
High Glycolytic Activity Enhances Stem Cell Reprogramming of Fahd1-KO Mouse Embryonic Fibroblasts

 ,
,  , , , and
, , , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Generation of iPSCs
2.2. iPSC Differentiation
2.3. MEF Growth Curve
2.4. qRT-PCR
2.5. Immunocytochemistry
2.6. Mitochondrial and Glycolysis Stress Tests
2.7. Statistical Analysis
3. Results
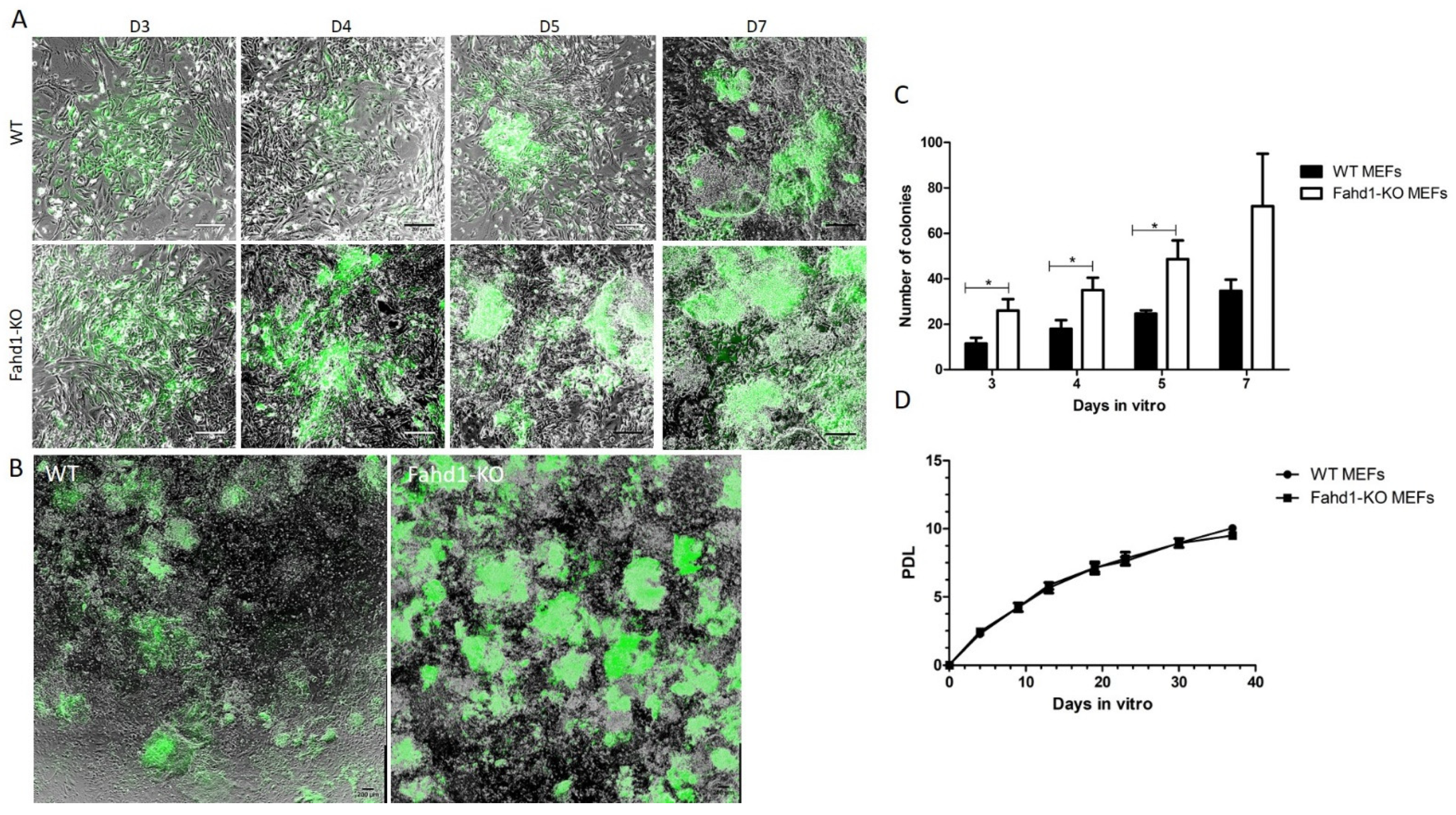
3.1. Reprogramming of WT and Fahd1-KO MEFs
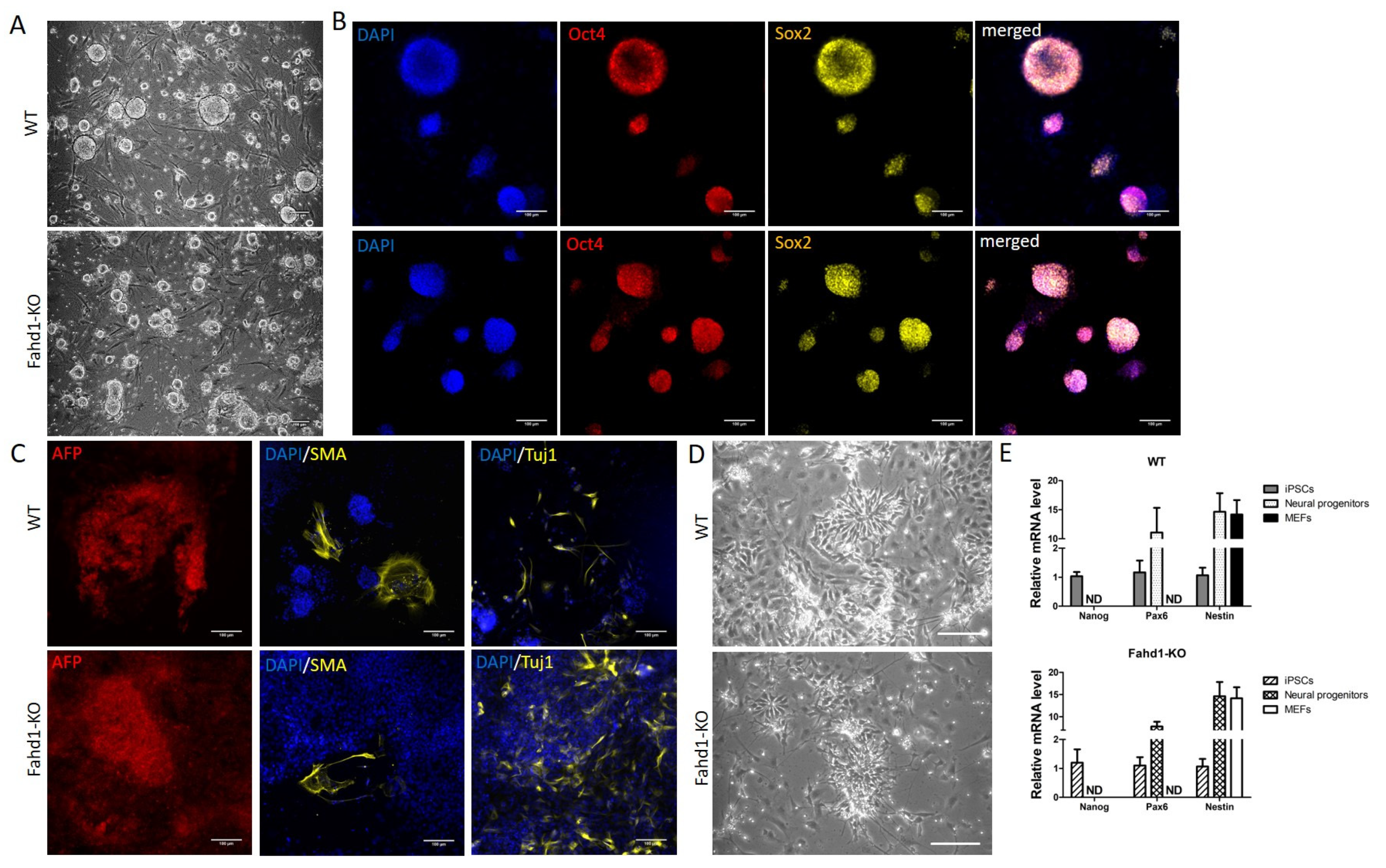
3.2. Germ Layer and Neural Differentiation
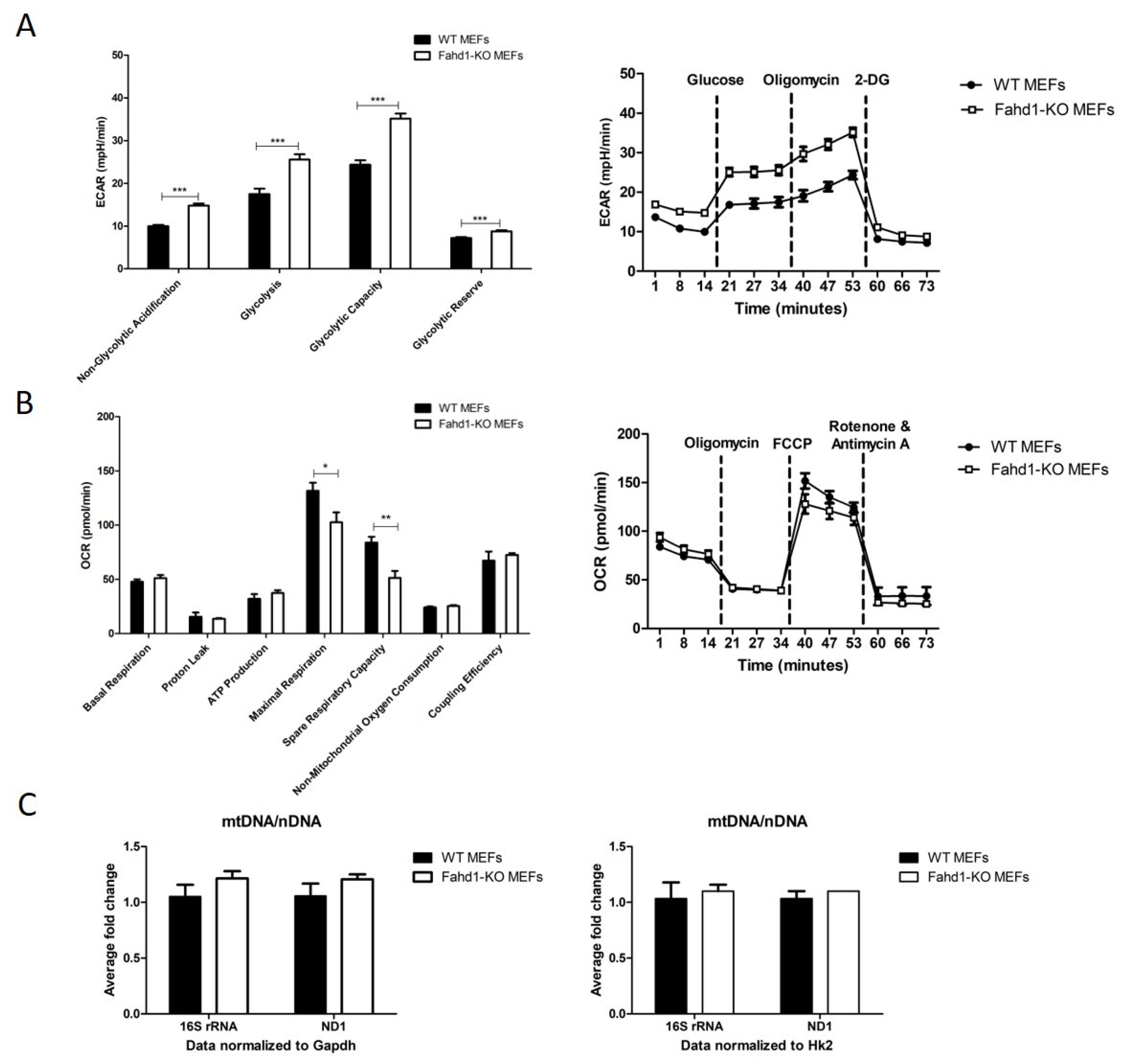
3.3. Glycolytic Activity Is Enhanced in Fahd1-Deficient Mouse Embryonic Fibroblasts
3.4. Mitochondrial Remodeling during Reprogramming Is Affected by Fahd1 Deficiency
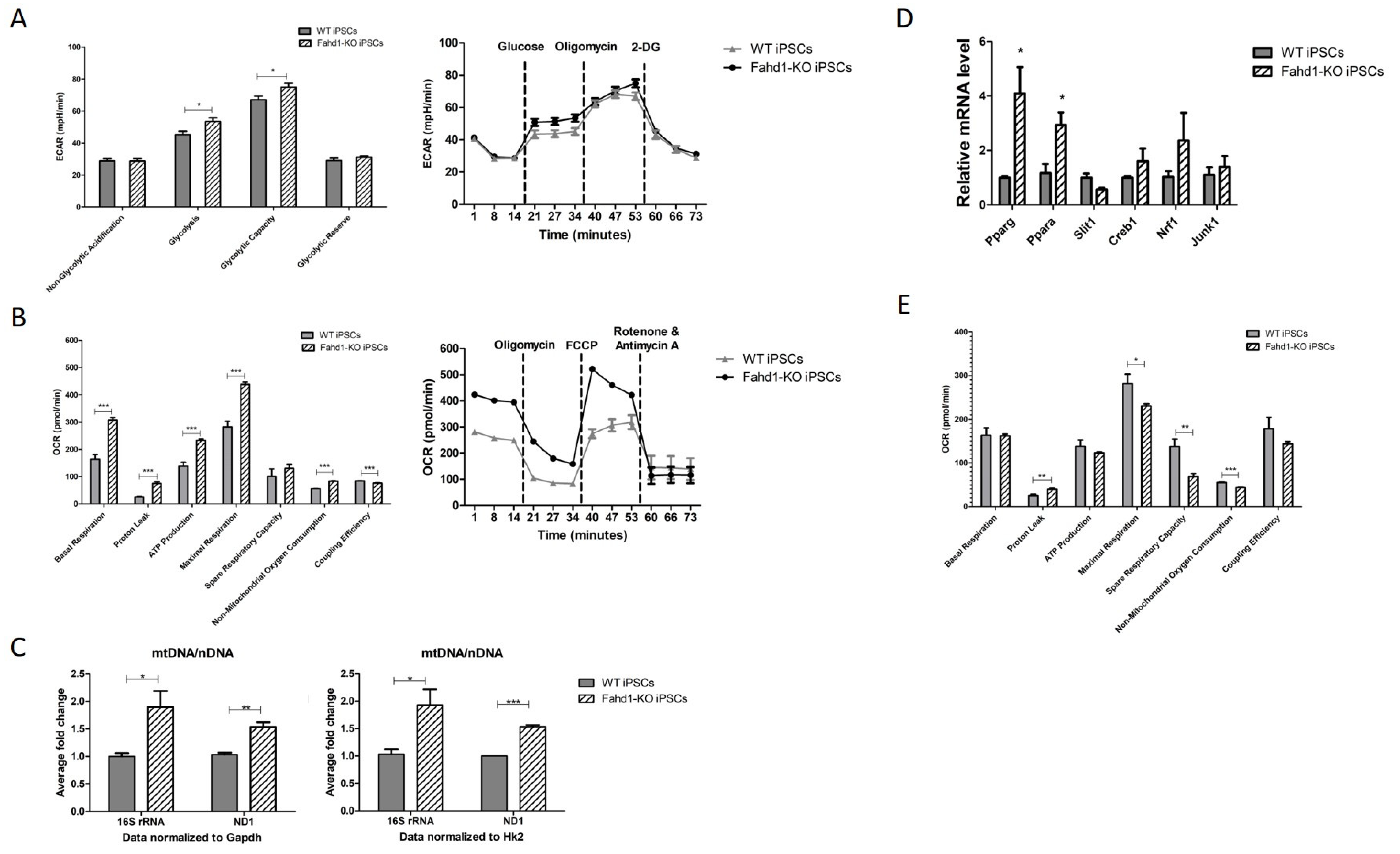
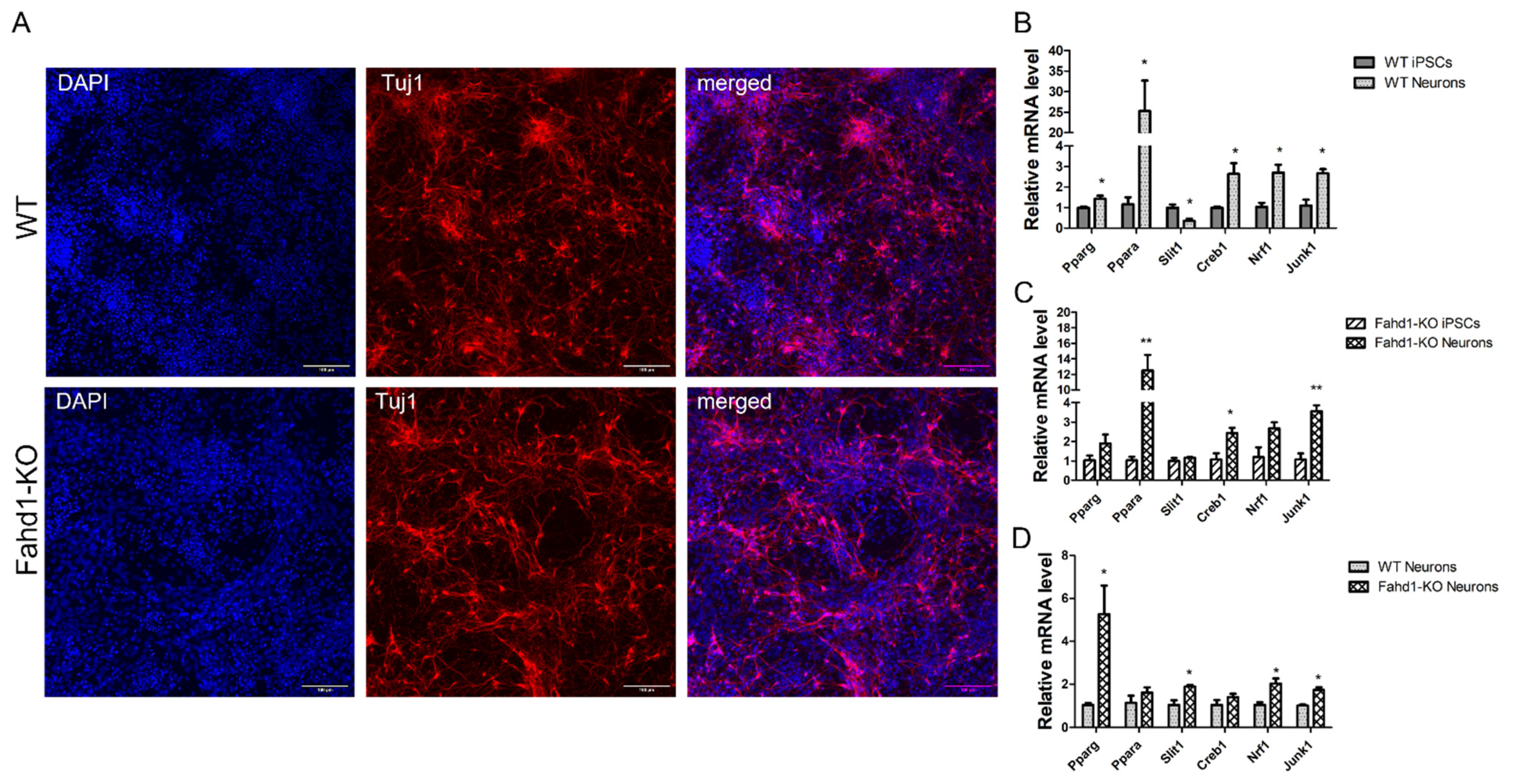
3.5. Higher Mitochondrial Biogenesis Is Maintained in iPSC-Derived Fahd1-KO Neurons Relative to WT
4. Discussion
4.1. Increased Reprogramming Efficiency of Fahd1-KO MEFs Correlates with Higher Glycolytic Activity
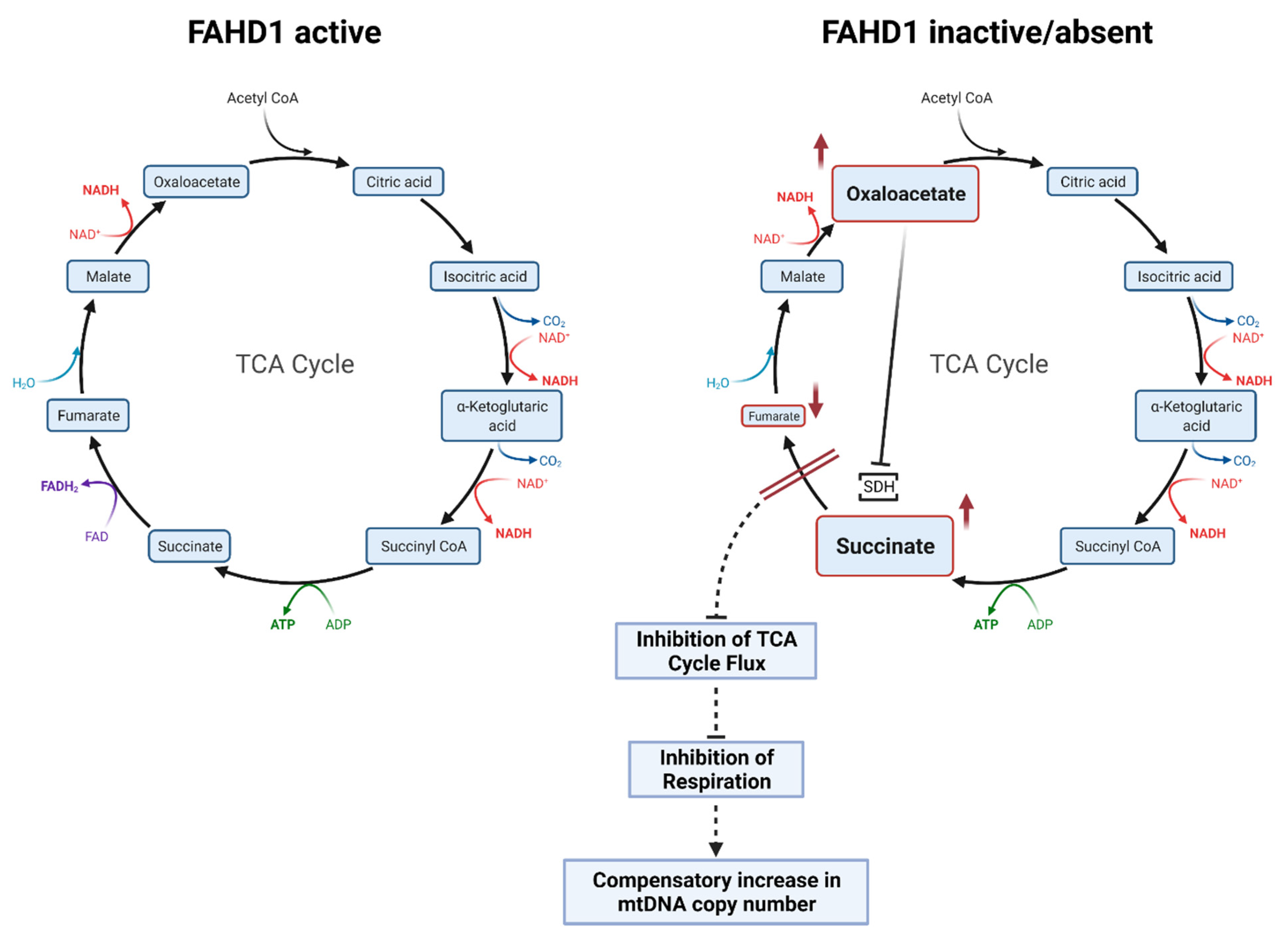
4.2. Fahd1 Deficiency Affects Mitochondrial Remodeling during Reprogramming to iPSCs
4.3. Fahd1 Deficiency Does Not Grossly Affect Pluripotency of iPSCs after Reprogramming nor Their Differentiation to Neurons
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sercel, A.J.; Carlson, N.M.; Patananan, A.N.; Teitell, M.A. Mitochondrial DNA Dynamics in Reprogramming to Pluripotency. Trends Cell Biol. 2021, 31, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Menzies, K.J.; Auwerx, J. The role of mitochondria in stem cell fate and aging. Development 2018, 145, dev143420. [Google Scholar] [CrossRef] [Green Version]
- Russell, O.M.; Fruh, I.; Rai, P.K.; Marcellin, D.; Doll, T.; Reeve, A.; Germain, M.; Bastien, J.; Rygiel, K.A.; Cerino, R.; et al. Preferential amplification of a human mitochondrial DNA deletion in vitro and in vivo. Sci. Rep. 2018, 8, 1799. [Google Scholar] [CrossRef]
- Pircher, H.; Straganz, G.D.; Ehehalt, D.; Morrow, G.; Tanguay, R.M.; Jansen-Dürr, P. Identification of human Fumarylacetoacetate Hydrolase Domain-containing Protein 1 (FAHD1) as a novel mitochondrial acylpyruvase. J. Biol. Chem. 2011, 286, 36500–36508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pircher, H.; Von Grafenstein, S.; Diener, T.; Metzger, C.; Albertini, E.; Taferner, A.; Unterluggauer, H.; Kramer, C.; Liedl, K.R.; Jansen-Dürr, P. Identification of FAH domain-containing protein 1 (FAHD1) as oxaloacetate decarboxylase. J. Biol. Chem. 2015, 290, 6755–6762. [Google Scholar] [CrossRef] [Green Version]
- Wiley, C.D.; Velarde, M.C.; Lecot, P.; Liu, S.; Sarnoski, E.A.; Freund, A.; Shirakawa, K.; Lim, H.W.; Davis, S.S.; Ramanathan, A.; et al. Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab. 2016, 23, 303–314. [Google Scholar] [CrossRef] [Green Version]
- Petit, M.; Koziel, R.; Etemad, S.; Pircher, H.; Jansen-Dürr, P. Depletion of oxaloacetate decarboxylase FAHD1 inhibits mitochondrial electron transport and induces cellular senescence in human endothelial cells. Exp. Gerontol. 2017, 92, 7–12. [Google Scholar] [CrossRef]
- Etemad, S.; Petit, M.; Weiss, A.K.H.; Schrattenholz, A.; Baraldo, G.; Jansen-Dürr, P. Oxaloacetate decarboxylase FAHD1—A new regulator of mitochondrial function and senescence. Mech. Ageing Dev. 2019, 177, 22–29. [Google Scholar] [CrossRef]
- Salti, A.; Nat, R.; Neto, S.; Puschban, Z.; Wenning, G.; Dechant, G. Expression of Early Developmental Markers Predicts the Efficiency of Embryonic Stem Cell Differentiation into Midbrain Dopaminergic Neurons. Stem Cells Dev. 2012, 22, 397–411. [Google Scholar] [CrossRef] [Green Version]
- Jovanovic, V.M.; Salti, A.; Tilleman, H.; Zega, K.; Jukic, M.M.; Zou, H.; Friedel, R.H.; Prakash, N.; Blaess, S.; Edenhofer, F.; et al. BMP/SMAD pathway promotes neurogenesis of midbrain dopaminergic neurons in vivo and in human induced pluripotent and neural stem cells. J. Neurosci. 2018, 38, 1662–1676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thier, M.C.; Hommerding, O.; Panten, J.; Pinna, R.; García-González, D.; Berger, T.; Wörsdörfer, P.; Assenov, Y.; Scognamiglio, R.; Przybylla, A.; et al. Identification of Embryonic Neural Plate Border Stem Cells and Their Generation by Direct Reprogramming from Adult Human Blood Cells. Cell Stem Cell 2019, 24, 166–182.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thier, M.; Münst, B.; Edenhofer, F. Exploring refined conditions for reprogramming cells by recombinant Oct4 protein. Int. J. Dev. Biol. 2011, 54, 1713–1721. [Google Scholar] [CrossRef]
- Urabe, M.; Kume, A.; Tobita, K.; Ozawa, K. DNA/Calcium phosphate precipitates mixed with medium are stable and maintain high transfection efficiency. Anal. Biochem. 2000, 278, 91–92. [Google Scholar] [CrossRef]
- Hutter, E.; Renner, K.; Pfister, G.; Stöckl, P.; Jansen-Dürr, P.; Gnaiger, E. Senescence-associated changes in respiration and oxidative phosphorylation in primary human fibroblasts. Biochem. J. 2004, 380, 919–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quiros, P.M.; Goyal, A.; Jha, P.; Auwerx, J. Analysis of mtDNA/nDNA Ratio in Mice. Curr. Protoc. Mouse Biol. 2017, 7, 47–54. [Google Scholar] [CrossRef] [Green Version]
- Tanhauser, S.M.; Laipis, P.J. Multiple deletions are detectable in mitochondrial DNA of aging mice. J. Biol. Chem. 1995, 270, 24769–24775. [Google Scholar] [CrossRef] [Green Version]
- Jamwal, S.; Blackburn, J.K.; Elsworth, J.D. PPARγ/PGC1α signaling as a potential therapeutic target for mitochondrial biogenesis in neurodegenerative disorders. Pharmacol. Ther. 2021, 219, 107705. [Google Scholar] [CrossRef]
- Büeler, H. Mitochondrial and autophagic regulation of adult neurogenesis in the healthy and diseased brain. Int. J. Mol. Sci. 2021, 22, 3342. [Google Scholar] [CrossRef] [PubMed]
- Maffezzini, C.; Calvo-Garrido, J.; Wredenberg, A.; Freyer, C. Metabolic regulation of neurodifferentiation in the adult brain. Cell. Mol. Life Sci. 2020, 77, 2483–2496. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Boyer, L.; Jin, M.; Mertens, J.; Kim, Y.; Ma, L.; Ma, L.; Hamm, M.; Gage, F.H.; Hunter, T. Metabolic reprogramming during neuronal differentiation from aerobic glycolysis to neuronal oxidative phosphorylation. Elife 2016, 5, e13374. [Google Scholar] [CrossRef]
- Calvo-Garrido, J.; Maffezzini, C.; Schober, F.A.; Clemente, P.; Uhlin, E.; Kele, M.; Stranneheim, H.; Lesko, N.; Bruhn, H.; Svenningsson, P.; et al. SQSTM1/p62-Directed Metabolic Reprogramming Is Essential for Normal Neurodifferentiation. Stem Cell Reports 2019, 12, 696–711. [Google Scholar] [CrossRef] [Green Version]
- Wanet, A.; Arnould, T.; Najimi, M.; Renard, P. Connecting Mitochondria, Metabolism, and Stem Cell Fate. Stem Cells Dev. 2015, 24, 1957–1971. [Google Scholar] [CrossRef] [Green Version]
- Folmes, C.D.L.; Nelson, T.J.; Martinez-Fernandez, A.; Arrell, D.K.; Lindor, J.Z.; Dzeja, P.P.; Ikeda, Y.; Perez-Terzic, C.; Terzic, A. Somatic oxidative bioenergetics transitions into pluripotency-dependent glycolysis to facilitate nuclear reprogramming. Cell Metab. 2011, 14, 264–271. [Google Scholar] [CrossRef] [Green Version]
- Panopoulos, A.D.; Yanes, O.; Ruiz, S.; Kida, Y.S.; Diep, D.; Tautenhahn, R.; Herrerías, A.; Batchelder, E.M.; Plongthongkum, N.; Lutz, M.; et al. The metabolome of induced pluripotent stem cells reveals metabolic changes occurring in somatic cell reprogramming. Cell Res. 2012, 22, 168–177. [Google Scholar] [CrossRef] [Green Version]
- Zhu, S.; Li, W.; Zhou, H.; Wei, W.; Ambasudhan, R.; Lin, T.; Kim, J.; Zhang, K.; Ding, S. Reprogramming of human primary somatic cells by OCT4 and chemical compounds. Cell Stem Cell 2010, 7, 651–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auger, C.; Vinaik, R.; Appanna, V.D.; Jeschke, M.G. Beyond mitochondria: Alternative energy-producing pathways from all strata of life. Metabolism 2021, 118, 154733. [Google Scholar] [CrossRef]
- Rafalski, V.A.; Mancini, E.; Brunet, A. Energy metabolism and energy-sensing pathways in mammalian embryonic and adult stem cell fate. J. Cell Sci. 2012, 125, 5597–5608. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Choi, M.; Margineantu, D.; Margaretha, L.; Hesson, J.; Cavanaugh, C.; Blau, C.A.; Horwitz, M.S.; Hockenbery, D.; Ware, C.; et al. HIF1α induced switch from bivalent to exclusively glycolytic metabolism during ESC-to-EpiSC/hESC transition. EMBO J. 2012, 31, 2103–2116. [Google Scholar] [CrossRef]
- Takehara, T.; Teramura, T.; Onodera, Y.; Hamanishi, C.; Fukuda, K. Reduced Oxygen Concentration Enhances Conversion of Embryonic Stem Cells to Epiblast Stem Cells. Stem Cells Dev. 2012, 21, 1239–1249. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Khvorostov, I.; Hong, J.S.; Oktay, Y.; Vergnes, L.; Nuebel, E.; Wahjudi, P.N.; Setoguchi, K.; Wang, G.; Do, A.; et al. UCP2 regulates energy metabolism and differentiation potential of human pluripotent stem cells. EMBO J. 2011, 30, 4860–4873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, S.L.; Grãos, M.; Rodrigues, A.S.; Anjo, S.I.; Carvalho, R.A.; Oliveira, P.J.; Arenas, E.; Ramalho-Santos, J. Inhibition of mitochondrial complex III blocks neuronal differentiation and maintains embryonic stem cell pluripotency. PLoS ONE 2013, 8, e82095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, Y.; Takahashi, K.; Okita, K.; Ichisaka, T.; Yamanaka, S. Hypoxia Enhances the Generation of Induced Pluripotent Stem Cells. Cell Stem Cell 2009, 5, 237–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prigione, A.; Fauler, B.; Lurz, R.; Lehrach, H.; Adjaye, J. The Senescence-Related Mitochondrial/Oxidative Stress Pathway is Repressed in Human Induced Pluripotent Stem Cells. Stem Cells 2010, 28, 721–733. [Google Scholar] [CrossRef]
- Prigione, A.; Rohwer, N.; Hoffmann, S.; Mlody, B.; Drews, K.; Bukowiecki, R.; Blümlein, K.; Wanker, E.E.; Ralser, M.; Cramer, T.; et al. HIF1α modulates cell fate reprogramming through early glycolytic shift and upregulation of PDK1-3 and PKM2. Stem Cells 2014, 32, 364–376. [Google Scholar] [CrossRef] [Green Version]
- Kida, Y.S.; Kawamura, T.; Wei, Z.; Sogo, T.; Jacinto, S.; Shigeno, A.; Kushige, H.; Yoshihara, E.; Liddle, C.; Ecker, J.R.; et al. ERRs mediate a metabolic switch required for somatic cell reprogramming to pluripotency. Cell Stem Cell 2015, 16, 547–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varum, S.; Rodrigues, A.S.; Moura, M.B.; Momcilovic, O.; Easley IV, C.A.; Ramalho-Santos, J.; van Houten, B.; Schatten, G. Energy metabolism in human pluripotent stem cells and their differentiated counterparts. PLoS ONE 2011, 6, e20914. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Osenbroch, P.; Skinnes, R.; Esbensen, Y.; Bjørås, M.; Eide, L. Mitochondrial DNA Integrity Is Essential For Mitochondrial Maturation During Differentiation of Neural Stem Cells. Stem Cells 2010, 28, 2195–2204. [Google Scholar] [CrossRef]
- Zhang, H.; Ryu, D.; Wu, Y.; Gariani, K.; Wang, X.; Luan, P.; D’Amico, D.; Ropelle, E.R.; Lutolf, M.P.; Aebersold, R.; et al. NAD+ repletion improves mitochondrial and stem cell function and enhances life span in mice. Science 2016, 352, 1436–1443. [Google Scholar] [CrossRef] [Green Version]
- Khacho, M.; Clark, A.; Svoboda, D.S.; Azzi, J.; MacLaurin, J.G.; Meghaizel, C.; Sesaki, H.; Lagace, D.C.; Germain, M.; Harper, M.E.; et al. Mitochondrial Dynamics Impacts Stem Cell Identity and Fate Decisions by Regulating a Nuclear Transcriptional Program. Cell Stem Cell 2016, 19, 232–247. [Google Scholar] [CrossRef] [Green Version]
- Haupt, S.; Edenhofer, F.; Peitz, M.; Leinhaas, A.; Brüstle, O. Stage-Specific Conditional Mutagenesis in Mouse Embryonic Stem Cell-Derived Neural Cells and Postmitotic Neurons by Direct Delivery of Biologically Active Cre Recombinase. Stem Cells 2007, 25, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Prowse, A.B.J.; Chong, F.; Elliott, D.A.; Elefanty, A.G.; Stanley, E.G.; Gray, P.P.; Munro, T.P.; Osborne, G.W. Analysis of Mitochondrial Function and Localisation during Human Embryonic Stem Cell Differentiation In Vitro. PLoS ONE 2012, 7, e52214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villena, J.A. New insights into PGC-1 coactivators: Redefining their role in the regulation of mitochondrial function and beyond. FEBS J. 2015, 282, 647–672. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.I.; Chou, Y.C.; Chang, Y.L.; Chien, Y.; Chen, K.H.; Song, W.S.; Peng, C.H.; Chang, C.H.; Lee, S.D.; Lu, K.H.; et al. Enhanced differentiation of three-gene-reprogrammed induced pluripotent stem cells into adipocytes via adenoviral-mediated PGC-1α over expression. Int. J. Mol. Sci. 2011, 12, 7554–7568. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salti, A.; Etemad, S.; Cubero, M.S.; Albertini, E.; Kovacs-Szalka, B.; Holzknecht, M.; Cappuccio, E.; Cavinato, M.; Edenhofer, F.; Jansen Dürr, P. High Glycolytic Activity Enhances Stem Cell Reprogramming of Fahd1-KO Mouse Embryonic Fibroblasts. Cells 2021, 10, 2040. https://doi.org/10.3390/cells10082040
Salti A, Etemad S, Cubero MS, Albertini E, Kovacs-Szalka B, Holzknecht M, Cappuccio E, Cavinato M, Edenhofer F, Jansen Dürr P. High Glycolytic Activity Enhances Stem Cell Reprogramming of Fahd1-KO Mouse Embryonic Fibroblasts. Cells. 2021; 10(8):2040. https://doi.org/10.3390/cells10082040
Chicago/Turabian StyleSalti, Ahmad, Solmaz Etemad, Marta Suarez Cubero, Eva Albertini, Beata Kovacs-Szalka, Max Holzknecht, Elia Cappuccio, Maria Cavinato, Frank Edenhofer, and Pidder Jansen Dürr. 2021. "High Glycolytic Activity Enhances Stem Cell Reprogramming of Fahd1-KO Mouse Embryonic Fibroblasts" Cells 10, no. 8: 2040. https://doi.org/10.3390/cells10082040
APA StyleSalti, A., Etemad, S., Cubero, M. S., Albertini, E., Kovacs-Szalka, B., Holzknecht, M., Cappuccio, E., Cavinato, M., Edenhofer, F., & Jansen Dürr, P. (2021). High Glycolytic Activity Enhances Stem Cell Reprogramming of Fahd1-KO Mouse Embryonic Fibroblasts. Cells, 10(8), 2040. https://doi.org/10.3390/cells10082040