
CDK4/6 Inhibitors in Melanoma: A Comprehensive Review


 , , ,
, , , 
Abstract
1. Introduction
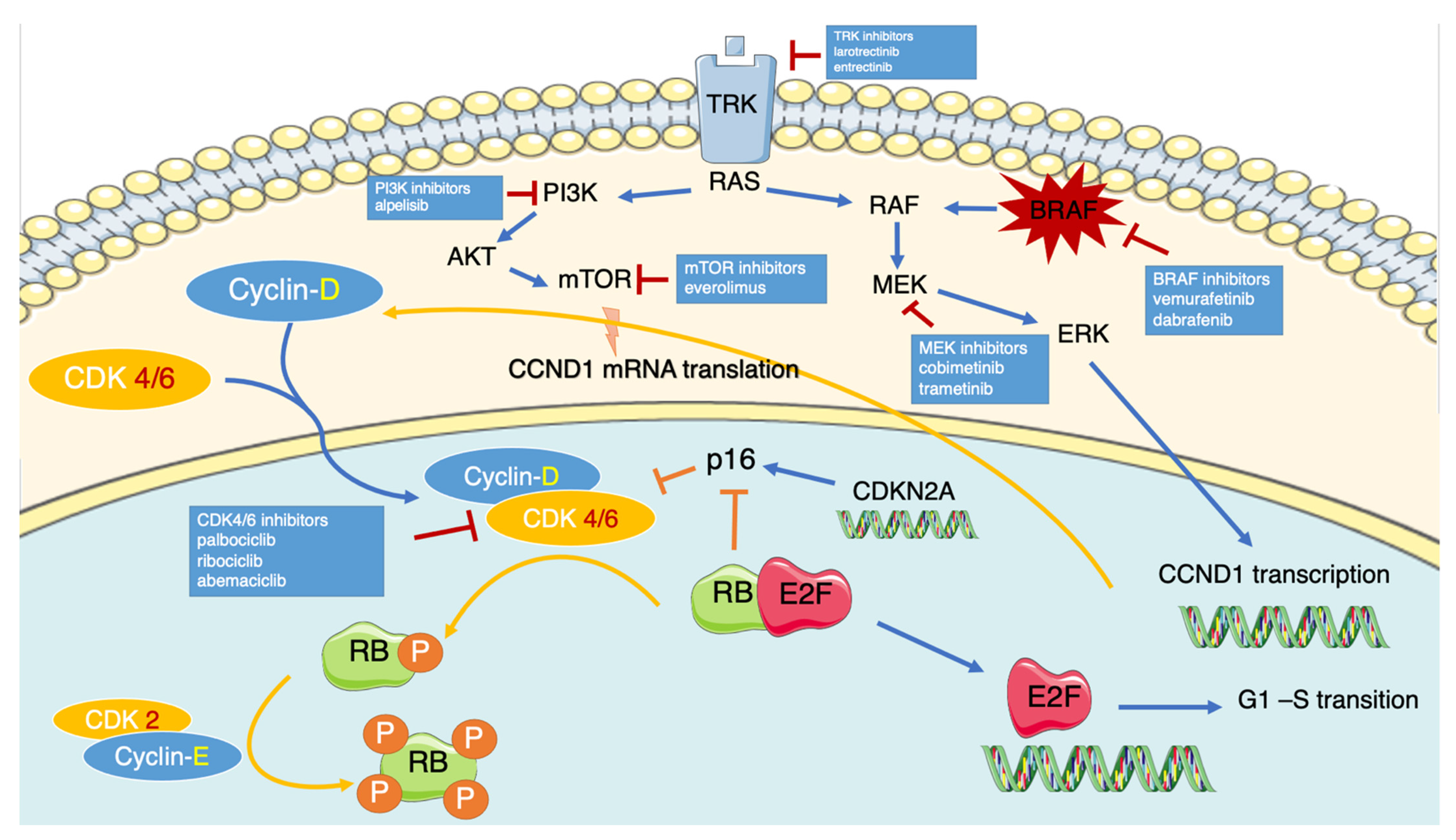
2. CDK4/6 Pathway
3. CDK4/6 Pathway Dysregulation in Melanoma
4. Pharmacology of CDK4/6 Inhibitors
5. Preclinical Activity of CDK4/6 Inhibitors in Melanoma
5.1. CDK4/6 Inhibitors as Single Agents
Resistance Mechanisms to CDK4/6 Inhibitors
5.2. CDK4/6 Inhibitors Combined with Immunotherapy
5.3. CDK4/6 Inhibitors Combined with BRAF/MEK Inhibitors
6. Clinical Activity of CDK4/6 Inhibitors in Melanoma
6.1. CDK4/6 Inhibitors as Single Agents
6.2. CDK4/6 Combined with Other Agents
7. Ongoing Clinical Trials with CDK4/6 Inhibitors in Melanoma
8. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Pollack, L.A.; Li, J.; Berkowitz, Z.; Weir, H.K.; Wu, X.-C.; Ajani, U.A.; Ekwueme, D.U.; Li, C.; Pollack, B.P. Melanoma Survival in the United States, 1992 to 2005. J. Am. Acad. Dermatol. 2011, 65, S78–S86. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Ribas, A.; Schachter, J.; Arance, A.; Grob, J.-J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.M.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma (KEYNOTE-006): Post-Hoc 5-Year Results from an Open-Label, Multicentre, Randomised, Controlled, Phase 3 Study. Lancet Oncol. 2019, 20, 1239–1251. [Google Scholar] [CrossRef]
- Dummer, R.; Ascierto, P.A.; Gogas, H.J.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; Gutzmer, R.; et al. Encorafenib plus Binimetinib versus Vemurafenib or Encorafenib in Patients with BRAF-Mutant Melanoma (COLUMBUS): A Multicentre, Open-Label, Randomised Phase 3 Trial. Lancet Oncol. 2018, 19, 603–615. [Google Scholar] [CrossRef]
- Dummer, R.; Hauschild, A.; Santinami, M.; Atkinson, V.; Mandalà, M.; Kirkwood, J.M.; Chiarion Sileni, V.; Larkin, J.; Nyakas, M.; Dutriaux, C.; et al. Five-Year Analysis of Adjuvant Dabrafenib plus Trametinib in Stage III Melanoma. N. Engl. J. Med. 2020, 383, 1139–1148. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, K.E.; McArthur, G.A. The Cell-Cycle Regulator CDK4: An Emerging Therapeutic Target in Melanoma. Clin. Cancer Res. 2013, 19, 5320–5328. [Google Scholar] [CrossRef]
- Simmons Kovacs, L.A.; Mayhew, M.B.; Orlando, D.A.; Jin, Y.; Li, Q.; Huang, C.; Reed, S.I.; Mukherjee, S.; Haase, S.B. Cyclin-Dependent Kinases Are Regulators and Effectors of Oscillations Driven by a Transcription Factor Network. Mol. Cell 2012, 45, 669–679. [Google Scholar] [CrossRef] [PubMed]
- Coudreuse, D.; Nurse, P. Driving the Cell Cycle with a Minimal CDK Control Network. Nature 2010, 468, 1074–1079. [Google Scholar] [CrossRef] [PubMed]
- Foster, D.A.; Yellen, P.; Xu, L.; Saqcena, M. Regulation of G1 Cell Cycle Progression: Distinguishing the Restriction Point from a Nutrient-Sensing Cell Growth Checkpoint(s). Genes Cancer 2010, 1, 1124–1131. [Google Scholar] [CrossRef]
- Morgan, D.O. Principles of CDK Regulation. Nature 1995, 374, 131–134. [Google Scholar] [CrossRef]
- Gao, X.; Leone, G.W.; Wang, H. Cyclin D-CDK4/6 functions in cancer. In Advances in Cancer Research; Elsevier: Amsterdam, The Netherlands, 2020; Volume 148, pp. 147–169. ISBN 978-0-12-820327-9. [Google Scholar]
- Tchakarska, G.; Sola, B. The Double Dealing of Cyclin D1. Cell Cycle 2020, 19, 163–178. [Google Scholar] [CrossRef]
- Poon, R.Y.C. Cell Cycle Control: A System of Interlinking Oscillators. In Cell Cycle Oscillators; Coutts, A.S., Weston, L., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2016; Volume 1342, pp. 3–19. ISBN 978-1-4939-2956-6. [Google Scholar]
- Giacinti, C.; Giordano, A. RB and Cell Cycle Progression. Oncogene 2006, 25, 5220–5227. [Google Scholar] [CrossRef]
- Lai, L.; Shin, G.Y.; Qiu, H. The Role of Cell Cycle Regulators in Cell Survival—Dual Functions of Cyclin-Dependent Kinase 20 and P21Cip1/Waf1. Int. J. Mol. Sci. 2020, 21, 8504. [Google Scholar] [CrossRef]
- Ortega, S.; Malumbres, M.; Barbacid, M. Cyclin D-Dependent Kinases, INK4 Inhibitors and Cancer. Biochim. Biophys. Acta BBA Rev. Cancer 2002, 1602, 73–87. [Google Scholar] [CrossRef]
- Asghar, U.; Witkiewicz, A.K.; Turner, N.C.; Knudsen, E.S. The History and Future of Targeting Cyclin-Dependent Kinases in Cancer Therapy. Nat. Rev. Drug Discov. 2015, 14, 130–146. [Google Scholar] [CrossRef] [PubMed]
- Hayward, N.K.; Wilmott, J.S.; Waddell, N.; Johansson, P.A.; Field, M.A.; Nones, K.; Patch, A.-M.; Kakavand, H.; Alexandrov, L.B.; Burke, H.; et al. Whole-Genome Landscapes of Major Melanoma Subtypes. Nature 2017, 545, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, E.; Infante, J.R. Targeting CDK4/6 in Patients with Cancer. Cancer Treat. Rev. 2016, 45, 129–138. [Google Scholar] [CrossRef]
- Young, R.J.; Waldeck, K.; Martin, C.; Foo, J.H.; Cameron, D.P.; Kirby, L.; Do, H.; Mitchell, C.; Cullinane, C.; Liu, W.; et al. Loss of CDKN2A Expression Is a Frequent Event in Primary Invasive Melanoma and Correlates with Sensitivity to the CDK4/6 Inhibitor PD0332991 in Melanoma Cell Lines. Pigment Cell Melanoma Res. 2014, 27, 590–600. [Google Scholar] [CrossRef]
- Sanki, A.; Li, W.; Colman, M.; Karim, R.Z.; Thompson, J.F.; Scolyer, R.A. Reduced Expression of P16 and P27 Is Correlated with Tumour Progression in Cutaneous Melanoma. Pathology (Phila.) 2007, 39, 551–557. [Google Scholar] [CrossRef]
- Tsao, H.; Chin, L.; Garraway, L.A.; Fisher, D.E. Melanoma: From Mutations to Medicine. Genes Dev. 2012, 26, 1131–1155. [Google Scholar] [CrossRef]
- Chan, S.H.; Chiang, J.; Ngeow, J. CDKN2A Germline Alterations and the Relevance of Genotype-Phenotype Associations in Cancer Predisposition. Hered. Cancer Clin. Pract. 2021, 19, 21. [Google Scholar] [CrossRef]
- Goldstein, A.M. Prospective Risk of Cancer in CDKN2A Germline Mutation Carriers. J. Med. Genet. 2004, 41, 421–424. [Google Scholar] [CrossRef]
- Rane, S.G.; Cosenza, S.C.; Mettus, R.V.; Reddy, E.P. Germ Line Transmission of the Cdk4R24C Mutation Facilitates Tumorigenesis and Escape from Cellular Senescence. Mol. Cell. Biol. 2002, 22, 644–656. [Google Scholar] [CrossRef]
- Sotillo, R.; Garcia, J.F.; Ortega, S.; Martin, J.; Dubus, P.; Barbacid, M.; Malumbres, M. Invasive Melanoma in Cdk4-Targeted Mice. Proc. Natl. Acad. Sci. USA 2001, 98, 13312–13317. [Google Scholar] [CrossRef]
- Montalto, F.I.; De Amicis, F. Cyclin D1 in Cancer: A Molecular Connection for Cell Cycle Control, Adhesion and Invasion in Tumor and Stroma. Cells 2020, 9, 2648. [Google Scholar] [CrossRef]
- Vízkeleti, L.; Ecsedi, S.; Rákosy, Z.; Orosz, A.; Lázár, V.; Emri, G.; Koroknai, V.; Kiss, T.; Ádány, R.; Balázs, M. The Role of CCND1 Alterations during the Progression of Cutaneous Malignant Melanoma. Tumor Biol. 2012, 33, 2189–2199. [Google Scholar] [CrossRef] [PubMed]
- González-Ruiz, L.; González-Moles, M.Á.; González-Ruiz, I.; Ruiz-Ávila, I.; Ayén, Á.; Ramos-García, P. An Update on the Implications of Cyclin D1 in Melanomas. Pigment Cell Melanoma Res. 2020, 33, 788–805. [Google Scholar] [CrossRef]
- Kong, Y.; Sheng, X.; Wu, X.; Yan, J.; Ma, M.; Yu, J.; Si, L.; Chi, Z.; Cui, C.; Dai, J.; et al. Frequent Genetic Aberrations in the CDK4 Pathway in Acral Melanoma Indicate the Potential for CDK4/6 Inhibitors in Targeted Therapy. Clin. Cancer Res. 2017, 23, 6946–6957. [Google Scholar] [CrossRef] [PubMed]
- Broit, N.; Johansson, P.A.; Rodgers, C.B.; Walpole, S.T.; Newell, F.; Hayward, N.K.; Pritchard, A.L. Meta-Analysis and Systematic Review of the Genomics of Mucosal Melanoma. Mol. Cancer Res. 2021, 34, 1541–7786. [Google Scholar] [CrossRef]
- Elefanti, L.; Zamuner, C.; Del Fiore, P.; Stagni, C.; Pellegrini, S.; Dall’Olmo, L.; Fabozzi, A.; Senetta, R.; Ribero, S.; Salmaso, R.; et al. The Molecular Landscape of Primary Acral Melanoma: A Multicenter Study of the Italian Melanoma Intergroup (IMI). Int. J. Mol. Sci. 2021, 22, 3826. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, K.V.; Spofford, L.S.; Aram, G.; McMullen, M.; Pumiglia, K.; Aplin, A.E. Adhesion Control of Cyclin D1 and P27Kip1 Levels Is Deregulated in Melanoma Cells through BRAF-MEK-ERK Signaling. Oncogene 2005, 24, 3459–3471. [Google Scholar] [CrossRef]
- Curtin, J.A.; Patel, H.N.; Cho, K.-H.; LeBoit, P.E. Distinct Sets of Genetic Alterations in Melanoma. N. Engl. J. Med. 2005, 353, 2135–2147. [Google Scholar] [CrossRef]
- Guo, L.; Qi, J.; Wang, H.; Jiang, X.; Liu, Y. Getting under the Skin: The Role of CDK4/6 in Melanomas. Eur. J. Med. Chem. 2020, 204, 112531. [Google Scholar] [CrossRef]
- Al-Mohanna, M.A.; Manogaran, P.S.; Al-Mukhalafi, Z.; Al-Hussein, K.A.; Aboussekhra, A. The tumor suppressor P16INK4a gene is a regulator of apoptosis induced by ultraviolet light and cisplatin. Oncogene 2004, 23, 201–212. [Google Scholar] [CrossRef]
- Al-Khalaf, H.H.; Mohideen, P.; Nallar, S.C.; Kalvakolanu, D.V.; Aboussekhra, A. The cyclin-dependent kinase inhibitor P16INK4a physically interacts with transcription factor Sp1 and cyclin-dependent kinase 4 to transactivate microRNA-141 and microRNA-146b-5p spontaneously and in response to ultraviolet light-induced DNA damage. J. Biol. Chem. 2013, 288, 35511–35525. [Google Scholar] [CrossRef]
- Kannan, K.; Sharpless, N.E.; Xu, J.; O’Hagan, R.C.; Bosenberg, M.; Chin, L. Components of the Rb Pathway Are Critical Targets of UV Mutagenesis in a Murine Melanoma Model. Proc. Natl. Acad. Sci. USA 2003, 100, 1221–1225. [Google Scholar] [CrossRef]
- Abd Elmageed, Z.Y.; Gaur, R.L.; Williams, M.; Abdraboh, M.E.; Rao, P.N.; Raj, M.H.G.; Ismail, F.M.; Ouhtit, A. Characterization of Coordinated Immediate Responses by P16INK4A and P53 Pathways in UVB-Irradiated Human Skin Cells. J. Invest. Dermatol. 2009, 129, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Poratti, M.; Marzaro, G. Third-Generation CDK Inhibitors: A Review on the Synthesis and Binding Modes of Palbociclib, Ribociclib and Abemaciclib. Eur. J. Med. Chem. 2019, 172, 143–153. [Google Scholar] [CrossRef] [PubMed]
- VanArsdale, T.; Boshoff, C.; Arndt, K.T.; Abraham, R.T. Molecular Pathways: Targeting the Cyclin D-CDK4/6 Axis for Cancer Treatment. Clin. Cancer Res. 2015, 21, 2905–2910. [Google Scholar] [CrossRef]
- Food and Drug Administration (FDA). Summary of Product Characteristics Palbociclib. Available online: https://www.fda.gov/drugs/informationondrugs/approveddrugs/Ucm549978.htm (accessed on 27 May 2021).
- Food and Drug Administration (FDA). Summary of Product Characteristics Ribociclib. Available online: https://www.fda.gov/drugs/informationondrugs/approveddrugs/Ucm546438.Htm (accessed on 27 May 2021).
- Food and Drug Administration (FDA). Summary of Product Characteristics Abemaciclib. Available online: https://www.fda.gov/drugs/informationondrugs/approveddrugs/Ucm578081.Htm (accessed on 27 May 2021).
- Roncato, R.; Angelini, J.; Pani, A.; Cecchin, E.; Sartore-Bianchi, A.; Siena, S.; De Mattia, E.; Scaglione, F.; Toffoli, G. Cdk4/6 Inhibitors in Breast Cancer Treatment: Potential Interactions with Drug, Gene, and Pathophysiological Conditions. Int. J. Mol. Sci. 2020, 21, 6350. [Google Scholar] [CrossRef] [PubMed]
- Gelbert, L.M.; Cai, S.; Lin, X.; Sanchez-Martinez, C.; Del Prado, M.; Lallena, M.J.; Torres, R.; Ajamie, R.T.; Wishart, G.N.; Flack, R.S.; et al. Preclinical Characterization of the CDK4/6 Inhibitor LY2835219: In-Vivo Cell Cycle-Dependent/Independent Anti-Tumor Activities Alone/in Combination with Gemcitabine. Invest. New Drugs 2014, 32, 825–837. [Google Scholar] [CrossRef]
- Spring, L.M.; Wander, S.A.; Andre, F.; Moy, B.; Turner, N.C.; Bardia, A. Cyclin-Dependent Kinase 4 and 6 Inhibitors for Hormone Receptor-Positive Breast Cancer: Past, Present, and Future. Lancet 2020, 395, 817–827. [Google Scholar] [CrossRef]
- Thein, K.Z.; Htut, T.W.; Ball, S.; Swarup, S.; Sultan, A.; Hlaing, O.T. Venous thromboembolism risk in patients with hormone receptor-positive HER2-negative metastatic breast cancer treated with combined CDK 4/6 inhibitors plus endocrine therapy versus endocrine therapy alone: A systematic review and meta-analysis of randomized controlled trials. Breast Cancer Res. Treat. 2020, 183, 479–487. [Google Scholar] [CrossRef]
- Tripathy, D.; Hortobagyi, G.N.; Chan, A.; Im, S.-A.; Chia, S.; Yardley, D.; Esteva, F.J.; Hurvitz, S.A.; Ridolfi, A.; Slamon, D. Pooled Safety Analysis of First-Line Ribociclib (RIB) plus Endocrine Therapy (ET) in HR+/HER2– Advanced Breast Cancer (ABC). Ann. Oncol. 2019, 30, iii53. [Google Scholar] [CrossRef]
- Infante, J.R.; Cassier, P.A.; Gerecitano, J.F.; Witteveen, P.O.; Chugh, R.; Ribrag, V.; Chakraborty, A.; Matano, A.; Dobson, J.R.; Crystal, A.S.; et al. A Phase I Study of the Cyclin-Dependent Kinase 4/6 Inhibitor Ribociclib (LEE011) in Patients with Advanced Solid Tumors and Lymphomas. Clin. Cancer Res. 2016, 22, 5696–5705. [Google Scholar] [CrossRef]
- Sammons, S.L.; Topping, D.L.; Blackwell, K.L. HR+, HER2– Advanced Breast Cancer and CDK4/6 Inhibitors: Mode of Action, Clinical Activity, and Safety Profiles. Curr. Cancer Drug Targets 2017, 17, 637–649. [Google Scholar] [CrossRef]
- Tate, S.C.; Sykes, A.K.; Kulanthaivel, P.; Chan, E.M.; Turner, P.K.; Cronier, D.M. A Population Pharmacokinetic and Pharmacodynamic Analysis of Abemaciclib in a Phase I Clinical Trial in Cancer Patients. Clin. Pharmacokinet. 2018, 57, 335–344. [Google Scholar] [CrossRef]
- Patnaik, A.; Rosen, L.S.; Tolaney, S.M.; Tolcher, A.W.; Goldman, J.W.; Gandhi, L.; Papadopoulos, K.P.; Beeram, M.; Rasco, D.W.; Hilton, J.F.; et al. Efficacy and Safety of Abemaciclib, an Inhibitor of CDK4 and CDK6, for Patients with Breast Cancer, Non–Small Cell Lung Cancer, and Other Solid Tumors. Cancer Discov. 2016, 6, 740–753. [Google Scholar] [CrossRef] [PubMed]
- Braal, C.L.; Jongbloed, E.M.; Wilting, S.M.; Mathijssen, R.H.J.; Koolen, S.L.W.; Jager, A. Inhibiting CDK4/6 in Breast Cancer with Palbociclib, Ribociclib, and Abemaciclib: Similarities and Differences. Drugs 2020, 81, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Hayes, T.K.; Luo, F.; Cohen, O.; Goodale, A.B.; Lee, Y.; Pantel, S.; Bagul, M.; Piccioni, F.; Root, D.E.; Garraway, L.A.; et al. A Functional Landscape of Resistance to MEK1/2 and CDK4/6 Inhibition in NRAS-Mutant Melanoma. Cancer Res. 2019, 79, 2352–2366. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, A.; Bu, Y.; Qie, S.; Wrangle, J.; Camp, E.R.; Hazard, E.S.; Hardiman, G.; de Leeuw, R.; Knudsen, K.E.; Diehl, J.A. SLC36A1-MTORC1 Signaling Drives Acquired Resistance to CDK4/6 Inhibitors. Sci. Adv. 2019, 5, eaax6352. [Google Scholar] [CrossRef]
- Vilgelm, A.E.; Saleh, N.; Shattuck-Brandt, R.; Riemenschneider, K.; Slesur, L.; Chen, S.-C.; Johnson, C.A.; Yang, J.; Blevins, A.; Yan, C.; et al. MDM2 Antagonists Overcome Intrinsic Resistance to CDK4/6 Inhibition by Inducing P21. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Klein, M.E.; Dickson, M.A.; Antonescu, C.; Qin, L.-X.; Dooley, S.J.; Barlas, A.; Manova, K.; Schwartz, G.K.; Crago, A.M.; Singer, S.; et al. PDLIM7 and CDH18 Regulate the Turnover of MDM2 during CDK4/6 Inhibitor Therapy-Induced Senescence. Oncogene 2018, 37, 5066–5078. [Google Scholar] [CrossRef]
- AbuHammad, S.; Cullinane, C.; Martin, C.; Bacolas, Z.; Ward, T.; Chen, H.; Slater, A.; Ardley, K.; Kirby, L.; Chan, K.T.; et al. Regulation of PRMT5-MDM4 Axis Is Critical in the Response to CDK4/6 Inhibitors in Melanoma. Proc. Natl. Acad. Sci. USA 2019, 116, 17990–18000. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zou, W.; Zhang, J.; Zhang, Y.; Xu, Q.; Li, S.; Chen, C. Mechanisms of CDK4/6 Inhibitor Resistance in Luminal Breast Cancer. Front. Pharmacol. 2020, 11. [Google Scholar] [CrossRef]
- Yang, C.; Li, Z.; Bhatt, T.; Dickler, M.; Giri, D.; Scaltriti, M.; Baselga, J.; Rosen, N.; Chandarlapaty, S. Acquired CDK6 Amplification Promotes Breast Cancer Resistance to CDK4/6 Inhibitors and Loss of ER Signaling and Dependence. Oncogene 2017, 36, 2255–2264. [Google Scholar] [CrossRef]
- Witkiewicz, A.K.; Knudsen, K.E.; Dicker, A.P.; Knudsen, E.S. The Meaning of P16(Ink4a) Expression in Tumors: Functional Significance, Clinical Associations and Future Developments. Cell Cycle 2011, 10, 2497–2503. [Google Scholar] [CrossRef]
- DeMichele, A.; Clark, A.S.; Tan, K.S.; Heitjan, D.F.; Gramlich, K.; Gallagher, M.; Lal, P.; Feldman, M.; Zhang, P.; Colameco, C.; et al. CDK 4/6 Inhibitor Palbociclib (PD0332991) in Rb+ Advanced Breast Cancer: Phase II Activity, Safety, and Predictive Biomarker Assessment. Clin. Cancer Res. 2015, 21, 995–1001. [Google Scholar] [CrossRef]
- Formisano, L.; Stauffer, K.M.; Young, C.D.; Bhola, N.E.; Guerrero-Zotano, A.L.; Jansen, V.M.; Estrada, M.M.; Hutchinson, K.E.; Giltnane, J.M.; Schwarz, L.J.; et al. Association of FGFR1 with ERα Maintains Ligand-Independent ER Transcription and Mediates Resistance to Estrogen Deprivation in ER+ Breast Cancer. Clin. Cancer Res. 2017, 23, 6138–6150. [Google Scholar] [CrossRef]
- Formisano, L.; Lu, Y.; Servetto, A.; Hanker, A.B.; Jansen, V.M.; Bauer, J.A.; Sudhan, D.R.; Guerrero-Zotano, A.L.; Croessmann, S.; Guo, Y.; et al. Aberrant FGFR Signaling Mediates Resistance to CDK4/6 Inhibitors in ER+ Breast Cancer. Nat. Commun. 2019, 10, 1373. [Google Scholar] [CrossRef]
- Li, Z.; Razavi, P.; Li, Q.; Toy, W.; Liu, B.; Ping, C.; Hsieh, W.; Sanchez-Vega, F.; Brown, D.N.; Da Cruz Paula, A.F.; et al. Loss of the FAT1 Tumor Suppressor Promotes Resistance to CDK4/6 Inhibitors via the Hippo Pathway. Cancer Cell 2018, 34, 893.e8–905.e8. [Google Scholar] [CrossRef]
- Caldon, C.E.; Sergio, C.M.; Schütte, J.; Boersma, M.N.; Sutherland, R.L.; Carroll, J.S.; Musgrove, E.A. Estrogen Regulation of Cyclin E2 Requires Cyclin D1 but Not C-Myc. Mol. Cell. Biol. 2009, 29, 4623–4639. [Google Scholar] [CrossRef]
- Turner, N.C.; Liu, Y.; Zhu, Z.; Loi, S.; Colleoni, M.; Loibl, S.; DeMichele, A.; Harbeck, N.; André, F.; Bayar, M.A.; et al. Cyclin E1 Expression and Palbociclib Efficacy in Previously Treated Hormone Receptor-Positive Metastatic Breast Cancer. J. Clin. Oncol. 2019, 37, 1169–1178. [Google Scholar] [CrossRef] [PubMed]
- Vijayaraghavan, S.; Karakas, C.; Doostan, I.; Chen, X.; Bui, T.; Yi, M.; Raghavendra, A.S.; Zhao, Y.; Bashour, S.I.; Ibrahim, N.K.; et al. CDK4/6 and Autophagy Inhibitors Synergistically Induce Senescence in Rb Positive Cytoplasmic Cyclin E Negative Cancers. Nat. Commun. 2017, 8, 15916. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Lim, B.; Pearson, T.; Tripathy, D.; Ordentlich, P.; Ueno, N.T. Abstract P5-21-15: The Synergistic Antitumor Activity of Entinostat (MS-275) in Combination with Palbociclib (PD 0332991) in Estrogen Receptor-Positive and Triple-Negative Breast Cancer. Cancer Res. 2018, 78, P5-21. [Google Scholar] [CrossRef]
- Cornell, L.; Wander, S.A.; Visal, T.; Wagle, N.; Shapiro, G.I. MicroRNA-Mediated Suppression of the TGF-β Pathway Confers Transmissible and Reversible CDK4/6 Inhibitor Resistance. Cell Rep. 2019, 26, 2667.e7–2680.e7. [Google Scholar] [CrossRef]
- Zhang, J.; Bu, X.; Wang, H.; Zhu, Y.; Geng, Y.; Nihira, N.T.; Tan, Y.; Ci, Y.; Wu, F.; Dai, X.; et al. Cyclin D-CDK4 Kinase Destabilizes PD-L1 via Cullin 3-SPOP to Control Cancer Immune Surveillance. Nature 2018, 553, 91–95. [Google Scholar] [CrossRef]
- Yu, J.; Yan, J.; Guo, Q.; Chi, Z.; Tang, B.; Zheng, B.; Yu, J.; Yin, T.; Cheng, Z.; Wu, X.; et al. Genetic Aberrations in the CDK4 Pathway Are Associated with Innate Resistance to PD-1 Blockade in Chinese Patients with Non-Cutaneous Melanoma. Clin. Cancer Res. 2019, 25, 6511–6523. [Google Scholar] [CrossRef]
- Jerby-Arnon, L.; Shah, P.; Cuoco, M.S.; Rodman, C.; Su, M.-J.; Melms, J.C.; Leeson, R.; Kanodia, A.; Mei, S.; Lin, J.-R.; et al. A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade. Cell 2018, 175, 984.e24–997.e24. [Google Scholar] [CrossRef]
- Goel, S.; DeCristo, M.J.; Watt, A.C.; BrinJones, H.; Sceneay, J.; Li, B.B.; Khan, N.; Ubellacker, J.M.; Xie, S.; Metzger-Filho, O.; et al. CDK4/6 Inhibition Triggers Anti-Tumour Immunity. Nature 2017, 548, 471–475. [Google Scholar] [CrossRef]
- Stopfer, L.E.; Mesfin, J.M.; Joughin, B.A.; Lauffenburger, D.A.; White, F.M. Multiplexed Relative and Absolute Quantitative Immunopeptidomics Reveals MHC I Repertoire Alterations Induced by CDK4/6 Inhibition. Nat. Commun. 2020, 11, 2760. [Google Scholar] [CrossRef]
- Deng, J.; Wang, E.S.; Jenkins, R.W.; Li, S.; Dries, R.; Yates, K.; Chhabra, S.; Huang, W.; Liu, H.; Aref, A.R.; et al. CDK4/6 Inhibition Augments Antitumor Immunity by Enhancing T-Cell Activation. Cancer Discov. 2018, 8, 216–233. [Google Scholar] [CrossRef]
- Cheng, L.; Lopez-Beltran, A.; Massari, F.; MacLennan, G.T.; Montironi, R. Molecular Testing for BRAF Mutations to Inform Melanoma Treatment Decisions: A Move toward Precision Medicine. Mod. Pathol. 2018, 31, 24–38. [Google Scholar] [CrossRef]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved Survival with Vemurafenib in Melanoma with BRAF V600E Mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef]
- Kakadia, S.; Yarlagadda, N.; Awad, R.; Kundranda, M.; Niu, J.; Naraev, B.; Mina, L.; Dragovich, T.; Gimbel, M.; Mahmoud, F. Mechanisms of Resistance to BRAF and MEK Inhibitors and Clinical Update of US Food and Drug Administration-Approved Targeted Therapy in Advanced Melanoma. OncoTargets Ther. 2018, 11, 7095–7107. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Jung, M.; Kang, H.N.; Kim, H.; Park, C.-W.; Kim, S.-M.; Shin, S.J.; Kim, S.H.; Kim, S.G.; Kim, E.K.; et al. Oncogenic BRAF Fusions in Mucosal Melanomas Activate the MAPK Pathway and Are Sensitive to MEK/PI3K Inhibition or MEK/CDK4/6 Inhibition. Oncogene 2017, 36, 3334–3345. [Google Scholar] [CrossRef] [PubMed]
- Appenzeller, S.; Gesierich, A.; Thiem, A.; Hufnagel, A.; Jessen, C.; Kneitz, H.; Regensburger, M.; Schmidt, C.; Zirkenbach, V.; Bischler, T.; et al. The Identification of Patient-Specific Mutations Reveals Dual Pathway Activation in Most Patients with Melanoma and Activated Receptor Tyrosine Kinases in BRAF/NRAS Wild-Type Melanomas. Cancer 2019, 125, 586–600. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, A.; Lee, E.K.; Diehl, J.A. Induction of Therapeutic Senescence in Vemurafenib-Resistant Melanoma by Extended Inhibition of CDK4/6. Cancer Res. 2016, 76, 2990–3002. [Google Scholar] [CrossRef]
- Kale, A.; Sharma, A.; Stolzing, A.; Desprez, P.-Y.; Campisi, J. Role of Immune Cells in the Removal of Deleterious Senescent Cells. Immun. Ageing A 2020, 17, 16. [Google Scholar] [CrossRef] [PubMed]
- Teh, J.L.F.; Erkes, D.A.; Cheng, P.F.; Tiago, M.; Wilski, N.A.; Field, C.O.; Chervoneva, I.; Levesque, M.P.; Xu, X.; Dummer, R.; et al. Activation of CD8+ T Cells Contributes to Antitumor Effects of CDK4/6 Inhibitors plus MEK Inhibitors. Cancer Immunol. Res. 2020, 8, 1114–1121. [Google Scholar] [CrossRef] [PubMed]
- Wan, L.; Chen, M.; Cao, J.; Dai, X.; Yin, Q.; Zhang, J.; Song, S.-J.; Lu, Y.; Liu, J.; Inuzuka, H.; et al. The APC/C E3 Ligase Complex Activator FZR1 Restricts BRAF Oncogenic Function. Cancer Discov. 2017, 7, 424–441. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.A.; Cullinane, C.; Kirby, L.; Abuhammad, S.; Lelliott, E.J.; Waldeck, K.; Young, R.J.; Brajanovski, N.; Cameron, D.P.; Walker, R.; et al. Palbociclib Synergizes with BRAF and MEK Inhibitors in Treatment Naïve Melanoma but Not after the Development of BRAF Inhibitor Resistance. Int. J. Cancer 2018, 142, 2139–2152. [Google Scholar] [CrossRef] [PubMed]
- Kwong, L.N.; Costello, J.C.; Liu, H.; Jiang, S.; Helms, T.L.; Langsdorf, A.E.; Jakubosky, D.; Genovese, G.; Muller, F.L.; Jeong, J.H.; et al. Oncogenic NRAS Signaling Differentially Regulates Survival and Proliferation in Melanoma. Nat. Med. 2012, 18, 1503–1510. [Google Scholar] [CrossRef] [PubMed]
- Teh, J.L.F.; Cheng, P.F.; Purwin, T.J.; Nikbakht, N.; Patel, P.; Chervoneva, I.; Ertel, A.; Fortina, P.M.; Kleiber, I.; HooKim, K.; et al. In Vivo E2F Reporting Reveals Efficacious Schedules of MEK1/2-CDK4/6 Targeting and MTOR-S6 Resistance Mechanisms. Cancer Discov. 2018, 8, 568–581. [Google Scholar] [CrossRef]
- Schwartz, G.K.; LoRusso, P.M.; Dickson, M.A.; Randolph, S.S.; Shaik, M.N.; Wilner, K.D.; Courtney, R.; O’Dwyer, P.J. Phase I Study of PD 0332991, a Cyclin-Dependent Kinase Inhibitor, Administered in 3-Week Cycles (Schedule 2/1). Br. J. Cancer 2011, 104, 1862–1868. [Google Scholar] [CrossRef]
- Pant, S.; Bendell, J.C.; Sullivan, R.J.; Shapiro, G.; Millward, M.; Mi, G.; Yuen, E.; Willard, M.D.; Wang, D.; Joseph, S.; et al. A Phase I Dose Escalation (DE) Study of ERK Inhibitor, LY3214996, in Advanced (Adv) Cancer (CA) Patients (Pts). J. Clin. Oncol. 2019, 37, 3001. [Google Scholar] [CrossRef]
- Sahebjam, S.; Rhun, E.L.; Queirolo, P.; Jerusalem, G.; Subramaniam, D.; Bear, M.; Yang, Z.; Chen, Y.; Conte, P.F. A Phase II Study of Abemaciclib in Patients (Pts) with Brain Metastases (BM) Secondary to Non-Small Cell Lung Cancer (NSCLC) or Melanoma (MEL). Ann. Oncol. 2019, 30, v117. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Lorusso, P.M.; DeMichele, A.; Abramson, V.G.; Courtney, R.; Randolph, S.S.; Shaik, M.N.; Wilner, K.D.; O’Dwyer, P.J.; Schwartz, G.K. Phase I, dose-escalation trial of the oral cyclin-dependent kinase 4/6 inhibitor PD 0332991, administered using a 21-day schedule in patients with advanced cancer. Clin. Cancer Res. 2012. [Google Scholar] [CrossRef]
- Mao, L.; Cao, Y.; Sheng, X.; Bai, X.; Chi, Z.; Cui, C.; Wang, X.; Tang, B.; Lian, B.; Yan, X.; et al. Palbociclib (P) in Advanced Acral Lentiginous Melanoma (ALM) with CDK4 Pathway Gene Aberrations. J. Clin. Oncol. 2019, 37, 9528. [Google Scholar] [CrossRef]
- Tang, B.; Sheng, X.; Kong, Y.; Chi, Z.; Si, L.; Cui, C.; Yan, X.; Mao, L.; Lian, B.; Li, S.; et al. Palbociclib for Treatment of Metastatic Melanoma with Copy Number Variations of CDK4 Pathway: Case Report. Chin. Clin. Oncol. 2018, 7, 62. [Google Scholar] [CrossRef]
- Taylor, M.; Sosman, J.; Gonzalez, R.; Carlino, M.S.; Kittaneh, M.; Lolkema, M.P.; Miller, W.; Marino, A.; Zhang, V.; Bhansali, S.G.; et al. 1086O—Phase Ib/Ii Study of Lee011 (Cdk4/6 Inhibitor) and Lgx818 (Braf Inhibitor) in Braf-Mutant Melanoma. Ann. Oncol. 2014, 25, iv374. [Google Scholar] [CrossRef]
- Sosman, J.A.; Kittaneh, M.; Lolkema, M.P.J.K.; Postow, M.A.; Schwartz, G.; Franklin, C.; Matano, A.; Bhansali, S.; Parasuraman, S.; Kim, K. A phase 1b/2 study of LEE011 in combination with binimetinib (MEK162) in patients with NRAS-mutant melanoma: Early encouraging clinical activity. J. Clin. Oncol. 2014. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Bechter, O.; Wolter, P.; Lebbe, C.; Elez, E.; Miller, W.H.; Long, G.V.; Omlin, A.G.; Siena, S.; Calvo, E.; et al. A Phase Ib/II Dose-Escalation Study Evaluating Triple Combination Therapy with a BRAF (Encorafenib), MEK (Binimetinib), and CDK 4/6 (Ribociclib) Inhibitor in Patients (Pts) with BRAF V600-Mutant Solid Tumors and Melanoma. J. Clin. Oncol. 2017, 35, 9518. [Google Scholar] [CrossRef]
- Louveau, B.; Resche-Rigon, M.; Lesimple, T.; Pracht, M.; Baroudjian, B.; Delyon, J.; Jouenne, F.; Amini-Adle, M.; Dutriaux, C.; Da Meda, L.; et al. Phase I-II Open Label Multicenter Study of PD0332991 in BRAFV600mut Metastatic Melanoma Patients Harboring CDKN2A Loss and RB1 Expression and Treated with Vemurafenib. J. Clin. Oncol. 2019, 37, 9545. [Google Scholar] [CrossRef]
- Janku, F.; Iyer, G.; Spreafico, A.; Yamamoto, N.; Bang, Y.-J.; Elez, E.; De Jonge, M.J.; Groen, H.J.M.; Marmé, F.; Gollmer, K.; et al. A Phase I Study of LXH254 in Patients (Pts) with Advanced Solid Tumors Harboring MAPK Pathway Alterations. J. Clin. Oncol. 2018. [Google Scholar] [CrossRef]


{kind=link}
| Palbociclib (IBRANCE®) [42] | Ribociclib (KISQUALI®) [43] | Abemaciclib (VERZENIOS®) [44] | |
|---|---|---|---|
| Chemistry |  |  |  |
| Dose and schedule | 125 mg daily three weeks on/one week off | 600 mg daily three weeks on/one week off | 150 mg twice daily continuously |
| Administration | Oral | Oral | Oral |
| Form | Capsule | Tablet | Tablet |
| Activity IC50 CDK4 IC50 CDK6 IC50 CDK9 | CDK4 and CDK6 CDK4 ≈ CDK6 11 nM 16 nM NR | CDK4 and CDK6 CDK4 > CDK6 10 nM 39 nM NR | CDK4, CDK6, CDK1, CDK2, CDK7 and CDK9 CDK4 >> CDK6 2 nM 10 nM 57 nM |
| PK Tmax (h) T1/2 (h) Vd (L) | 4–12 24–34 2583 | 1–4 30–55 1090 | 8 17–38 690.3 |
| Lipophilicity (cLog P) | 2.7 | 2.3 | 5.5 |
| Protein binding | 85% | 70% | 96–98% |
| Bioavailability | 46% | NR | 45% |
| Metabolism | Hepatic (CYP3A4 and SULT2A1) | Hepatic (CYP3A4) | Hepatic (CYP3A4) |
| Metabolites | Glucuronide conjugate: 1.5% | M13 (CCI284, N-hydroxylation): 22% M4 (LEQ803, N-demethylation): 20% M1 (secondary glucuronide): 18% | M2 (N-desethylabemaciclib): 25% M18 (hydroxy-N-desethylabemaciclib): 13% M20 (hydroxyabemaciclib): 26% |
| Excretion | Feces: 74% Urine: 17% | Feces: 69.1% Urine: 22.6% | Feces: 81% Urine: 3.4% |
| Drug | Evidence | N° Patients | NCT Number |
|---|---|---|---|
| Abemaciclib | Phase I dose escalation and tumor-specific cohort study. Primary objective: safety and tolerability. Secondary objectives: pharmacokinetics, evaluate biomarkers, antitumor activity, and establish a recommended dose range | 225 total (26 melanoma) | NCT01394016 |
| Abemaciclib | Phase 2, non-randomized study. Primary end point: objective intracranial response rate. Secondary end point: intracranial clinical benefit, PFS, OS and safety. | 51 total (23 melanoma) | NCT02308020 |
| Palbociclib | Phase I, dose-finding, non-comparative study. Primary objectives: safety, identifying dose-limiting toxicities (DLT), the maximum administered dose and the maximum tolerated dose (MTD), and to establish the recommended dose for Phase II studies (RP2D). Secondary objectives: characterization of single-dose and steady-state pharmacokinetics (PK) and evaluation of preliminary anti-tumor activity. | 33 total (4 melanoma) | [90] |
| Palbociclib | Phase I, dose-finding, non-comparative study. Primary objectives: safety, DLT, maximum administered dose, MTD. Secondary objectives: characterization of single-dose and PK and evaluation of preliminary anti-tumor activity | 41 total (6 melanoma) | NCT00141297 |
| Palbociclib | Phase II, open-labeled study. Primary end point: ORR. Secondary end point: OS, PFS, safety | 15 (melanoma) | NCT03454919 |
| Palbociclib | Case report | 2 (melanoma) | [91] |
| Ribociclib | Phase I, dose-escalation study. Primary end point: MTD/recommended dose for expansion (RDE), and DLT. Secondary end point: safety, PK, pharmacodynamics (PD), and preliminary activity of ribociclib | 132 total (3 melanoma) | NCT01237236 |
| Ribociclib and Binimetinib | Phase 1b/2 study of LEE011 + binimetinib. Primary objective: estimate MTD/RP2D. Secondary objectives: safety, PK and efficacy. | 14 (melanoma) | NCT01781572 |
| Ribociclib and Encorafenib | Phase 1b/2. Primary objective safety and efficacy | 18 (melanoma) | NCT01777776 |
| Ribociclib, Encorafenib and Binimetinib | Phase 1b, multicenter study, primary objective: MTD, DLT and ORR | 63 (melanoma) | NCT01543698 |
| Palbociclib and Vemurafenib | Phase I–II, multicenter study. Primary objective DLT, secondary objective: efficacy, tolerance and one year survival rate | 99 (melanoma) | NCT02202200 |
| Study | Phase | Setting | Investigated Drug | N. | Primary Outcome | Current Status | Estimated End |
|---|---|---|---|---|---|---|---|
| Melanoma specific studies | |||||||
| NCT04720768 (CELEBRATE) | Ib/II | Metastatic or unresectable untreated or previously treated melanoma BRAF V600 mutant | Encorafenib + Binimetinib + Palbociclib | 78 | DLTs | Recruiting | December 2023 |
| NCT02974725 * | Ib | Metastatic or advanced cutaneous previously treated melanoma NRAS mutant | LXH254 + Ribociclib | 331 | DLTs and safety | Recruiting | May 2022 |
| NCT02159066 (LOGIC-2) | II | Metastatic or unresectable melanoma BRAF V600 mutant progressed to prior Encorafenib + Binimetinib | Encorafenib + Binimetinib + Ribociclib | 160 | ORR | Active, not recruiting | January 2022 |
| NCT04417621 | II | Metastatic or unresectable previously treated melanoma BRAF V600 or NRAS mutant | LXH254 + Ribociclib | 320 | ORR | Recruiting | April 2023 |
| NCT03484923 (PLATforM) | II | Metastatic or unresectable previously treated melanoma | Spartalizumab + Ribociclib | 195 | ORR | Recruiting | June 2022 |
| NCT02645149 (MatchMel) | II | Metastatic or unresectable melanoma | Trametinib + Ribociclib | 1000 | Type and frequency of genetic aberrations in BRAF/NRAS wt metastatic melanoma and proportion of BRAF/NRAS wt receiving target therapy | Not yet recruiting (May 2021) | December 2028 |
| Non-melanoma specific studies | |||||||
| NCT02465060 (MATCH) | II | Metastatic or recurrent previously treated melanoma | Palbociclib | 6452 | ORR | Recruiting | June 2022 |
| NCT02857270 | I | Metastatic melanoma NRAS or BRAF mutant progressed to target therapy | LY3214996 + Abemaciclib | 245 | LY3214996 DLTs | Recruiting (not yet recruiting for melanoma cohort) | September 2021 |
| NCT02791334 (PACT) | I | Metastatic cutaneous melanoma | LY3300054 + Abemaciclib | 215 | LY3300054 DLTs | Active, not recruiting | December 2021 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garutti, M.; Targato, G.; Buriolla, S.; Palmero, L.; Minisini, A.M.; Puglisi, F. CDK4/6 Inhibitors in Melanoma: A Comprehensive Review. Cells 2021, 10, 1334. https://doi.org/10.3390/cells10061334
Garutti M, Targato G, Buriolla S, Palmero L, Minisini AM, Puglisi F. CDK4/6 Inhibitors in Melanoma: A Comprehensive Review. Cells. 2021; 10(6):1334. https://doi.org/10.3390/cells10061334
Chicago/Turabian StyleGarutti, Mattia, Giada Targato, Silvia Buriolla, Lorenza Palmero, Alessandro Marco Minisini, and Fabio Puglisi. 2021. "CDK4/6 Inhibitors in Melanoma: A Comprehensive Review" Cells 10, no. 6: 1334. https://doi.org/10.3390/cells10061334
APA StyleGarutti, M., Targato, G., Buriolla, S., Palmero, L., Minisini, A. M., & Puglisi, F. (2021). CDK4/6 Inhibitors in Melanoma: A Comprehensive Review. Cells, 10(6), 1334. https://doi.org/10.3390/cells10061334