
What Guides Peripheral Immune Cells into the Central Nervous System?



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Inflammation in Multiple Sclerosis
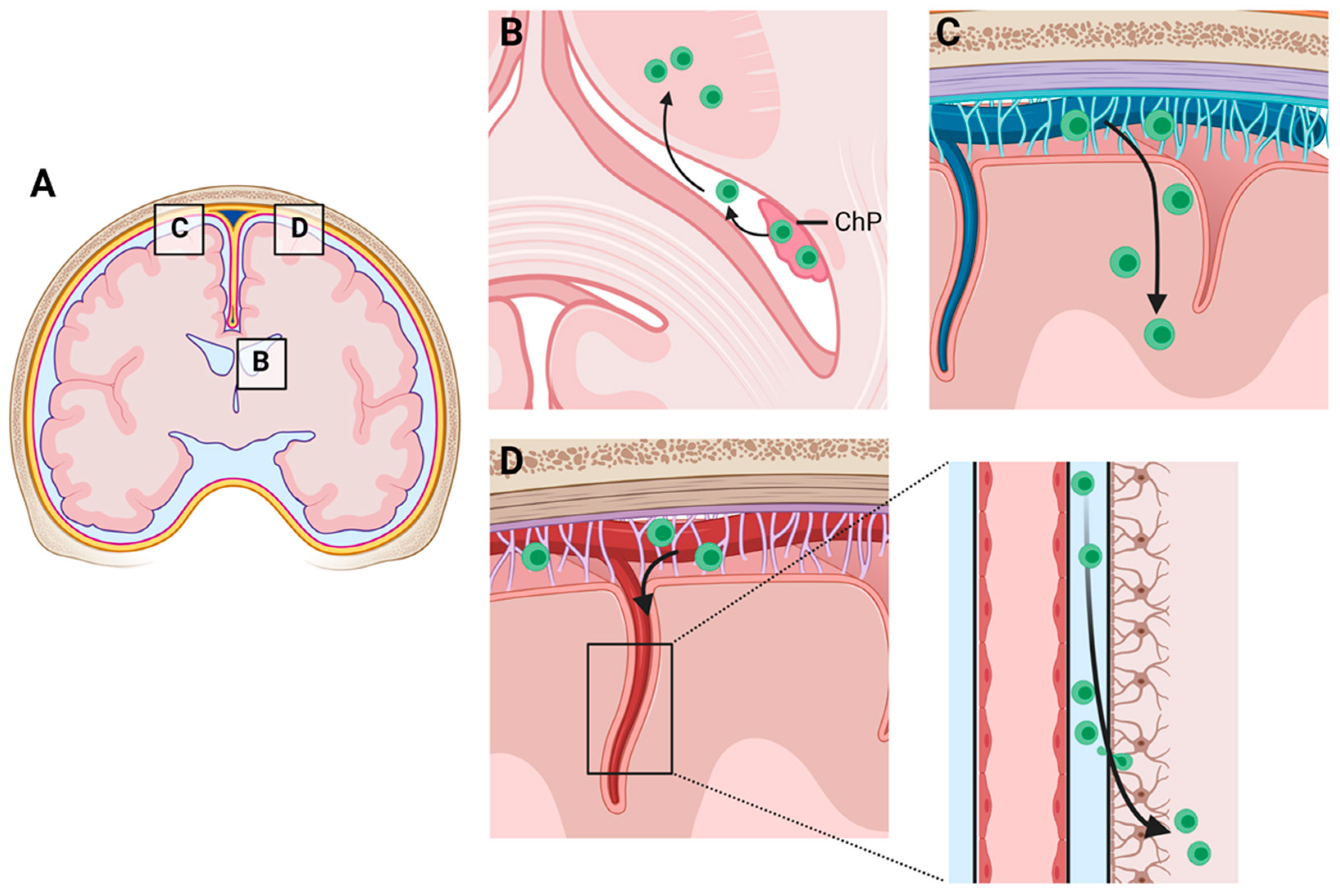
3. Along Which Neuroanatomical Pathways Do Peripheral Immune Cells Travel inside the CNS?
4. What Triggers Peripheral Immune Cell Recruitment?
5. Does a Degenerative Process in the CNS Trigger Autoimmunity?
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Steenwijk, M.D.; Geurts, J.J.; Daams, M.; Tijms, B.M.; Wink, A.M.; Balk, L.J.; Tewarie, P.K.; Uitdehaag, B.M.; Barkhof, F.; Vrenken, H.; et al. Cortical atrophy patterns in multiple sclerosis are non-random and clinically relevant. J. Neurol. 2016, 139, 115–126. [Google Scholar] [CrossRef]
- Tallantyre, E.C.; Bø, L.; Al-Rawashdeh, O.; Owens, T.; Polman, C.H.; Lowe, J.S.; Evangelou, N. Clinico-pathological evidence that axonal loss underlies disability in progressive multiple sclerosis. Mult. Scler. 2010, 16, 406–411. [Google Scholar] [CrossRef]
- Frischer, J.M.; Bramow, S.; Dal-Bianco, A.; Lucchinetti, C.F.; Rauschka, H.; Schmidbauer, M.; Laursen, H.; Sorensen, P.S.; Lassmann, H. The relation between inflammation and neurodegeneration in multiple sclerosis brains. J. Neurol. 2009, 132, 1175–1189. [Google Scholar] [CrossRef] [Green Version]
- Howell, O.W.; Reeves, C.A.; Nicholas, R.; Carassiti, D.; Radotra, B.; Gentleman, S.M.; Serafini, B.; Aloisi, F.; Roncaroli, F.; Magliozzi, R.; et al. Meningeal inflammation is widespread and linked to cortical pathology in multiple sclerosis. J. Neurol. 2011, 134, 2755–2771. [Google Scholar] [CrossRef] [PubMed]
- Serafini, B.; Rosicarelli, B.; Magliozzi, R.; Stigliano, E.; Aloisi, F. Detection of ectopic B-cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol. 2004, 14, 164–174. [Google Scholar] [CrossRef]
- Magliozzi, R.; Howell, O.; Vora, A.; Serafini, B.; Nicholas, R.; Puopolo, M.; Reynolds, R.; Aloisi, F. Meningeal B-cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. J. Neurol. 2007, 130, 1089–1104. [Google Scholar] [CrossRef] [PubMed]
- Roos, I.; Leray, E.; Casey, R.; Horakova, D.; Havrdova, E.; Izquierdo, G.; Madueño, S.E.; Patti, F.; Edan, G.; Debouverie, M.; et al. Effects of High and Low Efficacy Therapy in Secondary Progressive Multiple Sclerosis. Neurology 2021, 96. [Google Scholar] [CrossRef]
- Kapoor, R.; Ho, P.R.; Campbell, N.; Chang, I.; Deykin, A.; Forrestal, F.; Lucas, N.; Yu, B.; Arnold, D.L.; Freedman, M.S.; et al. Effect of natalizumab on disease progression in secondary progressive multiple sclerosis (ASCEND): A phase 3, randomised, double-blind, placebo-controlled trial with an open-label extension. Lancet Neurol. 2018, 17, 405–415. [Google Scholar] [CrossRef]
- Häusler, D.; Akgün, K.; Stork, L.; Lassmann, H.; Ziemssen, T.; Brück, W.; Metz, I. CNS inflammation after natalizumab therapy for multiple sclerosis: A retrospective histopathological and CSF cohort study. Brain Pathol. 2021, e12969. [Google Scholar] [CrossRef]
- Aboul-Enein, F.; Lassmann, H. Mitochondrial damage and histotoxic hypoxia: A pathway of tissue injury in inflammatory brain disease? Acta Neuropathol. Commun. 2005, 109, 49–55. [Google Scholar] [CrossRef]
- Smith, K.J.; Lassmann, H. The role of nitric oxide in multiple sclerosis. Lancet Neurol. 2002, 1, 232–241. [Google Scholar] [CrossRef]
- Funfschilling, U.; Supplie, L.M.; Mahad, D.; Boretius, S.; Saab, A.S.; Edgar, J.; Brinkmann, B.G.; Kassmann, C.M.; Tzvetanova, I.D.; Mobius, W.; et al. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature 2012, 485, 517–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, J.M.; McLaughlin, M.; Yool, D.; Zhang, S.C.; Fowler, J.H.; Montague, P.; Barrie, J.A.; McCulloch, M.C.; Duncan, I.D.; Garbern, J.; et al. Oligodendroglial modulation of fast axonal transport in a mouse model of hereditary spastic paraplegia. J. Cell Biol. 2004, 166, 121–131. [Google Scholar] [CrossRef]
- Uschkureit, T.; Sporkel, O.; Stracke, J.; Bussow, H.; Stoffel, W. Early onset of axonal degeneration in double (plp-/-mag-/-) and hypomyelinosis in triple (plp-/-mbp-/-mag-/-) mutant mice. J. Neurosci. 2000, 20, 5225–5233. [Google Scholar] [CrossRef] [Green Version]
- Thompson, A.J.; Kermode, A.G.; Wicks, D.; MacManus, D.G.; Kendall, B.E.; Kingsley, D.P.; McDonald, W.I. Major differences in the dynamics of primary and secondary progressive multiple sclerosis. Ann. Neurol. 1991, 29, 53–62. [Google Scholar] [CrossRef] [PubMed]
- van Walderveen, M.A.; Barkhof, F.; Tas, M.W.; Polman, C.; Frequin, S.T.; Hommes, O.R.; Thompson, A.J.; Valk, J. Patterns of brain magnetic resonance abnormalities on T2-weighted spin echo images in clinical subgroups of multiple sclerosis: A large cross-sectional study. Eur. Neurol. 1998, 40, 91–98. [Google Scholar] [CrossRef]
- Stevenson, V.L.; Miller, D.H.; Rovaris, M.; Barkhof, F.; Brochet, B.; Dousset, V.; Dousset, V.; Filippi, M.; Montalban, X.; Polman, C.H.; et al. Primary and transitional progressive MS: A clinical and MRI cross-sectional study. Neurology 1999, 52, 839–845. [Google Scholar] [CrossRef]
- Revesz, T.; Kidd, D.; Thompson, A.J.; Barnard, R.O.; McDonald, W.I. A comparison of the pathology of primary and secondary progressive multiple sclerosis. J. Neurol. 1994, 117, 759–765. [Google Scholar] [CrossRef] [PubMed]
- Prat, A.; Pelletier, D.; Duquette, P.; Arnold, D.L.; Antel, J.P. Heterogeneity of T-lymphocyte function in primary progressive multiple sclerosis: Relation to magnetic resonance imaging lesion volume. Ann. Neurol. 2000, 47, 234–237. [Google Scholar]
- Haider, L.; Simeonidou, C.; Steinberger, G.; Hametner, S.; Grigoriadis, N.; Deretzi, G.; Kovacs, G.G.; Kutzelnigg, A.; Lassmann, H.; Frischer, J.M. Multiple sclerosis deep grey matter: The relation between demyelination, neurodegeneration, inflammation and iron. J. Neurol. Neurosurg. Psychiatry 2014, 85, 1386–1395. [Google Scholar] [CrossRef] [Green Version]
- Eden, D.; Gros, C.; Badji, A.; Dupont, S.M.; De Leener, B.; Maranzano, J.; Zhuoquiong, R.; Liu, Y.; Granberg, T.; Ouellette, R.; et al. Spatial distribution of multiple sclerosis lesions in the cervical spinal cord. J. Neurol. 2019, 142, 633–646. [Google Scholar] [CrossRef]
- Gross, C.C.; Schulte-Mecklenbeck, A.; Hanning, U.; Posevitz-Fejfár, A.; Korsukewitz, C.; Schwab, N.; Meuth, S.G.; Wiendl, H.; Klotz, L. Distinct pattern of lesion distribution in multiple sclerosis is associated with different circulating T-helper and helper-like innate lymphoid cell subsets. Mult. Scler. 2017, 23, 1025–1030. [Google Scholar] [CrossRef]
- Kubes, P.; Ward, P.A. Leukocyte recruitment and the acute inflammatory response. Brain Pathol. 2000, 10, 127–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engelhardt, B.; Ransohoff, R.M. The ins and outs of T-lymphocyte trafficking to the CNS: Anatomical sites and molecular mechanisms. Trends Immunol. 2005, 26, 485–495. [Google Scholar] [CrossRef]
- Berlin, C.; Berg, E.L.; Briskin, M.J.; Andrew, D.P.; Kilshaw, P.J.; Holzmann, B.; Weissman, I.L.; Hamann, A.; Butcher, E.C. Alpha 4 beta 7 integrin mediates lymphocyte binding to the mucosal vascular addressin MAdCAM-1. Cell 1993, 74, 185–195. [Google Scholar] [CrossRef]
- Yednock, T.A.; Cannon, C.; Fritz, L.C.; Sanchez-Madrid, F.; Steinman, L.; Karin, N. Prevention of experimental autoimmune encephalomyelitis by antibodies against alpha 4 beta 1 integrin. Nature 1992, 356, 63–66. [Google Scholar] [CrossRef]
- Greenwood, J.; Wang, Y.; Calder, V.L. Lymphocyte adhesion and transendothelial migration in the central nervous system: The role of LFA-1, ICAM-1, VLA-4 and VCAM-1. off. Immunology 1995, 86, 408–415. [Google Scholar]
- Kramann, N.; Menken, L.; Pförtner, R.; Schmid, S.N.; Stadelmann, C.; Wegner, C.; Brück, W. Glial fibrillary acidic protein expression alters astrocytic chemokine release and protects mice from cuprizone-induced demyelination. Glia 2019, 67, 1308–1319. [Google Scholar] [CrossRef] [PubMed]
- Clarner, T.; Janssen, K.; Nellessen, L.; Stangel, M.; Skripuletz, T.; Krauspe, B.; Hess, F.M.; Denecke, B.; Beutner, C.; Linnartz-Gerlach, B.; et al. CXCL10 triggers early microglial activation in the cuprizone model. J. Immunol. 2015, 194, 3400–3413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cepok, S.; Jacobsen, M.; Schock, S.; Omer, B.; Jaekel, S.; Böddeker, I.; Oertel, W.H.; Sommer, N.; Hemmer, B. Patterns of cerebrospinal fluid pathology correlate with disease progression in multiple sclerosis. J. Neurol. 2001, 124, 2169–2176. [Google Scholar] [CrossRef] [Green Version]
- Reboldi, A.; Coisne, C.; Baumjohann, D.; Benvenuto, F.; Bottinelli, D.; Lira, S.; Uccelli, A.; Lanzavecchia, A.; Engelhardt, B.; Sallusto, F. C-C chemokine receptor 6-regulated entry of TH-17 cells into the CNS through the choroid plexus is required for the initiation of EAE. Nat. Immunol. 2009, 10, 514–523. [Google Scholar] [CrossRef]
- Vercellino, M.; Votta, B.; Condello, C.; Piacentino, C.; Romagnolo, A.; Merola, A.; Capello, E.; Mancardi, G.L.; Mutani, R.; Giordana, M.T.; et al. Involvement of the choroid plexus in multiple sclerosis autoimmune inflammation: A neuropathological study. J. Neuroimmunol. 2008, 199, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Lorenzo, S.; Konings, J.; van der Pol, S.; Kamermans, A.; Amor, S.; van Horssen, J.; Witte, M.E.; Kooij, G.; de Vries, H.E. Inflammation of the choroid plexus in progressive multiple sclerosis: Accumulation of granulocytes and T cells. Acta Neuropathol. Commun. 2020, 8, 9. [Google Scholar] [CrossRef]
- Kooij, G.; Kopplin, K.; Blasig, R.; Stuiver, M.; Koning, N.; Goverse, G.; van der Pol, S.M.; van Het Hof, B.; Gollasch, M.; Drexhage, J.A.; et al. Disturbed function of the blood-cerebrospinal fluid barrier aggravates neuro-inflammation. Acta Neuropathol. 2014, 128, 267–277. [Google Scholar] [CrossRef] [PubMed]
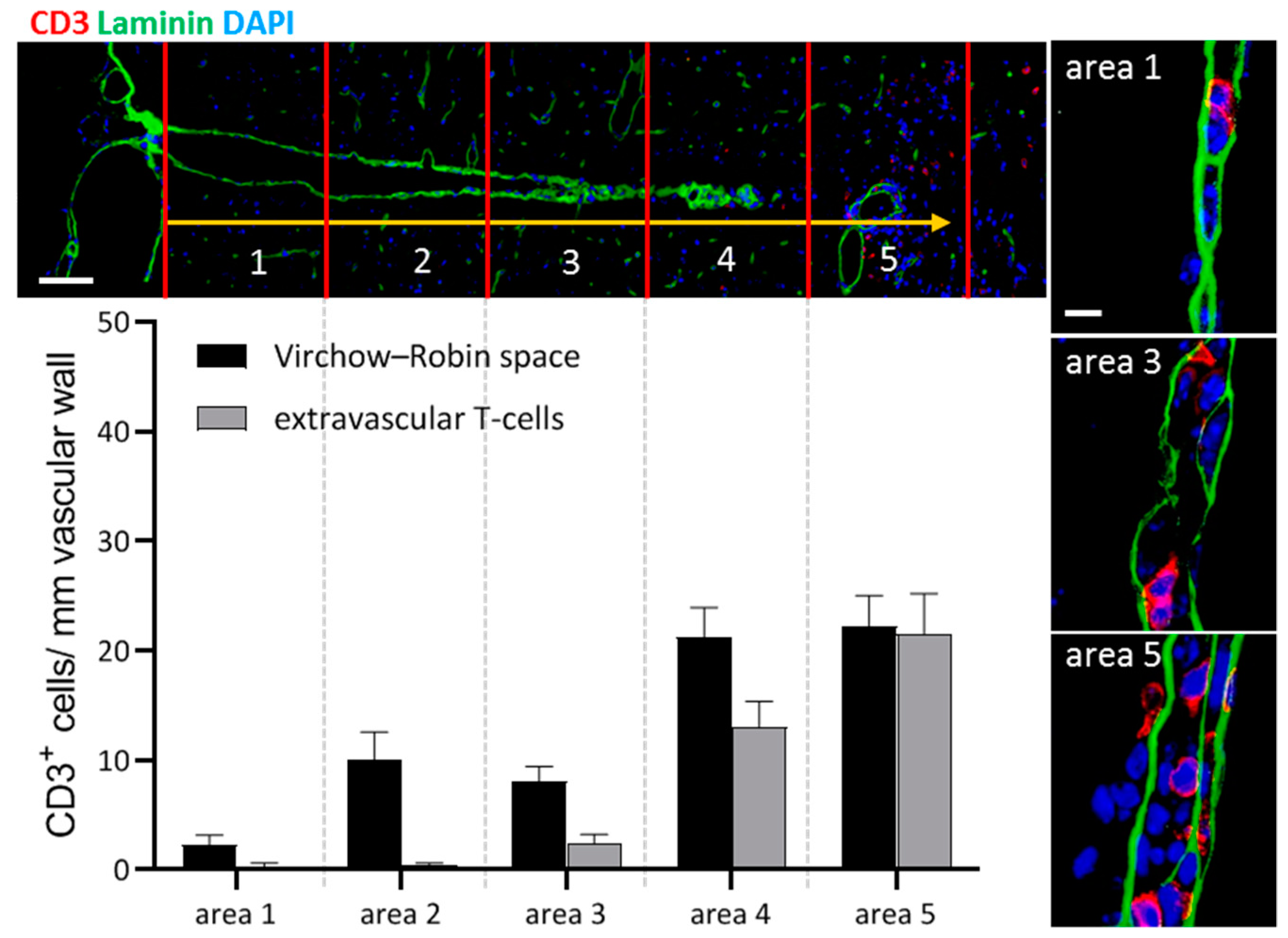
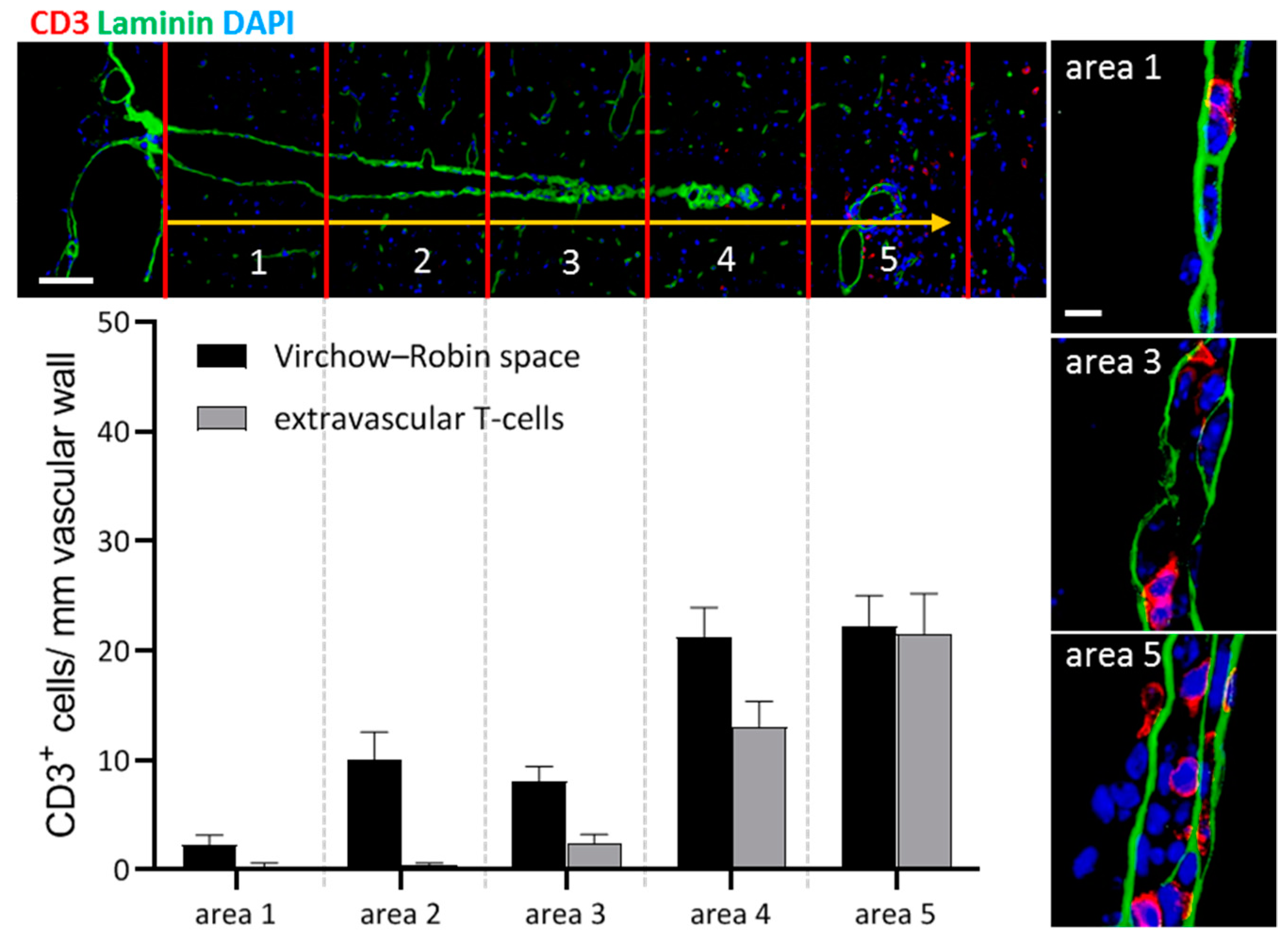
- Bartholomäus, I.; Kawakami, N.; Odoardi, F.; Schläger, C.; Miljkovic, D.; Ellwart, J.W.; Klinkert, W.E.; Flügel-Koch, C.; Issekutz, T.B.; Wekerle, H.; et al. Effector T cell interactions with meningeal vascular structures in nascent autoimmune CNS lesions. Nature 2009, 462, 94–98. [Google Scholar] [CrossRef]
- Schläger, C.; Körner, H.; Krueger, M.; Vidoli, S.; Haberl, M.; Mielke, D.; Brylla, E.; Issekutz, T.; Cabañas, C.; Nelson, P.J.; et al. Effector T-cell trafficking between the leptomeninges and the cerebrospinal fluid. Nature 2016, 530, 349–353. [Google Scholar] [CrossRef]
- Barnett, M.H.; Prineas, J.W. Relapsing and remitting multiple sclerosis: Pathology of the newly forming lesion. Ann. Neurol. 2004, 55, 458–468. [Google Scholar] [CrossRef]
- De Groot, C.J.; Bergers, E.; Kamphorst, W.; Ravid, R.; Polman, C.H.; Barkhof, F.; van der Valk, P. Post-mortem MRI-guided sampling of multiple sclerosis brain lesions: Increased yield of active demyelinating and (p)reactive lesions. Brain 2001, 124, 1635–1645. [Google Scholar] [CrossRef] [Green Version]
- Stys, P.K.; Zamponi, G.W.; van Minnen, J.; Geurts, J.J. Will the real multiple sclerosis please stand up? Nat. Rev. Neurosci. 2012, 13, 507–514. [Google Scholar] [CrossRef] [PubMed]
- van Noort, J.M.; van den Elsen, P.J.; van Horssen, J.; Geurts, J.J.; van der Valk, P.; Amor, S. Preactive multiple sclerosis lesions offer novel clues for neuroprotective therapeutic strategies. CNS Neurol. Disord. Drug Targets 2011, 10, 68–81. [Google Scholar] [CrossRef]
- van der Valk, P.; Amor, S. Preactive lesions in multiple sclerosis. Curr. Opin. Neurol. 2009, 22, 207–213. [Google Scholar] [CrossRef]
- van Noort, J.M.; Bsibsi, M.; Gerritsen, W.H.; van der Valk, P.; Bajramovic, J.J.; Steinman, L.; Amor, S. Alphab-crystallin is a target for adaptive immune responses and a trigger of innate responses in preactive multiple sclerosis lesions. J. Neuropathol. Exp. Neurol. 2010, 69, 694–703. [Google Scholar] [CrossRef] [Green Version]
- Filippi, M.; Rocca, M.A.; Martino, G.; Horsfield, M.A.; Comi, G. Magnetization transfer changes in the normal appearing white matter precede the appearance of enhancing lesions in patients with multiple sclerosis. Ann. Neurol. 1998, 43, 809–814. [Google Scholar] [CrossRef] [PubMed]
- Maggi, P.; Macri, S.M.; Gaitan, M.I.; Leibovitch, E.; Wholer, J.E.; Knight, H.L.; Ellis, M.; Wu, T.; Silva, A.C.; Massacesi, L.; et al. The formation of inflammatory demyelinated lesions in cerebral white matter. Ann. Neurol. 2014, 76, 594–608. [Google Scholar] [CrossRef] [PubMed]
- Werring, D.J.; Brassat, D.; Droogan, A.G.; Clark, C.A.; Symms, M.R.; Barker, G.J.; MacManus, D.G.; Thompson, A.J.; Miller, D.H. The pathogenesis of lesions and normal-appearing white matter changes in multiple sclerosis: A serial diffusion MRI study. J. Neurol. 2000, 123, 1667–1676. [Google Scholar] [CrossRef] [Green Version]
- Laule, C.; Vavasour, I.M.; Whittall, K.P.; Oger, J.; Paty, D.W.; Li, D.K.; MacKay, A.L.; Arnold, D.L. Evolution of focal and diffuse magnetisation transfer abnormalities in multiple sclerosis. J. Neurol. 2003, 250, 924–931. [Google Scholar] [CrossRef]
- Fischbach, F.; Nedelcu, J.; Leopold, P.; Zhan, J.; Clarner, T.; Nellessen, L.; Beissel, C.; van Heuvel, Y.; Goswami, A.; Weis, J.; et al. Cuprizone-induced graded oligodendrocyte vulnerability is regulated by the transcription factor DNA damage-inducible transcript 3. Glia 2019, 67, 263–276. [Google Scholar] [CrossRef]
- Clarner, T.; Diederichs, F.; Berger, K.; Denecke, B.; Gan, L.; van der Valk, P.; Beyer, C.; Amor, S.; Kipp, M. Myelin debris regulates inflammatory responses in an experimental demyelination animal model and multiple sclerosis lesions. Glia 2012, 60, 1468–1480. [Google Scholar] [CrossRef]
- Skripuletz, T.; Lindner, M.; Kotsiari, A.; Garde, N.; Fokuhl, J.; Linsmeier, F.; Trebst, C.; Stangel, M. Cortical demyelination is prominent in the murine cuprizone model and is strain-dependent. Am. J. Pathol. 2008, 172, 1053–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norkute, A.; Hieble, A.; Braun, A.; Johann, S.; Clarner, T.; Baumgartner, W.; Beyer, C.; Kipp, M. Cuprizone treatment induces demyelination and astrocytosis in the mouse hippocampus. J. Neurosci. Res. 2009, 87, 1343–1355. [Google Scholar] [CrossRef]
- Herder, V.; Hansmann, F.; Stangel, M.; Skripuletz, T.; Baumgartner, W.; Beineke, A. Lack of cuprizone-induced demyelination in the murine spinal cord despite oligodendroglial alterations substantiates the concept of site-specific susceptibilities of the central nervous system. Neuropathol. Appl. Neurobiol. 2011, 37, 676–684. [Google Scholar] [CrossRef]
- Iglesias, A.; Bauer, J.; Litzenburger, T.; Schubart, A.; Linington, C. T- and B-cell responses to myelin oligodendrocyte glycoprotein in experimental autoimmune encephalomyelitis and multiple sclerosis. Glia 2001, 36, 220–234. [Google Scholar] [CrossRef] [PubMed]
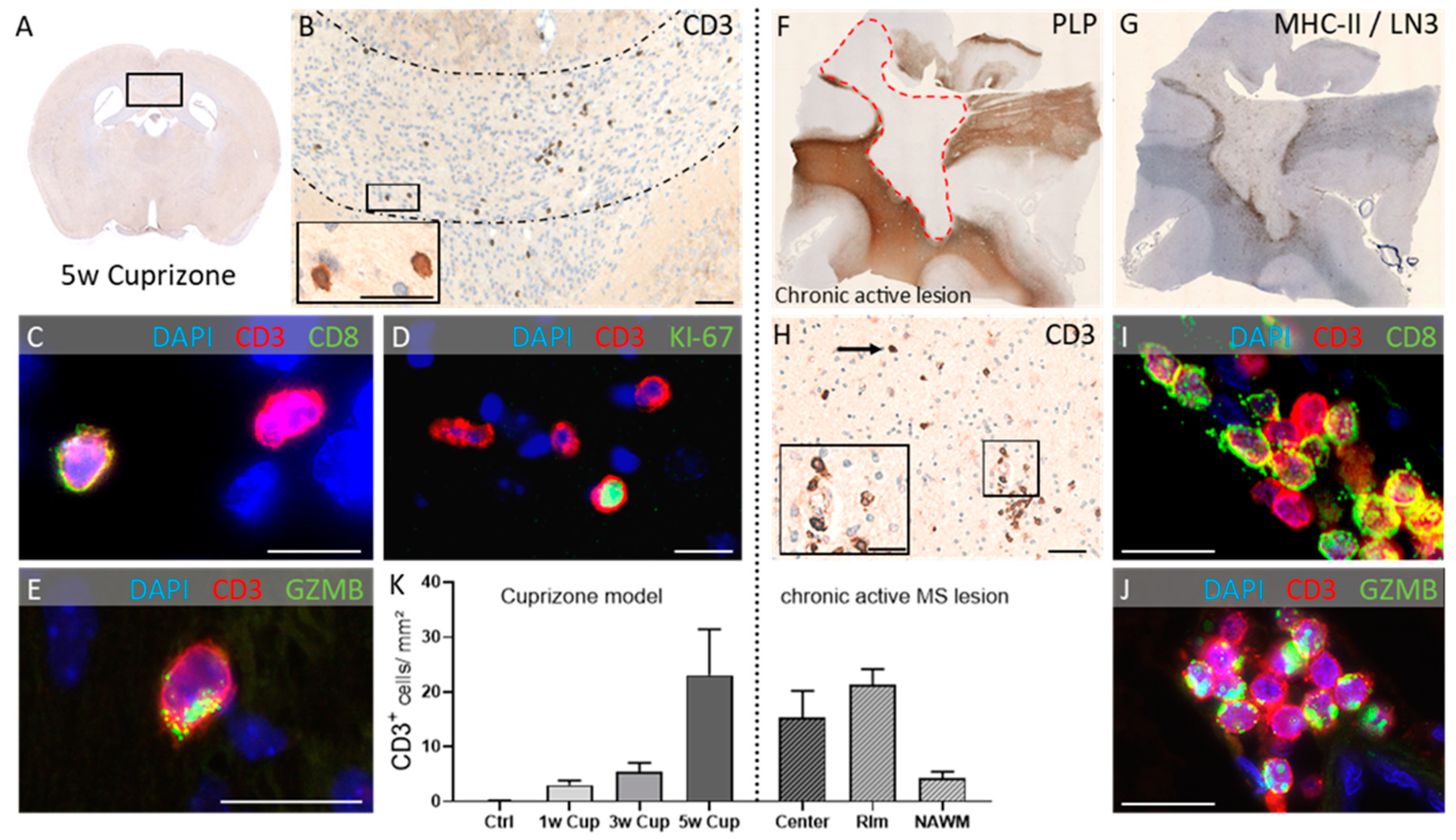
- Scheld, M.; Ruther, B.J.; Grosse-Veldmann, R.; Ohl, K.; Tenbrock, K.; Dreymuller, D.; Fallier-Becker, P.; Zendedel, A.; Beyer, C.; Clarner, T.; et al. Neurodegeneration Triggers Peripheral Immune Cell Recruitment into the Forebrain. J. Neurosci. 2016, 36, 1410–1415. [Google Scholar] [CrossRef] [PubMed]
- Ruther, B.J.; Scheld, M.; Dreymueller, D.; Clarner, T.; Kress, E.; Brandenburg, L.O.; Swartenbroekx, T.; Hoornaert, C.; Ponsaerts, P.; Fallier-Becker, P.; et al. Combination of cuprizone and experimental autoimmune encephalomyelitis to study inflammatory brain lesion formation and progression. Glia 2017, 65, 1900–1913. [Google Scholar] [CrossRef] [PubMed]
- Marik, C.; Felts, P.A.; Bauer, J.; Lassmann, H.; Smith, K.J. Lesion genesis in a subset of patients with multiple sclerosis: A role for innate immunity? J. Neurol. 2007, 130, 2800–2815. [Google Scholar] [CrossRef] [Green Version]
- Van der Valk, P.; De Groot, C.J. Staging of multiple sclerosis (MS) lesions: Pathology of the time frame of MS. Neuropathol. Appl. Neurobiol. 2000, 26, 2–10. [Google Scholar] [CrossRef]
- Chrzanowski, U.; Bhattarai, S.; Scheld, M.; Clarner, T.; Fallier-Becker, P.; Beyer, C.; Rohr, S.O.; Schmitz, C.; Hochstrasser, T.; Schweiger, F.; et al. Oligodendrocyte degeneration and concomitant microglia activation directs peripheral immune cells into the forebrain. Neurochem. Int. 2019, 126, 139–153. [Google Scholar] [CrossRef]
- Baxi, E.G.; DeBruin, J.; Tosi, D.M.; Grishkan, I.V.; Smith, M.D.; Kirby, L.A.; Strasburger, H.J.; Fairchild, A.N.; Calabresi, P.A.; Gocke, A.R. Transfer of myelin-reactive th17 cells impairs endogenous remyelination in the central nervous system of cuprizone-fed mice. J. Neurosci. 2015, 35, 8626–8639. [Google Scholar] [CrossRef]
- Kneussel, M.; Friese, M.A. SnapShot: Neuronal dysfunction in inflammation. Neuron 2021, 109, 1754. [Google Scholar] [CrossRef] [PubMed]
- Zarrouk, A.; Nury, T.; El Hajj, H.I.; Gondcaille, C.; Andreoletti, P.; Moreau, T.; Cherkaoui-Malki, M.; Berger, J.; Hammami, M.; Lizard, G.; et al. Potential Involvement of Peroxisome in Multiple Sclerosis and Alzheimer’s Disease : Peroxisome and Neurodegeneration. Adv. Exp. Med. Biol. 2020, 1299, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Linnerbauer, M.; Wheeler, M.A.; Quintana, F.J. Astrocyte Crosstalk in CNS Inflammation. Neuron 2020, 108, 608–622. [Google Scholar] [CrossRef]
- Colonna, M.; Brioschi, S. Neuroinflammation and neurodegeneration in human brain at single-cell resolution. Nat. Rev. Immunol. 2020, 20, 81–82. [Google Scholar] [CrossRef] [PubMed]
- Sommer, A.; Marxreiter, F.; Krach, F.; Fadler, T.; Grosch, J.; Maroni, M.; Graef, D.; Eberhardt, E.; Riemenschneider, M.J.; Yeo, G.W.; et al. Th17 Lymphocytes Induce Neuronal Cell Death in a Human iPSC-Based Model of Parkinson’s Disease. Cell Stem Cell 2019, 24, 1006. [Google Scholar] [CrossRef] [Green Version]
- Nitsch, R.; Pohl, E.E.; Smorodchenko, A.; Infante-Duarte, C.; Aktas, O.; Zipp, F. Direct impact of T cells on neurons revealed by two-photon microscopy in living brain tissue. J. Neurosci. 2004, 24, 2458–2464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siffrin, V.; Radbruch, H.; Glumm, R.; Niesner, R.; Paterka, M.; Herz, J.; Leuenberger, T.; Lehmann, S.M.; Luenstedt, S.; Rinnenthal, J.L.; et al. In vivo imaging of partially reversible th17 cell-induced neuronal dysfunction in the course of encephalomyelitis. Immunity 2010, 33, 424–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dombrowski, Y.; O’Hagan, T.; Dittmer, M.; Penalva, R.; Mayoral, S.R.; Bankhead, P.; Fleville, S.; Eleftheriadis, G.; Zhao, C.; Naughton, M.; et al. Regulatory T cells promote myelin regeneration in the central nervous system. Nat. Neurosci. 2017, 20, 674–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rook, G.A.; Lowry, C.A.; Raison, C.L. Lymphocytes in neuroprotection, cognition and emotion: Is intolerance really the answer? Brain Behav. Immun. 2011, 25, 591–601. [Google Scholar] [CrossRef]
- Chiu, I.M.; Chen, A.; Zheng, Y.; Kosaras, B.; Tsiftsoglou, S.A.; Vartanian, T.K.; Brown, R.H., Jr.; Carroll, M.C. T lymphocytes potentiate endogenous neuroprotective inflammation in a mouse model of ALS. Proc. Natl. Acad. Sci. USA 2008, 105, 17913–17918. [Google Scholar] [CrossRef] [Green Version]
- Caprariello, A.V.; Rogers, J.A.; Morgan, M.L.; Hoghooghi, V.; Plemel, J.R.; Koebel, A.; Tsutsui, S.; Dunn, J.F.; Kotra, L.P.; Ousman, S.S.; et al. Biochemically altered myelin triggers autoimmune demyelination. Proc. Natl. Acad. Sci. USA 2018, 115, 5528–5533. [Google Scholar] [CrossRef] [Green Version]
- Raivich, G.; Jones, L.L.; Kloss, C.U.; Werner, A.; Neumann, H.; Kreutzberg, G.W. Immune surveillance in the injured nervous system: T-lymphocytes invade the axotomized mouse facial motor nucleus and aggregate around sites of neuronal degeneration. J. Neurosci. 1998, 18, 5804–5816. [Google Scholar] [CrossRef] [Green Version]
- Konno, H.; Yamamoto, T.; Suzuki, H.; Yamamoto, H.; Iwasaki, Y.; Ohara, Y.; Terunuma, H.; Harata, N. Targeting of adoptively transferred experimental allergic encephalitis lesion at the sites of wallerian degeneration. Acta Neuropathol. 1990, 80, 521–526. [Google Scholar] [CrossRef] [PubMed]
- Graves, M.C.; Fiala, M.; Dinglasan, L.A.; Liu, N.Q.; Sayre, J.; Chiappelli, F.; van Kooten, C.; Vinters, H.V. Inflammation in amyotrophic lateral sclerosis spinal cord and brain is mediated by activated macrophages, mast cells and T cells. Amyotroph. Lateral Scler. Other Mot. Neuron Disord. 2004, 5, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Kawamata, T.; Akiyama, H.; Yamada, T.; McGeer, P.L. Immunologic reactions in amyotrophic lateral sclerosis brain and spinal cord tissue. Am. J. Pathol. 1992, 140, 691–707. [Google Scholar]
- Troost, D.; Van den Oord, J.J.; Vianney de Jong, J.M. Immunohistochemical characterization of the inflammatory infiltrate in amyotrophic lateral sclerosis. Neuropathol. Appl. Neurobiol. 1990, 16, 401–410. [Google Scholar] [CrossRef]
- Engelhardt, J.I.; Tajti, J.; Appel, S.H. Lymphocytic infiltrates in the spinal cord in amyotrophic lateral sclerosis. Arch. Neurol. 1993, 50, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Kaddatz, H.; Joost, S.; Nedelcu, J.; Chrzanowski, U.; Schmitz, C.; Gingele, S.; Gudi, V.; Stangel, M.; Zhan, J.; Santrau, E.; et al. Cuprizone-induced demyelination triggers a CD8-pronounced T cell recruitment. Glia 2021, 69, 925–942. [Google Scholar] [CrossRef] [PubMed]
- Duarte, J.; Carrie, N.; Oliveira, V.G.; Almeida, C.; Agua-Doce, A.; Rodrigues, L.; Simas, J.P.; Mars, L.T.; Graca, L. T cell apoptosis and induction of Foxp3+ regulatory T cells underlie the therapeutic efficacy of CD4 blockade in experimental autoimmune encephalomyelitis. J. Immunol. 2012, 189, 1680–1688. [Google Scholar] [CrossRef] [Green Version]
- Hickey, W.F.; Hsu, B.L.; Kimura, H. T-lymphocyte entry into the central nervous system. J. Neurosci. Res. 1991, 28, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhou, Y.; Jia, H.; Qi, Y.; Tu, S.; Shao, A. Affective Immunology: The Crosstalk Between Microglia and Astrocytes Plays Key Role? Front. Immunol. 2020, 11, 1818. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Barres, B.A. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef] [Green Version]
- Yun, S.P.; Kam, T.I.; Panicker, N.; Kim, S.; Oh, Y.; Park, J.S.; Kwon, S.H.; Park, Y.J.; Karuppagounder, S.S.; Park, H.; et al. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat. Med. 2018, 24, 931–938. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Greiner, T.; Kipp, M. What Guides Peripheral Immune Cells into the Central Nervous System? Cells 2021, 10, 2041. https://doi.org/10.3390/cells10082041
Greiner T, Kipp M. What Guides Peripheral Immune Cells into the Central Nervous System? Cells. 2021; 10(8):2041. https://doi.org/10.3390/cells10082041
Chicago/Turabian StyleGreiner, Theresa, and Markus Kipp. 2021. "What Guides Peripheral Immune Cells into the Central Nervous System?" Cells 10, no. 8: 2041. https://doi.org/10.3390/cells10082041
APA StyleGreiner, T., & Kipp, M. (2021). What Guides Peripheral Immune Cells into the Central Nervous System? Cells, 10(8), 2041. https://doi.org/10.3390/cells10082041