silencing of MAPK13 in primary cutaneous melanoma. Pigment. Cell Melanoma Res. 2013, 26, 542–554. [Google Scholar] [CrossRef]
- Tellez, C.S.; Shen, L.; Estecio, M.; Jelinek, J.; Gershenwald, J.E.; Issa, J.-P. CpG island methylation profiling in human melanoma cell lines. Melanoma Res. 2009, 19, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Koga, Y.; Pelizzola, M.; Cheng, E.; Krauthammer, M.; Sznol, M.; Ariyan, S.; Narayan, D.; Molinaro, A.M.; Halaban, R.; Weissman, S.M. Genome-wide screen of promoter methylation identifies novel markers in melanoma. Genome Res. 2009, 19, 1462–1470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conway, K.; Edmiston, S.N.; Khondker, Z.S.; Groben, P.A.; Zhou, X.; Chu, H.; Kuan, P.F.; Hao, H.; Carson, C.; Berwick, M.; et al. DNA-methylation profiling distinguishes malignant melanomas from benign nevi. Pigment. Cell Melanoma Res. 2011, 24, 352–360. [Google Scholar] [CrossRef] [PubMed]
- Muthusamy, V.; Duraisamy, S.; Bradbury, C.M.; Hobbs, C.; Curley, D.P.; Nelson, B.; Bosenberg, M. Epigenetic silencing of novel tumor suppressors in malignant melanoma. Cancer Res. 2006, 66, 11187–11193. [Google Scholar] [CrossRef] [Green Version]
- Falahat, R.; Berglund, A.; Putney, R.M.; Perez-Villarroel, P.; Aoyama, S.; Pilon-Thomas, S.; Barber, G.N.; Mulé, J.J. Epigenetic reprogramming of tumor cell–intrinsic STING function sculpts antigenicity and T cell recognition of melanoma. Proc. Natl. Acad. Sci. USA 2021, 118, 2013598118. [Google Scholar] [CrossRef]
- Al Emran, A.; Chatterjee, A.; Rodger, E.J.; Tiffen, J.C.; Gallagher, S.; Eccles, M.R.; Hersey, P. Targeting DNA methylation and EZH2 activity to overcome melanoma resistance to immunotherapy. Trends Immunol. 2019, 40, 328–344. [Google Scholar] [CrossRef] [Green Version]
- Goltz, D.; Gevensleben, H.; Vogt, T.J.; Dietrich, J.; Golletz, C.; Bootz, F.; Kristiansen, G.; Landsberg, J.; Dietrich, D. CTLA4 methylation predicts response to anti–PD-1 and anti–CTLA-4 immunotherapy in melanoma patients. JCI Insight 2018, 3, e96793. [Google Scholar] [CrossRef] [Green Version]
- Micevic, G.; Thakral, D.; McGeary, M.; Bosenberg, M.W. PD-L1 methylation regulates PD-L1 expression and is associated with melanoma survival. Pigment. Cell Melanoma Res. 2019, 32, 435–440. [Google Scholar] [CrossRef]
- Hoffmann, F.; Zarbl, R.; Niebel, D.; Sirokay, J.; Fröhlich, A.; Posch, C.; Holderried, T.A.W.; Brossart, P.; Saavedra, G.; Kuster, P.; et al. Prognostic and predictive value of PD-L2 DNA methylation and mRNA expression in melanoma. Clin. Epigenetics 2020, 12, 1–12. [Google Scholar] [CrossRef]
- Lian, C.; Xu, Y.; Ceol, C.; Wu, F.; Larson, A.; Dresser, K.; Xu, W.; Tan, L.; Hu, Y.; Zhan, Q.; et al. Loss of 5-hydroxymethylcytosine is an epigenetic hallmark of melanoma. Cell 2012, 150, 1135–1146. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, K.D.; Helin, K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. 2016, 30, 733–750. [Google Scholar] [CrossRef] [PubMed]
- Micevic, G.; Theodosakis, N.; Bosenberg, M. Aberrant DNA methylation in melanoma: Biomarker and therapeutic opportunities. Clin. Epigenetics 2017, 9, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.D.; Gillespie, S.K.; Borrow, J.M.; Hersey, P. The histone deacetylase inhibitor suberic bishydroxamate: A potential sensitizer of melanoma to TNF-related apoptosis-inducing ligand (TRAIL) induced apoptosis. Biochem. Pharmacol. 2003, 66, 1537–1545. [Google Scholar] [CrossRef]
- Bachmann, I.M.; Halvorsen, O.J.; Collett, K.; Stefansson, I.M.; Straume, O.; Haukaas, S.A.; Salvesen, H.B.; Otte, A.P.; Akslen, L.A. EZH2 expression is associated with high proliferation rate and aggressive tumor subgroups in cutaneous melanoma and cancers of the endometrium, prostate, and breast. J. Clin. Oncol. 2006, 24, 268–273. [Google Scholar] [CrossRef]
- Ceol, C.J.; Houvras, Y.; Jane-Valbuena, J.; Bilodeau, S.; Orlando, D.A.; Battisti, V.; Fritsch, L.; Lin, W.M.; Hollmann, T.J.; Ferre’, F.; et al. The histone methyltransferase SETDB1 is recurrently amplified in melanoma and accelerates its onset. Nat. Cell Biol. 2011, 471, 513–517. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Weng, Q.Y.; Insco, M.L.; Chen, K.Y.; Muralidhar, S.; Pozniak, J.; Diaz, J.M.S.; Drier, Y.; Nguyen, N.; Lo, J.A.; et al. Gain-of-function genetic alterations of G9a drive oncogenesis. Cancer Discov. 2020, 10, 980–997. [Google Scholar] [CrossRef] [Green Version]
- Bossi, D.; Cicalese, A.; Dellino, G.I.; Luzi, L.; Riva, L.; D’Alesio, C.; Diaferia, G.R.; Carugo, A.; Cavallaro, E.; Piccioni, R.; et al. In vivo genetic screens of patient-derived tumors revealed unexpected frailty of the transformed phenotype. Cancer Discov. 2016, 6, 650–663. [Google Scholar] [CrossRef] [Green Version]
- Roesch, A.; Fukunaga-Kalabis, M.; Schmidt, E.C.; Zabierowski, S.E.; Brafford, P.A.; Vultur, A.; Basu, D.; Gimotty, P.; Vogt, T.; Herlyn, M. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell 2010, 141, 583–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segura, M.F.; Fontanals-Cirera, B.; Gaziel-Sovran, A.; Guijarro, M.V.; Hanniford, D.; Zhang, G.; González-Gomez, P.; Morante, M.; Jubierre, L.; Zhang, W.; et al. BRD4 sustains melanoma proliferation and represents a new target for epigenetic therapy. Cancer Res. 2013, 73, 6264–6276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallagher, S.; Mijatov, B.; Gunatilake, D.; Tiffen, J.C.; Gowrishankar, K.; Jin, L.; Pupo, G.M.; Cullinane, C.; Prinjha, R.K.; Smithers, N.; et al. The epigenetic regulator I-BET151 induces BIM-dependent apoptosis and cell cycle arrest of human melanoma cells. J. Investig. Dermatol. 2014, 134, 2795–2805. [Google Scholar] [CrossRef] [Green Version]
- Gallagher, S.J.; Mijatov, B.; Gunatilake, D.; Gowrishankar, K.; Tiffen, J.; James, W.; Jin, L.; Pupo, G.; Cullinane, C.; McArthur, G.; et al. Control of NF-kB activity in human melanoma by bromodomain and extra-terminal protein inhibitor I-BET151. Pigment. Cell Melanoma Res. 2014, 27, 1126–1137. [Google Scholar] [CrossRef]
- Hodis, E.; Watson, I.; Kryukov, G.; Arold, S.T.; Imielinski, M.; Theurillat, J.-P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A landscape of driver mutations in melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qadeer, Z.A.; Harcharik, S.; Valle-Garcia, D.; Chen, C.; Birge, M.B.; Vardabasso, C.; Duarte, L.F.; Bernstein, E. Decreased expression of the chromatin remodeler ATRX associates with melanoma progression. J. Investig. Dermatol. 2014, 134, 1768–1772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koludrovic, D.; Laurette, P.; Strub, T.; Keime, C.; Le Coz, M.; Coassolo, S.; Mengus, G.; LaRue, L.; Davidson, I. Chromatin-remodelling complex NURF is essential for differentiation of adult melanocyte stem cells. PLoS Genet. 2015, 11, e1005555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Tetzlaff, M.T.; Cui, R.; Xu, X. miR-200c inhibits melanoma progression and drug resistance through down-regulation of BMI-1. Am. J. Pathol. 2012, 181, 1823–1835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satzger, I.; Mattern, A.; Kuettler, U.; Weinspach, D.; Niebuhr, M.; Kapp, A.; Gutzmer, R. microRNA-21 is upregulated in malignant melanoma and influences apoptosis of melanocytic cells. Exp. Dermatol. 2012, 21, 509–514. [Google Scholar] [CrossRef]
- Jin, L.; Hu, W.L.; Jiang, C.C.; Wang, J.X.; Han, C.C.; Chu, P.; Zhang, L.J.; Thorne, R.F.; Wilmott, J.; Scolyer, R.A.; et al. MicroRNA-149*, a p53-responsive microRNA, functions as an oncogenic regulator in human melanoma. Proc. Natl. Acad. Sci. USA 2011, 108, 15840–15845. [Google Scholar] [CrossRef] [Green Version]
- Pencheva, N.; Tran, H.; Buss, C.; Huh, D.; Drobnjak, M.; Busam, K.; Tavazoie, S.F. Convergent multi-miRNA targeting of ApoE drives LRP1/LRP8-dependent melanoma metastasis and angiogenesis. Cell 2012, 151, 1068–1082. [Google Scholar] [CrossRef] [Green Version]
- Tang, L.; Zhang, W.; Su, B.; Yu, B. Long noncoding RNA HOTAIR is associated with motility, invasion, and metastatic potential of metastatic melanoma. BioMed Res. Int. 2013, 2013, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Zheng, H.; Tse, G.; Chan, M.; Wu, W.K. Long non-coding RNAs in melanoma. Cell Prolif. 2018, 51, e12457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strub, T.; Ballotti, R.; Bertolotto, C. The “ART” of epigenetics in melanoma: From histone “alterations, to resistance and therapies”. Theranostics 2020, 10, 1777–1797. [Google Scholar] [CrossRef]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, C.T.; Weisenberger, D.J.; Velicescu, M.; A Gonzales, F.; Lin, J.C.Y.; Liang, G.; A Jones, P. Histone H3-lysine 9 methylation is associated with aberrant gene silencing in cancer cells and is rapidly reversed by 5-aza-2’-deoxycytidine. Cancer Res. 2002, 62, 6456–6461. [Google Scholar]
- Seligson, D.B.; Horvath, S.; Shi, T.; Yu, H.; Tze, S.; Grunstein, M.; Kurdistani, S. Global histone modification patterns predict risk of prostate cancer recurrence. Nat. Cell Biol. 2005, 435, 1262–1266. [Google Scholar] [CrossRef]
- Ye, Y.; Jin, L.; Wilmott, J.; Hu, W.L.; Yosufi, B.; Thorne, R.F.; Liu, T.; Rizos, H.; Yan, X.G.; Dong, L.; et al. PI(4,5)P2 5-phosphatase A regulates PI3K/Akt signalling and has a tumour suppressive role in human melanoma. Nat. Commun. 2013, 4, 1508. [Google Scholar] [CrossRef] [Green Version]
- McHugh, J.B.; Fullen, D.R.; Ma, L.; Kleer, C.G.; Su, L.D. Expression of polycomb group protein EZH2 in nevi and melanoma. J. Cutan. Pathol. 2007, 34, 597–600. [Google Scholar] [CrossRef]
- Hjelmeland, A.B.; Wu, Q.; Heddleston, J.M.; Choudhary, G.S.; MacSwords, J.; Lathia, J.D.; McLendon, R.; Lindner, D.; Sloan, A.; Rich, J.N. Acidic stress promotes a glioma stem cell phenotype. Cell Death Differ. 2011, 18, 829–840. [Google Scholar] [CrossRef] [Green Version]
- Pang, M.-F.; Siedlik, M.J.; Han, S.; Stallings-Mann, M.; Radisky, D.C.; Nelson, C.M. Tissue stiffness and hypoxia modulate the integrin-linked kinase ILK to control breast cancer stem-like cells. Cancer Res. 2016, 76, 5277–5287. [Google Scholar] [CrossRef] [Green Version]
- Lagadec, C.; Vlashi, E.; Della Donna, L.; Dekmezian, C.; Pajonk, F. Radiation-induced reprogramming of breast cancer cells. Stem Cells 2012, 30, 833–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Molley, T.G.; Seward, C.H.; Abdeen, A.A.; Zhang, H.; Wang, X.; Gandhi, H.; Yang, J.-L.; Gaus, K.; Kilian, K.A. Geometric regulation of histone state directs melanoma reprogramming. Commun. Biol. 2020, 3, 1–9. [Google Scholar] [CrossRef]
- Mashtalir, N.; D’Avino, A.; Michel, B.C.; Luo, J.; Pan, J.; Otto, J.E.; Zullow, H.J.; McKenzie, Z.M.; Kubiak, R.L.; Pierre, R.S.; et al. Modular organization and assembly of SWI/SNF family chromatin remodeling complexes. Cell 2018, 175, 1272–1288.e20. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Allis, C.D. SWI/SNF complex in cancer. Nat. Genet. 2017, 49, 178–179. [Google Scholar] [CrossRef] [PubMed]
- Vardabasso, C.; Hasson, D.; Ratnakumar, K.; Chung, C.-Y.; Duarte, L.F.; Bernstein, E. Histone variants: Emerging players in cancer biology. Cell. Mol. Life Sci. 2014, 71, 379–404. [Google Scholar] [CrossRef] [Green Version]
- Zingg, D.; Debbache, J.; Schaefer, S.M.; Tuncer, E.; Frommel, S.C.; Cheng, P.; Arenas-Ramirez, N.; Haeusel, J.; Zhang, Y.; Bonalli, M.; et al. The epigenetic modifier EZH2 controls melanoma growth and metastasis through silencing of distinct tumour suppressors. Nat. Commun. 2015, 6, 6051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapoor, A.; Goldberg, M.S.; Cumberland, L.K.; Ratnakumar, K.; Segura, M.F.; Emanuel, P.O.; Menendez, S.; Vardabasso, C.; LeRoy, G.; Vidal, C.I.; et al. The histone variant macroH2A suppresses melanoma progression through regulation of CDK8. Nat. Cell Biol. 2010, 468, 1105–1109. [Google Scholar] [CrossRef]
- Vardabasso, C.; Gaspar-Maia, A.; Hasson, D.; Pünzeler, S.; Valle-Garcia, D.; Straub, T.; Keilhauer, E.C.; Strub, T.; Dong, J.; Panda, T.; et al. Histone variant H2A.Z.2 mediates proliferation and drug sensitivity of malignant melanoma. Mol. Cell 2015, 59, 75–88. [Google Scholar] [CrossRef] [Green Version]
- Costa, F.F. Non-coding RNAs: Lost in translation? Gene 2007, 386, 1–10. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wapinski, O.; Chang, H.Y. Long noncoding RNAs and human disease. Trends Cell Biol. 2011, 21, 354–361. [Google Scholar] [CrossRef]
- Streicher, K.L.; Zhu, W.; Lehmann, K.P.; Georgantas, R.W.; Morehouse, C.A.; Brohawn, P.; Carrasco, R.A.; Xiao, Z.; Tice, D.A.; Higgs, B.W.; et al. A novel oncogenic role for the miRNA-506-514 cluster in initiating melanocyte transformation and promoting melanoma growth. Oncogene 2012, 31, 1558–1570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gajos-Michniewicz, A.; Czyz, M. Role of miRNAs in melanoma metastasis. Cancers 2019, 11, 326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prachayasittikul, V.; Prathipati, P.; Pratiwi, R.; Phanus-Umporn, C.; Malik, A.A.; Schaduangrat, N.; Seenprachawong, K.; Wongchitrat, P.; Supokawej, A.; Prachayasittikul, V.; et al. Exploring the epigenetic drug discovery landscape. Expert Opin. Drug Discov. 2017, 12, 345–362. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; He, C.; Wang, M.; Ma, X.; Mo, F.; Yang, S.; Han, J.; Wei, X. Targeting epigenetic regulators for cancer therapy: Mechanisms and advances in clinical trials. Signal Transduct. Target. Ther. 2019, 4, 1–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woods, D.M.; Woan, K.; Cheng, F.; Wang, H.; Perez-Villarroel, P.; Lee, C.; Lienlaf, M.; Atadja, P.; Seto, E.; Weber, J.; et al. The antimelanoma activity of the histone deacetylase inhibitor panobinostat (LBH589) is mediated by direct tumor cytotoxicity and increased tumor immunogenicity. Melanoma Res. 2013, 23, 341–348. [Google Scholar] [CrossRef] [Green Version]
- McMillan, T.J.; Hart, I.R. Enhanced experimental metastatic capacity of a murine melanoma following pre-treatment with anticancer drugs. Clin. Exp. Metastasis 1986, 4, 285–292. [Google Scholar] [CrossRef]
- Rollins, R.A.; Kim, K.H.; Tsao, C.-C. The emerging epigenetic landscape in melanoma. In Human Skin Cancer, Potential Biomarkers and Therapeutic Targets; IntechOpen: London, UK, 2016. [Google Scholar]
- Cossío, F.P.; Esteller, M.; Berdasco, M. Towards a more precise therapy in cancer: Exploring epigenetic complexity. Curr. Opin. Chem. Biol. 2020, 57, 41–49. [Google Scholar] [CrossRef]
- Ottaviano, M.; De Placido, S.; Ascierto, P.A. Recent success and limitations of immune checkpoint inhibitors for cancer: A lesson from melanoma. Virchows Archiv 2019, 474, 421–432. [Google Scholar] [CrossRef]
- Gide, T.N.; Wilmott, J.S.; Scolyer, R.A.; Long, G.V. Primary and acquired resistance to immune checkpoint inhibitors in metastatic melanoma. Clin. Cancer Res. 2018, 24, 1260–1270. [Google Scholar] [CrossRef] [Green Version]
- Karantanos, T.; Christofides, A.; Bardhan, K.; Li, L.; Boussiotis, V.A. Regulation of T cell differentiation and function by EZH2. Front. Immunol. 2016, 7, 172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manning, C.; Hooper, S.J.; A Sahai, E. Intravital imaging of SRF and Notch signalling identifies a key role for EZH2 in invasive melanoma cells. Oncogene 2015, 34, 4320–4332. [Google Scholar] [CrossRef] [Green Version]
- Zingg, D.; Arenas-Ramirez, N.; Sahin, D.; Rosalia, R.A.; Antunes, A.T.; Haeusel, J.; Sommer, L.; Boyman, O. The histone methyltransferase Ezh2 controls mechanisms of adaptive resistance to tumor immunotherapy. Cell Rep. 2017, 20, 854–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheng, W.; LaFleur, M.; Nguyen, T.; Chen, S.; Chakravarthy, A.; Conway, J.; Li, Y.; Chen, H.; Yang, H.; Hsu, P.-H.; et al. LSD1 ablation stimulates anti-tumor immunity and enables checkpoint blockade. Cell 2018, 174, 549–563.e19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, D.; Kobayashi, A.; Jiang, P.; De Andrade, L.F.; Tay, R.E.; Luoma, A.M.; Tsoucas, D.; Qiu, X.; Lim, K.; Rao, P.; et al. A major chromatin regulator determines resistance of tumor cells to T cell–mediated killing. Science 2018, 359, 770–775. [Google Scholar] [CrossRef] [Green Version]
- Covre, A.; Coral, S.; Nicolay, H.; Parisi, G.; Fazio, C.; Colizzi, F.; Fratta, E.; Di Giacomo, A.M.; Sigalotti, L.; Natali, P.G.; et al. Antitumor activity of epigenetic immunomodulation combined with CTLA-4 blockade in syngeneic mouse models. OncoImmunology 2015, 4, e1019978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vo, D.D.; Prins, R.; Begley, J.L.; Donahue, T.R.; Morris, L.F.; Bruhn, K.W.; De La Rocha, P.; Yang, M.-Y.; Mok, S.; Garban, H.; et al. Enhanced antitumor activity induced by adoptive T-cell transfer and adjunctive use of the histone deacetylase inhibitor LAQ824. Cancer Res. 2009, 69, 8693–8699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamm, S.; Wulff, T.; Kronthaler, K.; Schrepfer, S.; Parnitzke, U.; Bretz, A.C.; Bartz, R. Abstract 4722: 4SC-202 increases immunogenicity of tumor cells, induces infiltration of tumor microenvironment with cytotoxic T cells, and primes tumors for combinations with different cancer immunotherapy approaches. Immunology 2018, 78, 4722. [Google Scholar] [CrossRef]
- Khushalani, N.I.; Markowitz, J.; Eroglu, Z.; Giuroiu, I.; Ladanova, V.; Reiersen, P.; Rich, J.; Thapa, R.; Schell, M.J.; Sotomayor, E.M.; et al. A phase I trial of panobinostat with ipilimumab in advanced melanoma. J. Clin. Oncol. 2017, 35, 9547. [Google Scholar] [CrossRef]
- Hassel, J.; Berking, C.; Eigentler, T.; Gutzmer, R.; Ascierto, P.; Schilling, B.; Hermann, F.; Bartz, R.; Schadendorf, D. Phase Ib/II study (SENSITIZE) assessing safety, pharmacokinetics (PK), pharmacodynamics (PD), and clinical outcome of domatinostat in combination with pembrolizumab in patients with advanced melanoma refractory/non-responding to prior checkpoint inhibitor therapy. Ann. Oncol. 2019, 30, v559. [Google Scholar] [CrossRef]
- Efficacy and Safety of Entinostat (ENT) and Pembrolizumab (PEMBRO) in Patients with Melanoma Previously Treated with Anti-PD1 Therapy. Available online: https://www.abstractsonline.com/pp8/#!/6812/presentation/9829 (accessed on 16 June 2021).
- Echevarría-Vargas, I.M.; I Reyes-Uribe, P.; Guterres, A.; Yin, X.; Kossenkov, A.V.; Liu, Q.; Zhang, G.; Krepler, C.; Cheng, C.; Wei, Z.; et al. Co-targeting BET and MEK as salvage therapy for MAPK and checkpoint inhibitor-resistant melanoma. EMBO Mol. Med. 2018, 10, e8446. [Google Scholar] [CrossRef] [Green Version]
- Maertens, O.; Kuzmickas, R.; Manchester, H.E.; Emerson, C.E.; Gavin, A.G.; Guild, C.J.; Wong, T.C.; De Raedt, T.; Bowman-Colin, C.; Hatchi, E.; et al. MAPK pathway suppression unmasks latent DNA repair defects and confers a chemical synthetic vulnerability in BRAF-, NRAS-, and NF1-mutant melanomas. Cancer Discov. 2019, 9, 526–545. [Google Scholar] [CrossRef] [Green Version]
- Zakharia, Y.; Monga, V.; Swami, U.; Bossler, A.; Freesmeier, M.; Frees, M.; Khan, M.; Frydenlund, N.; Srikantha, R.; Vanneste, M.; et al. Targeting epigenetics for treatment of BRAF mutated metastatic melanoma with decitabine in combination with vemurafenib: A phase lb study. Oncotarget 2017, 8, 89182–89193. [Google Scholar] [CrossRef] [Green Version]
- Tawbi, H.A.; Beumer, J.; Tarhini, A.A.; Moschos, S.; Buch, S.C.; Egorin, M.J.; Lin, Y.; Christner, S.; Kirkwood, J.M. Safety and efficacy of decitabine in combination with temozolomide in metastatic melanoma: A phase I/II study and pharmacokinetic analysis. Ann. Oncol. 2013, 24, 1112–1119. [Google Scholar] [CrossRef]
- Daud, A.I.; Dawson, J.; DeConti, R.C.; Bicaku, E.; Marchion, D.; Bastien, S.; Hausheer, F.A.; Lush, R.; Neuger, A.; Sullivan, D.M.; et al. Potentiation of a topoisomerase I inhibitor, karenitecin, by the histone deacetylase inhibitor valproic acid in melanoma: Translational and phase I/II clinical trial. Clin. Cancer Res. 2009, 15, 2479–2487. [Google Scholar] [CrossRef] [Green Version]
- Ottaviano, M.; Giunta, E.; Tortora, M.; Curvietto, M.; Attademo, L.; Bosso, D.; Cardalesi, C.; Rosanova, M.; De Placido, P.; Pietroluongo, E.; et al. BRAF gene and melanoma: Back to the future. Int. J. Mol. Sci. 2021, 22, 3474. [Google Scholar] [CrossRef]
- Hugo, W.; Shi, H.; Sun, L.; Piva, M.; Song, C.; Kong, X.; Moriceau, G.; Hong, A.; Dahlman, K.B.; Johnson, D.B.; et al. Non-genomic and immune evolution of melanoma acquiring MAPKi resistance. Cell 2015, 162, 1271–1285. [Google Scholar] [CrossRef] [Green Version]
- Valpione, S.; Carlino, M.S.; Mangana, J.; Mooradian, M.J.; McArthur, G.; Schadendorf, D.; Hauschild, A.; Menzies, A.; Arance, A.; Ascierto, P.A.; et al. Rechallenge with BRAF-directed treatment in metastatic melanoma: A multi-institutional retrospective study. Eur. J. Cancer 2018, 91, 116–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.; Tian, C.; Hoffman, T.E.; Jacobsen, N.K.; Spencer, S.L. Melanoma subpopulations that rapidly escape MAPK pathway inhibition incur DNA damage and rely on stress signalling. Nat. Commun. 2021, 12, 1–14. [Google Scholar] [CrossRef]
- Khaliq, M.; Fallahi-Sichani, M. Epigenetic mechanisms of escape from BRAF oncogene dependency. Cancers 2019, 11, 1480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khaliq, M.; Manikkam, M.; Martinez, E.D.; Fallahi-Sichani, M. Epigenetic modulation reveals differentiation state specificity of oncogene addiction. Nat. Commun. 2021, 12, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Al Emran, A.; Marzese, D.M.; Menon, D.R.; Stark, M.; Torrano, J.; Hammerlindl, H.; Zhang, G.; Brafford, P.; Salomon, M.P.; Nelson, N.; et al. Distinct histone modifications denote early stress-induced drug tolerance in cancer. Oncotarget 2018, 9, 8206–8222. [Google Scholar] [CrossRef] [Green Version]
- Roesch, A.; Vultur, A.; Bogeski, I.; Wang, H.; Zimmermann, K.M.; Speicher, D.; Körbel, C.; Laschke, M.W.; Gimotty, P.A.; Philipp, S.E.; et al. Overcoming intrinsic multidrug resistance in melanoma by blocking the mitochondrial respiratory chain of slow-cycling JARID1Bhigh cells. Cancer Cell 2013, 23, 811–825. [Google Scholar] [CrossRef] [Green Version]
- Menon, D.R.; Das, S.; Krepler, C.; Vultur, A.; Rinner, B.; Schauer, S.; Kashofer, K.; Wagner, K.; Zhang, G.; Rad, E.B.; et al. A stress-induced early innate response causes multidrug tolerance in melanoma. Oncogene 2015, 34, 4448–4459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strub, T.; Ghiraldini, F.G.; Carcamo, S.; Li, M.; Wroblewska, A.; Singh, R.; Goldberg, M.S.; Hasson, D.; Wang, Z.; Gallagher, S.; et al. SIRT6 haploinsufficiency induces BRAFV600E melanoma cell resistance to MAPK inhibitors via IGF signalling. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef]
- Zanconato, F.; Battilana, G.; Forcato, M.; Filippi, L.; Azzolin, L.; Manfrin, A.; Quaranta, E.; Di Biagio, D.; Sigismondo, G.; Guzzardo, V.; et al. Transcriptional addiction in cancer cells is mediated by YAP/TAZ through BRD4. Nat. Med. 2018, 24, 1599–1610. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; de Oliveira, R.L.; Huijberts, S.; Bosdriesz, E.; Pencheva, N.; Brunen, D.; Bosma, A.; Song, J.-Y.; Zevenhoven, J.; Vries, T.L.-D.; et al. An Acquired Vulnerability of Drug-Resistant Melanoma with Therapeutic Potential. Cell 2018, 173, 1413–1425.e14. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Schleich, K.; Yue, B.; Ji, S.; Lohneis, P.; Kemper, K.; Silvis, M.R.; Qutob, N.; Van Rooijen, E.; Werner-Klein, M.; et al. Targeting the senescence-overriding cooperative activity of structurally unrelated H3K9 demethylases in melanoma. Cancer Cell 2018, 33, 322–336.e8. [Google Scholar] [CrossRef] [Green Version]
- Perotti, V.; Baldassari, P.; Molla, A.; Nicolini, G.; Bersani, I.; Grazia, G.; Benigni, F.; Maurichi, A.; Santinami, M.; Anichini, A.; et al. An actionable axis linking NFATc2 to EZH2 controls the EMT-like program of melanoma cells. Oncogene 2019, 38, 4384–4396. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, G.; Di Lorenzo, G.; Ottaviano, M.; Damiano, V. The future of melanoma therapy: Developing new drugs and improving the use of old ones. Futur. Oncol. 2016, 12, 2531–2534. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.; Leon-Ferre, R.; Laux, D.; Deutsch, J.; Smith, B.J.; Frees, M.; Milhem, M. Treatment of resistant metastatic melanoma using sequential epigenetic therapy (decitabine and panobinostat) combined with chemotherapy (temozolomide). Cancer Chemother. Pharmacol. 2014, 74, 691–697. [Google Scholar] [CrossRef] [Green Version]
- Valentini, A.; Gravina, P.; Federici, G.; Bernardini, S. Valproic acid induces apoptosis, p16INK4 Aupregulation and sensitization to chemotherapy in human melanoma cells. Cancer Biol. Ther. 2007, 6, 185–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fulda, S.; Küfer, M.U.; Meyer, E.; Van Valen, F.; Dockhorn-Dworniczak, B.; Debatin, K.-M. Sensitization for death receptor- or drug-induced apoptosis by re-expression of caspase-8 through demethylation or gene transfer. Oncogene 2001, 20, 5865–5877. [Google Scholar] [CrossRef] [Green Version]
- Soengas, M.; Capodieci, P.; Polsky, D.; Mora, J.; Esteller, M.; Opitz-Araya, X.; McCombie, W.R.; Herman, J.G.; Gerald, W.L.; Lazebnik, Y.A.; et al. Inactivation of the apoptosis effector Apaf-1 in malignant melanoma. Nat. Cell Biol. 2001, 409, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-G.; Park, M.-T.; Heo, K.; Yang, K.-M.; Yi, J.M. Epigenetics meets radiation biology as a new approach in cancer treatment. Int. J. Mol. Sci. 2013, 14, 15059–15073. [Google Scholar] [CrossRef] [Green Version]
- Munshi, A.; Kurland, J.F.; Nishikawa, T.; Tanaka, T.; Hobbs, M.L.; Tucker, S.L.; Ismail, S.; Stevens, C.; Meyn, R.E. Histone deacetylase inhibitors radiosensitize human melanoma cells by suppressing DNA repair activity. Clin. Cancer Res. 2005, 11, 4912–4922. [Google Scholar] [CrossRef] [Green Version]
- Munshi, A.; Tanaka, T.; Hobbs, M.L.; Tucker, S.L.; Richon, V.M.; Meyn, R.E. Vorinostat, a histone deacetylase inhibitor, enhances the response of human tumor cells to ionizing radiation through prolongation of γ-H2AX foci. Mol. Cancer Ther. 2006, 5, 1967–1974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rieth, J.; Swami, U.; Mott, S.; Zanaty, M.; Henry, M.; Bossler, A.; Greenlee, J.; Zakharia, Y.; Vanneste, M.; Jennings, B.; et al. Melanoma brain metastases in the era of targeted therapy and checkpoint inhibitor therapy. Cancers 2021, 13, 1489. [Google Scholar] [CrossRef]
- Marzese, D.M.; Scolyer, R.A.; Roqué, M.; Vargas-Roig, L.M.; Huynh, J.L.; Wilmott, J.; Murali, R.; Buckland, M.E.; Barkhoudarian, G.; Thompson, J.; et al. DNA methylation and gene deletion analysis of brain metastases in melanoma patients identifies mutually exclusive molecular alterations. Neuro-Oncology 2014, 16, 1499–1509. [Google Scholar] [CrossRef] [Green Version]
- Orozco, J.I.J.; Knijnenburg, T.A.; Manughian-Peter, A.; Salomon, M.P.; Barkhoudarian, G.; Jalas, J.R.; Wilmott, J.; Hothi, P.; Wang, X.; Takasumi, Y.; et al. Epigenetic profiling for the molecular classification of metastatic brain tumors. Nat. Commun. 2018, 9, 1–14. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, Y. Role of epigenetic regulation in plasticity of tumor immune microenvironment. Front. Immunol. 2021, 12, 1013. [Google Scholar] [CrossRef]
- Mitra, S.; Lauss, M.; Cabrita, R.; Choi, J.; Zhang, T.; Isaksson, K.; Olsson, H.; Ingvar, C.; Carneiro, A.; Staaf, J.; et al. Analysis of DNA methylation patterns in the tumor immune microenvironment of metastatic melanoma. Mol. Oncol. 2020, 14, 933–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
| Class of Epigenetic Alteration | Type of Epigenetic Alteration | Involved Gene(s) | Evidence in Melanoma | Ref |
|---|---|---|---|---|
| DNA Methylation status modification | Promoter methylation | PTEN | Up to 60% of melanoma serum samples Independent poor prognostic factor | [24,25] |
| CDKN2A | Up to 25% of melanoma tissue samples Cell cycle deregulation | [26,27,28] | ||
| ARF | Up to 60% of melanoma tissue samples Cell cycle deregulation | [30] | ||
| Gene methylation | RASSF1A | Up to 55% of melanoma tissue samples | [31] | |
| cGAS and STING | Resistance to T-cell-based anticancer therapies | [37] | ||
| Gene hypomethylation | IDH-2 (?) | Disruption of TET ability to maintain DNA methylation fidelity | [43] | |
| Chromatin remodeling perturbation | Histone hypoacetylation | Bcl2 | Downregulation of antiapoptotic members | [45] |
| Histone hypermethylation | EZH2 | High proliferation rate and aggressive tumor subgroups | [46] | |
| SETDB1 | Acceleration of melanoma onset | [47] | ||
| EHMT2 | Up to 25% of melanoma tissue samples | [48] | ||
| KMT2D | Participation to melanomagenesis | [49] | ||
| JARID1B | Tumor growth and intrinsic drug resistance | [50] | ||
| Chromatin modification recognition | BRD2 and BRD4 | Apoptosis inhibition and cell cycle deregulation | [51,52,53] | |
| SWI/SNF complex regulation | ARID1A, ARID1B, ARID2, and SMARCA4 | Loss of ability to repair DNA double strand breaks and UV-induced pyrimidine dimers | [54] | |
| ATRX | Melanoma progression | [55] | ||
| NURF complex regulation | BPTF | Disruption of gene expression programs | [56] | |
| Non-coding RNA regulation | miRNA | MiR-200c | Reduced expression of adhesion molecules | [57] |
| MiR-149 and MiR-21 | Apoptosis inhibition | [58,59] | ||
| MiR-1908, miR-199a-5p, and miR-199a-3p | Promotion of invasion and metastasis formation Shorter overall survival | [60] | ||
| lncRNA | HOTAIR | Alteration of chromatin structure | [61] | |
| MALAT1 | Apoptosis inhibition, promotion of invasion and metastasis formation | [62] |
| Class of Therapeutic Partner | Partner Drug | Epigenetic Drug | Evidence in Melanoma | Ref |
|---|---|---|---|---|
| Immunotherapy | 9H10 (anti-CTLA-4 antibody) | 5-aza-2′-deoxycytidine (DNA hypomethylating agent) | Significant immune-related antitumor activity in syngeneic transplantable murine models | [97] |
| Gp100 melanoma antigen-specific pmel-1 T-cells (adoptive transfer) | LAQ824 (HDACi) | Improvement of antitumor activity in murine models | [98] | |
| Ipilimumab (anti-CTLA-4 antibody) | Panobinostat (pan-HDACi) | Phase I clinical trial: not increased responses in advanced melanoma patients respect to single-agent ipilimumab | [100] | |
| SGI-110: precursor of decitabine (DNMTi) | Phase 1 clinical trial (ongoing) | NCT02608437 | ||
| Pembrolizumab (anti-PD-1 antibody) | Domatinostat (class I HDACi) | Phase Ib/II clinical trial (SENSITIZE): safety and tolerability of combination and potential increase in antitumor activity in pretreated melanoma patients | [101] | |
| Entinostat (class I HDACi) | Ph1b/2 Dose-Escalation Study (ENCORE 601) of Entinostat with Pembrolizumab in NSCLC with Expansion Cohorts in NSCLC, Melanoma, and Colorectal Cancer | [102] | ||
| Entinostat (class I HDACi) | Phase II clinical trial (ongoing) | NCT03765229 | ||
| Azacytidine | Phase II clinical trial (ongoing) | NCT02816021 | ||
| Nivolumab (anti-PD-1 antibody) | Tinostamustine: fusion molecule of bendamustine (alkylating agent) + vorinostat (pan-HDACi) | Phase I clinical trial (ongoing) | NCT03903458 | |
| Ipilimumab (anti-CTLA-4 antibody) + nivolumab (anti-PD-1 antibody) | ACY-241 (HDAC6 inhibitor) | Phase I clinical trial (ongoing) | NCT02935790 | |
| Targeted therapy | PD901 (MEK inhibitor) | JQ-1 (BET inhibitor) | Reversion of therapeutic resistance in preclinical models | [103] |
| Dabrafenib (BRAF inhibitor) + trametinib (MEK inhibitor) | Vorinostat (pan-HDACi) | Enhancement of tumor regression | [104] | |
| Vemurafenib (BRAF inhibitor) | Decitabine (DNMTi) | Phase I clinical trial (interrupted) | [105] | |
| Chemotherapy | Temozolomide (alkylating agent) | Decitabine (DNMTi) | Phase I/II clinical trial | [106] |
| Karenitecin (topoisomerase 1 inhibitor) | Valproic acid (HDACi) | Translational study Phase I/II clinical trial | [107] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giunta, E.F.; Arrichiello, G.; Curvietto, M.; Pappalardo, A.; Bosso, D.; Rosanova, M.; Diana, A.; Giordano, P.; Petrillo, A.; Federico, P.; et al. Epigenetic Regulation in Melanoma: Facts and Hopes. Cells 2021, 10, 2048. https://doi.org/10.3390/cells10082048
Giunta EF, Arrichiello G, Curvietto M, Pappalardo A, Bosso D, Rosanova M, Diana A, Giordano P, Petrillo A, Federico P, et al. Epigenetic Regulation in Melanoma: Facts and Hopes. Cells. 2021; 10(8):2048. https://doi.org/10.3390/cells10082048
Chicago/Turabian StyleGiunta, Emilio Francesco, Gianluca Arrichiello, Marcello Curvietto, Annalisa Pappalardo, Davide Bosso, Mario Rosanova, Anna Diana, Pasqualina Giordano, Angelica Petrillo, Piera Federico, and et al. 2021. "Epigenetic Regulation in Melanoma: Facts and Hopes" Cells 10, no. 8: 2048. https://doi.org/10.3390/cells10082048
APA StyleGiunta, E. F., Arrichiello, G., Curvietto, M., Pappalardo, A., Bosso, D., Rosanova, M., Diana, A., Giordano, P., Petrillo, A., Federico, P., Fabozzi, T., Parola, S., Riccio, V., Mucci, B., Vanella, V., Festino, L., Daniele, B., Ascierto, P. A., Ottaviano, M., & On Behalf of SCITO YOUTH. (2021). Epigenetic Regulation in Melanoma: Facts and Hopes. Cells, 10(8), 2048. https://doi.org/10.3390/cells10082048